

Abstract
Clinical application of doxorubicin (DOX), an anthracycline antibiotic with potent anti- tumor effects, is limited because of its cardiotoxicity. However, its pathogenesis is still not entirely understood. The aim of this paper was to explore the mechanisms and new drug targets to treat DOX-induced cardiotoxicity. The in vitro model on H9C2 cells and the in vivo models on rats and mice were developed. The results showed that DOX markedly decreased H9C2 cell viability, increased the levels of CK, LDH, caused histopathological and ECG changes in rats and mice, and triggered myocardial oxidative damage via adjusting the levels of intracellular ROS, MDA, SOD, GSH and GSH-Px. Total of 18 differentially expressed microRNAs in rat heart tissue caused by DOX were screened out using microRNA microarray assay, especially showing that miR-140-5p was significantly increased by DOX which was selected as the target miRNA. Double-luciferase reporter assay showed that miR-140-5p directly targeted Nrf2 and Sirt2, as a result of affecting the expression levels of HO-1, NQO1, Gst, GCLM, Keap1 and FOXO3a, and thereby increasing DOX-caused myocardial oxidative damage. In addition, the levels of intracellular ROS were significantly increased or decreased in H9C2 cells treated with DOX after miR-140-5p mimic or miR-140-5p inhibitor transfection, respectively, as well as the changed expression levels of Nrf2 and Sirt2. Furthermore, DOX- induced myocardial oxidative damage was worsened in mice treated with miR-140-5p agomir, and however the injury was alleviated in the mice administrated with miR-140-5p antagomir. Therefore, miR-140-5p plays an important role in DOX-induced cardiotoxicity by promoting myocardial oxidative stress via targeting Nrf2 and Sirt2. Our data provide novel insights for investigating DOX-induced heart injury. In addition, miR-140-5p/ Nrf2 and miR-140-5p/Sirt2 may be the new targets to treat DOX-induced cardiotoxicity.
Abbreviations: ARE, antioxidant response element; CK, creatine kinase; DAPI, 4′,6′- Diamidino-2-phenylindole; DOX, doxorubicin; ECG, electrocardiograms; FOXO3a, Forkhead box O3; GCLM, glutamate-cysteine ligase modifier subunit; GSH, glutathione; GSH-Px, glutathione peroxidase; GST, glutathione-S- transferase; H&E, hematoxylin-eosin; HO-1, heme oxygenase-1; Keap1, kelch like ECH- associated protein 1; LDH, lactate dehydrogenase; MDA, malondialdehyde; MTT, 3-(4,5-Dimethylthiazol-2-yl)-2,5 -diphenyl tetrazolium bromide; ncRNA, non-coding RNAs; NQO1, NAD(P)H Quinone Dehydrogenase 1; Nrf2, nuclear erythroid factor 2-related factor 2; PMSF, phenylmethanesulfonyl fluoride; ROS, reactive oxygen species; RT-PCR, reverse transcripttion polymerase chain reaction; Sirt2, silent information regulator factor 2-related enzyme 2; SDS, sodium dodecyl sulfate; SOD, superoxide dismutase; Tris, hydroxymethyl aminomethane
Keywords: Cardiotoxicity, Doxorubicin, miR-140-5p, Nrf2, Sirt2, Oxidative damage
Graphical abstract
Highlights
-
•
DOX significantly increased the levels of miR-140-5p in vitro and in vivo.
-
•
MiR-140-5p aggravated DOX-induced cardiotoxicity.
-
•
MiR-140-5p promoted myocardial oxidative stress.
-
•
MiR-140-5p directly targeted Nrf2 and Sirt2.
-
•
MiR-140-5p may be the new target of DOX-induced cardiotoxicity.
1. Introduction
Doxorubicin (DOX), a kind of anthracycline antibiotics, is widely used in clinical to treat some kinds of tumors because of its high efficiency and wide-spectrum [1]. However, DOX can produce some serious side effects on body after long-term use, especially to cardiotoxicity [2], [3], [4], [5]. DOX can lead to left ventricular dysfunction, or even heart failure, and DOX-induced heart injury is closely related to the accumulation in the body [6]. In addition, the risk of DOX-induced cardiotoxicity will be exacerbated in patients as the cumulative dose of DOX is increased. When the accumulation dose of DOX is increased from 400 mg/m2 to 700 mg/m2, the incidence of heart failure will rise from 5% to 48% [7], [8], [9]. Once the heart failure occurs, the patients' mortality s will be significantly increased. Thus, DOX-caused cardiotoxicity limits its clinical application.
The pathogenesis and prevention of DOX-induced cardiotoxicity has become a hotspot in recent years. Many researches have revealed that DOX-induced myocardial injury may be related to oxidative stress, calcium overload, mitochondrial damage, cardiomyocyte apoptosis and autophagy [10], [11], [12], [13], [14], [15], [16], [17]. However, oxidative stress attracts the most attention during DOX-induced myocardial damage. Studies have shown that the semi-quinone DOX, resulting from metabolic process of DOX, can quickly transfer its unpaired electrons to the molecular oxygen, then produce a large number of superoxide anion free radicals (O2-), which can lead to cell damage, mitochondrial dysfunction and DNA injury. In addition, DOX can also combine with iron to destroy the iron homeostasis, and then produce a large number of reactive oxygen species (ROS) [18], [19], [20]. Besides, the interaction of ROS with phosphorlipid on the mitochondrial membrane of cells can also lead to mitochondrial dysfunction, and further affect the energy metabolism [21]. Thus, oxidative stress is one potential mechanism of DOX- induced heart damage, and inhibiting oxidative stress may be one effective prevention and treatment method against cardiotoxicity caused by DOX.
MicroRNA (miRNA), belonging to the small non-coding RNAs (ncRNAs), can regulate gene expression through binding with target mRNA [22]. There have growing evidences that miRNAs play key roles in the development of various diseases, suggesting that miRNAs can be considered as the potential drug targets to treat human diseases [23], [24], [25], [26]. Some reports have shown that the aberrant expression of miR-499 can lead to irreversible damage of heart via causing arrhythmia and myocardial hypertrophy, indicating that miR-499 may be a potential target for the treatment of heart diseases [27]. In addition, the works of DOX- induced cardiotoxicity have shown that miR-532-3p and miR-208a play important roles in heart diseases [28], [29].
Microarrays technology is an effective method for high-throughput screening miRNAs from biological samples, which has been widely used in disease diagnosis, target selection and new drug development. Lu Y et al. assessed the potential role of miRNAs in regulating experimental atrial fibrillation using microarray technology, and found that miR-328 is a potential therapeutic target for the disease [30]. Therefore, microarrays technology is of great importance to pick out differentially expressed miRNAs for deeply investigating molecular mechanisms and drug discovery.
In this study, the differentially expressed miRNAs in DOX-induced heart tissue of rats were tested by using microarrays technology, and then the biological functions of the aimed miRNAs on DOX-induced heart damage were examined, which will provide potential mechanisms and new drug targets for the treatment of DOX-induced cardiotoxicity.
2. Materials and methods
2.1. Chemicals and materials
DOX was purchased from Sigma (Santa Clara, CA, USA). Tissue Protein Extraction Kit was obtained from KEYGEN Biotech. Co., Ltd. (Nanjing, China). The bicinchoninic acid (BCA) protein assay kit, ROS assay kit, cell lysis buffer and phenylmethanesulfonyl fluoride (PMSF) were obtained from Beyotime Institute of Biotechnology (Jiangsu, China). Creatine kinase (CK), lactate dehydrogenase (LDH), malondialdehyde (MDA), superoxide dismutase (SOD), glutathione (GSH) and glutathione peroxidase (GSH-Px) detection kits were purchased from Nanjing Jiancheng Institute of Biotechnology (Nanjing, China). Sodium dodecyl sulfate (SDS), hydroxymethyl aminomethane (Tris) and 4′,6′-Diamidino-2-phenylindole (DAPI) were purchased from Sigma (St. Louis, MO, USA). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) was provided by Roche Diagnostics (Basel, Switzerland). Lipofectamin2000, TransZolTM, TransScript® All-in-One First-Strand cDNA Synthesis SuperMix for qPCR (One-Step gDNA Removal), TransStart® Top Green qPCR SuperMix were purchased from Beijing TransGen Biotech Co., Ltd. (Beijing, China). SanPrep Column MicroRNA Mini-Preps Kit, MicroRNA First Strand cDNA Synthesis Kit and MicroRNAs Quantitation PCR Kit were purchased from Sangon Biological Engineering Technology & Services Co., Ltd. (Shanghai, China).
2.2. Cell culture
The H9C2 cells were purchased from the Shanghai Institutes for Biological Sciences (Shanghai, China), and the cells were maintained in DMEM supplemented with 10% FBS and antibiotics (100 IU/mL penicillin and 100 mg/mL streptomycin) in a humidified atmosphere of 5% CO2 and 95% O2 at 37 °C.
2.3. DOX-induced cell injury
DOX was prepared to make a series of working dilutions in serum-free DMEM. The H9C2 cells were plated in 96-well plates at a density of 5 × 104 cells/mL per well for 24 h before challenged with various concentrations of DOX (0–10 μM). After incubation with DOX for 24 h, 10 μL of MTT (5 mg/mL) solution was added to each well. After incubation at 37 °C for 4 h, the medium with MTT was removed. Next, 150 μL of DMSO was added to each well to dissolve the formazan crystals, and the absorbance at 490 nm was measured using a POLARstar OPTIMA multi-detection microplate reader (BioRad, San Diego, CA, USA).
2.4. Measurement of intracellular ROS
The H9C2 cells were plated in 6-well culture plates at a density of 5 × 104 cells/ mL and then treated with DOX (5 μM) for 24 h. After removing the medium, 1.5 mL of DCFH-DA (10.0 μM) was added at 37 °C for 25 min, and then the samples were analyzed by fluorescence microscopy (Olympus, Tokyo, Japan) at 200× magnification.
2.5. Animals and ethical approval
Male SD rats weighing 200 ± 20 g and Male C57BL/6 J mice weighing 20 ± 2 g were obtained from the Experimental Animal Center at Dalian Medical University (Dalian, China) (SCXK: 2013-0003). All experimental procedures were performed in strict accordance with PR China Legislation Regarding the Use and Care of Laboratory Animals, and all experiments involving animals were approved by the Animal Care and Use Committee of Dalian Medical University (Approval number: 20150658). The animals were group-housed with 2–3 rats or mouse per cage on a 12 h light/dark cycle in a temperature-controlled (25 ± 2 °C) room with free access to water and food, and were allowed one week to acclimatize before experiment. Randomization was used to assign samples to the experimental groups and to collect and process data. The experiments were performed by investigators blinded to the groups for which each animal was assigned. Animal studies have been reported in compliance with the ARRIVE guidelines [31], [32].
2.6. DOX-induced myocardial damage injury in vivo
Twenty rats and twenty mice were all randomly divided into two groups (n = 10): control group (0.9% saline) and DOX group. The animals in DOX groups were intraperitoneally injected with DOX (15 mg/kg diluted with 0.9% saline for eight days [33], [34]), whereas the animals in control groups were intraperitoneally injected with equal volumes of 0.9% saline. After that, the electrocardiograms (ECG) of rats and mice were detected, then all animals were sacrificed and the blood samples were harvested. The serum samples were obtained by centrifugation (3000 r/min, 4 °C) for 10 min, and the heart samples were promptly removed. Part of heart tissue was fixed and the remaining samples were stored at −80 °C for further assay.
2.7. Measurement of electrocardiograms in rats and mice
According to the previous method [35], the animals were anaesthetized and fixed on the table with the supine position. Subcutaneous needle electrodes of the commercial computer-based ECG device (BL-420F Biological Function Experiment System, Chengdu Thai Union Technology Co, Ltd., China) were connected to the animals, and electrocardiograms were recorded.
2.8. Measurement of CK, LDH, MDA, SOD, GSH and GSH-Px levels
The serum levels of CK and LDH were detected using the commercial kits according to the instructions. The heart tissues were placed in cold saline (1: 10, w/v), homogenized with a homogenizer machine, and then centrifuged at 3000r/min for 15 min to produce the supernatant for detecting the levels of MDA, SOD, GSH and GSH-Px using the commercial kits according to the respective instructions.
2.9. Histopathologic assay
The heart tissues were fixed in 10% formalin and embedded in paraffin, and the 5-μm -thick sections were stained with hematoxylin-eosin (H&E). Images of the stained sections were obtained using a light microscope (Nikon Eclipse TE2000-U, Japan) with 200× magnification.
2.10. miRNA microarray assay
The total RNA samples from DOX group and control group of rats (n = 5) were isolated using TRIzol reagent (Invitrogen, USA) and purified using RNeasy mini kit (Qiagen, Germany) according to the manufacturer's instructions. RNA quality and quantity were measured using a Nanodrop spectrophotometer (ND-1000, Nanodrop Technologies, Rockland, DE, USA), and RNA integrity was assessed by gel electrophoresis. RNA labeling and array hybridization were carried out according to Exiqon's manual. After quality control, the miRCURY™ Hy3™/Hy5™ Power labeling kit (Exiqon, Vedbaek, Denmark) was used for miRNA labeling. After stopping the labeling procedure, the Hy3™-labeled samples were hybridized using the miRCURYTM LNA Array (v.18.0) (Exiqon, Vedbaek, Denmark) according to the array manual. Finally, the slides were scanned using an Axon GenePix 4000B microarray scanner (Axon Instruments, Foster City, CA). Scanned images were then imported into GenePix Pro 6.0 software (Axon) for grid alignment and data extraction. Replicated miRNAs were averaged and miRNAs that intensities ≥ 30 in all samples were chosen for calculating normalization factor. Expressed data were normalized using the Median normalization. After normalization, significant differentially expressed miRNAs between two groups were identified through Fold change and P-value. Differentially expressed miRNAs between two samples were filtered through Fold change. Finally, hierarchical clustering was performed to show distinguishable miRNA expression profiling among samples.
2.11. Validation of the differentially expressed miRNAs
The total miRNA samples were extracted from H9C2 cells or heart tissues using SanPrep Column MicroRNA Mini-Preps Kit following the manufacturer's protocol. The primers of miRNAs are given in Supplementary Table S1. The purity of the extracted miRNA was determined, and the reverse transcripttion polymerase chain reaction (RT-PCR) was carried out using a microRNA First Strand cDNA Synthesis Kit following the manufacturer's instructions with a TC-512 PCR system (TECHNE, UK). Quantitative real-time PCR assay was performed with a MicroRNAs Quantitation PCR Kit and ABI 7500 Real Time PCR System (Applied Biosystems, USA). For the data of each sample, the Ct value of the target gene was normalized to that of U6 (Sangon Biological Engineering Technology & Services Co., Ltd., China). The unknown template was calculated using the standard curve for quantitative analysis. Eventually, the expression level of each gene was performed normalization for controlling unwanted sources of variation.
2.12. Prediction of target genes
The target genes of the differentially expressed miRNAs validated above were screened by the “mirbase”, “mirdb” and “miranda” databases, and each target gene must be searched in at least two databases. Ultimately, we obtained the potential target gene tree.
2.13. Dual-luciferase reporter assay
The plasmids containing the wild-type miR-140-5p-Nrf2 (wtLuc-Nrf2) and miR- 140-5p- Sirt2 (wtLuc-Sirt2) response element and the corresponding mutant (mut-Luc-Nrf2 and mut- Luc-Sirt2) were purchased from RiboBio. Co., Ltd. (Guangzhou, China). Plasmid DNA (wt-Luc- Nrf2, wtLuc-Sirt2, mut-Luc-Nrf2 or mut-Luc-Sirt2) and miR-140-5p mimic or miR-140-5p negative controls were co-transfected into H9C2 cells. When appropriate, the cells were incubated with DOX (5 μM) or without DOX for 24 h after transfection. The luciferase activity was assessed with a Double-Luciferase Reporter Assay Kit, purchased from Promega Biotech Co., Ltd. (Beijing, China), using the Dual-Light Chemiluminescent Reporter Gene Assay System (Berthold, Germany), which was normalized to firefly luciferase activity.
2.14. Immunofluorescence assay
Immunofluorescence staining for Sirt2 in tissue sections or cells was performed using anti-Sirt2 antibody in a humidified box at 4 °C overnight, and followed by incubation with an Alexa fluorescein-labeled secondary antibody for 1 h at 37 °C. The cell nuclei were stained with DAPI (5.0 μg/mL). Immunostained samples were imaged by fluorescence microscopy (Olympus, Japan) at 200× magnification.
2.15. Quantitative real-time PCR assay
The total RNA samples were obtained from H9C2 cells and heart tissues of rats and mice using TransZol according to the manufacturer's protocol. Each RNA sample was reverse transcribed into cDNA using the kit of TransStart Top Green qPCR SuperMix. The forward (F) and reverse (R) primers of RNA are given in Supplementary Table S2. The CT value of genes among the data from each sample was normalized to GAPDH.
2.16. Western blotting assay
The total protein samples from the cells and heart tissues were homogenized using RIPA lysis buffer containing protease and phosphatase inhibitors. The protein concentrations of the samples were determined using a BCA Protein Assay Kit (Bio-Rad, Hercules, CA, USA). After determination of the contents, the proteins were separated by SDS-PAGE (8–12%), and then transferred to PVDF membranes (Millipore, Massachusetts, USA). After being blocked with 5% skim milk for 3 h at room temperature, the membranes were incubated with primary antibodies (listed in Supplementary Table S3) overnight at 4 °C. After addition of the anti-rabbit or anti-mouse secondary antibody for 2 h at room temperature, the protein bands on the membranes were detected using an enhanced chemiluminescence system and a Bio-Spectrum Gel Imaging System, respectively (UVP, California, USA). Intensity values of the relative protein levels were normalized to GAPDH.
2.17. MiR-140-5p mimic and inhibitor transfection tests in vitro
Transfection was performed to up-regulate or down-regulate the expression levels of miR-140-5p. Briefly, the miR-140-5p mimic, mimic negative control, miR-140-5p inhibitor or inhibitor negative control was separately dissolved in Opti- MEM. The solutions were then equilibrated for 5 min at room temperature. Each solution was combined with Lipofectamine 2000 transfection reagent according to the manufacturer's protocol. Then, the solution was mixed gently and allowed to form inhibitor liposomes for 20 min. The H9C2 cells were transfected with the transfection mixture in serum-free cell medium. The cell medium was replaced by fresh Medium after incubated at 37 °C for 6 h. Then, the expression levels of nuclear erythroid factor 2-related factor 2 (Nrf2), kelch like ECH-associated protein 1 (Keap1), heme oxygenase-1 (HO-1), NAD(P)H Quinone Dehydrogenase 1 (NQO1), silent information regulator factor 2-related enzyme 2 (Gst), glutamate-cysteine ligase modifier subunit (GCLM), silent information regulator factor 2-related enzyme 2 (Sirt2) and Forkhead box O3 (FOXO3a) were detected after 24 h of administration with DOX (5 μM) or without DOX.
2.18. Agomir and antagomir transfection of miR-140-5p in mice
To determine the functional role of miR-140-5p in DOX-induced cardiotoxicity, twenty- four mice were randomly divided into four groups (n = 6): agomir NC group, antagomir NC group, agomir (miR-140-5p agonist) group and antagomir (miR-140-5p inhibitor) group. miR-140-5p agomir and antagomir were synthesized by Ribobio Co. (Guangzhou, China). The animals in agomir group and its negative control group were injected via tail vein with miR-140-5p agomir (1 nmol/g/day) or same dosage of agomir NC for 3 consecutive days (on day1 to day 3). The mice in antagomir group and its negative control group were injected via tail vein with miR-140-5p antagomir (5 nmol/g/day) or same dosage of antagomir NC for 4 days (on day0, day3, day7 and day 11). On day 4, the animals in agomir and antagomir groups were intraperitoneally injected with DOX (15 mg/kg), whereas all remained mice were injected with equal volumes of 0.9% saline. On day 11, the ECGs of the animals were detected at first, and then all of them were sacrificed. The blood samples were collected and the serum samples were obtained by centrifugation (3000 r/min, 4 °C) for 10 min and stored at −20 °C until assay. After the animals were sacrificed, the heart samples were promptly removed. Part of heart was fixed and the remaining tissues were stored at −80 °C for further assay.
2.19. Data analysis
The data are expressed as the mean ± standard deviation (SD). Statistical analysis was performed with GraphPad Prism 5.0 software (San Diego, CA, USA). It was performed with one-way analysis of variance (ANOVA) followed by Tukey's post-hoc test when comparing multiple groups; with unpaired t-test when comparing two different groups. Statistical significance was considered to be P < 0.05 or P < 0.01. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology [36].
3. Results
3.1. DOX causes myocardial injury in vitro and in vivo
As shown in Fig. 1A, the viability of H9C2 cells treated with different concentrations of DOX for 24 h was significantly inhibited compared with control group. As shown in Fig. 1B, the numbers of H9C2 cells administrated with 5 μM of DOX for 24 h were significantly reduced as well as the remarkable changed morphology compared with control group. Based on above data, 5 μM of DOX for 24 h treatment was optimized for subsequent experiments. As shown in Fig. 1C-D, the levels of serum CK and LDH in rats and mice in DOX groups were obviously increased compared with control groups. As shown in Fig. 1E-F, the myocardial cells in heart tissues in control groups were arranged neatly, with no bleeding, edema and other abnormalities. However, the animals in DOX-treated groups showed myocardial cell damage including obvious myocardial tissue texture unclear, pyknosis and plasma dissolve. In addition, the most obvious abnormalities in ECG of DOX-treated groups were the changes in ST-segment compared with control groups (shown in Fig. 1G-H).
Fig. 1.

DOX causes myocardial injuryin vitroandin vivo. (A) The viability of H9C2 cells treated by DOX. (B) The morphology of H9C2 cells treated by DOX. (C) Serum levels of CK and LDH in rats caused by DOX. (D) Serum levels of CK and LDH in mice caused by DOX. (E) HE staining images of heart tissues in rats treated by DOX (×200 magnification). (F) HE staining images of heart tissues in mice treated by DOX (×200 magnification). (G) Electrocardiograms of rats treated by DOX. (H) Electrocardiograms of mice treated by DOX. All data are expressed as the mean ± SD (n = 5 for in vitro test and n = 10 for in vivo test). *p < 0.05, **p < 0.01 compared with control group.
3.2. DOX aggravates oxidative damage in vitro and in vivo
The intracellular ROS level in H9C2 cells in control group was low, which was markedly increased by 5 μM of DOX for 24 h (Fig. 2A). In addition, as shown in Fig. 2B-C, the levels of SOD, GSH and GSH-Px in heart tissues of DOX-treated rats and mice were markedly decreased, and MDA levels were significantly increased compared with control groups. These results indicated that DOX aggravated oxidative damage in vitro and in vivo.
Fig. 2.

DOX aggravates oxidative damagein vitroandin vivo. (A) Intracellular ROS level in H9C2 cells treated by DOX. (B) The levels of MDA, SOD, GSH and GSH-PX in heart tissues of rats caused by DOX. (C) The levels of MDA, SOD, GSH and GSH-PX in heart tissues of mice caused by DOX. All data are expressed as the mean ± SD (n = 10). **p < 0.01 compared with control group.
3.3. Differentially expressed miRNAs in heart tissues of rats induced by DOX
A total of 18 differentially expressed miRNAs with at least 1.5-fold changes and P-values less than 0.05 compared with control group were identified in heart tissue of rats caused by DOX using microRNA microarray analysis (Supplementary Table S4). As shown in Fig. 3A, the heat map indicated the results of a two-way hierarchical clustering of the samples and the 18 differentially expressed miRNAs, which displayed the relative expression levels identified by microarray assay. Then, six up-regulated and two down-regulated miRNAs were further validated by real-time PCR assay. As shown in Fig. 3B, DOX significantly increased the levels of miR-208b-3p, miR-34a-5p, miR-499-5p, miR-140-5p and miR-29a-5p, and markedly decreased the levels of miR-142-5p and miR-142-3p in heart tissues of rats compared with control group. The interaction networks between the differentially expressed miRNAs and their target genes were predicted by Mirbase (http://www.mirbase.org/), Mirdb (http://www.mirdb.org/) and MiRanda (http://www.microrna.org/microrna/home.do). The results in Fig. 3C-D revealed that miR-140-5p modulated by DOX can regulate multiple target genes. These findings prompted us to concentrate on exploring the role of miR-140-5p in DOX-induced myocardial injury.
Fig. 3.

Differentially expressed miRNAs in heart tissues of rats induced by DOX based on microarrays assay. (A) Hierarchical clustering analysis of the differentially expressed miRNAs between DOX and control samples in rats. (B) Validation of the eight differentially expressed miRNAs in rats caused by DOX based on real-time PCR assay. (C) A schematic diagram used to search the target genes of miRNAs in three databases. (D) Prediction of the target genes of miR-140-5p screened by the database. All data are expressed as the mean ± SD (n = 5). *p < 0.05, **p < 0.01 compared with control group.
3.4. MiR-140-5p targets with Nrf2 and Sirt2
MiR-140-5p, which plays a critical role in cardiovascular disease [37], [38], [39], was selected for investigating the possible mechanisms of DOX-induced heart injury. Based on the interaction network illustrated in Fig. 3D, we found that Nrf2 and Sirt2 should be the targets gene of miR-140-5p. As shown in Fig. 4A, the RNA sequence alignment showed that the 3′-UTR of Nrf2 and Sirt2 mRNA contained a complementary site for the seed region of miR-140-5p. As shown in Fig. 4B-C, the dual-luciferase reporter plasmid was obtained to perform the dual-luciferase reporter assay. In the groups of Nrf2-Wt and Sirt2-Wt, the luciferase activities were significantly repressed by miR-140-5p overexpression compared with mimic control group. However, these effects were not observed with the mutated Nrf2 and Sirt2 groups, suggesting that Nrf2 and Sirt2 are the target genes of miR-140-5p.
Fig. 4.

MiR-140-5p targets Nrf2 and Sirt2. (A) The diagram of miR-140-5p conservative seed binding sites on the 3′-UTRs of the target genes (Nrf2 and Sirt2). (B) The construction diagram of the target genes (Nrf2 and Sirt2) double-luciferase reporter genes (the mutant sequences used in the double luciferase reporter genes are in red). (C) The relative luciferase expression with Nrf2 or Sirt2 3′-UTR after co transfection with miR-140-5p mimic or NC in H9C2 cells. All data are expressed as the mean ± SD (n = 5). **p < 0.01 compared with N control group; ns, no significance.
3.5. DOX up-regulates the levels of miR-140-5p in vitro and in vivo
As shown in Fig. 5A, the results of real-time PCR assay indicated that DOX significantly up-regulated the expression levels of miR-140-5p by 2.09-fold in H9C2 cells, 2.15 -fold in heart tissue of rats, and 1.66-fold in heart tissue of mice, respectively, compared with control groups. These results were consistent with the data from microRNA microarray assay.
Fig. 5.

DOX up-regulates the levels of miR-140-5p and affects Nrf2 and Sirt2 signaling pathwaysin vitroandin vivo. (A) The expression levels of miR-140-5p in H9C2 cells, rats and mice treated by DOX based on real-time PCR assay. (B) The expression levels of Sirt2 in H9C2 cells, rats and mice treated by DOX based on immunofluorescence staining assay (×200 magnification). (C) The expression levels of Nrf2, keap1, HO-1, NQO1, Gst, GCLM, Sirt2 and FOXO3a in H9C2 cells, rats and mice treated by DOX based on western blotting assay. All data are expressed as the mean ± SD (n = 5). **p < 0.01 compared with control group.
3.6. DOX affects Nrf2 and Sirt2 signaling pathways in vitro and in vivo
As shown in Fig. 5B, the expression levels of Sirt2 were clearly suppressed in DOX groups based on immunofluorescence assays compared with control groups in vitro and in vivo. Then, the expression levels of Nrf2, Sirt2 and some downstream signaling molecules were assessed. As shown in Fig. 5C and Supplementary Figs. S1–3, the levels of Nrf2, HO-1, NQO1, Gst, GCLM, Sirt2 and FOXO3a were markedly decreased, and the levels of Keap1 were significantly increased by DOX compared with control groups in vitro and in vivo. In addition, our results also showed that DOX significantly decreased the mRNA levels of Nrf2, HO-1 and NQO1 compared with control groups (Supplementary Figs. S4).
3.7. MiR-140-5p overexpression aggravates DOX-induced oxidative damage in vitro
To investigate the role of miR-140-5p in DOX -induced cardiotoxicity, miR-140-5p mimic was transfected to H9C2 cells. As shown in Fig. 6A, miR-140-5p mimic significantly increased the miR-140-5p levels in H9C2 cells. As shown in Fig. 6B, transfection with miR- 140-5p mimic markedly aggravated H9C2 cells morphological damage compared with mimic negative control group. As shown in Fig. 6C, the intracellular ROS levels in model group transfected with miR-140-5p mimic was significantly higher than the un-transfected group. In addition, as shown in Fig. 6D-E and Supplementary Fig. S5, the levels of Nrf2, HO-1, NQO1, Gst, GCLM, Sirt2 and FOXO3a in H9C2 cells were notably decreased and the levels of Keap1 were significantly increased after treating with miR-140-5p mimic compared with negative control group, and the changes were more significant than that the un-transfected group, suggesting that miR-140-5p affected Nrf2 and Sirt2 signaling pathways. These results suggested that overexpression of miR-140-5p aggravated oxidative damage induced by DOX in cardiomyocytes.
Fig. 6.

MiR-140-5p overexpression aggravates DOX-induced oxidative damagein vitro. (A) MiR-140-5p level in DOX-treated H9C2 cells with transfection of miR-140-5p mimic. (B) The morphology of DOX-treated H9C2 cells with transfection of miR-140-5p mimic. (C) ROS level in DOX-treated H9C2 cells with transfection of miR-140-5p mimic. (D) The expression level of Sirt 2 in DOX-treated H9C2 cells with transfection of miR-140-5p mimic. (E) The expression levels of Nrf2, keap1, HO-1, NQO1, Gst, GCLM, Sirt2 and FOXO3a in DOX-treated H9C2 cells with transfection of miR-140-5p mimic. All data are expressed as the mean ± SD (n = 5). *p < 0.05, miR-140-5p mimic + DOX group compared with DOX group.
3.8. MiR-140-5p inhibition alleviates DOX-induced myocardial oxidative damage in vitro
MiR-140-5p inhibitor was transfected to H9C2 cells to further investigate the role of miR-140-5p on DOX-induced cardiotoxicity. As shown in Fig. 7A, the levels of miR-140-5p in H9C2 cells were significantly decreased after administrated with miR-140-5p inhibitor. As shown in Fig. 7B, transfection with miR-140-5p inhibitor markedly alleviated H9C2 cells morphological damage compared with negative control group. As shown in Fig. 7C, the intracellular ROS level in DOX group transfected with miR-140-5p inhibitor was significantly lower than the un-transfected group. In addition, as shown in Fig. 7D-E and Supplementary Fig. S6, the levels of Nrf2, HO-1, NQO1, Gst, GCLM, Sirt2 and FOXO3a in H9C2 cells were notably increased, and the levels of Keap1 were significantly decreased after treating with miR-140-5p inhibitor compared with DOX group, suggesting that miR-140-5p inhibition alleviated myocardial oxidative damage induced by DOX.
Fig. 7.

MiR-140-5p inhibition alleviates DOX-induced myocardial oxidative damagein vitro. (A) MiR-140-5p level in DOX-treated H9C2 cells with transfection of miR-140-5p inhibitor. (B) The morphology of DOX-treated H9C2 cells with transfection of miR-140-5p inhibitor. (C) ROS level in DOX-treated H9C2 cells with transfection of miR-140-5p inhibitor. (D) The expression level of Sirt 2 in DOX-treated H9C2 cells with transfection of miR-140-5p inhibitor. (E) The expression levels of Nrf2, keap1, HO-1, NQO1, Gst, GCLM, Sirt2 and FOXO3a in DOX-treated H9C2 cells with transfection of miR-140-5p inhibitor. All data are expressed as the mean ± SD (n = 5). *p < 0.05 miR-140-5p mimic + inhibitor group compared with DOX group.
3.9. MiR-140-5p promotes DOX-induced heart damage in vivo
To further determine whether increased miR-140-5p level can cause increased heart damage induced by DOX, we intraperitoneally administered DOX or saline (as control) into mice treated with miR-140-5p agomir. We found that the levels of CK and LDH in agomir group were obviously increased compared with agomir NC group (shown in Fig. 8A). The levels of CK and LDH in antagomir group were increased compared with antagomir NC group, but lower than those of in overexpression group (Fig. 8A). In addition, as shown in Fig. 8B, SOD level in agomir group was markedly decreased, and SOD level in antagomir group was as the same as antagomir NC group. As shown in Fig. 8C-D, administered with miR-140-5p agomir markedly aggravated abnormal histopathology of myocardium and EEG recording induced by DOX. However, miR-140-5p antagomir markedly alleviated abnormal histopathology of myocardium and EEG recording induced by DOX. These results indicated that overexpression of miR-140-5p aggravated oxidative damage, and miR-140-5p inhibition alleviated the oxidative damage of myocardium induced by DOX in vivo.
Fig. 8.

MiR-140-5p agomir or antagomir affects DOX-induced heart damagein vivo. (A) The serum levels of CK and LDH in DOX-treated mice after administered with agomir or antagomir of miR-140-5p. (B) The serum level of SOD in DOX-treated mice after administered with agomir or antagomir of miR-140-5p. (C) HE staining images of heart tissues in DOX- treated mice after administered with agomir or antagomir of miR-140-5p. (D) Electrocardiograms of DOX-treated mice after administered with agomir or antagomir of miR-140-5p. All data are expressed as the mean ± SD (n = 5). **p < 0.01 compared with agomir NC group; #p < 0.05 compared with antagomir NC group.
3.10. MiR140-5p adjusts Nrf2 and Sirt2 signaling pathways in vivo
As shown in Fig. 9A, the expression level of miR-140-5p was clearly increased in miR-140-5p agomir group, and obviously suppressed in miR-140-5p antagomir group. The results shown in Fig. 9B-C and Supplementary Fig. S7 indicated that miR-140-5p overexpression markedly decreased the protein levels of Nrf2, HO-1, NQO1, Gst, GCLM, Sirt2 and FOXO3a, and significantly increased the protein level of Keap1. However, the same results were not observed in miR-140-5p inhibition group.
Fig. 9.

MiR140-5p agomir or antagomir adjusts Nrf2 and Sirt2 signaling pathwaysin vivo. (A) The levels of miR-140-5p in DOX-treated mice after administered with agomir or antagomir of miR-140-5p. (B) The expression level of Sirt2 in DOX-treated mice after administered with agomir or antagomir of miR-140-5p. (C) The expression levels of Nrf2, keap1, HO-1, NQO1, Gst, GCLM, Sirt2 and FOXO3a in DOX-treated mice after administered with agomir or antagomir of miR-140-5p. All data are expressed as the mean ± SD (n = 5). **p < 0.01 compared with agomir NC group; ##p < 0.05 compared with antagomir NC group.
4. Discussion
Anticancer therapy can usually demolish the physiological homeostasis and affect multiple organ damages during treatment process. DOX, one of anthracyclines, has attracted much attention not only in its anti-tumor therapeutic effects with broad spectrum, but also in its serious side effects after long-term use [40], [41]. The clinical application of DOX is limited because of its toxicity to various organs especially to heart, which can affect the levels of CK, LDH, and aggravate ECG abnormality and histopathological changes of heart.
Many scientists have made efforts to reduce DOX-induced cardiotoxicity. However, encouraging progress has been hampered by the complex pathological mechanism of the damage caused by DOX. Some evidences have suggested that oxidative stress plays a critical role in the pathogenesis of DOX-induced myocardial damage [42], [43], [44]. In addition, some biological pointers have been verified to prompt oxidative stress. Among them, MDA, an indicator of ROS, is an end-product of lipid hydroperoxide. SOD is an enzyme that catalytically reduces O2- to hydrogen peroxide. GSH and GSH-Px can catalyze the reduction of hydrogen peroxide and other peroxides. In this paper, we found that DOX caused oxidative damage of myocardium as evidenced by the high levels of ROS in vitro, high levels of MDA and low levels of SOD, GSH, GSH-Px in vivo. Therefore, oxidative stress may be a key factor that can aggravate DOX - induced myocardial insult.
A large number of previous studies have proved that multiple molecular mechanisms and signaling pathways can regulate oxidative stress. The growing evidences have confirmed that miRNAs can be considered as the potential drug targets to treat human diseases [23], [24], [25], [26]. In the present study, total of 18 differentially expressed microRNAs in heart tissue of rat caused by DOX were screened out using microRNA microarray assay, in which the levels of miR- 140-5p were aberrantly up-regulated in H9C2 cells, and the heart tissues of rats and mice with the cardiotoxicity induced by DOX, as well as the elevated CK and LDH levels, histopathological changes and ECG abnormality. MiR-140-5p has also been confirmed to play fundamental roles in cardiovascular disease [37], [38], [39]. However, there have no papers to report the effects of miR-140-5p on DOX-induced cardiotoxicity, and thus miR-140-5p may be considered as one potential drug target to treat DOX-induced cardiotoxicity.
How did miR-140-5p promote DOX-induced cardiotoxicity? Based on the database and double-luciferase reporter assay, the results showed that miR-140-5p can directly target Nrf2 and Sirt2, which were the target genes of it. Thus, miR-140-5p may be through adjusting Nrf2 and Sirt2 signal pathways to aggravate DOX-induced myocardial injury.
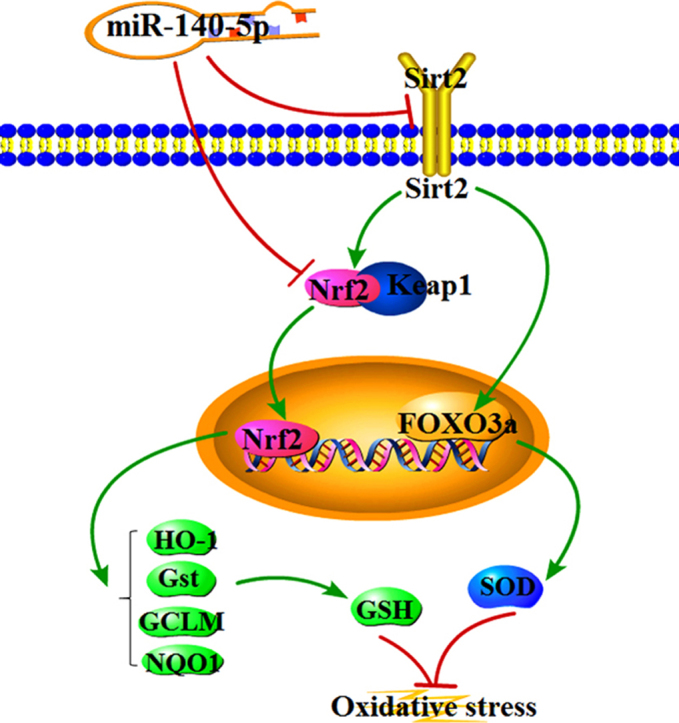
Nrf2, belongs to the transcription factor CNC family, can negatively regulate the polymerization or dissociation with Keap1, thereby control the expressions of various antioxidative genes and enzymes against oxidative stress [45], [46]. Briefly, Nrf2, an important member of the cap ‘n’ collar family of basic leucine zipper transcription factors, plays a part in the anti-oxidative defense system by upregulating the expression levels of anti-oxidant enzymes [47]. After activation, Nrf2 translocates to the nuclear area and binds with antioxidant response element (ARE) which is located in the promoter region of genes encoding the phase II antioxidant enzymes including such as HO-1 and Gst [48]. Sirt2, a member of deacetylation enzymes, can control oxidative stress through activating FOXO3a, up-regulating MnSOD, and then decreasing ROS levels [49]. In the present work, we found that miR-140-5p significantly decreased the expression levels of Nrf2 and Sirt2, then affected the expression levels of HO-1, NQO1, Gst, GCLM, Keap1 and FOXO3a, and thereby increased DOX-caused myocardial oxidative damage. In addition, the levels of intracellular ROS were significantly increased or decreased in H9C2 cells administrated with DOX after miR-140-5p mimic or miR-140-5p inhibitor transfection, as well as the changed expression levels of Nrf2 and Sirt2 signals. Furthermore, DOX-induced myocardial oxidative damage became more serious in mice treated with miR-140-5p agomir. The expression levels of the proteins associated Nrf2 and Sirt2 signal pathways were also controlled as well as the elevated CK and LDH levels, histopathological changes and ECG abnormality. However, the injury was alleviated in mice administrated with miR-140-5p antagomir. Thus, our data demonstrated that miR-140-5p directly targeted Nrf2 and Sirt2, and affected Nrf2 and Sirt2 signaling pathways to increase DOX- induced myocardial oxidative damage (Supplementary Fig. S8).
Therefore, our study provided novel insight into the pathological mechanisms of DOX- induced cardiotoxicity, and miR-140-5p/Nrf2 and miR-140-5p/Sirt2 may be considered as the new targets for the treatment of DOX-induced cardiotoxicity. Although our study confirmed miR- 140-5p as an important factor in controlling DOX-induced myocardial injury, other target genes of miR-140-5p or other differentially expressed miRNAs found in this work are also necessary to be carefully tested for DOX-induced cardiotoxicity.
Acknowledgments
This work was financially supported by the Project of Liaoning BaiQianWan Talents Program (2015-65) and Special Grant for Translational Medicine, Dalian Medical University (2015004).
Acknowledgments
Competing interests
The authors declare no competing financial interests.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2017.12.013.
Appendix A. Supplementary material
Supplementary material.
References
- 1.DeSantis C.E., Siegel R.L., Sauer A.G., Miller K.D., Fedewa S.A., Alcaraz K.I. Cancer statistics for african americans, 2016: progress and opportunities in reducing racial disparities. Cancer J. Clin. 2016;66:290–308. doi: 10.3322/caac.21340. [DOI] [PubMed] [Google Scholar]
- 2.Chung W.B., Youn H.J. Pathophysiology and preventive strategies of anthracycline - induced cardiotoxicity. Korean J. Intern. Med. 2016;31:625–633. doi: 10.3904/kjim.2016.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zagar T.M., Cardinale D.M., Marks L.B. Breast cancer therapy-associated cardiovascular disease. Nat. Rev. Clin. Oncol. 2015;13:172–184. doi: 10.1038/nrclinonc.2015.171. [DOI] [PubMed] [Google Scholar]
- 4.Brown S.-A., Sandhu N., Herrmann J. Systems biology approaches to adverse drug effects: the example of cardio-oncology. Nat. Rev. Clin. Oncol. 2015;12:718–731. doi: 10.1038/nrclinonc.2015.168. [DOI] [PubMed] [Google Scholar]
- 5.Varga Z.V., Ferdinandy P., Liaudet L., Pacher P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am. J. Physiol. Heart Circ. Physiol. 2015;309:H1453–H1467. doi: 10.1152/ajpheart.00554.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bloom M.W., Hamo C.E., Cardinale D., Ky B., Nohria A., Baer L. Cancer therapy–related cardiac dysfunction and heart failure. Circ.: Heart Fail. 2016;9:e002661. doi: 10.1161/CIRCHEARTFAILURE.115.002843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lipshultz S.E. Chronic progressive cardiac dysfunction years after doxorubicin therapy for childhood acute lymphoblastic leukemia. J. Clin. Oncol. 2004;23:2629–2636. doi: 10.1200/JCO.2005.12.121. [DOI] [PubMed] [Google Scholar]
- 8.Christiansen S., Autschbach R. Doxorubicin in experimental and clinical heart failure. Eur. J. Cardio-Thorac. Surg. 2006;30:611–616. doi: 10.1016/j.ejcts.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 9.Li D.L., Hill J.A. Cardiomyocyte autophagy and cancer chemotherapy. J. Mol. Cell. Cardiol. 2014;71:54–61. doi: 10.1016/j.yjmcc.2013.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghigo A., Li M., Hirsch E. New signal transduction paradigms in anthracycline-induced cardiotoxicity. Biochim. Biophys. Acta Cell Res. 2016;1863:1916–1925. doi: 10.1016/j.bbamcr.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 11.Pereira G.C.S.A., Diogo C.V., Carvalho F.S., Monteiro P., Oliveira P.J. Drug-induced cardiac mitochondrial toxicity and protection: from doxorubicin to carvedilol. Curr. Des. 2011;17:2113–2129. doi: 10.2174/138161211796904812. [DOI] [PubMed] [Google Scholar]
- 12.Pecoraro M., Del Pizzo M., Marzocco S., Sorrentino R., Ciccarelli M., Iaccarino G. Inflammatory mediators in a short-time mouse model of doxorubicin-induced cardiotoxicity. Toxicol. Appl. Pharmacol. 2016;293:44–52. doi: 10.1016/j.taap.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 13.Elsherbiny N.M., Salama M.F., Said E., El-Sherbiny M., Al-Gayyar M.M.H. Crocin protects against doxorubicin-induced myocardial toxicity in rats through down-regulation of inflammatory and apoptic pathways. Chemico-Biol. Interact. 2016;247:39–48. doi: 10.1016/j.cbi.2016.01.014. [DOI] [PubMed] [Google Scholar]
- 14.Cappetta D., Esposito G., Piegari E., Russo R., Ciuffreda L.P., Rivellino A. Sirt1 activation attenuates diastolic dysfunction by reducing cardiac fibrosis in a model of anthracycline cardiomyopathy. Int. J. Cardiol. 2016;205:99–110. doi: 10.1016/j.ijcard.2015.12.008. [DOI] [PubMed] [Google Scholar]
- 15.Bartlett J.J., Trivedi P.C., Pulinilkunnil T. Autophagic dysregulation in doxorubicin cardiomyopathy. J. Mol. Cell. Cardiol. 2017;104:1–8. doi: 10.1016/j.yjmcc.2017.01.007. [DOI] [PubMed] [Google Scholar]
- 16.Pillai V.B., Bindu S., Sharp W.W., Fang Y.H., Kim G.H., Gupta M. Sirt3 protects mitochondrial DNA damage and blocks the development of doxorubicin-induced cardiomyopathy in mice. Am. J. Physiol. Heart Circ. Physiol. 2016 doi: 10.1152/ajpheart.00832.2015. ajpheart.00832.02015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nitiss K.C., Nitiss J.L. Twisting and ironing: doxorubicin cardiotoxicity by mitochondrial DNA damage. Clin. Cancer Res. 2014;20:4737–4739. doi: 10.1158/1078-0432.CCR-14-0821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rochette L., Guenancia C., Gudjoncik A., Hachet O., Zeller M., Cottin Y. Anthracyclines/ trastuzumab: new aspects of cardiotoxicity and molecular mechanisms. Trends Pharmacol. Sci. 2015;36:326–348. doi: 10.1016/j.tips.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 19.Barteková M.Š.P., Fogarassyová M., Ivanová M., Okruhlicová Ľ., Tribulová N., Dovinová I. Quercetin improves postischemic recovery of heart function in doxorubicin -treated rats and prevents doxorubicin-induced matrix metalloproteinase-2 activation and apoptosis induction. Int. J. Mol. Sci. 2015;16:8168–8185. doi: 10.3390/ijms16048168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zordoky B.N., Anwar-Mohamed A., Aboutabl M.E., El-Kadi A.O. Acute doxorubicin toxicity differentially alters cytochrome p450 expression and arachidonic acid metabolism in rat kidney and liver. Drug Metab. Dispos. 2011;39:1440–1450. doi: 10.1124/dmd.111.039123. [DOI] [PubMed] [Google Scholar]
- 21.Damiani R.M., Moura D.J., Viau C.M., Caceres R.A., Henriques J.A.P., Saffi J. Pathways of cardiac toxicity: comparison between chemotherapeutic drugs doxorubicin and mitoxantrone. Arch. Toxicol. 2016;90:2063–2076. doi: 10.1007/s00204-016-1759-y. [DOI] [PubMed] [Google Scholar]
- 22.Mann D.L. The emerging role of small non-coding RNAs in the failing heart: hopes for small molecules. Drugs. 2011;25 doi: 10.1007/s10557-011-6292-x. (149–149) [DOI] [PubMed] [Google Scholar]
- 23.De Rosa S., Curcio A., Indolfi C. Emerging role of micrornas in cardiovascular diseases. Circ. J. 2014;78:567–575. doi: 10.1253/circj.cj-14-0086. [DOI] [PubMed] [Google Scholar]
- 24.Hackfort B.T., Mishra P.K. Emerging role of hydrogen sulfide-microrna cross-talk in cardiovascular diseases. Am. J. Physiol. Heart Circ. Physiol. 2016;310(7):H802–H812. doi: 10.1152/ajpheart.00660.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Rosa S., Indolfi C. Circulating microRNAs as biomarkers in cardiovascular diseases. EXS. 2015;106:139–149. doi: 10.1007/978-3-0348-0955-9_6. [DOI] [PubMed] [Google Scholar]
- 26.Boštjančič E.Z.N. Micrornas and cardiac sarcoplasmic reticulum calcium atpase-2 in human myocardial infarction: Expression and bioinformatic analysis. BMC Genom. 2012;13:552. doi: 10.1186/1471-2164-13-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang Y., Yu T., Jiang S., Zhang Y., Li M., Tang N. Mirnas as potential therapeutic targets and diagnostic biomarkers for cardiovascular disease with a particular focus on wo2010091204. Expert Opin. Therap. Pat. 2017:1–9. doi: 10.1080/13543776.2017.1344217. [DOI] [PubMed] [Google Scholar]
- 28.Hasahya Tony K.Y., Qiutang Zeng. Microrna-208a silencing attenuates doxorubicin induced myocyte apoptosis and cardiac dysfunction. Oxid. Med. Cell. Longev. 2015;2015:597032. doi: 10.1155/2015/597032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J.X., Zhang X.J., Feng C., Sun T., Wang K., Wang Y. Microrna-532-3p regulates mitochondrial fission through targeting apoptosis repressor with caspase recruitment domain in doxorubicin cardiotoxicity. Cell Death Dis. 2015;6:e1677. doi: 10.1038/cddis.2015.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu Y., Zhang Y., Wang N., Pan Z., Gao X., Zhang F. Microrna-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation. 2010;122:2378–2387. doi: 10.1161/CIRCULATIONAHA.110.958967. [DOI] [PubMed] [Google Scholar]
- 31.Kilkenny C., Browne W., Cuthill I.C., Emerson M., Altman D.G. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br. J. Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGrath J.C., Lilley E. Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br. J. Pharmacol. 2015;172:3189–3193. doi: 10.1111/bph.12955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elberry A.A., Abdel-Naim A.B., Abdel-Sattar E.A., Nagy A.A., Mosli H.A., Mohamadin A.M. Cranberry (Vaccinium macrocarpon) protects against doxorubicin- induced cardiotoxicity in rats. Food Chem. Toxicol. 2010;48:1178–1184. doi: 10.1016/j.fct.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 34.Yuan Y.P., Ma Z.G., Zhang X., Xu S.C., Zeng X.F., Yang Z. CTRP3 protected against doxorubicin-induced cardiac dysfunction, inflammation and cell death via activation of Sirt1. J. Mol. Cell. Cardiol. 2017;114:38–47. doi: 10.1016/j.yjmcc.2017.10.008. [DOI] [PubMed] [Google Scholar]
- 35.Li W.N., Qian Z.Y. Effects of crocetin on cardiotoxicity of doxorubicin in rats. Chin. J. New Drugs. 2005;14:1166–1169. [Google Scholar]
- 36.Curtis M.J., Bond R.A., Spina D., Ahluwalia A., Alexander S.P., Giembycz M.A. Experimental design and analysis and their reporting: new guidance for publication in BJP. Br. J. Pharmacol. 2015;172:3461–3471. doi: 10.1111/bph.12856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Joshi S.R., Dhagia V., Gairhe S., Edwards J.G., McMurtry I.F., Gupte S.A. MicroRNA-140 is elevated and mitofusin-1 is downregulated in the right ventricle of the Sugen5416 /hypoxia/normoxia model of pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 2016;311:689–698. doi: 10.1152/ajpheart.00264.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J., Li Y., Jiao J., Wang J., Li Y., Qin D. Mitofusin 1 is negatively regulated by MicroRNA -140 in cardiomyocyte apoptosis. Mol. Cell. Biol. 2014;34:1788–1799. doi: 10.1128/MCB.00774-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boštjančič E.Z.N. MicroRNAs and cardiac sarcoplasmic reticulum calcium ATPase-2 in human myocardial infarction: expression and bioinformatic analysis. BMC Genom. 2012;13:552. doi: 10.1186/1471-2164-13-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Minotti G. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 41.Carvalho F.S., Burgeiro A., Garcia R., Moreno A.J., Carvalho R.A., Oliveira P.J. Doxorubicin -induced cardiotoxicity: from bioenergetic failure and cell death to cardiomyopathy. Med. Res. Rev. 2014;34:106–135. doi: 10.1002/med.21280. [DOI] [PubMed] [Google Scholar]
- 42.Sun J., Sun G., Cui X., Meng X., Qin M., Sun X. Myricitrin protects against doxorubicin induced cardiotoxicity by counteracting oxidative stress and inhibiting mitochondrial apoptosis via ERK/P53 pathway. Evid. Based Complement. Altern. Med. 2016;2016:1–16. doi: 10.1155/2016/6093783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.El-Agamy D.S., Abo-Haded H.M., Elkablawy M.A. Cardioprotective effects of sitagliptin against doxorubicin-induced cardiotoxicity in rats. Exp. Biol. Med. 2016;241:1577–1587. doi: 10.1177/1535370216643418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Asensio-López M.C., Soler F., Sánchez-Más J., Pascual-Figal D., Fernández-Belda F., Lax A. Early oxidative damage induced by doxorubicin: source of production, protection by GKT137831 and effect on Ca2+ transporters in HL-1 cardiomyocytes. Arch. Biochem. Biophys. 2016;594:26–36. doi: 10.1016/j.abb.2016.02.021. [DOI] [PubMed] [Google Scholar]
- 45.Luo C., Urgard E., Vooder T., Metspalu A. The role of COX-2 and Nrf2/ARE in anti- inflammation and antioxidative stress: aging and anti-aging. Med. Hypotheses. 2011;77:174–178. doi: 10.1016/j.mehy.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 46.Wang F., Pu C., Zhou P., Wang P., Liang D., Wang Q. Cinnamal-dehyde prevents endothelial dysfunction induced by high glucose by activating Nrf2. Cell. Physiol. Biochem. 2015;36:315–324. doi: 10.1159/000374074. [DOI] [PubMed] [Google Scholar]
- 47.Chen J.Y., Zhu G.Y., Su X.H., Wang R., Liu J., Liao K. 7-deacetylgedunin suppresses inflammatory responses through activation of Keap1/Nrf2/HO-1 signaling. Oncotarget. 2017;8:55051–55063. doi: 10.18632/oncotarget.19017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith R.E., Tran K., Smith C.C., McDonald M., Shejwalkar P., Hara K. The role of the Nrf2/ARE antioxidant system in preventing cardiovascular diseases. Diseases. 2016;4 doi: 10.3390/diseases4040034. (pii: E34) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matsushima S., Sadoshima J. The role of sirtuins in cardiac disease. Am. J. Physiol. Heart Circ. Physiol. 2015;309:H1375–H1389. doi: 10.1152/ajpheart.00053.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material.