

Abstract
Aims
Endothelial cells (ECs) constitute the defensive barrier of vasculature, which maintains the vascular homeostasis. Mitochondrial oxidative stress (mitoOS) in ECs significantly affects the initiation and progression of vascular diseases. The higher serum thyroid stimulating hormone (TSH) level is being recognized as a nonconventional risk factor responsible for the increased risk of cardiovascular diseases in subclinical hypothyroidism (SCH). However, effects and underlying mechanisms of elevated TSH on ECs are still ambiguous. We sought to investigate whether cyclophilin D (CypD), emerging as a crucial mediator in mitoOS, regulates effects of TSH on ECs.
Methods and results
SCH patients with TSH > = 10 mIU/L showed a positive correlation between serum TSH and endothelin-1 levels. When TSH levels declined to normal in these subjects after levothyroxine therapy, serum endothelin-1 levels were significantly reduced. Supplemented with exogenous thyroxine to keep normal thyroid hormones, thyroid-specific TSH receptor (TSHR)-knockout mice with injection of exogenous TSH exhibited elevated serum TSH levels, significant endothelial oxidative injuries and disturbed endothelium-dependent vasodilation. However, Tshr-/- mice resisted to TSH-impaired vasotonia. We further confirmed that elevated TSH triggered excessive mitochondrial permeability transition pore (mPTP) opening and mitochondrial oxidative damages in mouse aorta, as well as in cultured ECs. Genetic or pharmacological inhibition of CypD (the key regulator for mPTP opening) attenuated TSH-induced mitochondrial oxidative damages and further rescued endothelial functions. Finally, we confirmed that elevated TSH could activate CypD by enhancing CypD acetylation via inhibiting adenosine monophosphate-activated protein kinase/sirtuin-3 signaling pathway in ECs.
Conclusions
These findings reveal that elevated TSH triggers mitochondrial perturbations in ECs and provide insights that blocking mitochondrial CypD enhances the defensive ability of ECs under TSH exposure.
Abbreviations: Ach, Acetylcholine; AMPK, Adenosine monophosphate-activated protein kinase; CsA, Cyclosporine A; CVD, Cardiovascular diseases; ECs, Endothelial cells; eNOS, Endothelial nitric oxide synthase; ET-1, Endothelin-1; HA-VSMC, Human aortic smooth muscle cell; HUVEC, Human umbilical vein endothelial cell; L-NAME, N-nitro-l-arginine methyl ester; mitoOS, Mitochondrial oxidative stress; mPTP, Mitochondrial permeability transition pore; OCR, Oxygen consumption rate; ROS, Reactive oxygen species; SIRT3, Sirtuin-3; SNP, Sodium nitroprusside; TSH, Thyroid stimulating hormone; TSHR, Thyroid stimulating hormone receptor
Keywords: Mitochondria, Endothelial cell, Oxidative stress, Cyclophilin D, Thyroid stimulating hormone
Graphical abstract
Highlights
-
•
Elevated TSH impairs endothelium-dependent vasodilation via mPTP -induced mitoOS.
-
•
Blocking CypD attenuates TSH-induced mitoOS and endothelial dysfunction.
-
•
Elevated TSH enhances CypD acetylation via inhibiting AMPK/SIRT3 signaling pathway.
1. Introduction
Endothelial cells (ECs) are in a dynamic equilibrium with their environments and constitute a defensive barrier in the vasculature, controlling vascular permeability, smooth muscle tone, inflammatory and immune responses, angiogenesis, and thromboresistance. In many vascular diseases, the endothelium is thus both origin and victim [1]. The interactions within the redox reactions are richly elaborated in an oxygen-dependent life and contribute to spatiotemporal organization for differentiation, development, and adaptation to the environment [2]. Disruption of the balance between oxidants and anti- oxidants in favor of the oxidants leads to the occurrence of oxidative stress [3], in which the damaging effects of reactive oxygen species (ROS) exceed the ability of biological systems to neutralize the oxidizing agents and to repair cellular damage [4]. Studies in human subjects and animals have demonstrated that oxidative stress and the associated endothelial dysfunction significantly correlate to the classic cardiovascular risk factors such as hypercholesterolemia, diabetes mellitus and chronic smoking, and also play important roles in the development of vasculopathies, including atherosclerosis, hypertension, restenosis after angioplasty, angiogenesis, cardiac injury associated with post-ischemic reperfusion and heart failure [4], [5], [6], [7], [8].
Subclinical hypothyroidism (SCH) is characterized by elevated serum thyroid stimulating hormone (TSH) levels with normal serum thyroid hormone concentrations [9], [10]. It is a common thyroid disease with a prevalence ranging from 4% to 20% in adults, and the prevalence is progressively increasing [11]. Although defined as an asymptomatic state, SCH is proposed to lead adverse consequences including systemic hypothyroid symptoms, neuromuscular dysfunction, progression to overt hypothyroidism, hypercholesterolemia and cardiovascular dysfunction [12], [13], [14]. The higher serum TSH level has been recognized as a nonconventional risk factor responsible for the increased risk of cardiovascular diseases (CVD) in SCH [15]. Although with strong positive relationship to increased markers of oxidative stress [16] and endothelial dysfunction [17] in serum, the effects and underlying mechanisms of elevated TSH on ECs still remain to be established.
Although with lower content, mitochondria are crucial in maintaining redox homeostasis and cellular functions of ECs [18]. Imbalance of redox state in mitochondria leads to excessive generation of mitochondrial ROS, which serve as kindling radicals to activate nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, xanthine oxido-reductase and uncoupled endothelial nitric oxide synthase (eNOS) [19], with endothelial dysfunction as a result [20].
The mitochondrial permeability transition pore (mPTP) is a key regulator of mitochondrial homeostasis. Transient opening of the pore is important for release of ions and metabolites to maintain mitochondrial health [21]. However, under extreme stress, excessive activation of mPTP can be triggered to aggravate mitochondrial degeneration, leading to matrix expansion and mitochondrial membrane rupture, which dissipates the mitochondrial membrane potential, deregulates Ca2+ homeostasis, generates and releases pathological mitochondrial ROS [22]. Cyclophilin D (CypD), a protein with peptidyl-prolyl cis-trans isomerase (PPIase) activity, is a critical regulator of the mPTP opening. It resides in the mitochondrial matrix in resting state. Once activated, CypD translocates to the mitochondrial inner membrane and binds to the mPTP constitutes, for instance ANT [23] or F1F0 ATP synthase [24], [25], [26], to facilitate mPTP opening. Although Marcu et al. [27] confirmed the effect of CypD on endothelial proliferation and angiogenesis, it remains to determine whether CypD-mediated mPTP opening and altered mitochondrial redox state are involved in the effects of TSH on ECs.
Here we report for the first time that elevated TSH triggers mitochondrial oxidative stress in ECs and shed light onto the crucial role of CypD in modulating TSH-induced mitochondrial and endothelial perturbations.
2. Materials and methods
2.1. Human subjects and data collection
Human subjects were recruited from Ningyang County, Shandong Province, China. Subjects under pregnancy or breast-feeding, taking medicines that affect thyroid status, and without good compliance were excluded.
Clinical assessments of the enrollments were performed at baseline and end-of-study. Venous blood samples were drawn between 8:00 a.m. and 10:00 a.m. after a minimum 10-h fasting, followed by the measurement of weight (kilograms) and standing height (meters). Body mass index (BMI) was calculated by dividing weight by the square of the height. The methods for determining waist circumference (WC) and blood pressure were according to the previous described [28].
The following serum variables were all completed at the clinical laboratory of the Shandong Provincial Hospital. Lipid profiles and fasting plasma glucose (FPG) were quantified using a BECKMAN Chemistry Analyzer AU5800 System (Beckman Coulter, Tokyo, Japan). Non HDL cholesterol (non HDL-C) was calculated by subtracting HDL-C from TC. Serum free triiodothyronine (FT3), free thyroxine (FT4), and TSH levels were measured by chemiluminescence methods (Cobas E601; Roche, Basel, Switzerland).
Euthyroidism was defined as serum TSH level between 0.27 and 4.2 mIU/L with normal serum FT4 levels. Subclinical hypothyroidism (SCH) was TSH ≥ 4.2 mIU/L with normal serum FT4 confirmed on the basis of at least two hormonal assays with a three-month interval [29]. All SCH patients underwent levothyroxine (LT4, Euthyrox, 50 μg per tablet, Merck Serono, Darmstadt, Germany) replacement therapy with the initial dosage 25 μg/day. The dosage of LT4 was adjusted according to serum TSH and FT4 levels. The dosage with which SCH patients achieved euthyroidism was subsequently maintained. All participants were followed up for 15 months.
SCH patients who didn’t achieve euthyroidism after LT4 therapy were also excluded in the present study. After being matched by age, sex, BMI, TC and LDL-C, 33 euthyroid subjects, 33 mild SCH patients (TSH of 4.2–10 mIU/L) and 33 significant SCH patients (TSH > = 10 mIU/L) were finally enrolled in the study. Serum endothelin-1 (ET-1) levels of these humans were measured using Elisa Kits (abcam, USA). Measurement processes followed strictly to the manufacture’s instructions.
The human study was performed according to the Declaration of Helsinki, approved by the Ethics Committee of Shandong Provincial Hospital, and was registered at ClinicalTrials.gov (NCT01848171). All participants signed an informed consent.
2.2. Animals and treatment
2.2.1. Generation of TT-KO mice
We utilized a Cre/LoxP strategy to yield thyroid-specific TSH receptor (TSHR)-knockout (TT-KO) mice. Tshrflox/+ mice with C57BL/6J background were obtained from Cyagen Biosciences (Guangzhou, China). For the generation of TT-KO mice, Tshrflox/flox mice were crossed with the heterozygous mice expressing TPO-driven Cre recombinase (kindly donated by Shioko Kimura, National Institutes of Health, Bethesda). Mice homozygous for the floxed gene and heterozygous for TPO-Cre (TT-KO) were used as experimental animals.
The TT-KO mice were genotyped by polymerase chain reaction. Primer pairs of 5-GAGGATTTCTGTTGGTGGCTGG-3/5-CACCCTTGATCCCCTTGACC-3 and 5-GTAAACTGCTGGAGTACATGA-3/5-AAAATTTAGCCTATGTGTAGCTT-3 were used to identify floxed allele. The primers of 5-TGC CACGACCAAGTGACAGCA ATG -3/5- AGAGACGGA AATCCATCGCTCG -3 were used to identify mice with TPO-Cre.
2.2.2. Treatments to TT-KO mice
After discontinuing breast feeding at 4 weeks old, all male TT-KO mice were supplemented with exogenous T4 (Sigma) to keep normal thyroid hormone levels. Mice aged 9–10 weeks were subcutaneously injected with freshly prepared TSH (TT-KO+TSH, 7 mIU/g·d, Sigma,) or solvent (TT-KO+solvent, same volume as TSH) for additional 2 weeks before sacrificed. To evaluate the consequences of pharmacological blocking of CypD in vivo, we injected cyclosporin A (CsA, 15 mg/kg·d, Sandimmune, Novartis) or PBS (same volume as CsA) to TT-KO mice for 4 weeks prior to TSH or solvent co-injection for another 2 weeks (TT-KO+solvent, TT-KO+TSH, TT-KO+CsA, TT-KO+CsA+TSH).
