

Abstract
The skin is highly sensitive to the chemical warfare agent in mustard gas, sulfur mustard (SM) that initiates a delayed injury response characterized by erythema, inflammation and severe vesication (blistering). Although SM poses a continuing threat, used as recently as in the Syrian conflict, no mechanism-based antidotes against SM are available.
Recent studies demonstrated that Transient Receptor Potential Ankyrin 1 (TRPA1), a chemosensory cation channel in sensory nerves innervating the skin, is activated by SM and 2-chloroethyl ethyl sulfide (CEES), an SM analog, in vitro, suggesting it may promote vesicant injury.
Here, we investigated the effects of TRPA1 inhibitors, and an inhibitor of Calcitonin Gene Related Peptide (CGRP), a neurogenic inflammatory peptide released upon TRPA1 activation, in a CEES-induced mouse ear vesicant model. TRPA1 inhibitors (HC-030031 and A-967079) and a CGRP inhibitor (MK-8825) reduced skin edema, pro-inflammatory cytokines (IL-1β, CXCL1/KC), MMP-9, a protease implicated in skin damage, and improved histopathological outcomes.
These findings suggest that TRPA1 and neurogenic inflammation contribute to the deleterious effects of vesicants in vivo, activated either directly by alkylation, or indirectly, by reactive intermediates or pro-inflammatory mediators. TRPA1 and CGRP inhibitors represent new leads that could be considered for validation and further development in other vesicant injury models.
Keywords: TRPA1, sulfur mustard, CEES, CGRP, medical countermeasures, skin vesicant injury, mouse ear vesicant model (MEVM)
Graphical abstract
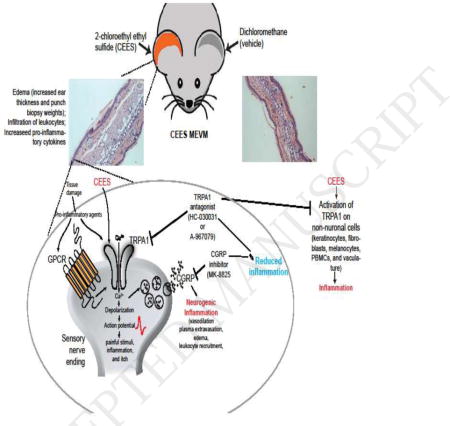
1. Introduction
Sulfur mustard (SM, bis-(2-chloroethyl)-sulfide), also known as mustard gas, is a chemical warfare agent first used in World War I (Buch et al., 2013a; Wattana and Bey, 2009). Although banned by the Geneva protocol and the Chemical Weapons Convention since 1925, several countries and militant organizations have used mustard gas in recent conflicts. In the 1980s Iraq used mustard gas in attacks against Iranian forces and its Kurdish population (Wattana and Bey, 2009). Mustard gas exposures likely contributed to Gulf War illness (Golomb et al., 1998). Most recently, mustard gas was used in chemical attacks in the ongoing Syrian and Iraqi conflicts.
SM causes injuries to the skin, the eyes, and the respiratory system, as well as systemic injuries. The initial dermal exposure to sulfur mustard is frequently unnoticed and injuries are delayed, developing over hours after exposure. Injuries first manifest as inflammation with erythema, followed by severe skin vesication (blistering). Blistering is preceded by significant morphological changes, including apoptosis and necrosis of skin cells that result in edema formation and separation of dermal and epidermal cell layers (Jones et al., 1997). While the mortality from SM skin injuries is relatively low, injuries are severe and incapacitating with long-term complications often requiring life-long treatment (Emadi et al., 2012). Skin wounds heal very slowly and patients continue to develop lesions and neoplasms years after the exposure (Emadi et al., 2012). Wound scarring is often associated with severe pruritus, further diminishing the quality of life of SM-exposed victims (Panahi et al., 2012).
At this time no mechanism-based antidote has received regulatory approval for the treatment of SM-induced skin injuries. The primary roadblock for countermeasures development are the complex toxicodynamics of SM (Kehe et al., 2009). The most plausible mechanism of toxicity of SM is that it acts as an alkylating agent, forming electrophilic intermediates that react with a gamut of biological matrices (Kehe et al., 2009). Decontamination approaches use chemical inactivation strategies either by oxidation, applying treatments containing iodine or bleach, or by reduction using agents such as glutathione or N-acetylcysteine, with some benefits (Jain et al., 2011; Jugg et al., 2013; Wormser et al., 1997). Limited preclinical and case studies suggest beneficial therapeutic benefits with calmodulin, oral antihistamines, anti-TNFα antibodies or antioxidant flavonones (Babin et al., 2000; Casillas et al., 2000; Graham and Schoneboom, 2013; Gu, 2014; Kadar et al., 2000; Kim et al., 1996; Poursaleh et al., 2012; Tewari-Singh et al., 2012). However, none of these potential therapeutics have been approved for clinical use. The current line of treatment for SM-induced injuries remains primarily symptomatic.
Transient receptor potential (TRP) ion channels play an important role in systemic and cellular chemical sensing. In peripheral sensory neurons, TRP channels serve as detectors for noxious chemical and physical stimuli, inducing pain and irritation as a warning signal for imminent injuries (Julius, 2013). TRPA1, also expressed in peripheral chemosensory neurons, was initially characterized as a target of noxious sulfur-containing natural products, including allyl isothiocyanate, the pungent ingredient in mustard, allicin (from garlic) and diallyl disulfide (in onions) (Bautista et al., 2005; Jordt et al., 2004). Subsequent studies revealed that TRPA1 is also a detector for reactive airborne environmental irritants, including acrolein, the major electrophilic smoke irritant, and related environmental and endogenous compounds (Bautista et al., 2006; Bessac and Jordt, 2008; Bessac et al., 2009; Chen and Chen, 1980; Dietrich et al., 2017). Acrolein was briefly used as a tear gas agent in World War I, leading to studies investigating the role of TRPA1 in pain and reflex responses elicited by modern tear gas agents and other warfare chemicals. Indeed, the tear gas agents, CN (2-chloroacetophenone), CS (2-chlorobenzalmalononitrile) and CR (dibenz[b,f][1,4]oxazepine) were found to be the most potent TRPA1 agonists (Bessac et al., 2009; Brone et al., 2008; Rothenberg et al., 2016). Mice deficient in TRPA1, or wild-type mice treated with a pharmacological TRPA1 inhibitor, lacked nocifensive behavioral responses to tear gas agent exposures (Bessac et al., 2009). Chlorine gas, also used as a warfare agent, and ozone were found to activate TRPA1, eliciting respiratory irritation and inflammation (Bessac and Jordt, 2008). Structure-function studies revealed that TRPA1 is a reactivity detector, activated by electrophiles and oxidants through covalent modification of specific cysteine residues located on the N-terminus of the channel (Hinman et al., 2006). In addition to their function in pain transduction, TRP channels in sensory neurons actively contribute to tissue inflammation by facilitating a mechanism known as neurogenic inflammation (Beard and Beard, 1989). TRP activation by noxious stimuli triggers the peripheral neurogenic release of pro-inflammatory neuropeptides such as CGRP (calcitonin gene-related peptide) and Substance P (SP) into the surrounding tissue. CGRP and Substance P activate immune cells and induce inflammatory tissue responses such as swelling, vasodilation, increase in blood flow and edema (Beard and Beard, 1989). In a mouse model of SM skin injury, pre-exposure treatment with olvanil, a TRPV1 ligand causing depletion of inflammatory neuropeptides, attenuated skin inflammation and edema and reduced Substance P immunoreactivity in the skin, suggesting that neurogenic inflammation contributes to the cutaneous toxicological effects of SM (Babin et al., 2000; Casbohm et al., 2004; Sabourin et al., 2003). Neurogenic inflammation mediated by CGRP has been implicated in trigeminal pain and migraine, leading to the development of CGRP receptor inhibitors that showed promising preclinical and clinical efficacy (Bell et al., 2012; Romero-Reyes et al., 2015; Voss et al., 2016).
Two recent studies reported that SM and CEES (2-chloroethyl-ethyl-sulfide), a widely used SM analog, activate human TRPA1 channels (Stenger et al., 2017; Stenger et al., 2015). Both compounds elicited Ca2+-influx into TRPA1-transfected HEK293 cells, leading to increased cellular toxicity (Stenger et al., 2017; Stenger et al., 2015). These effects were attenuated in the presence of a TRPA1 inhibitor, and by pre-incubation with N-acetyl-L-cysteine, a reducing agent (Stenger et al., 2017). The authors concluded that the alkylating effects of SM and CEES lead to TRPA1 activation, and suggest that TRPA1 activity contributes to the pathophysiological effects of SM and CEES vesicant injury. TRPA1 is present on cutaneous peptidergic sensory nerve endings and pharmacological inhibition of TRPA1 was reported to counteract chemically-induced swelling and inflammation of the mouse paw skin elicited by mustard oil and other electrophilic TRPA1 activators (Bautista et al., 2006; Bessac et al., 2009). TRPA1 inhibition also diminished skin inflammation and pruritus in allergic contact dermatitis elicited by environmental haptens such as urushiol, the poison ivy allergen known to elicit skin blistering (Buch et al., 2013b; Chen and Chen, 1980). Studies in rodents revealed that activation of TRPA1 by smoke irritants and tear gas agents triggered the release of neuropeptide from nerve endings in respiratory epithelia (Bessac et al., 2009; Kichko et al., 2015). Inhibition of TRPA1 attenuated asthmatic inflammation and diminished acute lung injury and mortality in acrolein exposure models, demonstrating the crucial role of TRPA1 in neurogenic inflammation and in the toxicological responses to severe chemical exposures (Achanta and Jordt, 2017; Conklin et al., 2016). However, in vivo evidence for a contribution of TRPA1 to vesicant injury mechanisms is lacking.
The objective of the present study was to investigate the effects of TRPA1 and CGRP inhibitors on cutaneous injury and inflammation in vivo in the CEES-induced mouse ear vesicant model (MEVM), a widely used model in vesicant research. Effects on skin pathology, edema formation and induction of inflammatory and mechanistic markers were studied, with the hope to discover novel leads for the development of mechanism-based countermeasures for the treatment of these severe chemical injuries.
2. Materials and Methods
2.1. Chemicals, animals, and drugs
2-chloroethyl ethyl sulfide (CEES), a monofunctional analog of sulfur mustard gas (SM; 2, 2′-dichloro diethyl sulfide) was purchased from Sigma-Aldrich Inc., MO, USA. C56BL/6 mice (male, 8-weeks old) were purchased from Charles River Laboratories, Wilmington, MA. Trpa1−/− mice were a gift from David Julius (University of California, San Francisco). The Trpa1-knock out allele was backcrossed into the C57BL/6NCrl background (>99.5%) by marker-assisted accelerated backcrossing (Charles River Laboratories). Mice were given at least 48 hr acclimation period before initiating studies. Mice were given access to mouse chow and water ad libitum. All animal protocols were approved by the IACUCs of Yale University, New Haven, CT and Duke University, Durham, NC. The TRPA1 antagonist, HC-030031, was a gift from Hydra Biosciences, Cambridge, MA; The TRPA1 antagonist, A-967079 was custom-synthesized by Medchem101 (Conshohocken, PA). Methylcellulose was purchased from Fluka (Buchs, Switzerland, Methocell MC, medium viscosity, 1200–1800 mPa.s.). MK-8825, a rodent CGRP receptor inhibitor, was a gift from Merck & Co., Inc.
2.2. Mouse-ear vesicant model (CEES MEVM)
Mice were anesthetized briefly with sevoflurane. Using a positive displacement pipette, 0.2 mg of 2-chloroethyl-ethyl-sulfide (CEES) in 20 μL dichloromethane (vehicle) were applied topically to the skin of the right ears, 10μL on each side of the ear, of male 8-week old mice. The left ears received 20 μL vehicle only, serving as controls. TRPA1 antagonist (HC-030031, intraperitoneally (10 μL/g BW) or A-967079, orally (10 μL/g BW) or vehicle (0.5% methylcellulose) was administered at a dose of 200 mg/kg body weight at 1 hr after topical exposure, and again at 8 and 16 hr post-CEES exposure, at 100 mg/kg each. MK-8825 was administered at 50 mg/kg body weight at 1, 8, and 16 hr post-CEES exposure by oral gavage, in 0.5% methylcellulose. Control mice were administered vehicles at the same time points in the respective route of administration. Animals were euthanized following American Veterinary Medical Association (AVMA) and IACUC guidelines 24 hr post-CEES exposure. Ear thickness was measured using a spring-loaded electronic calipers (Mitutoyo QUICKmini, Japan). Three 4 mm ear punch biopsies were excised (4 mm Biopsy Punch, Miltex Inc., York, PA, USA) and weighed to determine edema.
2.3. Determination of biomarkers
Ear punch biopsy samples were homogenized in lysis buffer (50 mM Tris-base, 150 mM NaCl, 5 mM EGTA supplemented with EDTA-free complete protease inhibitor (Roche Diagnostics GmbH, Mannheim, Germany) and 0.5% TritonX-100), using a Bullet Blender and Zirconium Oxide Beads (NextAdvance®, Averill Park, NY). Homogenization was performed for at least 20 minutes at full speed. Then, samples were centrifuged at 10,000g for 10 min at 4°C. Using the enzyme-linked immunosorbent assay (ELISA) (R&D Systems cytokine kits, Minneapolis, MN), we examined the concentrations of matrix metalloproteinase 9 (MMP-9), IL-1β (interleukin-1 beta), and KC/CXCL1(keratinocyte chemoattractant)/(chemokine (C-K-X motif) ligand 1) in homogenized supernatant protein extracts of ear punch biopsy samples. All samples were analyzed in at least in duplicate using an Infinite M200 Pro plate reader (Tecan, Germany). Following manufacturer’s instructions, the concentrations of cytokines were quantified using a standard curve or a 5-parameter logistic regression analysis. The concentrations of total protein in homogenate samples were determined using Pierce BCA protein assay (Thermo Scientific, Rockford, IL).
2.4. Histopathology
One punch biopsy was fixed in 10% formaldehyde, embedded in paraffin, sectioned at 5 μm thickness, and stained with hematoxylin and eosin (H&E) as per standard protocols. Images were obtained with a Zeiss Axio Imager Z1 microscope and analyzed by AxioVision Rel. 4.7 software (Zeiss, Munich, Germany). Cutaneous histopathology was assessed based on guidelines in Silny et al., 2005 (Silny et al., 2005).
2.5. Data Analysis and Statistics
Data were analyzed using GraphPad Prism 7 for Windows (GraphPad Software, Inc. La Jolla, CA, USA) software. Statistical difference was tested either by two-tailed t-test or by one-way ANOVA with Tukey’s Multiple Comparison Test. Error bars are standard error of mean estimates (SEM). Statistical significance is denoted by *p<0.05 or **p<0.01 or ***p<0.001 or ****p<0.0001.
3. Results
3.1. Skin edema and inflammation in the 2-chloroethyl ethyl sulfide (CEES)-induced mouse ear vesicant model (MEVM) in C57BL/6 mice
Previous studies used mice of the CD-1 or hairless (Cr1:SKH1-hrBR) strains to examine the effects of countermeasure candidate treatments on vesicant-induced inflammation and edema of the mouse skin (Mouse Ear Vesicant Model; MEVM) (Babin et al., 2000; Fisher and Sant’Ambrogio, 1982; Tewari-Singh et al., 2012). In the present study we investigated effects in C57BL/6 mice, a more widely used strain for which extensive pharmacological and pathophysiological datasets are available, including efficacy data for TRPA1- and neurogenic inflammation inhibitors. CEES (2-chloroethyl ethyl sulfide) was used to model the effects of SM. CEES is a monofunctional SM analog and, similar to SM, was demonstrated to activate TRPA1 channels in previous studies. While CEES does not cause the full spectrum of toxicological effects of SM, it produces major hallmarks of the vesicant injury process and is commonly used due to restricted availability of SM (Jain et al., 2011; Kehe et al., 2008; Stenger et al., 2015).
CEES (0.2 mg in 20 μL) was applied topically to the ear skin. To probe the role of TRPA1 in the cutaneous injury mechanism, the efficacies of two TRPA1 inhibitors were investigated, HC-030031 and A-967079, with different chemical scaffolds and binding sites within the target. Inhibitors were administered 1h (200mg/kg) after CEES exposure, and again at 8 hr and 16 hr (100mg/kg each), dosing regimens that proved efficacious in previous studies (Fig. 1A). Topical application of CEES to the ear skin of C57BL/6 mice resulted in an increase in ear thickness (39.37 ± 14.51 %) compared to vehicle (dichloromethane)-treated ears, measured 24h after exposure (Fig. 1B). The weights of circular ear punch biopsies excised from CEES-treated ears were higher (34.9 ± 16.8 %) than the weights of control biopsies (Fig. 1C). Exposure to CEES resulted in clear edema, desquamation of epidermis, increased leucocyte infiltration, and microvesication (Fig. 1D-ii).
Figure 1. Gross morphological parameters and histopathology in CEES-exposed mice, treated with TRPA1 inhibitors or vehicle.
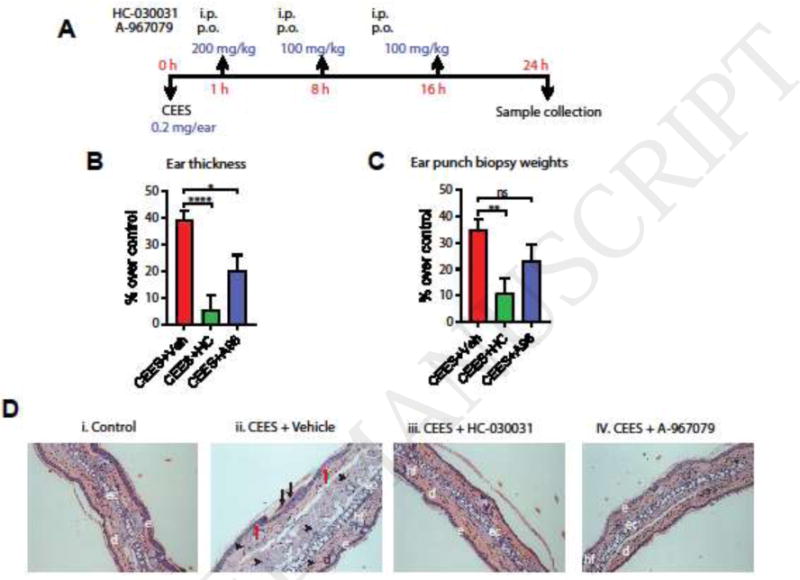
(A) CEES exposure and treatment regimen. TRPA1 inhibitors (HC-030031, i.p or A-967079, p.o) or vehicle (i.p or p.o) was administered at 1, 8, and 16 h after CEES exposure. (B and C) Ear thickness and ear punch biopsy weights. Ear thickness was measured by spring loaded calipers. Ear punch biopsy weights were measured from three 4 mm ear punch biopsy samples. Percent increase or decrease in ear thickness and ear punch biopsy weights over control is presented. (D) Representative H&E stained ear punch biopsy histopathology profiles are presented. e=epidermis; d=dermis; hf=hair follicle; ec=elastic cartilage; black arrows=desquamation and necrosis of epidermis; black arrow heads = infiltration of leucocytes; red arrows = microvesication; Data are presented as mean ± SEM. n=18/vehicle group; 10/treatment group. Statistical significance of the difference between the groups was determined by one-way ANOVA followed by Tukey’s multiple comparison post-hoc test. * p < 0.05; **p < 0.01; ****p<0.0001; ns=not significant.
3.2. Diminished skin inflammation and edema after treatment with TRPA1 inhibitors
Post-exposure treatment of CEES-exposed mice with TRPA1 inhibitors (Fig. 1A) reduced the increases in ear thickness and biopsy weights and diminished the histopathological signs of CEES injury (Fig. 1B–D). HC-030031 produced stronger effects (5.6 ± 16.86 %, p<0.001 ear thickness; 10.9 ± 17.9 %, p<0.01 ear punch biopsy weight) than A-967079 (20.3 ± 18.2 %, p<0.05 thickness, 23.45 ± 18.4 %, p>0.05 weight) at the dosages given. Corroborating previous studies in other mouse strains, CEES application increased skin protein levels of pro-inflammatory cytokine and chemokines markers such IL-1β and CXCL1/KC by approximately 6–10 fold (Fig. 2A)(Sabourin et al., 2002; Sabourin et al., 2000; Sabourin et al., 2003). We also measured levels of Matrix Metalloproteinase 9 (MMP-9) an enzyme implicated in vesication, promoting dermal-epidermal separation through its proteolytic activity (Shakarjian et al., 2006). MMP-9 skin protein levels were clearly increased in the CEES-exposed skin, and reduced by either TRPA1 inhibitor (Fig. 2B). TRPA1 inhibitors diminished the levels of pro-inflammatory cytokines and MMP-9 significantly compared to vehicle-treated animals.
Figure 2. Effects of TRPA1 inhibitor treatment on pro-inflammatory cytokines in ear punch biopsy samples of CEES-exposed mice.
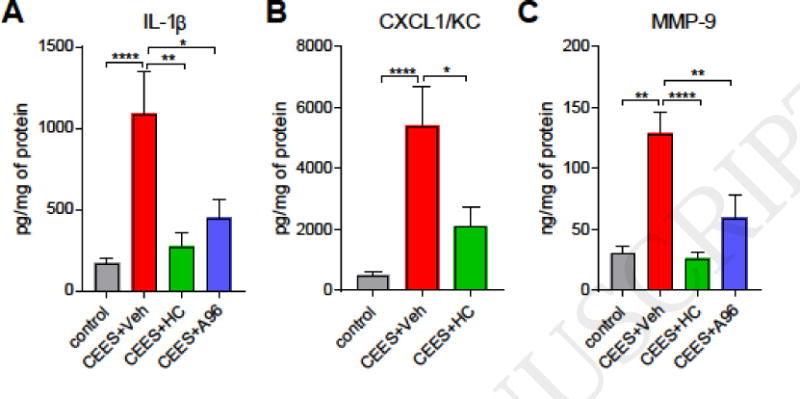
TRPA1 inhibitors (HC-030031, i.p or A-967079, p.o) or vehicle (i.p or p.o) were administered at 1, 8, and 16 h after CEES exposure. (A and B) Quantification of pro-inflammatory cytokines IL-1β and CXCL1/KC in tissue homogenates using ELISA. (C) Levels of MMP-9 in tissue homogenates determined by ELISA. Data are presented as mean ± SEM. n=41/control (dichloromethane only) measurments from all groups; 18/CEES+vehicle (0.5% methyl cellulose), and 9–10/treatment (CEES+HC or CEES+A96). Statistical significance of the difference between the groups was determined by one-way ANOVA followed by Tukey’s multiple comparison post-hoc test. *p < 0.05; **p < 0.01; ****p<0.0001.
We also investigated the response of TRPA1-deficient mice to CEES exposure of the ear skin. In Trpa1−/− mice CEES induced similar or exaggerated edema formation and MMP-9 expression as in WT mice (Supplementary Figure S1). Treatment of CEES-exposed Trpa1−/− mice with the TRPA1 inhibitor, HC-030031, did not reduce ear swelling (Supplementary Figure S1).
3.3. A small molecule CGRP inhibitor ameliorates CEES-induced skin injury and inflammation
To examine the role of CGRP, the neurogenic inflammatory neuropeptide, in CEES-induced skin injury a small molecule CGRP receptor inhibitor, MK-8825, was administered orally (Fig. 3A). MK-8825 was developed to specifically inhibit rodent CGRP receptors since human CGRP receptor antagonists have poor efficacies in rodents (Bell et al., 2012). Exposure to CEES resulted in elevated levels of CGRP in ear punch biopsy homogenate samples whereas CGRP levels were non-detectable in naïve and vehicle (dichloromethane) exposed samples (Figure 3B). MK-8825, administered at dosages within a range previously demonstrated to induce analgesia in mice, partially inhibited the CEES-induced increase in ear thickness and ear punch biopsy weights (Fig. 3C, D) (Daiutolo et al., 2016; Romero-Reyes et al., 2015). CGRP inhibitor treatment also resulted in a decrease in levels of MMP-9, IL-1β, but not in CXCL1/KC (Fig. 4).
Figure 3. Gross morphological parameters in CEES-exposed mice, treated with CGRP antagonist or vehicle.
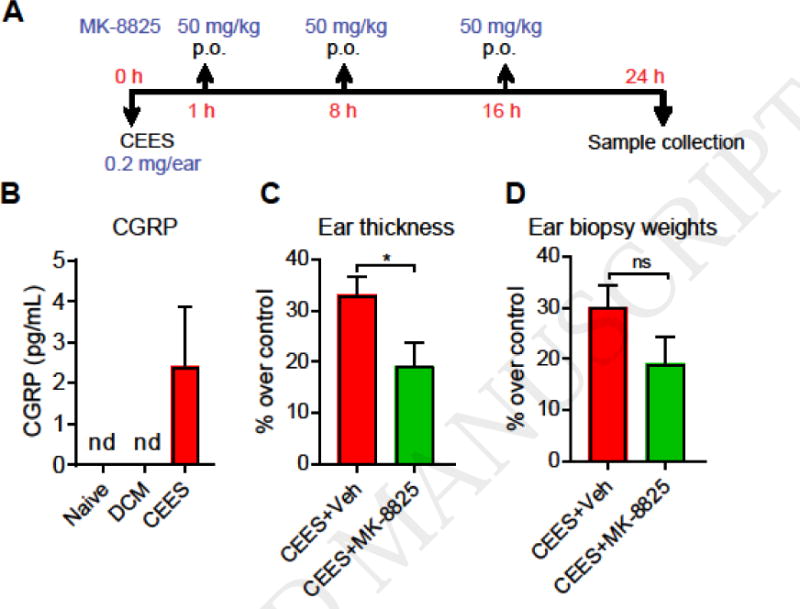
(A) CEES exposure and treatment regimen. CGRP antagonist (MK-8825, p.o) or vehicle (p.o) was administered at 1, 8, and 16 h after CEES exposure. (B) CGRP levels in ear punch biopsy homogenate samples, determined by ELISA (C) Ear thickness, and (D) ear punch biopsy weights. Ear thickness was measured by spring loaded calipers. Ear punch biopsy weights were measured from three 4 mm ear punch biopsy samples. Percent increase or decrease in ear thickness and ear punch biopsy weights over control is presented. Data are presented as mean ± SEM. n= 4–6/group (B), 8–9/group (C and D). Statistical significance of the difference between the groups was determined by two-tailed unpaired t-test. *p < 0.05; ns= non-significant; nd = non-detectable.
Figure 4. Effects of CGRP inhibition on levels of pro-inflammatory cytokines and injury mediators in ear skin of CEES-exposed mice, treated with CGRP antagonist or vehicle.
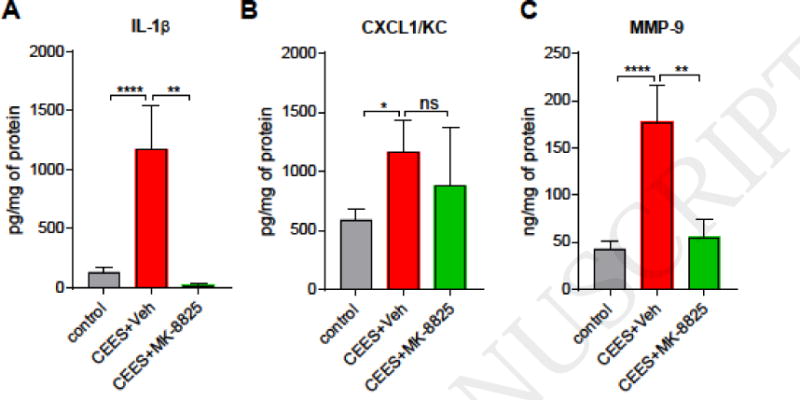
CGRP antagonist (MK-8825, p.o) or vehicle (p.o) was administered at 1, 8, and 16 h after CEES exposure. (A and B) Pro-inflammatory cytokines IL-1β and CXCL1/KC were quantified in tissue homogenates using ELISA (C) Levels of MMP-9 in tissue homogenates, determined by ELISA. Data are presented as mean ± SEM. For MMP-9 and IL-1β: n=20-22/control (dichloromethane only) ears from all groups, 9/CEES+vehicle (methyl cellulose) ears, and 6–8/treatment (CEES+MK-8825) ears; For KC: n=12/control (dichloromethane only) ears, 8/CEES+vehicle (0.5% methyl cellulose) ears, and 7/treatment (CEES+MK-8825) ears. Statistical significance of the difference between the groups was determined by one-way ANOVA followed by Tukey’s multiple comparison post-hoc test. *p < 0.05; **p < 0.01; ****p<0.0001; ns = non-significant.
4. Discussion and Conclusions
This study, using the CEES-induced mouse ear vesicant model, demonstrates that pharmacological inhibition of TRPA1, or inhibition of neurogenic inflammation with a CGRP inhibitor, counteract edema formation and inflammation of the mouse skin when administered after cutaneous vesicant exposure. Inhibition of these targets diminished gross tissue and histopathological hallmarks of skin vesicant injury, including swelling, microvesication, desquamation and leukocyte infiltration. On the molecular level, TRPA1- or CGRP inhibition reduced skin levels of key pro-inflammatory biomarkers, and levels of MMP-9, an enzyme promoting extracellular matrix degradation, a key mechanism of vesicant skin injury (Shakarjian et al., 2006).
These findings expand the role of TRPA1 as a key sensor of chemical threat agents and an important mediator of their pathophysiological effects. While the role of TRPA1 in the acute noxious effects of chlorine, acrolein and tear gas agents is firmly established the present study adds vesicants as another group of threats acting, at least in part, through TRPA1 and subsequent neurogenic inflammatory mechanisms in vivo. However, there are key differences between the in vivo actions of the former and those of vesicants. While chlorine, acrolein, and tear gas agents cause immediate severe pain and reflex responses such as cough, secretions, blepharospasm, retching and vomiting, the actions of vesicants are usually delayed and reports describing immediate noxious effects of SM exposures are scarce (Shakarjian et al., 2010). While CEES and SM were shown to activate TRPA1 in vitro, concentrations exceeding 4mM CEES were required to detect robust TRPA1-mediated Ca2+-influx into heterologous cells, with an EC50 of approximately 7.5mM, while SM was shown to activate TRPA1 at a concentration of 0.5mM (Stenger et al., 2015). In contrast, tear gas agents activate TRPA1 with much higher potencies, with EC50s in the upper picomolar or lower nanomolar range (Bessac et al., 2009; Brone et al., 2008). Acrolein, allyl isothiocyanate, and chlorine are also more potent TRPA1 agonists, with EC50s in the lower micromolar range (Bautista et al., 2006; Bessac and Jordt, 2008; Jordt et al., 2004). It can, therefore, be hypothesized that direct effects of vesicants on TRPA1 require a longer time to manifest in vivo and may strongly depend on the site of exposure, vesicant concentration, and distribution over the affected tissue area. TRPA1 activation may further depend on the kinetics of chemical conversions that vesicants undergo in the exposed tissue in vivo. SM forms electrophilic sulfonium- and carbenium-ions in tissue that very likely would react with the electrophile acceptor sites in the TRPA1 protein, leading to channel activation (Hinman et al., 2006; Kehe et al., 2009). The observation of inhibitory effects of N-acetylcysteine (NAC) on vesicant-induced TRPA1 activation supports the requirement for chemical conversion of vesicants to electrophilic chemical species prior to channel activation (Stenger et al., 2017). Finally, products of oxidative stress generated subsequent to vesicant exposures may also contribute to the TRPA1 activity. Vesicants induce lipid peroxidation and nitric oxide production in the exposed tissues (Kehe et al., 2009). Vesicant-exposed tissues contain elevated levels of MDA (malon dialdehyde) and 4-HNE (4-hydroxy-nonenal), a lipid peroxidation product validated as an endogenous agonist of TRPA1 (Alizadeh et al., 2008; O’Neill et al., 2010; Trevisani et al., 2007; Vijayaraghavan et al., 1991). Short-lived oxygen radicals and hypochlorite, produced by infiltrating neutrophils, also activate TRPA1 (Bessac and Jordt, 2008). Together, the delayed chemical conversion of vesicants and secondary effects through inflammatory mediators can explain the delayed onset of TRPA1-mediated pathobiological effects.
In the present study, we examined the therapeutic effects of two TRPA1 inhibitors, HC-030031 and A-967079, derived from two independent inhibitor screens and with different chemical scaffolds (McGaraughty et al., 2010; McNamara et al., 2007). While both inhibitors were administered at saturating levels, HC-030031 was more efficacious in reducing edema and levels of inflammatory mediators than A-967079. While this difference may be explained by divergent pharmacokinetics or molecular efficacies due to their different binding sites within the target, AP-18, a TRPA1 inhibitor structurally almost identical to A-967079, was shown to completely block TRPA1-mediated Ca2+-signals in heterologous cells (Stenger et al., 2015). HC-030031 was recently found to also inhibit TRPV1 channels, suggesting that this compound may be less selective than A-967079 (Iwasaki et al., 2009). TRPV1, also the target of olvanil, a compound shown to diminish SM-induced skin injury responses when administered pre-exposure, may, therefore, represent an additional anti-vesicant target worth examining as potent and selective inhibitors are now available (Babin et al., 2000).
In TRPA1-deficient mice responses to CEES were not attenuated. Paradoxically, these mice showed similar or exaggerated edema formation and MMP-9 production compared to WT mice. This response unmasked a potential anti-inflammatory role of TRPA1 in the onset of the chemical injury. In these mice, TRPA1 activity is absent at the onset and during CEES exposure, while inhibitor treatments in WT mice only started 1 hour after CEES exposure. It is possible that TRPA1 is crucial for the animals to develop a normal nociceptive and inflammatory response in the time immediately after exposure, with TRPA1 playing a balanced pro- and anti-inflammatory role, preventing an exaggerated response that becomes visible in the Trpa1−/− mice. Alternatively, this effect may be due to compensatory mechanisms in the Trpa1−/− strain. Indeed, compensation was observed in a prior study in Trpa1−/− mice in the Complete Freund Adjuvant (CFA) inflammatory pain model (Petrus et al., 2007). These mice showed a paradoxical increase in mechanical hyperalgesia, while a TRPA1 inhibitor, AP-18, effectively blocked mechanical hyperalgesia in WT mice (Petrus et al., 2007). This inhibitor was ineffective in Trpa1−/− mice, corroborating on-target selectivity (Petrus et al., 2007). HC-030031, the TRPA1 inhibitor used here, did not significantly diminish CEES-induced edema in TRPA1-deficient mice, also suggesting an on-target mechanism of action. A trend toward mild anti-inflammatory effects may be visible, possibly due to the reported effects of this inhibitor on TRPV1 (Iwasaki et al., 2009).
TRPA1 and TRPV1 activity trigger neurogenic inflammation that was implicated as a key injury mechanism following vesicant exposures. In the present study, inhibition of CGRP-signaling, a major component of the neurogenic inflammatory response, diminished vesicant-induced edema formation (ear thickness) and inflammation, including the production of IL-1β and MMP-9. The results from this study and previous reports suggest that IL-1β and MMP-9 are potential biomarkers to study CGRP-evoked pain and inflammation (Kawasaki et al., 2008; Opree and Kress, 2000; Ren and Torres, 2009). CGRP inhibitor treatment did not diminish levels of CXCL1/KC, suggesting that TRPA1 activity may engage additional neurogenic mechanisms, such as the release of Substance P. The effects of the neurokinin 1 receptor inhibitors in the CEES model remain to be explored. We detected increased levels of CGRP peptide in the CEES-exposed skin 24 hours after exposure, suggesting that CGRP not only mediates acute neurogenic inflammation, but may contribute to the sustained inflammatory response to the vesicant. It remains to be explored whether CEES exposures induce increased CGRP expression in the skin-innervating nerves, or whether CGRP in skin-resident or infiltrating cells contributes to the observed increased levels. In addition to sensory nerves, CGRP expression was detected in monocytes, including stimulated macrophages, that infiltrate injured tissues (Bracci-Laudiero et al., 2005; Granstein et al., 2015). CGRP-inhibitors, either small molecule antagonists of CGRP receptor or monoclonal antibodies targeting the CGRP peptide, are currently in late-stage clinical trials for pain indications, with so far promising outcomes (Schuster and Rapoport, 2016). The roles of TRPV1 and of Substance P in vesicant injury also need to be further explored using specific inhibitors and knockout mice, since these targets also contribute to neurogenic inflammation.
The initial cutaneous symptoms of vesicant exposure include itching sensations that begin even before erythema is visible and blisters are formed. The sensation of itch is initiated by excitation of a specialized subset of peripheral sensory neurons, the pruriceptors (Abara et al., 2014). Intriguingly, TRPA1, in addition to its role in pain, was identified as a major integrator of pruritic signals in pruriceptors (Abara et al., 2014). TRPA1 is activated downstream of pruritogen receptors for itch-inducing chemicals such as chloroquine, and for endogenous pruritogens (Wilson et al., 2011). Thus, the initial itching sensed by vesicant exposure victims may be the first indication of TRPA1 activity. TRPA1 was also found to be critical for chronic itch and inflammation in models of acute and chronic contact dermatitis and atopic dermatitis (Buch et al., 2013a; Wilson and Takata, 2013). Chronic debilitating itch originating from the injured skin region is a common complaint of vesicant victims, even decades after exposure (Panahi et al., 2012; Panahi et al., 2013). While some clinical interventions were reported to alleviate itch in patients, TRPA1 inhibition, and inhibition of neurogenic inflammation, may represent additional anti-pruritic strategies worth exploring (Panahi et al., 2012). It is likely that the slow and often incomplete wound repair of vesicant skin exposure sites leads to abnormal re-innervation, permanent neuropathic damage, and continuing inflammation. Whether TRPA1 inhibition confers neuroprotection and promotes wound repair remains to be explored.
Taken together, the present study suggests that inhibition of TRPA1 and neurogenic inflammation in vivo counteracts CEES-induced skin injury and inflammation. Future studies should examine whether the therapeutic effects of TRPA1 and CGRP inhibitors are sustained in extended observation models and whether they have similar effects in models of exposures to other vesicants, including SM, nitrogen mustard or Lewisite. The outcomes from this study pave the way for further development of these potential therapeutic agents under US FDA’s animal rule.
Supplementary Material
Highlights.
CEES, a sulfur mustard analog, induced mouse ear vesicant model was developed
TRPA1 inhibitors (HC-030031 and A-967079) ameliorated CEES-induced skin injuries
CGRP inhibitor (MK-8825) reduced skin edema and pro-inflammatory cytokines
Potential targets & countermeasures for sulfur mustard skin injuries were identified
Acknowledgments
This work was supported by cooperative agreement U01ES015674 to SEJ of the NIH Countermeasures Against Chemical Threats (CounterACT) program. The content is solely the responsibility of the author and does not necessarily represent the views of the NIH.
Abbreviations
- CEES
2-chloroethyl ethyl sulfide
- CGRP
calcitonin gene-related peptide
- ELISA
Enzyme-linked immunosorbent assay
- i.p
intraperitoneal
- IL-1β
Interleukin-1beta
- KC/CXCL1
Keratinocyte chemoattractant/Chemokine (C-X-C motif) ligand 1
- MMP-9
Matrix metalloproteinase 9
- MEVM
Mouse ear vesicant model
- NKA
neurokinin A
- p.o
per os (orally)
- SM
Sulfur mustard bis-(2-chloroethyl) sulfide
- SP
substance P
- TRP
Transient receptor potential
- TRPA1
Transient receptor potential Ankyrin 1 channel
- TRPV1
Transient receptor potential vanilloid 1 channel
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
Sven-Eric Jordt is serving on the Scientific Advisory Board of Hydra Biosciences Inc., a biopharmaceutical company developing TRP ion channel inhibitors for the treatment of pain and inflammation.
References
- Abara W, Wilson S, Vena J, Sanders L, Bevington T, Culley JM, Annang L, Dalemarre L, Svendsen E. Engaging a chemical disaster community: lessons from Graniteville. Int J Environ Res Public Health. 2014;11:5684–5697. doi: 10.3390/ijerph110605684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achanta S, Jordt SE. TRPA1: Acrolein meets its target. Toxicology and applied pharmacology. 2017;324:45–50. doi: 10.1016/j.taap.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alizadeh AH, Ranjbar M, Yadollahzadeh M. Patient concerns regarding chronic hepatitis B and C infection. East Mediterr Health J. 2008;14:1142–1147. [PubMed] [Google Scholar]
- Babin MC, Ricketts K, Skvorak JP, Gazaway M, Mitcheltree LW, Casillas RP. Systemic administration of candidate antivesicants to protect against topically applied sulfur mustard in the mouse ear vesicant model (MEVM) J Appl Toxicol. 2000;20(Suppl 1):S141–144. doi: 10.1002/1099-1263(200012)20:1+<::aid-jat666>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Bautista DM, Jordt SE, Nikai T, Tsuruda PR, Read AJ, Poblete J, Yamoah EN, Basbaum AI, Julius D. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell. 2006;124:1269–1282. doi: 10.1016/j.cell.2006.02.023. [DOI] [PubMed] [Google Scholar]
- Bautista DM, Movahed P, Hinman A, Axelsson HE, Sterner O, Hogestatt ED, Julius D, Jordt SE, Zygmunt PM. Pungent products from garlic activate the sensory ion channel TRPA1. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:12248–12252. doi: 10.1073/pnas.0505356102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard GB, Beard DM. Geriatric dentistry. Vet Clin North Am Small Anim Pract. 1989;19:49–74. doi: 10.1016/s0195-5616(89)50005-6. [DOI] [PubMed] [Google Scholar]
- Bell IM, Stump CA, Gallicchio SN, Staas DD, Zartman CB, Moore EL, Sain N, Urban M, Bruno JG, Calamari A, Kemmerer AL, Mosser SD, Fandozzi C, White RB, Zrada MM, Selnick HG, Graham SL, Vacca JP, Kane SA, Salvatore CA. MK-8825: a potent and selective CGRP receptor antagonist with good oral activity in rats. Bioorg Med Chem Lett. 2012;22:3941–3945. doi: 10.1016/j.bmcl.2012.04.105. [DOI] [PubMed] [Google Scholar]
- Bessac BF, Jordt SE. Breathtaking TRP channels: TRPA1 and TRPV1 in airway chemosensation and reflex control. Physiology (Bethesda) 2008;23:360–370. doi: 10.1152/physiol.00026.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessac BF, Sivula M, von Hehn CA, Caceres AI, Escalera J, Jordt SE. Transient receptor potential ankyrin 1 antagonists block the noxious effects of toxic industrial isocyanates and tear gases. FASEB J. 2009;23:1102–1114. doi: 10.1096/fj.08-117812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracci-Laudiero L, Aloe L, Caroleo MC, Buanne P, Costa N, Starace G, Lundeberg T. Endogenous NGF regulates CGRP expression in human monocytes, and affects HLA-DR and CD86 expression and IL-10 production. Blood. 2005;106:3507–3514. doi: 10.1182/blood-2004-10-4055. [DOI] [PubMed] [Google Scholar]
- Brone B, Peeters PJ, Marrannes R, Mercken M, Nuydens R, Meert T, Gijsen HJ. Tear gasses CN, CR, and CS are potent activators of the human TRPA1 receptor. Toxicology and applied pharmacology. 2008;231:150–156. doi: 10.1016/j.taap.2008.04.005. [DOI] [PubMed] [Google Scholar]
- Buch T, Schafer E, Steinritz D, Dietrich A, Gudermann T. Chemosensory TRP channels in the respiratory tract: role in toxic lung injury and potential as “sweet spots” for targeted therapies. Rev Physiol Biochem Pharmacol. 2013a;165:31–65. doi: 10.1007/112_2012_10. [DOI] [PubMed] [Google Scholar]
- Buch TR, Schafer EA, Demmel MT, Boekhoff I, Thiermann H, Gudermann T, Steinritz D, Schmidt A. Functional expression of the transient receptor potential channel TRPA1, a sensor for toxic lung inhalants, in pulmonary epithelial cells. Chem Biol Interact. 2013b;206:462–471. doi: 10.1016/j.cbi.2013.08.012. [DOI] [PubMed] [Google Scholar]
- Casbohm SL, Rogers JV, Stonerock MK, Martin JL, Ricketts-Kaminsky KM, Babin MC, Casillas RP, Sabourin CL. Localization of substance P gene expression for evaluating protective countermeasures against sulfur mustard. Toxicology. 2004;204:229–239. doi: 10.1016/j.tox.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Casillas RP, Kiser RC, Truxall JA, Singer AW, Shumaker SM, Niemuth NA, Ricketts KM, Mitcheltree LW, Castrejon LR, Blank JA. Therapeutic approaches to dermatotoxicity by sulfur mustard. I. Modulaton of sulfur mustard-induced cutaneous injury in the mouse ear vesicant model. J Appl Toxicol. 2000;20(Suppl 1):S145–151. doi: 10.1002/1099-1263(200012)20:1+<::aid-jat665>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Chen CH, Chen SC. Angiogenic activity of vitreous and retinal extract. Investigative ophthalmology & visual science. 1980;19:596–602. [PubMed] [Google Scholar]
- Conklin DJ, Haberzettl P, Jagatheesan G, Kong M, Hoyle GW. Role of TRPA1 in acute cardiopulmonary toxicity of inhaled acrolein. Toxicol Appl Pharmacol. 2016 doi: 10.1016/j.taap.2016.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daiutolo BV, Tyburski A, Clark SW, Elliott MB. Trigeminal Pain Molecules, Allodynia, and Photosensitivity Are Pharmacologically and Genetically Modulated in a Model of Traumatic Brain Injury. Journal of neurotrauma. 2016;33:748–760. doi: 10.1089/neu.2015.4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich A, Steinritz D, Gudermann T. Transient receptor potential (TRP) channels as molecular targets in lung toxicology and associated diseases. Cell calcium. 2017 doi: 10.1016/j.ceca.2017.04.005. [DOI] [PubMed] [Google Scholar]
- Emadi SN, Babamahmoodi F, Poursaleh Z, Sayad-Noori SS, Soroush MR, Maleki AR, Izadi M, Khodaei-Ardakan MR, Emadi SE. Sezary syndrome, Kaposi sarcoma and generalized dermatophytosis 15 years after sulfur mustard gas exposure. Journal of dermatological case reports. 2012;6:86–89. doi: 10.3315/jdcr.2012.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher JT, Sant’Ambrogio G. Effects of inhaled CO2 on airway stretch receptors in the newborn dog. J Appl Physiol. 1982;53:1461–1465. doi: 10.1152/jappl.1982.53.6.1461. [DOI] [PubMed] [Google Scholar]
- Golomb BA, Marshall GN, Harley NH, Spektor DM, Cecchine G, Hilborne LH, United States Department of Defense Office of the Secretary of Defense., National Defense Research Institute (U.S.) A review of the scientific literature as it pertains to Gulf War illnesses. Rand; Santa Monica, CA: 1998. <1-2, 4-8>. [Google Scholar]
- Graham JS, Schoneboom BA. Historical perspective on effects and treatment of sulfur mustard injuries. Chemico-biological interactions. 2013;206:512–522. doi: 10.1016/j.cbi.2013.06.013. [DOI] [PubMed] [Google Scholar]
- Granstein RD, Wagner JA, Stohl LL, Ding W. Calcitonin gene-related peptide: key regulator of cutaneous immunity. Acta physiologica (Oxford, England) 2015;213:586–594. doi: 10.1111/apha.12442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu TY. Mechanism and treatment of sulfur mustard-induced cutaneous injury. Chin J Traumatol. 2014;17:345–350. [PubMed] [Google Scholar]
- Hinman A, Chuang HH, Bautista DM, Julius D. TRP channel activation by reversible covalent modification. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:19564–19568. doi: 10.1073/pnas.0609598103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki Y, Tanabe M, Kayama Y, Abe M, Kashio M, Koizumi K, Okumura Y, Morimitsu Y, Tominaga M, Ozawa Y, Watanabe T. Miogadial and miogatrial with alpha, beta-unsaturated 1,4-dialdehyde moieties–novel and potent TRPA1 agonists. Life sciences. 2009;85:60–69. doi: 10.1016/j.lfs.2009.04.017. [DOI] [PubMed] [Google Scholar]
- Jain AK, Tewari-Singh N, Gu M, Inturi S, White CW, Agarwal R. Sulfur mustard analog, 2-chloroethyl ethyl sulfide-induced skin injury involves DNA damage and induction of inflammatory mediators, in part via oxidative stress, in SKH-1 hairless mouse skin. Toxicology letters. 2011;205:293–301. doi: 10.1016/j.toxlet.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BC, Tyman CA, Smith DA. Identification of the cytochrome P450 isoforms involved in the O-demethylation of 4-nitroanisole in human liver microsomes. Xenobiotica. 1997;27:1025–1037. doi: 10.1080/004982597240000. [DOI] [PubMed] [Google Scholar]
- Jordt SE, Bautista DM, Chuang HH, McKemy DD, Zygmunt PM, Hogestatt ED, Meng ID, Julius D. Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature. 2004;427:260–265. doi: 10.1038/nature02282. [DOI] [PubMed] [Google Scholar]
- Jugg B, Fairhall S, Smith A, Rutter S, Mann T, Perrott R, Jenner J, Salguero J, Shute J, Sciuto AM. N-acetyl-L-cysteine protects against inhaled sulfur mustard poisoning in the large swine. Clinical toxicology (Philadelphia, Pa) 2013;51:216–224. doi: 10.3109/15563650.2013.780208. [DOI] [PubMed] [Google Scholar]
- Julius D. TRP channels and pain. Annual review of cell and developmental biology. 2013;29:355–384. doi: 10.1146/annurev-cellbio-101011-155833. [DOI] [PubMed] [Google Scholar]
- Kadar T, Fishbeine E, Meshulam Y, Sahar R, Chapman S, Liani H, Barness I, Amir A. