

Abstract
Planctomycetes, a unique group of widespread and understudied bacteria, are known to be associated with macroalgae. The temporal dynamics and the host-specific association of planctomycetal communities on Fucus spiralis, Ulva sp. and Chondrus crispus from two locations in the North Coast of Portugal were assessed both by denaturing gradient gel electrophoresis with group-specific primers and 16S rDNA amplicon libraries. The epiphytic planctomycetal communities showed a significant association with the host macroalgal species independently of the geographical location and the season. This pattern was confirmed by clone libraries of winter and summer samples: we obtained 720 16S rRNA gene sequences that represented 44 operational taxonomic units (OTUs) within the phylum Planctomycetes. Most of the OTUs belonged to Blastopirellula, followed by Rhodopirellula, Planctomyces, the Pir4 lineage and the uncultured class OM190 (this last one nearly 30% of the OTUs). Ulva sp. and C. crispus had more diverse planctomycetal communities than F. spiralis. Analysis of beta diversity showed that the planctomycetal microbiome was host specific. We hypothesize that the specific association of Planctomycetes and their macroalgal hosts is likely determined by nutritional molecules provided by the algae and the set of sulfatases inherent to each Planctomycetes species.
Keywords: Planctomycetes, macroalgae, 16S rRNA gene clone libraries, DGGE
The epiphytic planctomycetal communities on three macroalgae are significantly host specific which may be determined by nutritional molecules provided by the alga.
INTRODUCTION
Bacteria are frequent colonizers of biotic surfaces in the marine environment, by a process referred as epibiosis (Wahl 2008; Wahl et al.2012). These surfaces provide rich habitats for settlement and development of different epibiotic bacterial communities. Many eukaryotic hosts, e.g. sponges, corals, ascidians, bryozoans and microalgae and macroalgae, have established a wide range of interactions with prokaryotes that have been particularly investigated because of the potential production of new bioactive products (Egan, Thomas and Kjelleberg 2008). Microbial biofilms associated with macroalgae have increasingly been studied in the last years, but the understanding of their ecology and interactions is still very limited.
Epiphytic bacteria can have positive effects on the host macroalgae through nutritional and protective roles (Wahl 2008; Goecke et al.2010). These include prevention against biofouling, invasion by pathogens and protection against toxic substances (Holmstrom et al.1996; Maximilien et al.1998; Egan, Thomas and Kjelleberg 2008; Wiese et al.2009). Bacteria isolated from macroalgae are known to produce growth factors and nutrients through matter mineralization and nitrogen fixation that are needed by the macroalgae (Dimitrieva, Crawford and Yuksel 2006; Goecke et al.2010). However, bacteria can also have noxious effects on macroalgae by inducing diseases and causing decomposition (Vairappan et al.2001; Ivanova et al.2002; Michel et al.2006).
Members of many bacterial phyla have been detected by molecular studies to be associated with macroalgae. The main groups are Alphaproteobacteria and Gammaproteobacteria, Bacteroidetes, Firmicutes and Actinobacteria (Goecke et al.2010; Friedrich 2012; Hollants et al.2013) but a great variety exists among the various macroalgae (Lage and Graça 2016). Firmicutes, Actinobacteria and Planctomycetes are more frequently found associated with red and brown algae, while Bacteroidetes and Alphaproteobacteria are more abundant on green macroalgae (Hollants et al.2013). Different parts of the macroalgal thallus from different parts of the alga (rhizoid, cauloid, meristem and phyloid) present different bacterial phylotypes as observed in Saccharina latissima (Staufenberger et al.2008). Seasonal differences in the bacterial community at phylum level were observed in biofilms of Fucus vesiculosus (brown), Gracilaria vermiculophylla (red) and Ulva intestinalis (green) living in close proximity (Lachnit et al.2011). Temporal and spatial variability in the epibiontic bacterial community of four red macroalgae was observed by Wu et al. (2014).
Due to their uncommon characteristics, Planctomycetes are an interesting group of bacteria and culture-dependent methods have proven a wide association of Planctomycetes with macroalgae (Lage and Bondoso 2011). Fluorescence in situ hybridization of biofilms on the kelp Laminaria hyperborea revealed that Planctomycetes accounted for 51%–53% of all bacteria (Bengtsson and Ovreås 2010). Several culture-independent studies also reported the presence of Planctomycetes in the microbial community of macroalgae (Meusnier et al.2001; Longford et al.2007; Bengtsson, Sjotun and Ovreås 2010; Hengst et al.2010; Liu et al.2010; Burke et al.2011; Lachnit et al.2011; Miranda et al.2013; Bondoso et al.2014). In addition, the novel order Phycisphaerales determined after an isolate obtained from the surface of the macroalgae Porphyra sp., was found in Japan (Fukunaga et al.2009). Moreover, studies in Norway and the Baltic Sea (Bengtsson and Ovreås 2010; Lachnit et al.2011) reported a temporal variation of Planctomycetes associated with macroalgae. In fact, the epibacterial communities on the surface of Fucus, Gracilaria and Ulva showed consistent seasonal differences on each algal species at the bacterial phyla level (Lachnit et al.2011).
Due to their capability to attach to surfaces, Planctomycetes are, in fact, good candidates to live in the biofilm community of macroalgae. Furthermore, genome sequencing of several Planctomycetes has revealed the existence of a high number of sulfatase genes, which are involved in the degradation of sulphated heteropolysaccharides (agars and carrageenans) commonly produced by macroalgae (Wegner et al.2013). This may constitute a clear advantage of Planctomycetes living on macroalgal surface.
Previous studies established Planctomycetes as members of biofilms on macroalgae, yet the specificity of this association between planctomycetal and macroalgal species has so far not been studied in detail. This study assessed the seasonality, geographical and host variation of Planctomycetes in epiphytic communities on the macroalgae Fucus spiralis (Ochrophyta), Ulva sp. (Chlorophyta) and Chondrus crispus (Rhodophyta) from two geographical locations (80 km in distance) of the North coast of Portugal applying the analysis of planctomycetal-specific PCR amplicons by denaturing gradient gel electrophoresis (DGGE) and 16S rRNA gene clone libraries.
MATERIAL AND METHODS
Macroalgae sampling
Fresh vegetative thalli of Fucus spiralis, Ulva sp. and Chondrus crispus were collected from two rocky beaches in the North coast of Portugal in Porto (41°09΄N, 8°40΄W) and Carreço (41°44΄N, 8°52΄W), between October 2010 and August 2011, at four different occasions, with 3 month intervals, corresponding to the annual seasonal cycle. The samples were transported under cold conditions in a thermal box and, once in the laboratory, three individuals of each alga were rinsed with sterile natural seawater to remove loosely attached bacteria and frozen at –20°C until DNA extraction was performed.
DNA extraction
Genomic DNA extractions from the macroalgae were performed using 10 circular cuts from three individuals of each specimen with a circular 0.5 cm diameter cork borer. DNA was extracted with the UltraClean® Soil DNA Isolation Kit (Mo Bio Laboratories, Inc., Solana Beach, CA, USA). The extractions were performed according to the manufacturer's instructions with a slight modification including an initial 15 min vortex of the macroalgae pieces in a Disruptor Cell Genie (Scientific Industries Inc., Springfield, MA, USA) in the bead solution tubes.
PCR-DGGE fingerprinting
Planctomycetal community structures were analyzed with a nested PCR for planctomycetal 16S rRNA genes and amplicon analysis by DGGE as previously described (Bondoso et al.2014). The samples were first amplified with the pair of primers 9bfm/1512R (Muhling et al.2008), and the resulting PCR products were re-amplified with the pair of primers PLA352F-GC/PLA920R (Muhling et al.2008). Eight hundred nanograms of each PCR amplicon were separated in a 50%–70% gradient (6% acrylamide) DGGE gel at 60°C and 120 V for 18 h using a CBS Scientific system (CBS Scientific Co., Del Mar, CA, USA) as previously described by Pollet, Tadonleke and Humbert (2011a). The gel was stained with SybrGold (Molecular Probes-Invitrogen, Carlsbad, CA, USA) as described previously (Bondoso et al.2014) and visualized with UV in a Chemidoc system (BioRad Laboratories, Hercules, CA).
16S rRNA clone libraries
In order to investigate the community diversity of Planctomycetes in the macroalgal biofilms, eleven 16S rRNA gene clone libraries from C. crispus, F. spiralis and Ulva sp. collected from Porto and Carreço in February 2011 (winter) and August 2011 (summer) were constructed with the Planctomycetes-specific pair of primers PLA46F/P1390R (Chouari et al.2003). The two different seasons, winter and summer, were chosen to cover the most diverse environmental conditions. PCR amplifications of the 16S rRNA genes were performed in 50 μL reactions with 4 μL of pooled DNA obtained from 10 cuts retrieved from three individuals of each of the three macroalgae. PCR reactions were performed in a MyCycler thermocycler (Bio-Rad Laboratories) and each consisted of 1x PCR Master Mix (Promega, Madison, WI, USA), 100 μmol of each primer and 40 μg of bovine serum albumin. Thermal cycling was as described by Pollet, Tadonleke and Humbert (2011b). In order to minimize PCR bias in cloning, PCR reactions were performed in triplicate and pooled for library construction. Cloning, plasmid transformation and clone picking were performed by Macrogen (www.macrogen.com) with pTOP TA V2 vector (Enzymonics, Inc., Korea). Ninety-six clones were selected from each library and both strands of each clone were sequenced with the universal primers M13F and M13R.
DATA ANALYSES AND STATISTICS
DGGE patterns
Digitized DGGE images were analyzed using the Chemidoc software (Bio-Rad Laboratories). To avoid variations among gels, only individual gels were analyzed. Similarity of resulting banding patterns was assessed by constructing a matrix taking into account the presence or absence of individual bands in each sample and their relative abundance (intensity). Based on this matrix, a Bray-Curtis similarity matrix was produced and used for beta-diversity cluster analysis grouping samples on a dendrogram. These analyses were performed using the software PRIMER6 (Plymouth Routines in Multivariate Ecological Research). Comparisons between the sampling sites and macroalgae hosts were made using analyses of similarity (ANOSIM) to determine significant differences between two or more groups of the macroalgal Planctomycetes communities.
16S rDNA clone libraires
Sequences derived from the 16S rRNA gene clone libraries were edited with Sequencing Analysis 5.2 (Applied Biosystems, Foster City, CA, USA), assembled with Vector NTI Advance 11.5 and checked manually for errors. Sequence analyses were done using the Quantitative Insights Into Microbial Ecology (QIIME) pipeline (Byrne et al.2003; Caporaso et al.2010), which performs standard microbial community analysis. Clustering into OTUs was done at 97% similarity (corresponding to the species level) using cd-hit (Li and Godzik 2006), and taxonomic assignment was done using the Silva reference database (Pruesse et al.2007) in May 2016 through the uclust algorithm with each cluster representing an OTU. Ambiguous and unclassified sequences were assigned taxonomically with the Greengenes database (DeSantis et al.2006). We constructed alpha diversity and richness plots (amount of diversity within each sample), and estimated the amount of diversity shared between two or more samples (beta diversity) using the UniFrac metric and plotting the samples dendrogram (Lozupone, Hamady and Knight 2006) as well as principal component analyses (PCoA). ANOVA significance testing was performed to compare richness and diversity of microbial communities among algal species and season. The OTU network of shared and unique OTUs was visualized using Cytoscape (Saito et al.2012). Permutated analysis of variance using the Adonis function was done in R (Team 2011). Heatmaps and histograms comparing bacterial profiles according to season and algal type were done using the R package phyloseq (McMurdie and Holmes 2013). The 16S rRNA gene sequences representatives of the OTUs were deposited in the NCBI database under GenBank accession numbers KX213849 to KX213892.
RESULTS
DGGE fingerprinting profiles of the planctomycetal communities
Planctomycetal biofilm communities were visualized in 24 DGGE profiles, on five different gels (Figs S1 and S2, Supporting Information) to compare samples from three macroalgae originated from two locations and four seasons. Replicates were not used since we had previously shown that the variability among individuals of the same algal species in DGGE profiles was low (Bondoso et al.2014). The visualization of differences in dendrograms (Fig. 1) showed that, in general, the DGGE band profiles of the Planctomycetes communities were grouped according to the algal host, independent of the season. The similarity of the communities within each alga varied between 40% and 60% through the year, with the exception of Chondrus crispus sampled in Carreço (20% similarity), while the differences between the three algae were higher than 80%. These findings were supported by an ANOSIM statistical test (Table S1, Supporting Information) in which R values higher than 0.75 indicated clearly separated groups (Clarke and Warwick 2001). Thus, planctomycetal communities are significantly different between each macroalgal host (R = 0.813, P = 0.001) while no significant differences according to annual seasons were observed (R < 0.021, P > 0.5). However, the ANOSIM subtest for each pair of algae showed that similar communities existed on Ulva sp. and C. crispus (R < 0.385), while Fucus spiralis communities were clearly different from both Ulva sp. and C. crispus (R = 1). The communities from these two macroalgae, with the exception of C. crispus sampled in summer from Carreço, formed a cluster (Fig. 1) with 40% similarity, in agreement with data obtained previously by Bondoso et al. (2014).
Figure 1.

Dendrograms of DGGE profiles based on Bray-Curtis similarity evidencing the clustering of the samples of the Planctomycetes communities associated with the macroalgae from Carreço (a) and Porto (b).
In the 1-year DGGE study, the planctomycetal communities tended to group according to the geographical site (Fig. S3, Supporting Information). An exception was the case in F. spiralis sampled in summer (Fig. S3a). In C. crispus, the communities from Carreço were separated from the ones of Porto and shared <30% bands (Figs S3e and f). In Ulva sp., the overall similarity between the communities was 60%, with the exception of the one sampled in winter at Porto (Figs S3c and d). These results were supported by an ANOSIM test (Table S2, Supporting Information), and concur with previous results (Bondoso et al.2014), suggesting that there is a stable community of Planctomycetes associated with each particular macroalga which is maintained regardless of the geographical location and the time of the year.
Microbiome profiles of the 16S rRNA libraries
Eleven 16S rDNA clone libraries were constructed from the macroalgae sampled in the two sites and seasons in order to gather in-depth information on the composition and diversity of the epiphytic planctomycetal communities. We lacked only a 16S rRNA gene clone library of the sample C. crispus-Carreço winter. A total of 720 good quality assembled sequences were related to the Planctomycetes and were binned into 44 OTUs at a 97% identity cut-off value on the over 1300-bp-long 16S rRNA gene sequences (representing potentially different species) (Table 1; Table S3, Supporting Information). The alpha-diversity metrics and predicted OTU richness (Chao1) (Table 1) revealed that the diversity was the highest for communities associated with the macroalgae C. crispus followed by Ulva sp. and both were significantly more diverse than the community associated with F. spiralis according to ANOVA testing (Table 1, Fig. S3). ANOVA tests showed no significant differences in diversity between winter and summer, although richness was higher in winter. Planctomycetal biofilm communities from Porto showed a higher diversity than those from Carreço. With the exception of F. spiralis, the observed rarefaction curves of clone libraries (Fig. S4, Supporting Information) were lower than the estimated ones.
Table 1.
Clone library diversity and richness estimates grouped by alga, site and season over the 720 sequences and 44 OTUs.
| No of | No of | ||||
|---|---|---|---|---|---|
| Group | sequences | OTUs | Shannon | Evenness | Chao1 |
| Algal Host | |||||
| Fucus spiralis | 271 | 21 | 1.17 | 0.44 | 5.2 |
| Ulva sp. | 222 | 50 | 2.42 | 0.74 | 13.9 |
| Chondrus crispus | 227 | 49 | 2.67 | 0.76 | 25.1 |
| Location | |||||
| Carreço | 327 | 27 | 2.02 | 0.61 | 8.8 |
| Porto | 393 | 39 | 2.80 | 0.76 | 15.3 |
| Season | |||||
| Winter | 347 | 58 | 2.55 | 0.69 | 15.7 |
| Summer | 373 | 62 | 2.50 | 0.71 | 10.8 |
Beta-diversity analyses performed by hierarchical clustering of samples and PCoA indicated that the microbiomes are host specific. The planctomycetal communities were similar in structure according to the algal species regardless of the location or season (Figs 2a and b). OTUs present in Ulva and Fucus biofilms showed in the PCoA a difference of ∼75% in the principal component 1. The communities of Ulva sp. and C. crispus were more related among themselves than to ones of F. spiralis in the permutated analysis of variance (ANOSIM), thus also demonstrating that the variability of community structure is best explained by the macroalgal host species (P = 0.009)).
Figure 2.

Beta-diversity plots: 2D-weighted PCoA using UniFrac (A) and UPGMA dendrogram (B) considering the relative abundance of each OTU). FC—Fucus spiralis, Carreço; FF—Fucus spiralis, Porto; UC—Ulva sp., Carreço; UF—Ulva sp., Porto; CC—Chondrus crispus, Carreço; CF—Chondrus crispus, Porto; W—winter, S–summer.
Taxonomic composition of the Planctomycetes community
The 16S rRNA gene sequences were distributed across two different orders of Planctomycetes within two main classes (Planctomycetia and the uncultured class OM190) (Fig. 3). The order Planctomycetales, which includes the genera Planctomyces, Rhodopirellula, Blastopirellula and the Pir4 lineage (Ward 2010), was the most abundant with 76.4% of the total clones. Their representatives were present in all the samples in proportions ranging from 47.1% to 100%. The macroalga F. spiralis from Carreço was the only one containing the Pir4 lineage. Class OM190 was more abundant in Ulva sp., decreased in dominance in C. crispus and was nearly absent in F. spiralis (Fig. 3). Comprising the orders CL500-15 and agg227, class OM190 represented 17.5% of all sequences, but was absent in F. spiralis sampled in Carreço. In fact, CL-500 only appeared in Porto and in summer (Fig. 4). Similarly to the distribution patterns of OM190, the taxon Rhodopirellula was more abundant in Ulva sp., had a lower contribution in C. crispus and had clearly less contribution in F. spiralis samples (Figs 3 and 4). Less abundant was the genus Planctomyces (5.7%), which appeared only in some clone libraries of Ulva sp. and C. crispus (Fig. 4).
Figure 3.
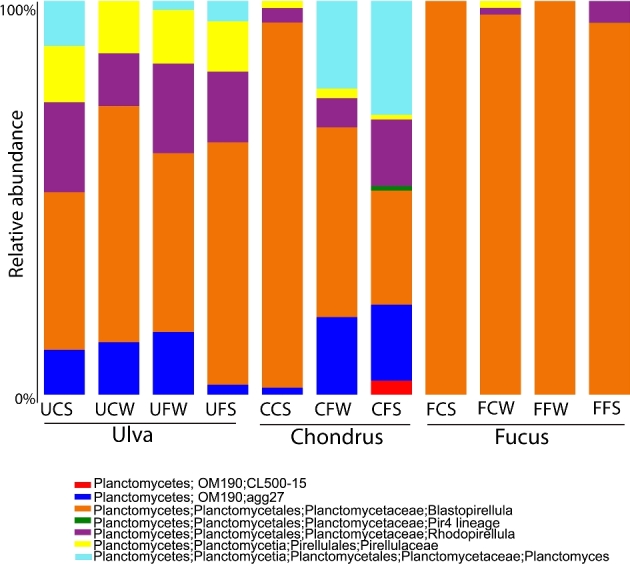
Taxonomic distribution of OTUs in the 16S rRNA gene clone libraries. FC—Fucus spiralis, Carreço; FF—Fucus spiralis, Porto; UC—Ulva sp., Carreço; UF—Ulva sp., Porto; CC—Chondrus crispus, Carreço; CF—Chondrus crispus, Porto; W—winter, S—summer.
Figure 4.

Genus-level OTUs according to algal type and season as shown by heatmaps (top panels).
The OTU taxonomic profiles were different according to the macroalgal species regardless of the location or season: Ulva sp. samples had ∼20%–30% unclassified OTUs from class OM190 which were more abundant than in other macroalgae; additionally, Ulva sp. had more abundant Rhodopirellula OTUs, as well as unclassified OTUs from family Pirellulaceae(Figs 3 and 4). In turn, Planctomyces OTUs were more abundant in C. crispus samples and Blastopirellula were more abundant in F. spiralis samples (Fig 3), in particular OTU 30 (Fig 4). The few differences found between sites include a decrease in Blastopirellula and an increase in Planctomyces and OM190 in Porto as compared to Carreço. As for the season, differences between summer and winter correspond to more unclassified OTUs, and OM190 in winter and Planctomyces OTUs in the summer samples.
The distribution of the 44 OTUs among the different macroalgae clone libraries was visualized using an OTU network that was constructed to highlight both shared and unique OTUs (Fig. 5). The macroalgae C. crispus had more exclusive OTUs (not shared with any other algae type), while F. spiralis had the lowest number of unique OTUs. Chondrus crispus and Ulva sp. shared more OTUs between them than with F. spiralis. None of the OTUs was found to be present in all the clone libraries indicating the absence of a common core community of Planctomycetes among macroalgae.
Figure 5.
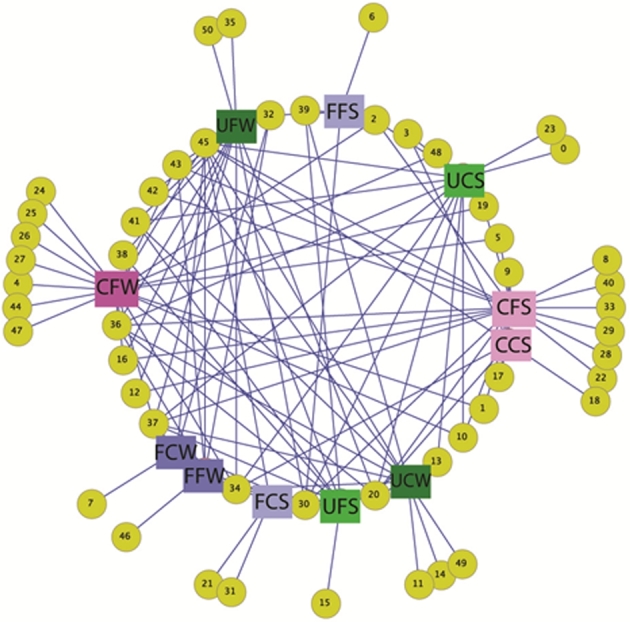
OTU network map showing the shared and unique OTUs (yellow circles) among the clone libraries. Fucus spiralis in blue squares, Ulva sp. in green and Chondrus crispus in pink. Darker colors represent winter and brighter squares represent summer. FC—Fucus spiralis, Carreço; FF—Fucus spiralis, Porto; UC—Ulva sp., Carreço; UF—Ulva sp., Porto; CC—Chondrus crispus, Carreço; CF—Chondrus crispus, Porto; W—winter, S—summer.
About 60% of the OTUs were phylogenetically related to 16S rRNA gene sequences of organisms known to be associated with macroalgae including F. vesiculosus, Gracilaria vermiculophylla, Laminaria hyperborea and Ulva australis. Among them 40% were unclassified taxa (Table S4, Supporting Information). Several of the OTUs were related to cultured strains previously isolated from the surface of several macroalgae, including Planctomyces sp. Pd1, planctomycete sp. FC18, Roseimaritima ulvae strains UC8 and UF2, Rubripirellula obstinata strain LF1, Rhodopirellula rubra strains LF2 and FC3, R. lusitana strain UC16 and R. baltica strain CcC1. Most of the 16S rRNA gene sequences similarities of the OTUs to cultured representatives of Planctomycetes were <96%, indicating the existence of an unknown diversity within this phylum inhabiting macroalgae. The most abundant OTU was OTU30 representing 30% of the total clones, followed by OTUs 34 (11%) and 45 (9%). These OTUs were taxonomically related to the planctomycete FC18 isolated from the surface of F. spiralis and showed similarities in the 16S rRNA gene ranging from 93.3% to 98.1%.
DISCUSSION
Previous research on the bacterial populations inhabiting the surface of macroalgae has shown that Planctomycetes are part of this community in proportions ranging from as low as 1% in Ulva prolifera to 53% in the kelp Laminaria hyperborea (Bengtsson and Ovreås 2010; Liu et al.2010). Planctomycetes present several characteristics that allow them to colonize these surfaces. These include a glycoproteic holdfast for attachment, sulfatase genes for the removal of sulfates from polysaccharides and the resistance to some antibiotics (Schlesner 1994; Cayrou, Raoult and Drancourt 2010; Lage and Bondoso 2011; Lage 2013). A previous study based only on the analysis of DGGE profiles of seven different algae sampled in the same localities of this study (Bondoso et al.2014) already suggested that the communities of Planctomycetes on macroalgal surfaces are host specific and are more similar between different geographical locations than between co-occurring macroalgae. Here, we analyzed the temporal and geographical variation of the Planctomycetes communities on three different macroalgae and the existence of a possible association of these communities to specific macroalgae.
DGGE fingerprinting showed that the planctomycetal communities of macroalgae did not vary significantly during the seasonal cycle. This finding is supported by the similar OTU diversities and richness obtained in winter and summer clone libraries. Furthermore, not a single OTU presented significant seasonal changes which is a novel finding compared to previous studies that indicated that planctomycetal communities associated with macroalgae could present significant temporal variations (Bengtsson and Ovreås 2010; Lachnit et al.2011). Our results also suggest a stable temporal and spatial planctomycetal community associated with Ulva sp. The planctomycetal community in Ulva sp. clone libraries was always very similar, with the exception of the one sampled in Carreço in summer. Indeed, clone libraries from all the macroalgae sampled in Carreço in summer were phylogenetically more distant from the other clone libraries. This result may be related with unusual non-determined sporadic environmental or biological factors that could have induced stress in the communities and changed their composition. Further temporal analyses performed in consecutive years would be necessary to determine if the Planctomycetes communities in summer are truly distinct from the other seasons in this location.
The results from this study also indicated that the geographical location did not influence the distribution of the planctomycetal communities associated with macroalgae. In general, DGGE profiles of the planctomycetal communities showed no significant differences between Porto (41°09΄N, 8°40΄W) and Carreço (41°44΄N, 8°52΄W) although these two sampling sites are about 80 km away. These results are in accordance with a previous DGGE fingerprinting study of six different algae sampled in the same locations (Lachnit et al.2009), which showed that the epibacterial communities from macroalgae sampled in the Baltic and North Seas were more similar between representatives of the same species than between macroalgae in the same location. Similar findings were also reported for Bonnemaisonia asparagoides (Nylund et al.2010), Saccharina latissima (Staufenberger et al.2008), Dyctyosphaeria ocellata (Sneed and Pohnert 2011), Ulva spp., Scytosiphon lomentaria and Lessonia nigrescens (Hengst et al.2010). Our clone sequences revealed that the planctomycetal community composition of each macroalgal species was more similar between the sampling sites than with other co-occurring macroalgal species. Furthermore, none of the sequenced OTUs showed a correlation with geographical location. However, the Planctomycetes populations from macroalgae sampled in Porto presented a higher diversity, richness and evenness, something that could be related with the number of clone sequences obtained from Porto (n = 400) that was higher than the values from Carreço (n = 321). The sampling site in Porto, which is the second largest city in Portugal, is much more affected by anthropogenic factors than the one in Carreço, an agricultural zone. Pollution usually leads to a decrease in bacterial diversity (Torsvik et al.1998) but Cho and Kim (2000) showed that aquifers with livestock wastewater input had higher diversity. Planctomycetes are able to utilize a wide range of organic substrates, and strains previously isolated from macroalgae have shown a large range of physiological tolerance, for example, to heavy metals (Lage, Bondoso and Catita 2012) and UV (Viana, Lage and Oliveira 2013), which could give them the ability to inhabit these polluted environments. These characteristics are typical of bacteria usually found in disturbed microbial assemblages (Atlas et al.1991).
The existence of specific communities of Planctomycetes associated with each macroalga was demonstrated both by DGGE fingerprinting and clone sequencing. Host-specificity of microbial communities has been shown in other macroalgae (Longford et al.2007; Lachnit et al.2009; Hengst et al.2010; Nylund et al.2010, Barott et al.2011; Trias et al.2012; Vega Thurber et al.2012). This highly specific association of bacteria with macroalgae is mostly due to a combination of physiological and biochemical properties of the macroalgae (Goecke et al.2010) that probably also influence the community of planctomycetes. In this study, we also showed that there is a clear separation of the planctomycetal communities of Fucus spiralis relatively to the other two macroalgae that presented more similar DGGE profiles and taxonomic composition. In our previous DGGE study (Bondoso et al.2014), planctomycetal DGGE profiles obtained from Rhodophyta were more similar to the ones of Ulva sp. (Chlorophyta) than to the ones of Ochrophyta macroalgae. Similar findings were also reported by Lachnit et al. (2009) for the whole bacterial community, which may indicate that these macroalgal groups present similar chemical surface characteristics and intrinsic mechanisms that modulate the epibacterial community on their surfaces. The absence of a core community of Planctomycetes among the three macroalgae, shown by the clone libraries analyses, supports the hypothesis of host-specificity. Furthermore, statistical analyses showed the existence of planctomycete OTUs specifically associated with each macroalgal species. Hollants et al. (2013) reported that in many cases the differences among different macroalgae species and similarities within the same species are evident at the phylum or class level but not at the genera or species level. With this work, we demonstrate the existence of particular planctomycetal OTUs associated with each species of macroalgae. Goecke et al. (2010) suggested that the specific associations existent between bacteria and macroalgae are mainly due to substrate preferences of the bacterial strains and to antifouling and antimicrobial metabolites produced by the macroalgae (Goecke et al.2010). Macroalgae are known to produce a wide range of secondary metabolites in order to chemically control the epibiosis on their surface and therefore modulate their bacterial communities (Hellio et al.2001; Nylund et al.2005; Wahl et al.2010; Sneed and Pohnert 2011). A study investigating the antibacterial and antifungal activity of 82 macroalgae from the Iberian Peninsula showed that F. spiralis was effective in inhibiting the growth of several Gram-positive and Gram-negative bacteria while U. rigida was not (Salvador et al.2007). Although in the same study several Rhodophyta algae showed the highest activity against bacteria, C. crispus has been reported to have low antimicrobial activity (Hellio et al.2001; Nylund et al.2005; Cox, Abu-Ghannam and Gupta 2010). These data could explain why F. spiralis showed the lowest Planctomycetes diversity and richness of all the three macroalgae studied here.
The ability to utilize the macroalgae exudates as well as the structural polysaccharides of their cells walls by the associated bacteria is also an important factor that modulates the communities on macroalgae (Goecke et al.2010; Lachnit, Wahl and Harder 2010). The composition of the macroalgal cell walls and the polysaccharides produced varies among the main groups of these organisms. In fact, the green and red macroalgae share with glaucophyte algae and plants a monophyletic eukaryotic origin after a single event of a primary endosymbiosis of a cyanobacterium (Popper et al.2011). On the other end, the brown algae were originated through a secondary endosymbiosis of a red alga. Although the cell wall of the three macroalgal groups has evolved independently, these groups share wall components with common ancestry. The closeness of the planctomycetal community associated with green and red macroalgae observed in our study may somehow be related to the closer evolutionary history of these two groups and their cell walls. Brown algae are producers of alginates, fucoidan and laminarin; red algae produce agar and carrageenans; and green algae contain ulvans (Popper et al.2011). The majority of these polymers are sulphated polysaccharides and Planctomycetes are known to contain a high abundance of different sulfatases (Wegner et al.2013), which would allow them to utilize these substrates. Furthermore, the number of genes coding for sulfatases differs in species and genera of planctomycetes: 104–196 in Rhodopirellula species, 40 in Blastopirellula marina, 83 in Planctomyces maris and 101 in Planctomyces brasiliensis (Wegner et al.2013). The various Rhodopirellula species only share ca. 60 sulfatase genes, which means that each species harbors a specific set of sulfatases that is probably related to their ecological niche. In fact, planctomycetal diversity is reflected in an enzymatic diversity besides sulfatases (e.g. glycoside hydrolases or polysaccharide lyases) that allows these groups to access a range of polysaccharides in macroalgal cell walls. Phylogenetic analyses on planctomycetal genomes indicate augmentation of gene functions through gene duplications and horizontal gene transfers in genes involved in cell wall degradation (e.g. κ-carrageenase, alginate lyase, fucosidase) (Kim et al.2016). Based on our results and data from the literature, we propose that the specific Planctomycetes association to the macroalgal host is likely determined by the excreted polymers from the algae and by the set of sulfatases genes of each planctomycete.
CONCLUSIONS
The results presented in this study support for Planctomycetes the existence of host-specific communities associated with macroalgae, as revealed by genus-level taxonomic profiles. No Planctomycetes core community was associated with the three macroalgae, which are representatives of the main groups of macroalgae. Our data showed that the planctomycetal communities associated with macroalgae do not present significant temporal or geographical variations, with the exception of the epibiontic communities of samples from Porto that had more diverse Planctomycetes communities.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary data are available at FEMSEC online.
FUNDING
This work was partially supported by the Strategic Funding UID/Multi/04423/2013 through national funds provided by FCT – Foundation for Science and Technology and European Regional Development Fund (ERDF), in the framework of the programme PT2020. The first authorwas financed by FCT [PhD grant SFRH/BD/35933/2007] and by a Marie Curie short term fellowship Ph.D. stipend in MarMic EST at Max Planck Institute for Marine Microbiology, Bremen. The work was also partially supported by a US NIH National Institute of General Medical Sciences INBRE award grant P20 GM103475 attributed to F. G-V.
Conflict of interest. None declared.
REFERENCES
- Atlas R, Horowitz A, Krichevsky M et al. . Response of microbial populations to environmental disturbance. Microbial Ecol 1991;22:249–56 [DOI] [PubMed] [Google Scholar]
- Barott KL, Rodriguez-Brito B, Janouškovec J et al. . Microbial diversity associated with four functional groups of benthic reef algae and the reef-building coral Montastraea annularis. Environ Microbiol 2011;13:1192–204 [DOI] [PubMed] [Google Scholar]
- Bengtsson MM, Ovreås L. Planctomycetes dominate biofilms on surfaces of the kelp Laminaria hyperborea. BMC Microbiol 2010;10:261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson MM, Sjotun K, Ovreås L. Seasonal dynamics of bacterial biofilms on the kelp Laminaria hyperborea. Aquat Microbial Ecol 2010;60:71–83 [Google Scholar]
- Bondoso J, Balagué V, Gasol JM et al. . Community composition of the Planctomycetes associated with different macroalgae. FEMS Microbiol Ecol 2014;88:445–56 [DOI] [PubMed] [Google Scholar]
- Burke C, Thomas T, Lewis M et al. . Composition, uniqueness and variability of the epiphytic bacterial community of the green alga Ulva australis. ISME J 2011;5:590–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne M, Hart MW, Cerra A et al. . Reproduction and larval morphology of broadcasting and viviparous species in the Cryptasterina species complex. Biol Bull 2003;205:285–94 [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J et al. . QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010;7:335–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrou C, Raoult D, Drancourt M. Broad-spectrum antibiotic resistance of Planctomycetes organisms determined by Etest. J Antimicrob Chemoth 2010;65:2119–22 [DOI] [PubMed] [Google Scholar]
- Cho JC, Kim SJ. Increase in bacterial community diversity in subsurface aquifers receiving livestock wastewater input. Appl Environ Microb 2000;66:956–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouari R, Le Paslier D, Daegelen P et al. . Molecular evidence for novel planctomycete diversity in a municipal wastewater treatment plant. Appl Environ Microb 2003;69:7354–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke K, Warwick R. Change in Marine Communities: An Approach to Statistical Analysis and Interpretation. 2nd edn, Plymouth:PRIMER-E Ltd, 2001 [Google Scholar]
- Cox S, Abu-Ghannam N, Gupta S. An assessment of the antioxidant and antimicrobial activity of six species of edible Irish seaweeds. Int Food Res J 2010;17:05–220 [Google Scholar]
- DeSantis TZ, Hugenholtz P, Larsen N et al. . Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microb 2006;72:5069–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrieva GY, Crawford RL, Yuksel GU. The nature of plant growth-promoting effects of a pseudoalteromonad associated with the marine algae Laminaria japonica and linked to catalase excretion. J Appl Microbiol 2006;100:1159–69 [DOI] [PubMed] [Google Scholar]
- Egan S, Thomas T, Kjelleberg S. Unlocking the diversity and biotechnological potential of marine surface associated microbial communities. Curr Opin Microbiol 2008;11:219–25 [DOI] [PubMed] [Google Scholar]
- Friedrich MW. Bacterial communities on macroalgae. Wiencke C, Bischof K. Seaweed Biology Berlin Heidelberg:Springer, 2012, 189–201 [Google Scholar]
- Fukunaga Y, Kurahashi M, Sakiyama Y et al. . Phycisphaera mikurensis gen. nov., sp. nov., isolated from a marine alga, and proposal of Phycisphaeraceae fam. nov., Phycisphaerales ord. nov. and Phycisphaerae classis nov. in the phylum Planctomycetes. J Gen Appl Microbiol 2009;55:267–75 [DOI] [PubMed] [Google Scholar]
- Goecke F, Labes A, Wiese J et al. . Chemical interactions between marine macroalgae and bacteria. Mar Ecol Prog Ser 2010;409:267–99 [Google Scholar]
- Hellio C, De La Broise D, Dufossé L et al. . Inhibition of marine bacteria by extracts of macroalgae: potential use for environmentally friendly antifouling paints. Mar Environ Res 2001;52:231–47 [DOI] [PubMed] [Google Scholar]
- Hengst MB, Andrade S, González B et al. . Changes in epiphytic bacterial communities of intertidal seaweeds modulated by host, temporality, and copper enrichment. Microbial Ecol 2010;60:282–90 [DOI] [PubMed] [Google Scholar]
- Hollants J, Leliaert F, De Clerck O et al. . What we can learn from sushi: a review on seaweed-bacterial associations. FEMS Microbiol Ecol 2013;83:1–16 [DOI] [PubMed] [Google Scholar]
- Holmstrom C, James S, Egan S et al. . Inhibition of common fouling organisms by marine bacterial isolates with special reference to the role of pigmented bacteria. Biofouling 1996;10:251–9 [DOI] [PubMed] [Google Scholar]
- Ivanova EP, Sawabe T, Alexeeva YV et al. . Pseudoalteromonas issachenkonii sp. nov., a bacterium that degrades the thallus of the brown alga Fucus evanescens. Int J Syst Evol Microbiol 2002;52:229–34 [DOI] [PubMed] [Google Scholar]
- Kim JW, Brawley SH, Prochnik S et al. . Genome analysis of Planctomycetes inhabiting blades of the red alga Porphyra umbilicalis. PLoS One 2016;11:e0151883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachnit T, Blümel M, Imhoff JF et al. . Specific epibacterial communities on macroalgae: phylogeny matters more than habitat. Aquat Biol 2009;5:181–6 [Google Scholar]
- Lachnit T, Meske D, Wahl M et al. . Epibacterial community patterns on marine macroalgae are host-specific but temporally variable. Environ Microbiol 2011;13:655–65 [DOI] [PubMed] [Google Scholar]
- Lachnit T, Wahl M, Harder T. Isolated thallus-associated compounds from the macroalga Fucus vesiculosus mediate bacterial surface colonization in the field similar to that on the natural alga. Biofouling 2010;26:247–55 [DOI] [PubMed] [Google Scholar]
- Lage O. Characterization of a planctomycete associated with the marine dinoflagellate Prorocentrum micans. Anton Leeuw 2013;104:499–508 [DOI] [PubMed] [Google Scholar]
- Lage OM, Bondoso J. Planctomycetes diversity associated with macroalgae. FEMS Microbiol Ecol 2011;78:66–375 [DOI] [PubMed] [Google Scholar]
- Lage OM, Bondoso J, Catita JA. Determination of zeta potential in Planctomycetes and its application in heavy metals toxicity assessment. Arch Microbiol 2012;194:847–55 [DOI] [PubMed] [Google Scholar]
- Lage OM, Graça AP. Biofilms: an extra coat on macroalgae. Dharumadurai D. Algae - Organisms for Imminent Biotechnology. InTech, 2016, 183–210 [Google Scholar]
- Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006;22:1658–9 [DOI] [PubMed] [Google Scholar]
- Liu M, Dong Y, Zhao Y et al. . Structures of bacterial communities on the surface of Ulva prolifera and in seawaters in an Ulva blooming region in Jiaozhou Bay, China. World J Microb Biot 2010;27:1703–12 [Google Scholar]
- Longford SR, Tujula NA, Crocetti GR et al. . Comparisons of diversity of bacterial communities associated with three sessile marine eukaryotes. Aquat Microbial Ecol 2007;48:217–29 [Google Scholar]
- Lozupone C, Hamady M, Knight R. UniFrac–an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics 2006;7:371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 2013;8:e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximilien R, de Nys R, Holmström C et al. . Chemical mediation of bacterial surface colonisation by secondary metabolites from the red alga Delisea pulchra. Aqua Microb Ecol 1998;15:233–46 [Google Scholar]
- Meusnier I, Olsen JL, Stam WT et al. . Phylogenetic analyses of Caulerpa taxifolia (Chlorophyta) and of its associated bacterial microflora provide clues to the origin of the Mediterranean introduction. Mol Ecol 2001;10:931–46 [DOI] [PubMed] [Google Scholar]
- Michel G, Nyval-Collen P, Barbeyron T et al. . Bioconversion of red seaweed galactans: a focus on bacterial agarases and carrageenases. Appl Microbiol Biot 2006;71:23–33 [DOI] [PubMed] [Google Scholar]
- Miranda LN, Hutchison K, Grossman AR et al. . Diversity and abundance of the bacterial community of the red macroalga Porphyra umbilicalis: did bacterial farmers produce macroalgae?. PLoS One 2013;8:e58269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhling M, Woolven-Allen J, Murrell JC et al. . Improved group-specific PCR primers for denaturing gradient gel electrophoresis analysis of the genetic diversity of complex microbial communities. ISME J 2008;2:379–92 [DOI] [PubMed] [Google Scholar]
- Nylund GM, Cervin G, Hermansson M et al. . Chemical inhibition of bacterial colonization by the red alga Bonnemaisonia hamifera. Mar Ecol Prog Ser 2005;302:27–36 [Google Scholar]
- Nylund GM, Persson F, Lindegarth M et al. . The red alga Bonnemaisonia asparagoides regulates epiphytic bacterial abundance and community composition by chemical defence. FEMS Microbiol Ecol 2010;71:84–93 [DOI] [PubMed] [Google Scholar]
- Pollet T, Tadonleke RD, Humbert JF. Comparison of primer sets for the study of Planctomycetes communities in lentic freshwater ecosystems. Environ Microbiol Rep 2011a;3:254–61 [DOI] [PubMed] [Google Scholar]
- Pollet T, Tadonleke RD, Humbert JF. Spatiotemporal changes in the structure and composition of a less-abundant bacterial phylum (Planctomycetes) in two perialpine lakes. Appl Environ Microb 2011b;77:4811–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popper ZA, Michel G, Hervé C et al. . Evolution and diversity of plant cell walls: from algae to flowering plants. Annu Rev Plant Biol 2011;62:567–90 [DOI] [PubMed] [Google Scholar]
- Pruesse E, Quast C, Knittel K et al. . SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 2007;35:7188–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito R, Smoot ME, Ono K et al. . A travel guide to Cytoscape plugins. Nat Methods 2012;11:1069–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvador N., Gómez Garreta A, Lavelli L et al. . Antimicrobial activity of Iberian macroalgae. Sci Mar 2007;71:101–13 [Google Scholar]
- Schlesner H. The development of media suitable for the microorganisms morphologically resembling Planctomyces spp., Pirellula spp., and other Planctomycetales from various aquatic habitats using dilute media. Syst Appl Microbiol 1994;17:135–45 [Google Scholar]
- Sneed JM, Pohnert G. The green macroalga Dictyosphaeria ocellata influences the structure of the bacterioplankton community through differential effects on individual bacterial phylotypes. FEMS Microbiol Ecol 2011;75:242–54 [DOI] [PubMed] [Google Scholar]
- Staufenberger T, Thiel V, Wiese J et al. . Phylogenetic analysis of bacteria associated with Laminaria saccharina. FEMS Microbiol Ecol 2008;64:65–77 [DOI] [PubMed] [Google Scholar]
- Team RDC. R: A Language and Environment for Statistical Computing. Vienna, Austria:The R Foundation for Statistical Computing, 2011, ISBN: 3-900051-07-0. http://www.R-project.org/ (2 January 2016, date last accessed) [Google Scholar]
- Torsvik V, Daae FL, Sandaa RA et al. . Novel techniques for analysing microbial diversity in natural and perturbed environments. J Biotechnol 1998;64:53–62 [DOI] [PubMed] [Google Scholar]
- Trias R, García-Lledó A, Sánchez N et al. . Abundance and composition of epiphytic bacterial and archaeal ammonia oxidizers of marine red and brown macroalgae. Appl Environ Microb 2012;78:318–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vairappan CS, Suzuki M, Motomura T et al. . Pathogenic bacteria associated with lesions and thallus bleaching symptoms in the Japanese kelp Laminaria religiosa Miyabe (Laminariales, Phaeophyceae). Hydrobiologia 2001;445:183–91 [Google Scholar]
- Vega Thurber R, Burkepile DE, Correa AM et al. . Macroalgae decrease growth and alter microbial community structure of the reef-building coral, Porites astreoides. PLoS One 2012;7:e44246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viana F, Lage OM, Oliveira R. High ultraviolet C resistance of marine Planctomycetes. Anton Leeuw 2013;104:585–95 [DOI] [PubMed] [Google Scholar]
- Wahl M. Ecological lever and interface ecology: epibiosis modulates the interactions between host and environment. Biofouling 2008;24:427–38 [DOI] [PubMed] [Google Scholar]
- Wahl M, Goecke F, Labes A et al. . The second skin: ecological role of epibiotic biofilms on marine organisms. Front Microbiol 2012;3:292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl M, Shahnaz L, Dobretsov S et al. . Ecology of antifouling resistance in the bladder wrack Fucus vesiculosus: patterns of microfouling and antimicrobial protection. Mar Ecol Prog Ser 2010;411:33–48 [Google Scholar]
- Ward NL. Family I. Planctomycetaceae Schlesner and Stackebrandt 1987, 179VP (Effective publication: Schlesner and Stackebrandt 1986, 175) emend. Ward (this volume). Krieg NR, et al. The Bacteroidetes, Spirochaetes, Tenericutes (Mollicutes), Acidobacteria, Fibrobacteres, Fusobacteria, Dictyoglomi, Gemmatimonadetes, Lentisphaerae, Verrucomicrobia, Chlamydiae, and Planctomycetes. New York:Springer, 2010, 879–925 [Google Scholar]
- Wegner CE, Richter-Heitmann T, Klindworth A et al. . Expression of sulfatases in Rhodopirellula baltica and the diversity of sulfatases in the genus Rhodopirellula. Mar Genomics 2013;9:51–61 [DOI] [PubMed] [Google Scholar]
- Wiese J, Thiel V, Nagel K et al. . Diversity of antibiotic-active bacteria associated with the brown alga Laminaria saccharina from the Baltic Sea. Mar Biotechnol 2009;11:287–300 [DOI] [PubMed] [Google Scholar]
- Wu H, Liu M, Zhang W et al. . Phylogenetic analysis of epibacterial communities on the surfaces of four red macroalgae. J Ocean Univ China 2014;13:1025–32 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.