2.2.3. Tshr-/- mice
Tshr-/- mice were obtained from Jackson Laboratory (USA). All information about these mice could be achieved from our previous research [30]. Mice were backcrossed > 10 times onto the C57BL/6J background before use. The primer pair 5-AAG TTC ATC TGC ACC ACC G-3 and 5-TCC TTG AAG AAG ATG GTG CG-3 were used to identify the mutant type. The primer pair 5- CAG GGT GGA GAC GCA CAC TC-3 and 5-AGA GAG TCC CAC AAC AGT C-3 were used to identify the wild type. After discontinuing breast feeding at 4 weeks old, male Tshr-/- mice were supplemented with exogenous T4 to keep normal thyroid hormone levels. The littermate male Tshr+/+ mice were used as controls. Mice were sacrificed for experiments at the age of 11–12 weeks old.
2.2.4. CypD KO mice
CypD KO mice were kindly donated by professor Heng Du (The University of Texas, Dallas). Mice were backcrossed > 10 times onto the C57BL/6J background before use. The primer pairs 5-CTC TTC TGG GCA AGA ATT GC-3, 5-ATT GTG GTT GGT GAA GTC GCC-3 and 5- GGC TGC TAA AGC GCA TGC TCC-3 were used to identify wild-type allele and CypD-null allele. The male wild type mice in the same litter were used as controls. Mice aged 11–12 weeks were sacrificed for experiments.
Male mice aged 11–12 weeks were sacrificed for experiments with sodium pentobarbital anaesthesia (100 mg/kg, intraperitoneal) [31]. All animal experiments were conformed to the Guide for the Care and Use of Laboratory Animals, Eighth Edition, updated by the US National Research Council Committee in 2011. All animal experimental procedures were approved by the Animal Ethics Committee of Shandong Provincial Hospital (Jinan, China), with the approval number 2015-003.
2.3. Cell culture and treatment
Human umbilical vein endothelial cells (HUVECs, ATCC) were cultured according to manufacturer’s instructions. Cells of passage 6–8 were used for experiments. Before treatment, HUVECs were serum deprived in 0.5% FBS medium for 6 h. When subjected to TSH (Sigma), CsA (Solarbio), AICAR(Sigma) or Apocynin (MedChem Express), cells were cultured in 2.5% FBS medium.
HUVECs were treated with TSH (2 μmol/L) or vehicle for 24 h. After the incubation period, the medium was collected as conditioned medium (CM). Human aortic smooth muscle cells (HA-VSMCs, ATCC) of passage 5–7 were allowed to adhere for 24 h. After reaching 70–80% confluence, the cells were synchronized in serum free medium for 6 h and then cultured in 2.5% FBS medium, 2.5% FBS medium containing 2 μmol/L TSH or CM for 24 h.
2.4. Genetic knockdown of CypD
For the genetic knockdown of CypD, HUVECs were transfected with PPIF-shRNA, and ADV1-NC was used as control. Adenoviruses were bought from GenePharma (Shanghai, China). The adenovirus transfection was according to manufacturer’s instructions.
2.5. Mouse thyroid function determination
Serum total T4 levels and TSH levels were respectively measured using a total T4 RIA Kit (Tian jin jiu ding, Tian jin, China) and a TSH ELISA Kit (MyBioSource, San Diego, California, USA). All processes were according to manufacturer’s instructions.
2.6. The whole body metabolic status of the TT-KO mice
Male mice aged 11–12 weeks were kept in individual metabolic chambers (PhenoMaster, TSE Systems, Germany) maintaining 24 °C with a 12 h dark-light cycle. Mice accommodated to the chambers for 24 h. Whole body metabolic status, indicated by oxygen consumption (VO2), carbon dioxide production (VCO2) and heat production, were measured during the subsequent 24 h and adjusted by the body weight of each mouse. Respiratory exchange ratio (RER) was calculated by dividing VCO2 by VO2. Physical activity and food intake were recorded every 27 min.
2.7. Vascular reactivity experiments
All processes were according to the previously described [31] with modification.
2.7.1. Extraction of mouse mesenteric artery
Briefly, second-order mesenteric arteries (2–3 mm long) were carefully dissected from male mice aged 11–12 weeks, and connective tissues surrounding the arteries were removed. The individual vessel segment was mounted to a vessel chamber filled with Krebs’ solution (mmol/L: NaCl 118.3, KCl 4.7, CaCl2 2.5, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25, and dextrose 11.1, pH 7.4), and placed on the stage of an inverted microscope ( CK40, Olympus) that was coupled to a CCD camera (OLY-105, Olympus) and a video micrometer (VIA-100, Boeckeler Instruments). The vessel lumen was maintained at constant intralumia pressure of 80 mmHg using a pressure-servo controller (MODEL PS-200-S, Living Systems Instrumentation).
2.7.2. Measurement of endothelial dependent and independent vasodilation
Vessels were equilibrated for 40 min in oxygenated (95% O2, 5% CO2, balanced with N2, 37 °C) Krebs’ solution. Endothelin-1 (ET-1, maximum to 10−8 mmol/L, Sigma) was applied to pre-contract the vessels to 50–70% of the passive diameter. Subsequently, vessels were given cumulative addition of acetylcholine (Ach, Sigma) or sodium nitroprusside (SNP, Sigma) for the measurement of endthelium dependent or independent vasodilation respectively. To inhibit eNOS activity, vessels were preincubated with N-nitro-l-arginine methyl ester (L-NAME, 10−4 mmol/L, Sigma) for 25 min before ET-1 administration. At the end of each experiment, vessels were constricted by addition of 60 mmol/L KCl and then dilated by a Ca2+-free solution for 20 min to achieve maximal relaxation.
After the first equilibration, mesenteric arteries from Tshr-/- mice, CypD KO mice and their relevant controls were given an acute stimulation of freshly prepared TSH (2 μmol/L,Sigma) for another 2 h before ET-1 administration. The diameter of the vessel at each specific point was recorded.
Vessels were not acceptable for experiments if they showed leaks, failed to constrict by 50% to 10−8 mmol/L ET-1or to 60 mmol/L KCl, or failed to dilate to Ca2+-free solution. The vasodilation was calculated by the diameters recorded during the measurement process. The vasorelaxation was calculated as a percentage of the maximum diameter as defined by the following equation:
where DAch and DSNP are the vessel diameter at the specific level of Ach and SNP, respectively, DET is the vessel diameter after application of ET-1, and DMAX is the vessel diameter in Ca2+-free Krebs’ solution.
2.8. Isolation of mitochondria
The process was according to the previously described [32] with modifications. Samples were placed in a 9× volume of ice-cold mitochondria isolation buffer (mmol/L: mannitol 225, sucrose 75, K2HPO4 2, EGTA 1, pH 7.2) and homogenized until no visible particles. The homogenate was centrifuged at 1300×g, 5 min, 4 °C (Eppendorf, Germany) and the supernatant was then layered on 15% (vol/vol) Percoll (Solarbio) and subjected to a centrifugation at 36,500×g, 14 min, 4 °C (HITACHI, Japan). Decant the supernatant, re-suspend the pellet with mitochondria isolation buffer and centrifuge at 10,000×g, 10 min, 4 °C. Decant the supernatant, re-suspend the pellet with mitochondria isolation buffer and centrifuge at 8000×g, 10 min, 4 °C.
Mitochondrial protein concentration was determined at 540 nm by NanoDrop 2000c (Thermo Scientific).
2.9. Assessment of mPTP opening
The mPTP opening was assessed according to a previous method [32] with modifications. 20 µg freshly isolated mitochondria were suspended in 200 μl swelling assay buffer (mmol/L: KCl 150, HEPES 5, K2HPO4 2, glutamate 5, malate 5, pH 7.2). Mitochondrial swelling was immediately recorded with addition of calcium (1 μmol/mg protein) at 540 nm for 11 min on SpectraMax M2/M2e (Molecular Devices). The vehicle was determined for 11 min without addition of calcium.
2.10. Mitochondrial membrane potential and superoxide production
Mitochondrial membrane potential and superoxide were measured by tetramethylrhodamine methyl ester (TMRM, 50 nmol/L, Invitrogen) and MitoSox Red (5 μmol/L, Invitrogen) respectively. Staining processes were according to manufacture’s instructions. The fluorescence was detected by a fluorescence microscopy (Leica, Germany, excitation: 515–560 nm; emission: 580–620 nm).
2.11. Assessment of mitochondrial respiratory chain enzymatic activities
Mitochondrial complex I-III and citrate synthase activities were measured as previously described [33] with modifications.
For enzymatic assays of respiratory chain complexes I–III, sufficient mitochondria of tissues or cells were isolated. The spectrophotometric kinetic assays for mitochondrial respiratory chain enzymatic activities were performed by using a microplate reader (Molecular Devices, USA).
Complex I activity was determined by measuring oxidation of NADH at 340 nm in a reaction mixture of potassium phosphate buffer (0.5 mol/L, pH 7.5), BSA (50 mg ml−1), KCN (10 mmol/L) and NADH (10 mmol/L). The reaction was started by adding ubiquinone (10 mmol/L). Complex II activity determined by measuring reduction of 2,6-dichlorophenol-indophenol at 600 nm in a reaction mixture of potassium phosphate buffer (0.5 M, pH 7.5), fatty acid–free BSA (50 mg ml−1), KCN (10 mmol/L), succinate (400 mmol/L) and 2,6-Dichlorophenolindophenol sodium salt hydrate (DCPIP, 0.015% (wt/vol)). The reaction was started by adding decylubiquinone (12.5 mmol/L). Complex III activity was measured by the reduction of cytochrome c at 550 nm in reaction mixture of potassium phosphate buffer (0.5 mol/L, pH 7.5), oxidized cytochrome c (1 mmol/L), KCN (10 mmol/L), EDTA (5 mmol/L, pH7.5) and Tween-20 (2.5%(vol/vol)). The reaction was started by adding decylubiquinol (10 mmol/L). Citrate synthase activity was determined by measuring the reduction of acetyl-CoA in the presence of oxaloacelate at 412 nm in the reaction mixture of Tris (200 mmol/L, pH 8.0) with Triton X-100 (0.2%(vol/vol)), DTNB (1 mmol/L) and Ac CoA (10 mmol/L). The reaction was started by adding oxaloacetic acid (10 mmo/L). All regents were bought from Sigma. Complex activities were adjusted to the corresponding citrate synthase activity.
2.12. Measurement of cellular oxygen consumption rate
Oxygen consumption rate (OCR) was measured using the XF96 Analyzer (Seahorse Bioscience). All procedures were according to the manufacture’s instructions. Briefly, HUVEC were seeded overnight at 6 × 103 cells per well on Seahorse XF96 cell culture microplate (Seahorse Bioscience). After TSH stimulation, the plate was incubated in low-buffered non-bicarbonated assay medium (XF base medium with 2 mmo/L Glutamine, 1 mmol/L Sodium Pyruvate and 25 mmol/L Glucose) for 1 h in a non-CO2 incubator at 37 °C before measuring in an XFe 96 extracellular flux analyzer (Seahorse Bioscience). Oxygen consumption rate was measured for 3 periods with a mixing of 3 min in each cycle. Inhibitors and activators were used at the following concentrations: Oligomycin (2 µmol/L), FCCP (1 µmol/L), Antimycin A and Rotenone (0.5 µmol/L). Using these agents, we determined the mitochondrial OCR in basal respiration, ATP production, maximal and spare respiration. Results were normalized to the corresponding total protein content per well. For the assessment of OCR in HUVEC with CypD knockdown, cells were seeded overnight at 2–3 × 103 cells per well and then transfected with PPIF-shRNA (CypD knockdown) or ADV1-NC (control) for 20 h before stimulation with TSH. Following steps were the same as the aboved.
2.13. Measurement of adenine nucleotide levels
Adenine nucleotide levels were determined by high-performance liquid chromatography (HPLC) as described before [34]. Briefly, HUVEC were transferred to an ice-cold 0.6 mol/L HClO4 and immediately homogenized and centrifuged at 10,000g, 10 min, 4 °C (Eppendorf, Germany). The supernatant was neutralized with equivoluminal Na2HPO4 (1 mol/L) and centrifuged at 10,000g, 10 min, 4 °C again. The supernatant was filtered with 0.22 µm membrane and an aliquot of 50 μl were transferred into HPLC vials. Adenine nucleotides in samples were separated on a C18 column (Beckman, 5 µm, 250 mm × 4.6 mm) at a flow rate of 0.5 ml/min for 30 min and were detected at 254 nm with a ultraviolet detector (SPD-10A). The mobile phase was 96% 0.05 mol/L KH2PO4 (PH = 6.5) with 4% methanol (vol/vol). Results were adjusted by the corresponding cell numbers. Energy charge = [ATP + ADP/2]/[ATP + ADP + AMP]. Standard ATP, ADP and AMP were bought from Sigma.
2.14. Measurement of intracellualr oxidative stress
Intracellular oxidative stress was measured by 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA, 10 μmol/L, Sigma) staining. The process was according to manufactures’ instructions. The fluorescence was detected by a fluorescence microscopy (Leica, Germany, excitation: 450–490 nm; emission: 500–550 nm).
2.15. Measurement of 8-OHdG
Immunofluorescence staining for 8-hydroxydeoxyguanosine (8-OHdG, abcam) was used for the determination of DNA oxidative damage [35] in cultured HUVECs. Briefly, cells were fixed by 4% paraformaldehyde (pH 7.4, BOSTER). Fixed cells were incubated with the goat polyclonal anti-8-OHdG antibody (1:200) overnight, followed by the secondary antibody (ZSGB-BIO, Beijing, China) and then mounted with DAPI (Invitrogen). The fluorescence was detected by a fluorescence microscopy (Leica, Germany).
2.16. Immunofluorescence staining for ET-1
The frozen section of mouse aorta (5 µm thick) and 4% paraformaldehyde fixed HUVECs were incubated with primary antibody overnight, followed by the secondary antibody, and then mounted with DAPI. The primary antibodies included rabbit polyclonal anti-endothelin 1 (1:100, proteintech) and rat monoclonal anti-CD31 (1:50, abcam).
2.17. Nitric oxide assays
Nitrate reduction was used as an indirect measure of nitric oxide formation [36], [37]. Briefly, HUVECs were treated with TSH (2 μmol/L) or vehicle for 24 h. After the incubation period, the supernatant was collected to detect NO secreted by HUVECs via nitrate reduction according to the instructions of the NO detection kit (Nanjing Jiancheng, Nanjing, China). A microplate reader (Molecular Devices, USA) was used to examine the OD values at 550 nm. The calculation of NO content was according to the manufacture’s instruction.
2.18. Statistical analysis
All statistical analyses were performed using SPSS version 22.0 for Windows (Chicago, IL, USA). Values for quantitative data are expressed as mean ± standard deviation, or median (inter-quartile range). The relationship between TSH and ET-1 levels was performed by simple linear regression analysis. Differences between means were compared using One-Way ANOVA (Dunnett’s t or LSD test) or Independent-Samples T Test. Differences were considered significant at p < 0.05.
3. Results
3.1. Serum TSH levels positively correlated to ET-1 levels in humans with SCH
We firstly enrolled human subjects with euthyroidism and SCH to evaluate the correlation between TSH and endothelial perturbation. As shown in Table 1, no significant difference was observed in demographic, anthropometric, biochemical and clinical variables at baseline. Thyroid functions of enrollments throughout the study were shown in Table 2. At the end-of-study, serum TSH levels decreased to normal in all SCH patients.
Table 1.
Baseline characteristics of participants.
| Variables | Euthyroidism (n = 33) | Mild SCH (n = 33) | Significant SCH (n = 33) | p value |
|---|---|---|---|---|
| Age (year) | 57.03 ± 8.22 | 57.85 ± 8.20 | 57.79 ± 8.51 | 0.906 |
| BMI (kg/m2) | 15.55 ± 5.31 | 26.02 ± 3.03 | 25.96 ± 3.15 | 0.402 |
| WC (cm) | 89.33 ± 8.60 | 89.30 ± 9.65 | 91.67 ± 9.06 | 0.484 |
| TC (mmol/L) | 5.34 ± 0.89 | 5.41 ± 0.98 | 5.68 ± 0.93 | 0.295 |
| HDL-C (mmol/L) | 1.40 ± 0.37 | 1.38 ± 0.30 | 1.35 ± 0.27 | 0.798 |
| LDL-C (mmol/L) | 3.12 ± 0.66 | 3.11 ± 0.73 | 3.37 ± 0.91 | 0.292 |
| Non HDL-C (mmol/L) | 3.94 ± 0.78 | 4.03 ± 0.90 | 4.33 ± 0.92 | 0.162 |
| TG (mmol/L) | 1.22 (0.62) | 1.18 (0.89) | 1.35 (0.78) | 0.357 |
| SBP (mmHg) | 142.42 ± 16.36 | 146.06 ± 19.01 | 149.73 ± 19.61 | 0.277 |
| DBP (mmHg) | 83.52 ± 9.20 | 81.52 ± 10.60 | 82.55 ± 10.42 | 0.724 |
| FPG (mmol/ L) | 6.59 ± 2.48 | 6.42 ± 2.25 | 6.98 ± 2.92 | 0.666 |
Values for quantitative data are expressed as mean ± standard deviation, or median (inter-quartile range).
p value for comparing variables among euthyroidism, mild SCH and significant SCH groups.
SCH, subclinical hypothyroidism; BMI, body mass index; WC, waist circumference; TC, total cholesterol; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; Non HDL-C, non high-density lipoprotein cholesterol; TG, triglyceride; SBP, systolic blood pressure; DBP, diastolic blood pressure; FPG, fasting plasma glucose.
Table 2.
Thyroid functions of participants throughout the study.
| Variables | Euthyroidism (n = 33) | Mild SCH (n = 33) | Significant SCH (n = 33) |
|---|---|---|---|
| FT4 (p mol/L) | |||
| Baseline | 16.44 ± 2.05 | 15.35 ± 2.07 | 14.15 ± 1.37 |
| End point | 15.49 ± 1.86 | 17.26 ± 2.44 | 19.01 ± 2.63 |
| p value | 0.010* | 0.000* | 0.000* |
| FT3 (pmol/L) | |||
| Baseline | 4.94 ± 0.48 | 5.02 ± 0.57 | 4.88 ± 0.54 |
| End point | 4.76 ± 0.44 | 4.62 ± 0.54 | 4.75 ± 0.74 |
| p value | 0.095 | 0.172 | 0.364 |
| TSH (mIU/L) | |||
| Baseline | 2.05 (1.55) | 6.14 (1.65) | 11.58 (1.96) |
| End point | 2.32 (1.93) | 2.73 (1.42) | 1.75 (2.54) |
| p value | 0.077 | 0.000* | 0.000* |
Values for quantitative data are expressed as mean ± standard deviation, or median (inter-quartile range).
p value for comparing variables between baseline and end point in each group. * p < 0.05.
SCH, subclinical hypothyroidism; FT3, free triiodothyronine; FT4, free thyroxine; TSH, thyroid-stimulating hormone.
ET-1 is the most abundant isoform of endothelins in the human cardiovascular system and is the most potent vasoconstrictor identified to date. The primary source of ET-1 is thought to be vascular endothelial cells, although the peptide is produced by other cell types [38]. Under physiological conditions, its synthesis and release were inhibited by endothelium-derived nitric oxide [39]. Increased ET-1 can disrupt the equilibrium within the vascular wall [40]. In our experiment, no significant correlation between TSH and ET-1 was observed in subjects with euthyroidism (Fig. 1A) or mild SCH (Fig. 1B). However, in patients with TSH > = 10 mIU/L, serum TSH levels positively correlated to ET-1 levels at baseline (Fig. 1C). Moreover, when TSH declined to normal levels in these subjects, serum ET-1 levels were significantly reduced (Fig. 1D). Our results indicated that elevated serum TSH positively correlated to endothelial perturbation.
Fig. 1.

Serum TSH levels positively correlated to ET-1 levels in humans with SCH. (A-C) Relations between serum TSH and ET-1 in the enrollments at baseline were represented by the regression line. R2 indicates the correlation coefficient derived from simple linear regression analysis. (D) Serum ET-1 levels in significant SCH patients at baseline and after levothyroxine (LT4) therapy (n = 33). * p < 0.05.
3.2. TSH impaired endothelial functions with enhanced oxidative stress
Supplemented with exogenous T4 to keep normal thyroid hormones, thyroid-specific TSH receptor (TSHR)-knockout mice with injection of exogenous TSH (TT-KO+TSH) or solvent (TT-KO+solvent) were generated to evaluate the effects of elevated TSH on endothelium. As expected, serum TSH levels were significantly increased in TT-KO+TSH group (Fig. 2A). Serum total T4 (TT4) levels and body weight were comparable in the two groups (Figs. 2B–2C).
Fig. 2.

TSH impaired endothelial functionsin vivo. Serum TSH (A), serum total T4 (B) and body weight (C) of male TT-KO mice (n = 7–16). (D) Representative immunofluorescence staining for ET-1 (red), with ECs stained with anti-CD31 (green), nuclei stained with DAPI (blue). Endothelium are depicted by arrows (n = 6–7). Quantitative analysis of aortic ET-1 generation was shown in the right. The fluorescence IntDen was adjusted by total aorta area. (E) Quantitative analysis of DCFH-DA staining for aortic oxidative stress, expressed as fluorescence IntDen relative to total aorta area (n = 6). (F) Endothelium-dependent vasodilation in mouse mesenteric arteries. L-NAME was used to inhibit eNOS activity. Mode pattern (left), results in the indicated mouse models (middle and right, n = 4–7). Data were shown as mean ± SD. * p < 0.05 vs. TT-KO + solvent mice (middle) or Tshr+/+ mice without TSH stimulation (right).
We confirmed increased ET-1 expression (Fig. 2D) and oxidative stress (measured by DCFH-DA staining, Fig. 2E) in aorta of TT-KO+TSH mice. Next, we adopted sodium nitroprusside (SNP) and acetylcholine (Ach) to observe endothelium-independent (Fig. S1) and endothelium-dependent (Fig. 2F) vasodilation, respectively. Mode patterns were respectively shown in the left of each panel. As we can see, there was no difference in SNP-induced vasodilation between TT-KO groups, but vessels of TT-KO+TSH mice were less responsive to Ach, indicating that TSH inhibited endothelium-dependent vasodilation. Interestingly, Ach elicited similar relaxation response in TT-KO groups when arteries were pretreated with N-nitro-l-arginine methyl ester (L-NAME, the inhibitor of eNOS). These results indicated that the impaired vasorelaxation in TT-KO+TSH mice might be eNOS-mediated [41].
TSH exerts its biological functions via TSH receptor (TSHR), which is functionally expressed in ECs as in thyroid [42]. As shown in the right of Fig. S1 and 2F, endothelium-independent vasodilation was comparable in Tshr+/+ and Tshr-/- mice. While compared to Tshr+/+ mice, Tshr-/- mice significantly resisted to TSH-induced decline of endothelium-dependent vasodilation. Therefore, depending on its receptor, elevated serum TSH impaired endothelial functions in vivo.
Moreover, we confirmed that elevated TSH per se significantly impaired endothelial functions in vitro. TSH dose or time dependently increased ET-1 expression (Fig. 3A) and decreased eNOS phosphorylation at Ser1177, the phosphorylation of which stimulates eNOS activity [43] (Fig. 3B). Akt phosphorylation is well-known to facilitate eNOS activation [43]. In our result, we found that elevated TSH time dependently decreased Akt phosphorylation at Thr308 (Fig. 3C). Besides, NO release was also inhibited with TSH exposure (Fig. 3D). Endothelial dysfunction closely correlates to excessive oxidative stress, which refers to elevated intracellular ROS that cause damages to lipids, proteins and DNA [44]. Catalase is an important enzyme hydrolyzing H2O2 to H2O and O2 [45]. Our data demonstrated that in cultured HUVECs, TSH reduced catalase expression (Fig. 3E) and increased intracellular oxidative stress (Fig. 3F) in a dose-dependent manner. Besides, we confirmed that elevated TSH increased DNA oxidative injury via using immunofluorescence staining for 8-OHdG (Fig. 3G).
Fig. 3.

Cellular dysfunction and oxidative stress in HUVECs with TSH exposure. HUVECs were exposed to TSH in a dose- or time-dependent manner. All experiments were repeated at least 3 times. (A) Representative immunofluorescence staining for ET-1 (green), nuclei stained with DAPI (blue). (B) Western blot analysis of p-eNOS (Ser1177) and total eNOS, normalized to GAPDH. (C) Western blot analysis of p-Akt (Thr308) and total Akt, normalized to GAPDH. (D) NO release from cultured HUVECs was measured via nitrate reduction in the indicated groups. (E) Western blot analysis of catalase, normalized to GAPDH. (F) Representative DCFH-DA staining for oxidative stress in HUVECs. (G) DNA oxidative injury was stained by 8-OHdG (red), nuclei stained with DAPI (blue).
In summary, we confirmed that elevated TSH per se, depending on its receptor TSHR, could induce oxidative injury in ECs.
3.3. Elevated TSH induced excessive mPTP opening and mitochondrial oxidative damages in ECs
Excessive mitochondrial ROS generation triggered and aggravated endothelial oxidative damages [46]. We proposed mitochondrial oxidative stress (mitoOS) be one underlying mechanism for TSH-induced endothelial dysfunction. The tricarboxylic acid cycle (TCA) and oxidative phosphorylation in mitochondria are final metabolic pathways for all classes of nutrients under most cases [47]. To some extent, changes in mitochondrial function influence the whole body metabolism [21]. Therefore, we firstly evaluated the metabolic status of TT-KO mice. With exogenous TSH injection, their whole body metabolisms were slightly decreased in the light phase. Whereas in the dark phase, metabolic status of the two groups were almost the same (Fig. S2). These results indicated that elevated TSH slightly reduced mouse basal metabolism [48].
Excessive opening of mPTP has been shown to result in mitochondrial dysfunction under many pathological status [49], [50]. As shown in Fig. 4A, aortic mitochondria from TT-KO+TSH mice showed a greater mitochondrial swelling than the ones from TT-KO+solvent mice, indicating excessive mPTP opening [32] in TT-KO+TSH mouse aorta. Excessive opening of mPTP leads to the dissipation of mitochondrial membrane potential [51]. To assess the mitochondrial membrane potential in aorta in situ, we loaded fresh aortic slices of mice with tetramethylrhodamine methyl ester (TMRM). Mitochondrial depolarization results in a decrease in fluorescence intensity of TMRM [32]. As we can see in Fig. 4B, the intensity of TMRM staining significantly decreased in TT-KO+TSH mouse aorta. Meanwhile, aortic mitochondrial ROS generation notably increased in TT-KO+TSH mice, indicated by the increment in the intensity of MitoSox Red staining (Fig. 4C).
Fig. 4.

Elevated TSH induced excessive mPTP opening and mitoOS in ECs. (A-C) Effects of TSH on mitochondrial function and ROS production were analyzed in TT-KO mouse aorta. (A) Ca2+-overload induced mitochondrial swelling (mean ± SD, n = 5). Data were shown as the percentage change relative to the corresponding initial OD at 540 nm.* p < 0.05 vs. TT-KO mice without TSH injection. (B) TMRM staining (red) for mitochondrial membrane potential in freshly frozened aorta slices, with nuclei stained by Hochest 33342 (blue) (n = 5–6). (C) MitoSox Red staining for mitochondrial ROS generation in freshly frozened aortic slices, with nuclei stained DAPI (n = 5–6). (D-G) Dose- or time-dependent effects of TSH on mitochondrial function and ROS production in HUVECs. * p < 0.05 vs. HUVECs without TSH treatment. † p < 0.05 vs. HUVECs treated with TSH for 6 h. (D) Ca2+-overload induced mitochondrial swelling in the indicated groups (mean ± SD, n = 3), expressed as percentage decrease of the corresponding initial OD at 540 nm. (E) Mitochondrial complex I-III (CI-CIII) activities, normalized to the corresponding citrate synthase (CS) activity (mean ± SD, n = 3). (F) MitoSox Red staining for mitochondrial ROS production in the indicated groups, with mitochondria located by MitoTracker Green and nuclei by Hochest 33342 (blue) (n = 4). (G) Oxygen consumption rates (OCR) were measured by Seahorse XF96 analyzer (mean ± SD, n = 7–8). OCR relating to mitochondrial basal respiration, ATP production, maximal respiration and spare capacity were respectively analyzed, normalized to the corresponding total protein content per well.
In cultured HUVECs, elevated TSH significantly increased sensitivities of mPTP to Ca2+ overload, especially when treated for 24 h (Fig. 4D). Mitochondrial respiratory complex I-III activities significantly reduced (Fig. 4E), paralled with notable increment in mitochondrial ROS production (Fig. 4F). Besides, mitochondrial oxygen consume rates (OCR) relating to basal respiration, ATP production and maximal respiration were markedly blunted with TSH exposure (Fig. 4G). Interestingly, Fig. S3 showed that with direct TSH exposure, HA-VSMCs exhibited no difference in mitochondrial ROS generation but significant enhancement in the intensity of TMRM staining. However, conditioned medium (CM) from HUVECs with TSH exposure significantly promoted mitochondrial ROS generation and potential depolarization in HA-VSMCs, indicating that TSH-induced mitochondrial dysfunction in VSMCs was initiated by ECs.
Therefore, excessive mPTP opening and mitochondrial oxidative damages could be motivated by elevated TSH in ECs.
3.4. Blocking CypD attenuated TSH-induced mitoOS and endothelial dysfunction
Although several proteins have been proposed to constitute mPTP, only CypD is the genetically proven regulator [52]. To confirm the specificity of CypD inhibition in ameliorating TSH-induced mitochondrial and endothelial perturbations, we reduced CypD expression in HUVECs with PPIF shRNA transfection (Fig. 5A) and cells transfected by ADV1-NC were used as controls. As shown in Fig. 5B, CypD deficient ECs displayed significant resistance to TSH-induced mitochondrial ROS over-production. Besides, the impaired mitochondrial OCR were markedly ameliorated (Fig. 5C). Moreover, CypD deficiency attenuated TSH-triggered endothelial perturbations, exhibiting as lowered ET-1 expression (Fig. 5D) and increased eNOS phosphorylation at Ser1177 (Fig. 5E). Mesenteric arteries from CypD KO mice exhibited similar endothelium-independent vasodilation compared to those from their WT littermates (Fig. S4A), but significantly resisted to TSH-induced decline of endothelium-dependent vasodilation (Fig. 5F).
Fig. 5.

Genetic CypD knockdown attenuated TSH-induced mitoOS and endothelial dysfunction. (A-E) HUVECs transfected with PPIF shRNA or ADV1-NC (control) were exposed to 2 μM TSH or vehicle (none TSH) for 24 h. All experiments were repeated at least 3 times. (A) Virus infection efficiency was exhibited by the intensity of GFP (up). CypD reduction was analyzed by Western blot (down). (B) MitoSox Red staining for mitochondrial ROS generation. (C) Mitochondrial OCR were measured by Seahorse XF96 analyzer. OCR relating to mitochondrial basal respiration, ATP production, maximal respiration and spare capacity were respectively analyzed, normalized to the corresponding total protein content per well (mean ± SD, n = 6–9). * p < 0.05 vs. HUVECs transfected with ADV1-NC. † p < 0.05 vs. HUVECs with both PPIF shRNA transfection and TSH exposure. ‡ p < 0.05 vs. HUVEC with PPIF shRNA transfection. (D) Representative immunofluorescence staining for ET-1 (red), nuclei stained with DAPI (blue). (E) Western blot analysis of p-eNOS (Ser1177) and total eNOS, normalized to GAPDH. (F) Endothelium-dependent vasodilation in CypD KO mice and their WT littermate controls with or without acute TSH stimulation (mean ± SD, n = 4–7). * p < 0.05 vs. WT mice without TSH stimulation.
Initially used to inhibit the immune response following organ transplantations, cyclosporine A (CsA) is emerging as the most notable inhibitor of mPTP opening by binding to a special domain of CypD, which is adjacent to the acetylation site [53]. We adopted 1 μmol/L CsA to the cultured HUVECs 1 h before TSH co-incubation for 24 h. As expected, CsA pretreatment not only attenuated TSH-triggered excessive mPTP opening and mitochondrial ROS generation (Figs. 6A–6B), but also ameliorated mitochondrial OCR (Fig. 6C). Adenine nucleotide levels were detected by HPLC (Fig. S5). Results showed that ATP production and energy charge were reduced by TSH, both of which were reversed by CsA pretreatment. Functionally, CsA reversed TSH-induced catalase reduction (Fig. 6D) and ET-1 increment (Fig. 6E). Interestingly, we found that inhibition of NADPH oxidase with Apocynin also attenuated TSH-induced oxidative stress in ECs (Fig. S6), indicating at least part of TSH-triggered intracellular ROS generation came from NADPH oxidase activation.
Fig. 6.

Pharmacological blocking of CypD ameliorated TSH-impaired mitochondrial and endothelial perturbationsin vitro. HUVECs were pretreated with 1 μM CsA for 1 h before 2 μM TSH co-incubation for 24 h. * p < 0.05 vs. HUVECs with 0 μM TSH. † p < 0.05 vs. HUVECs treated with both CsA and TSH. ‡ p < 0.05 vs. HUVECs treated with CsA. (A) Ca2+-overload induced mitochondrial swelling in the indicated groups (mean ± SD, n = 6). Data were shown as percentage changes relative to the corresponding initial OD at 540 nm. (B) MitoSox Red staining for mitochondrial ROS production in the indicated groups, with mitochondria located by MitoTracker Green and nuclei by Hochest 33342 (blue) (n = 4). (C) Mitochondrial OCR were measured by Seahorse XF96 analyzer (mean ± SD, n = 9–12). OCR relating to mitochondrial basal respiration, ATP production, maximal respiration and spare capacity were respectively analyzed, normalized to the corresponding total protein content per well. (D) Western blot analysis of catalase expression, normalized to β-actin (n = 4). (E) Representative immunofluorescence staining for ET-1 (red) of 3 independent experiments, nuclei stained with DAPI (blue).
To evaluate the consequences of pharmacological blocking of CypD in vivo, we injected CsA (15 mg/kg d) or PBS (the same volume as CsA) to TT-KO mice for 4 weeks prior to TSH or solvent co-injection for another 2 weeks (TT-KO+solvent, TT-KO+TSH, TT-KO+CsA, TT-KO+CsA+TSH). We found that pretreatment with CsA attenuated TSH-triggered mitochondrial swelling in mouse aorta (Fig. 7A). Besides, TSH-induced decreased aortic mitochondrial complex I and III activities were reversed by CsA (Fig. 7B) and the excessive ROS production in aortic mitochondria was accordingly reduced (Fig. 7C). Furthermore, pretreatment with CsA inhibited TSH-induced excessive ET-1 expression in aorta (Fig. 7D). Pharmacological inhibition of CypD didn’t significantly affect SNP-induced vasodilation (Fig. S4B) but did reverse TSH-induced decline of endothelium-dependent vasorelaxation (Fig. 7E).
Fig. 7.

CsA pretreatment ameliorated TSH-impaired mitochondrial and endothelial perturbationsin vivo. (A) Ca2+-overload induced mitochondrial swelling in the indicated groups (mean ± SD, n = 5–6). Data were shown as percentage changes relative to the corresponding initial OD at 540 nm. * p < 0.05 vs. TT-KO+solvent mice. † p < 0.05 vs. TT-KO+CsA+TSH mice. (B) Mitochondrial complex I and III activities of the indicated mouse aorta (mean ± SD, n = 5–8), normalized to the corresponding CS activity. (C) Representative MitoSox Red staining for mitochondrial ROS generation in the indicated mouse aorta (n = 6–7). (D) Representative immunofluorescence staining for ET-1 (red) in the indicated mouse models, with endothelium stained by anti-CD31 (green) and nuclei by DAPI (blue) (n = 6). Aortic ET-1 fluorescence IntDen was adjusted by total aorta area. (E) Endothelium-dependent vasodilation of the indicated groups (mean ± SD, n = 4–5, * p < 0.05).
3.5. TSH enhanced CypD acetylation via inhibiting AMPK/SIRT3 signaling pathway
Based on our results, we proposed that TSH sensitized mPTP opening by modulating CypD in ECs. In comparison with the control group, TSH exposure did’t significantly increase CypD expression (Fig. 8A), but did markedly elevate CypD acetylation (Fig. 8B), an important form for CypD activation [54].
Fig. 8.

TSH enhanced CypD acetylation via inhibiting AMPK/SIRT3 signaling pathway in HUVECs. (A) Western blot analysis of CypD in HUVECs under TSH treatment in different doses (n = 6). (B) CypD was immunoprecipitated from HUVECs treated with 0 or 2 μM TSH for 24 h. The western blots of the immunoprecipitates were probed with antibody against acetylated lysine, stripped and then re-probed with antibody against CypD (n = 3). (C) Western blot analysis of p-AMPK (Thr-172), total AMPK and SIRT3 in HUVECs exposed to TSH in different doses (n = 3–5). (D-E) HUVECs were exposed to 2 μM TSH for 24 h with 1 μM AICAR co-incubation for the last 6 h. (D) Western blot analysis of p-AMPK (Thr-172), total AMPK and SIRT3 (n = 3). (E) CypD was immunoprecipitated from HUVEC of the indicated groups. The western blots of the immunoprecipitates were probed with antibody against acetylated lysine, stripped and then re-probed with antibody against CypD (n = 3).
Sirtuins are main deacetylases that regulate protein acetylation. Studies suggest that sirtuin-3 (SIRT3), but not other mitochondrial sirtuins, deacetylates CypD [55]. Adenosine monophosphate-activated protein kinase (AMPK) is a known positive modulator of sirtuin activity [56]. As we found, TSH inhibited AMPK phorphorylation at Thr-172 and decreased SIRT3 expression in dose-dependent manner (Fig. 8C). To confirm whether AMPK/SIRT3 signaling pathway was involved in TSH-induced CypD acetylation, HUVECs were exposed to TSH for 24 h with AICAR ( the activator for AMPK activation) co-incubation for the last 6 h. As shown in Fig. 8D, AICAR exposure stimulated AMPK phosphorylation (Thr-172) and SIRT3 expression, leading to a reversal in the increment of CypD acetylation caused by TSH exposure (Fig. 8E).
4. Discussion
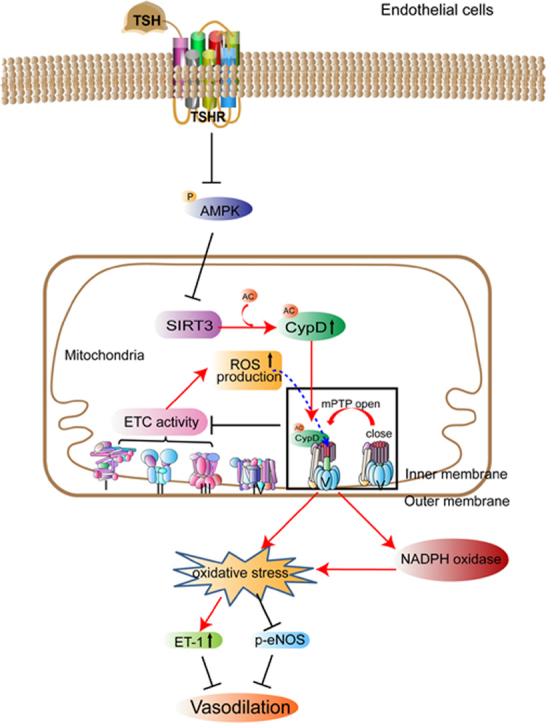
In this study, we confirmed that CypD mediated TSH-induced mitochondrial and endothelial perturbations. Elevated TSH triggered CypD acetylation via inhibiting AMPK/SIRT3 signaling pathway, leading to excessive mPTP opening, decreased electron transport chain activities and elevated mitochondrial ROS generation. With mPTP opening, excessive mitochondrial ROS were released to the cytoplasm. Meanwhile, through the cross-talk with mitochondria, NADPH oxidase was activated and synergetically evoked the intracellular oxidative stress. The equilibrium between ET-1 and eNOS was disturbed and functionally, endothelium-dependent vasodilation was perturbed as a result. Our study provided the first evidence that elevated TSH per se impaired endothelium-dependent vasodilation and the excessive mitoOS, triggered by excessive mPTP opening, was the underlying mechanism. Either genetic or pharmacological inhibition of CypD rescued TSH-impaired mitochondrial and endothelial functions.
To evaluate the correlation between TSH and endothelial perturbation in human subjects, we enrolled patients with SCH. Increased cholesterol levels are conventional risk factors for higher risk of CVD in SCH [57]. In order to eliminate the effects of dyslipidemia, we matched serum TC and LDL-C levels of the enrollments. Our results demonstrated that in patients with TSH > = 10 mIU/L, serum TSH positively correlated to the ET-1 expression. However, we didn’t find obvious correlation between TSH and ET-1 when TSH < 10 mIU/L, which was inconsistent with the previous results from patients with metabolic syndrome [17]. Metabolic syndrome is a cluster of metabolic disorders that collectively increase the risk of CVD [58]. The correlation between TSH and endothelial dysfunctions in SCH with metabolic syndrome might be amplified by other CVD risk factors.
In order to explore effects of elevated TSH on mitochondria and ECs in vivo, we adopted flox/cre system to generate a mouse model in which TSHR is specifically knocked out from thyroid. As a result, endogenous thyroid hormones (THs) couldn’t be synthesized. THs has been shown to have effect on metabolism [59]. Besides, changes in THs adversely influence the cardiovascular system [60]. In order to exclude the influence of THs, we supplied exogenous T4 to the TT-KO mice to maintain normal serum THs levels and stable endogenous TSH levels. With this approach, the elevation of serum TSH level could be controlled by injection of exogenous TSH (Fig. 2A) without altering serum THs levels (Fig. 2B). Besides, the comparable serum TT4 levels and body weights in TT-KO and WT male mice with same ages (data were not shown) proved that we had supplied the TT-KO mice with a suitable dose of exogenous T4.
HUVECs are accepted and widely used in vitro investigations for endothelial disorders of arteries [61], [62], [63], [64]. Although HUVECs exhibit phenotypic and functional differences versus human arterial endothelial cells (HAECs) [65], considering the limited lifespan and the unstable characteristics of the primary HAECs [66], we adopted the immortalized HUVEC cell line [64] in our study that are generally better characterized and more stable in their endothelial traits [66].
Previous studies have suggested that both ECs and VSMCs play important roles in maintaining vasorelaxant activity [67]. No difference in SNP-induced vasodilation in our study excluded the effect of TSH on the diastolic and systolic functions of VSMCs. The decline of Ach-mediated relaxation in TT-KO+TSH mice indicated that elevated TSH perturbed the vasotonia by disturbing endothelial functions. Besides, with pretreatment of L-NAME, the similar vascular responses to Ach in TT-KO groups indicated that the impaired vasorelaxation in TT-KO+TSH mice was eNOS-mediated [41] and we further provided the evidence that elevated TSH inhibited eNOS activation and NO release in cultured ECs. Moreover, we confirmed that elevated TSH triggered ET-1 increment in ECs both in vivo and in vitro. Our data showed that with TSHR knockout, mice resisted to TSH-impaired endothelium-dependent vasodilation. The results indicated that the detrimental effects of TSH on ECs were TSHR dependent. Meanwhile, our data confirmed that the perturbed endothelial function was accompanied by oxidative stress, indicated by reduced catalase expression, elevated fluorescence in DCFH-DA staining and increased DNA oxidative injury. Notablely, we identified that TSH not only exerted its effects on thyroid but also had substantial endothelial-specific effects. Depending on its receptor, elevated TSH triggered oxidative damages in ECs and the disturbed equilibrium between ET-1 and eNOS accounted for the perturbed endothelium-dependent vasodilation.
Based on the findings that very few small organic molecules stoichiometrically react with H2O2 to form a detectable intracellular fluorescent product and that the few molecules may still cause underlying problems [68], we adopted the widely used probe DCFH-DA for the detection of the oxidative stress in HUVECs. Of note, it should be noted that the DCFH-DA indirectly probes oxidative stress but not directly reflect the ROS levels [68], [69]. Therefore, our results indicate that elevated TSH triggers oxidative stress in ECs, which might be related to increased ROS production to some degree.
MitoOS has been implicated as a critical resource for oxidative stress in ECs. However, whether TSH induces mitoOS in ECs and the potent signaling events linking TSH with mitochondrial function remains unknown. Our study provided a link between elevated TSH and endothelial dysfunction via a previously unrecognized signaling pathway, which involved CypD activation signaling cascade and the subsequent mitoOS.
In addition to its role in cellular apoptosis, mPTP is generally considered as a key modulator for mitochondrial ROS generation and cellular oxidative stress. Studies confirmed that mPTP opening-induced mitochondrial ROS participates in the pathological process of vascular disease. Hana A. Itani et al have reported that loss of CypD, the key regulator of mPTP opening, prevents mitochondrial ROS generation in angiotensin II–infused mice, thus improves vascular relaxation and reduces hypertension [70]. In our study, we found that elevated TSH induced excessive mPTP opening in ECs, indicated by enhanced sensitivity to Ca2+-overload induced mitochondrial swelling. Excessive mPTP opening leads to the collapse of the mitochondrial proton motive force, resulting in decreased mitochondrial membrane potential [71]. Our study demonstrated that concomitant with the mPTP opening, mitochondrial membrane potential was reduced by TSH. Normal functions of complexes CI-III [72] are known for redox homeostasis in mitochondria. We confirmed that with excessive mPTP opening, activities of mitochondrial respiratory chain complexes were also blunted by TSH. As a result, excessive ROS were generated in mitochondria.
Of note, excessive and prolonged mPTP activation leads to mitochondrial outer membrane rupture, thus resulting in the release of pathological ROS from mitochondria to cytoplasm [22] and further cellular dysfunction. We proposed that the increased cellular oxidative stress observed in our study was strongly associated with the release of mitochondrial ROS from mPTP. As we found that inhibition of NADPH oxidase attenuated TSH-induced oxidative stress in ECs, we attributed part of the increased intracellular ROS coming from NADPH oxidase activation which has been demonstrated to have a crosstalk with mPTP-associated mitochondrial ROS generation. Given the facts that inhibition of mPTP by CypD deficiency or sanglifehrin A reduced angiotensin II-triggered NADPH oxidase activation and further rescued endothelial function [73], the cross-talk between mitochondria and NADPH oxidases, therefore, may represent a feed-forward vicious cycle of ROS production [74], [75]. In this regard we could not exclude the possibility that the increased mitochondrial ROS and perturbed mitochodrial functions is at least in part due to TSH induced intracellular oxidative stress through NADPH oxidase activation.
CypD is the well-accepted regulator of mPTP opening. Traditional concept is that CypD mainly acts as Ca2+ sensitizer for mPTP opening, leading to mitochondrial swelling and cell death [70]. It is notable that unconventional effects of CypD on regulating redox status and vascular homeostasis have been uncovered in recent years. Studies have shown that CypD regulated mitochondrial and intracellular ROS generation via a crosstalk with NADPH oxidase in isolated leukocytes [73]. Besides, CypD deficiency prevented overproduction of mitochondrial ROS in aorta isolated from angiotensin II–infused mice, improved endothelium-dependent vascular relaxation and further attenuated angiotensin II–induced hypertension [70]. All these results demonstrate the role of CypD in regulating mitochodrial and intracellular ROS generation, as well as its specific contribution in regulation of endothelial dysfunction.
In order to ellucidate the potent molecular mechanism for TSH-induced mitoOS, we reduced CypD expression in HUVECs. No significant difference in mitochondrial and endothelial functions were observed between cells with only CypD inhibition and their vehicle controls. However, CypD deficiency in HUVECs did reverse TSH-induced excessive mitochondrial ROS generation and ameliorate the mitochondrial OCR. Functionally, the increased ET-1 expression and reduced eNOS activation under TSH exposure were also reversed. Under normal conditions, we found that CypD KO mice exhibited a similar vasotonia compared to their littermate controls. But once exposed to the acute TSH stimulation, CypD KO mice exhibited significant resistance to TSH-impaired endothelium-dependent vasodilation. All indicated that CypD was of crucial role in TSH-induced mitoOS and endothelial dysfunctions. Besides, the protective role of blocking CypD could only be triggered under pathological conditions. CsA is an inhibitor of CypD activation, whose binding pocket is adjacent to the acetylation site of CypD [55]. In our studies, we found that pharmacological inhibition of CypD by CsA not only attenuated TSH-induced mitochondrial swelling, but also reduced mitoOS and further ameliorated TSH-induced endothelial dysfunction. Acetylation is an important post-translational modification of CypD, which modulates the translocation of CypD from matrix to the inner membrane to facilitate the mPTP opening [51], [76]. In our study, the obvious increment in CypD acetylation under TSH exposure demonstrated that elevated TSH stimulated mPTP opening mainly by enhancing CypD activity.
SIRT3 is a class 1 sirtuin with deacetylase activity for not only histones, but also other proteins. A previous study has confirmed that SIRT3 depletion increases mitochondrial SOD2 acetylation and mitochondrial ROS generation in ECs, further diminishing endothelial nitric oxide and promoting hypertension [77]. The results indicate that protein deacetylation regulated by SIRT3 plays an important role in regulating mitochondrial and vascular oxidative stress. Conceivably, the increased CypD acetylation under TSH exposure in our study seems to suggest that SIRT3 might be down regulated by elevated TSH in HUVECs. AMPK is a known positive modulator of sirtuin activity [56]. In agreement with our previous study that TSH inhibited AMPK activity in liver [78], we have found that elevated TSH reduced AMPK activity, as well as SIRT3 expression in HUVECs. Furthermore, AICAR not only rescued AMPK activity and SIRT3 expression, but also reversed CypD acetylation under TSH exposure. Therefore, our findings have demonstrated that TSH-induced CypD acetylation in HUVECs is at least partially dependent on AMPK/SIRT3 signaling pathway. Indeed, the results do not exclude the involvement of other CypD over-acetylation-independent TSH pathways. Such effect will be examined in our future study.
Previous studies confirmed that CypD interaction with mPTP could be regulated by redox status. For example, H2O2 as a strong oxidizer induces CypD-dependent mPTP opening. Furthermore, nitric oxide donor could induce CypD to undergo protein S-nitrosylation, thus reducing CypD activation and inhibiting mPTP opening [79]. However, the underlying mechanism by which redox status regulates CypD activation is still unclear. Acetylation is an important post-translational modification for CypD activation and mPTP opening [51], [76]. We confirmed that inhibition of AMPK/SIRT3 signaling pathway, to some extent, accounted for TSH-induced CypD acetylation. As TSH triggered imbalance of intracellular redox status and the latter has been demonstrated to modulate CypD interaction with mPTP, further studies are needed to investigate that whether the intracellular redox status regulates TSH-induced CypD acetylation and by which mechanism CypD over-acetylation promotes mPTP opening in ECs.
Recent studies notice that using generalized ROS scavengers in clinical trials to reduce the oxidative burden is ineffective in the context of cardiovascular pathology [80] because normal physiological functions depend on a basal production of ROS [81]. Besides, anti-oxidants can undergo auto-oxidation before use [82]. Targeting ROS elimination is proposed to overcome the limitations of generalized ROS scavengers. Our results suggested that targeting inhibition of mitochondrial CypD might be a potential approach not only for reducing excessive ROS generation, but also for prevention and treatment of CVD correlating to elevated TSH.
A critical issue for the evaluation of CsA therapy merits discussion is its specificity. Although CsA is a well-documented inhibitor of CypD resulting in the suppression of excessive mPTP formation [32],previous studies have shown that CsA exerts an immunosuppressive effect through complicated yet not-fully-elucidated mechanisms [83]. In addition, CsA is a known inhibitor of calcineurin [84]. In this context, we cannot fully exclude the potential involvement of these side-effects of CsA. However, based on our observation of the protective effects of low dose CsA on mitochondrial function, we propose that this protection of CsA is, to a certain degree, largely associated with the attenuation of excessive mPTP opening. Of interest, in addition to the concerns about the specificity of CsA, the procedure of CsA preparation may also influence the function of CsA. Previous studies have reported that CsA prepared by polyoxyethylated castor oil (Sandimmune, Novartis) improved myocardial infarction damage in patients by targeting mPTP, which was in sharp contrast with the little effect of CsA prepared by lipid emulsion (CicloMulsion) [85], [86]. In this regard, we adopted CsA from Sandimmune, Novartis in our study. Moreover, the development of more specific CypD inhibitors is called for.
In conclusion, our results implicate that blocking mitochondrial CypD improves the defensive ability of ECs under TSH exposure. Although our data are preliminary, these findings might lead to new and promising methods for targeting ROS elimination to prevent and treat CVD in SCH patients.
Acknowledgments
We thank professor Lin Li for suggestions in the preparation of the manuscript.
Acknowledgments
Source of funding
This study was supported by National Key Research and Development Program of China (2017YFC1309800), the National Natural Science Foundation (81230018, 81430020, 81500595, 81470489), the Clinical Research Fund of Chinese Medical Association (15010010589), and Key Research and Department of Shandong Province (2016GSF201013).
Disclosures
None.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2018.01.004
Contributor Information
Ling Gao, Email: gaoling1@medmail.com.cn.
Jiajun Zhao, Email: jjzhao@medmail.com.cn.
Appendix A. Supplementary material
Supplementary material
References
- 1.Roumenina L.T., Rayes J., Frimat M., Fremeaux-Bacchi V. Endothelial cells: source, barrier, and target of defensive mediators. Immunol. Rev. 2016;274:307–329. doi: 10.1111/imr.12479. [DOI] [PubMed] [Google Scholar]
- 2.Jones D.P., Sies H. The redox code. Antioxid. Redox Signal. 2015;23:734–746. doi: 10.1089/ars.2015.6247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sies H. Oxidative stress: a concept in redox biology and medicine. Redox Biol. 2015;4:180–183. doi: 10.1016/j.redox.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Egea J., Fabregat I., Frapart Y.M., Ghezzi P., Gorlach A., Kietzmann T. European contribution to the study of ROS: a summary of the findings and prospects for the future from the cost action BM1203 (EU-ROS) Redox Biol. 2017;13:94–162. doi: 10.1016/j.redox.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Griendling K.K., FitzGerald G.A. Oxidative stress and cardiovascular injury: Part I: basic mechanisms and in vivo monitoring of ROS. Circulation. 2003;108:1912–1916. doi: 10.1161/01.CIR.0000093660.86242.BB. [DOI] [PubMed] [Google Scholar]
- 6.Griendling K.K., FitzGerald G.A. Oxidative stress and cardiovascular injury: Part II: animal and human studies. Circulation. 2003;108:2034–2040. doi: 10.1161/01.CIR.0000093661.90582.c4. [DOI] [PubMed] [Google Scholar]
- 7.Daiber A., Steven S., Weber A., Shuvaev V.V., Muzykantov V.R., Laher I. Targeting vascular (endothelial) dysfunction. Br. J. Pharmacol. 2017;174:1591–1619. doi: 10.1111/bph.13517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li J.M., Shah A.M. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004;287:R1014–R1030. doi: 10.1152/ajpregu.00124.2004. [DOI] [PubMed] [Google Scholar]
- 9.Hak A.E., Pols H.A., Visser T.J., Drexhage H.A., Hofman A., Witteman J.C. Subclinical hypothyroidism is an independent risk factor for atherosclerosis and myocardial infarction in elderly women: the Rotterdam study. Ann. Intern. Med. 2000;132:270–278. doi: 10.7326/0003-4819-132-4-200002150-00004. [DOI] [PubMed] [Google Scholar]
- 10.Tseng F.Y., Lin W.Y., Lin C.C., Lee L.T., Li T.C., Sung P.K. Subclinical hypothyroidism is associated with increased risk for all-cause and cardiovascular mortality in adults. J. Am. Coll. Cardiol. 2012;60:730–737. doi: 10.1016/j.jacc.2012.03.047. [DOI] [PubMed] [Google Scholar]
- 11.Cooper D.S., Biondi B. Subclinical thyroid disease. Lancet. 2012;379:1142–1154. doi: 10.1016/S0140-6736(11)60276-6. [DOI] [PubMed] [Google Scholar]
- 12.Biondi B., Cooper D.S. The clinical significance of subclinical thyroid dysfunction. Endocr. Rev. 2008;29:76–131. doi: 10.1210/er.2006-0043. [DOI] [PubMed] [Google Scholar]
- 13.Razvi S., Weaver J.U., Pearce S.H. Subclinical thyroid disorders: significance and clinical impact. J. Clin. Pathol. 2010;63:379–386. doi: 10.1136/jcp.2008.057414. [DOI] [PubMed] [Google Scholar]
- 14.Fatourechi V. Subclinical hypothyroidism: an update for primary care physicians. Mayo Clin. Proc. 2009;84:65–71. doi: 10.4065/84.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodondi N., den Elzen W.P., Bauer D.C., Cappola A.R., Razvi S., Walsh J.P. Subclinical hypothyroidism and the risk of coronary heart disease and mortality. JAMA. 2010;304:1365–1374. doi: 10.1001/jama.2010.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haribabu A., Reddy V.S., Pallavi C., Bitla A.R., Sachan A., Pullaiah P. Evaluation of protein oxidation and its association with lipid peroxidation and thyrotropin levels in overt and subclinical hypothyroidism. Endocrine. 2013;44:152–157. doi: 10.1007/s12020-012-9849-y. [DOI] [PubMed] [Google Scholar]
- 17.Ahirwar A.K., Singh A., Jain A., Patra S.K., Goswami B., Bhatnagar M.K. Raised TSH is associated with endothelial dysfunction in metabolic syndrome: a case control study. Rom. J. Intern. Med. = Rev. Roum. De. Med. Interne. 2017 doi: 10.1515/rjim-2017-0023. [DOI] [PubMed] [Google Scholar]
- 18.Dromparis P., Michelakis E.D. Mitochondria in vascular health and disease. Annu. Rev. Physiol. 2013;75:95–126. doi: 10.1146/annurev-physiol-030212-183804. [DOI] [PubMed] [Google Scholar]
- 19.Daiber A., Di Lisa F., Oelze M., Kroller-Schon S., Steven S., Schulz E. Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. Br. J. Pharmacol. 2015 doi: 10.1111/bph.13403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mikhed Y., Daiber A., Steven S. Mitochondrial oxidative stress, mitochondrial DNA damage and their role in age-related vascular dysfunction. Int. J. Mol. Sci. 2015;16:15918–15953. doi: 10.3390/ijms160715918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laker R.C., Taddeo E.P., Akhtar Y.N., Zhang M., Hoehn K.L., Yan Z. The Mitochondrial permeability transition pore regulator cyclophilin D exhibits tissue-specific control of metabolic homeostasis. PLoS One. 2016;11:e0167910. doi: 10.1371/journal.pone.0167910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morciano G., Giorgi C., Bonora M., Punzetti S., Pavasini R., Wieckowski M.R. Molecular identity of the mitochondrial permeability transition pore and its role in ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 2015;78:142–153. doi: 10.1016/j.yjmcc.2014.08.015. [DOI] [PubMed] [Google Scholar]
- 23.Vyssokikh M.Y., Katz A., Rueck A., Wuensch C., Dorner A., Zorov D.B. Adenine nucleotide translocator isoforms 1 and 2 are differently distributed in the mitochondrial inner membrane and have distinct affinities to cyclophilin D. Biochem. J. 2001;358:349–358. doi: 10.1042/0264-6021:3580349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giorgio V., Bisetto E., Soriano M.E., Dabbeni-Sala F., Basso E., Petronilli V. Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J. Biol. Chem. 2009;284:33982–33988. doi: 10.1074/jbc.M109.020115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alavian K.N., Beutner G., Lazrove E., Sacchetti S., Park H.A., Licznerski P. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. USA. 2014;111:10580–10585. doi: 10.1073/pnas.1401591111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gauba E., Guo L., Du H. Cyclophilin D promotes brain mitochondrial F1FO ATP synthase dysfunction in aging mice. J. Alzheimer's Dis.: JAD. 2017;55:1351–1362. doi: 10.3233/JAD-160822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marcu R., Kotha S., Zhi Z., Qin W., Neeley C.K., Wang R.K. The mitochondrial permeability transition pore regulates endothelial bioenergetics and angiogenesis. Circ. Res. 2015;116:1336–1345. doi: 10.1161/CIRCRESAHA.116.304881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu Y., Wang L., He J., Bi Y., Li M., Wang T. Prevalence and control of diabetes in Chinese adults. JAMA. 2013;310:948–959. doi: 10.1001/jama.2013.168118. [DOI] [PubMed] [Google Scholar]
- 29.Garber J.R., Cobin R.H., Gharib H., Hennessey J.V., Klein I., Mechanick J.I. Clinical practice guidelines for hypothyroidism in adults: cosponsored by the American Association of clinical endocrinologists and the American Thyroid Association. Endocr. Pract.: Off. J. Am. Coll. Endocrinol. Am. Assoc. Clin. Endocrinol. 2012;18:988–1028. doi: 10.4158/EP12280.GL. [DOI] [PubMed] [Google Scholar]
- 30.Wang T., Xu J., Bo T., Zhou X., Jiang X., Gao L. Decreased fasting blood glucose is associated with impaired hepatic glucose production in thyroid-stimulating hormone receptor knockout mice. Endocr. J. 2013;60:941–950. doi: 10.1507/endocrj.ej12-0462. [DOI] [PubMed] [Google Scholar]
- 31.Chai Q., Wang X.L., Zeldin D.C., Lee H.C. Role of caveolae in shear stress-mediated endothelium-dependent dilation in coronary arteries. Cardiovasc. Res. 2013;100:151–159. doi: 10.1093/cvr/cvt157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Du H., Guo L., Fang F., Chen D., Sosunov A.A., McKhann G.M. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat. Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spinazzi M., Casarin A., Pertegato V., Salviati L., Angelini C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 2012;7:1235–1246. doi: 10.1038/nprot.2012.058. [DOI] [PubMed] [Google Scholar]
- 34.Jing Y., Liu W., Cao H., Zhang D., Yao X., Zhang S. Hepatic p38alpha regulates gluconeogenesis by suppressing AMPK. J. Hepatol. 2015;62:1319–1327. doi: 10.1016/j.jhep.2014.12.032. [DOI] [PubMed] [Google Scholar]
- 35.Kumar N., Rosy, Goyal R.N. A melamine based molecularly imprinted sensor for the determination of 8-hydroxydeoxyguanosine in human urine. Talanta. 2017;166:215–222. doi: 10.1016/j.talanta.2017.01.058. [DOI] [PubMed] [Google Scholar]
- 36.Cao Z.H., Gao L., Jiang L., Zhang P., Ning H.Y., Zhang H. Effect of beta-arrestin on damage of human umbilical vein endothelial cell induced by angiotensin II. Eur. Rev. Med. Pharmacol. Sci. 2017;21:5821–5826. doi: 10.26355/eurrev_201712_14030. [DOI] [PubMed] [Google Scholar]
- 37.Wang Y., Narsinh K., Zhao L., Sun D., Wang D., Zhang Z. Effects and mechanisms of ghrelin on cardiac microvascular endothelial cells in rats. Cell Biol. Int. 2011;35:135–140. doi: 10.1042/CBI20100139. [DOI] [PubMed] [Google Scholar]
- 38.Davenport A.P., Hyndman K.A., Dhaun N., Southan C., Kohan D.E., Pollock J.S. Endothelin. Pharmacol. Rev. 2016;68:357–418. doi: 10.1124/pr.115.011833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bassenge E. Endothelial function in different organs. Prog. Cardiovasc. Dis. 1996;39:209–228. doi: 10.1016/s0033-0620(96)80002-8. [DOI] [PubMed] [Google Scholar]
- 40.Finch J., Conklin D.J. Air pollution-induced vascular dysfunction: potential role of endothelin-1 (ET-1) system. Cardiovasc. Toxicol. 2016;16:260–275. doi: 10.1007/s12012-015-9334-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang W., Wang Q., Wu Y., Moriasi C., Liu Z., Dai X. Endothelial cell-specific liver kinase B1 deletion causes endothelial dysfunction and hypertension in mice in vivo. Circulation. 2014;129:1428–1439. doi: 10.1161/CIRCULATIONAHA.113.004146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tian L., Zhang L., Liu J., Guo T., Gao C., Ni J. Effects of TSH on the function of human umbilical vein endothelial cells. J. Mol. Endocrinol. 2014;52:215–222. doi: 10.1530/JME-13-0119. [DOI] [PubMed] [Google Scholar]
- 43.Szabo C. Hydrogen sulfide, an enhancer of vascular nitric oxide signaling: mechanisms and implications. Am. J. Physiol. Cell Physiol. 2017;312:C3–C15. doi: 10.1152/ajpcell.00282.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schieber M., Chandel N.S. ROS function in redox signaling and oxidative stress. Curr. Biol.: CB. 2014;24:R453–R462. doi: 10.1016/j.cub.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cui H., Kong Y., Zhang H. Oxidative stress, mitochondrial dysfunction, and aging. J. Signal Transduct. 2012;2012:646354. doi: 10.1155/2012/646354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davidson S.M., Duchen M.R. Endothelial mitochondria: contributing to vascular function and disease. Circ. Res. 2007;100:1128–1141. doi: 10.1161/01.RES.0000261970.18328.1d. [DOI] [PubMed] [Google Scholar]
- 47.Amigo I., da Cunha F.M., Forni M.F., Garcia-Neto W., Kakimoto P.A., Luevano-Martinez L.A. Mitochondrial form, function and signalling in aging. Biochem. J. 2016;473:3421–3449. doi: 10.1042/BCJ20160451. [DOI] [PubMed] [Google Scholar]
- 48.Sunagawa G.A., Takahashi M. Hypometabolism during daily torpor in mice is dominated by reduction in the sensitivity of the thermoregulatory system. Sci. Rep. 2016;6:37011. doi: 10.1038/srep37011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taddeo E.P., Laker R.C., Breen D.S., Akhtar Y.N., Kenwood B.M., Liao J.A. Opening of the mitochondrial permeability transition pore links mitochondrial dysfunction to insulin resistance in skeletal muscle. Mol. Metab. 2014;3:124–134. doi: 10.1016/j.molmet.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lingan J.V., Alanzalon R.E., Porter G.A., Jr. Preventing permeability transition pore opening increases mitochondrial maturation, myocyte differentiation and cardiac function in the neonatal mouse heart. Pediatr. Res. 2017;81:932–941. doi: 10.1038/pr.2017.19. [DOI] [PubMed] [Google Scholar]
- 51.Li P., Meng X., Bian H., Burns N., Zhao K.S., Song R. Activation of sirtuin 1/3 improves vascular hyporeactivity in severe hemorrhagic shock by alleviation of mitochondrial damage. Oncotarget. 2015;6:36998–37011. doi: 10.18632/oncotarget.6076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lam C.K., Zhao W., Liu G.S., Cai W.F., Gardner G., Adly G. HAX-1 regulates cyclophilin-D levels and mitochondria permeability transition pore in the heart. Proc. Natl. Acad. Sci. USA. 2015;112:E6466–E6475. doi: 10.1073/pnas.1508760112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gutierrez-Aguilar M., Baines C.P. Structural mechanisms of cyclophilin D-dependent control of the mitochondrial permeability transition pore. Biochim. Biophys. Acta. 2015;1850:2041–2047. doi: 10.1016/j.bbagen.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Elrod J.W., Molkentin J.D. Physiologic functions of cyclophilin D and the mitochondrial permeability transition pore. Circ. J.: Off. J. Jpn. Circ. Soc. 2013;77:1111–1122. doi: 10.1253/circj.cj-13-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hafner A.V., Dai J., Gomes A.P., Xiao C.Y., Palmeira C.M., Rosenzweig A. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging. 2010;2:914–923. doi: 10.18632/aging.100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morigi M., Perico L., Rota C., Longaretti L., Conti S., Rottoli D. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Investig. 2015;125:715–726. doi: 10.1172/JCI77632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ochs N., Auer R., Bauer D.C., Nanchen D., Gussekloo J., Cornuz J. Meta-analysis: subclinical thyroid dysfunction and the risk for coronary heart disease and mortality. Ann. Intern. Med. 2008;148:832–845. doi: 10.7326/0003-4819-148-11-200806030-00225. [DOI] [PubMed] [Google Scholar]
- 58.Litvinova L., Atochin D.N., Fattakhov N., Vasilenko M., Zatolokin P., Kirienkova E. Nitric oxide and mitochondria in metabolic syndrome. Front. Physiol. 2015;6:20. doi: 10.3389/fphys.2015.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sinha R., Yen P.M. In: Cellular Action of Thyroid Hormone. De Groot L.J., Chrousos G., Dungan K., Feingold K.R., Grossman A., Hershman J.M., editors. Endotext; South Dartmouth (MA): 2000. [Google Scholar]
- 60.Jabbar A., Pingitore A., Pearce S.H., Zaman A., Iervasi G., Razvi S. Thyroid hormones and cardiovascular disease. Nat. Rev. Cardiol. 2017;14:39–55. doi: 10.1038/nrcardio.2016.174. [DOI] [PubMed] [Google Scholar]
- 61.Di M., Wang L., Li M., Zhang Y., Liu X., Zeng R. Dickkopf1 destabilizes atherosclerotic plaques and promotes plaque formation by inducing apoptosis of endothelial cells through activation of ER stress. Cell Death Dis. 2017;8:e2917. doi: 10.1038/cddis.2017.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yan F.X., Li H.M., Li S.X., He S.H., Dai W.P., Li Y. The oxidized phospholipid POVPC impairs endothelial function and vasodilation via uncoupling endothelial nitric oxide synthase. J. Mol. Cell. Cardiol. 2017;112:40–48. doi: 10.1016/j.yjmcc.2017.08.016. [DOI] [PubMed] [Google Scholar]
- 63.Liu D., Ding Z., Wu M., Xu W., Qian M., Du Q. The apolipoprotein A-I mimetic peptide, D-4F, alleviates ox-LDL-induced oxidative stress and promotes endothelial repair through the eNOS/HO-1 pathway. J. Mol. Cell. Cardiol. 2017;105:77–88. doi: 10.1016/j.yjmcc.2017.01.017. [DOI] [PubMed] [Google Scholar]
- 64.An R., Xi C., Xu J., Liu Y., Zhang S., Wang Y. Intramyocardial injection of recombinant adeno-associated viral vector coexpressing PR39/adrenomedullin enhances angiogenesis and reduces apoptosis in a rat myocardial infarction model. Oxid. Med. Cell. Longev. 2017;2017:1271670. doi: 10.1155/2017/1271670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chi J.T., Chang H.Y., Haraldsen G., Jahnsen F.L., Troyanskaya O.G., Chang D.S. Endothelial cell diversity revealed by global expression profiling. Proc. Natl. Acad. Sci. USA. 2003;100:10623–10628. doi: 10.1073/pnas.1434429100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bouis D., Hospers G.A., Meijer C., Molema G., Mulder N.H. Endothelium in vitro: a review of human vascular endothelial cell lines for blood vessel-related research. Angiogenesis. 2001;4:91–102. doi: 10.1023/a:1012259529167. [DOI] [PubMed] [Google Scholar]
- 67.Sun T., Liu R., Cao Y.X. Vasorelaxant and antihypertensive effects of formononetin through endothelium-dependent and -independent mechanisms. Acta Pharmacol. Sin. 2011;32:1009–1018. doi: 10.1038/aps.2011.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kalyanaraman B., Darley-Usmar V., Davies K.J., Dennery P.A., Forman H.J., Grisham M.B. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic. Biol. Med. 2012;52:1–6. doi: 10.1016/j.freeradbiomed.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wrona M., Wardman P. Properties of the radical intermediate obtained on oxidation of 2′,7′-dichlorodihydrofluorescein, a probe for oxidative stress. Free Radic. Biol. Med. 2006;41:657–667. doi: 10.1016/j.freeradbiomed.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 70.Itani H.A., Dikalova A.E., McMaster W.G., Nazarewicz R.R., Bikineyeva A.T., Harrison D.G. Mitochondrial cyclophilin D in vascular oxidative stress and hypertension. Hypertension. 2016;67:1218–1227. doi: 10.1161/HYPERTENSIONAHA.115.07085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Song R., Bian H., Huang X., Zhao K.S. Atractyloside induces low contractile reaction of arteriolar smooth muscle through mitochondrial damage. J. Appl. Toxicol. : JAT. 2012;32:402–408. doi: 10.1002/jat.1688. [DOI] [PubMed] [Google Scholar]
- 72.Lanciano P., Khalfaoui-Hassani B., Selamoglu N., Ghelli A., Rugolo M., Daldal F. Molecular mechanisms of superoxide production by complex III: a bacterial versus human mitochondrial comparative case study. Biochim. Biophys. Acta. 2013;1827:1332–1339. doi: 10.1016/j.bbabio.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kroller-Schon S., Steven S., Kossmann S., Scholz A., Daub S., Oelze M. Molecular mechanisms of the crosstalk between mitochondria and NADPH oxidase through reactive oxygen species-studies in white blood cells and in animal models. Antioxid. Redox Signal. 2014;20:247–266. doi: 10.1089/ars.2012.4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011;51:1289–1301. doi: 10.1016/j.freeradbiomed.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Daiber A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim. Biophys. Acta. 2010;1797:897–906. doi: 10.1016/j.bbabio.2010.01.032. [DOI] [PubMed] [Google Scholar]
- 76.Bochaton T., Crola-Da-Silva C., Pillot B., Villedieu C., Ferreras L., Alam M.R. Inhibition of myocardial reperfusion injury by ischemic postconditioning requires sirtuin 3-mediated deacetylation of cyclophilin D. J. Mol. Cell. Cardiol. 2015;84:61–69. doi: 10.1016/j.yjmcc.2015.03.017. [DOI] [PubMed] [Google Scholar]
- 77.Dikalova A.E., Itani H.A., Nazarewicz R.R., McMaster W.G., Flynn C.R., Uzhachenko R. Sirt3 Impairment and SOD2 hyperacetylation in vascular oxidative stress and hypertension. Circ. Res. 2017;121:564–574. doi: 10.1161/CIRCRESAHA.117.310933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang X., Song Y., Feng M., Zhou X., Lu Y., Gao L. Thyroid-stimulating hormone decreases HMG-coa reductase phosphorylation via amp-activated protein kinase in the liver. J. Lipid Res. 2015;56:963–971. doi: 10.1194/jlr.M047654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nguyen T.T., Stevens M.V., Kohr M., Steenbergen C., Sack M.N., Murphy E. Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. J. Biol. Chem. 2011;286:40184–40192. doi: 10.1074/jbc.M111.243469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sugamura K., Keaney J.F., Jr. Reactive oxygen species in cardiovascular disease. Free Radic. Biol. Med. 2011;51:978–992. doi: 10.1016/j.freeradbiomed.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kornfeld O.S., Hwang S., Disatnik M.H., Chen C.H., Qvit N., Mochly-Rosen D. Mitochondrial reactive oxygen species at the heart of the matter: new therapeutic approaches for cardiovascular diseases. Circ. Res. 2015;116:1783–1799. doi: 10.1161/CIRCRESAHA.116.305432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chan E.D., Riches D.W., White C.W. Redox paradox: effect of N-acetylcysteine and serum on oxidation reduction-sensitive mitogen-activated protein kinase signaling pathways. Am. J. Respir. Cell Mol. Biol. 2001;24:627–632. doi: 10.1165/ajrcmb.24.5.4280. [DOI] [PubMed] [Google Scholar]
- 83.Sarzi-Puttini P., Cazzola M., Panni B., Turiel M., Fiorini T., Belai-Beyene N. Long-term safety and efficacy of low-dose cyclosporin A in severe psoriatic arthritis. Rheumatol. Int. 2002;21:234–238. doi: 10.1007/s00296-001-0166-7. [DOI] [PubMed] [Google Scholar]
- 84.Ponticelli C., Cucchiari D., Graziani G. Hypertension in kidney transplant recipients. Transplant. Int. : Off. J. Eur. Soc. Organ Transplant. 2011;24:523–533. doi: 10.1111/j.1432-2277.2011.01242.x. [DOI] [PubMed] [Google Scholar]
- 85.Piot C., Croisille P., Staat P., Thibault H., Rioufol G., Mewton N. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 2008;359:473–481. doi: 10.1056/NEJMoa071142. [DOI] [PubMed] [Google Scholar]
- 86.Cung T.T., Morel O., Cayla G., Rioufol G., Garcia-Dorado D., Angoulvant D. Cyclosporine before PCI in patients with acute myocardial infarction. N. Engl. J. Med. 2015;373:1021–1031. doi: 10.1056/NEJMoa1505489. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material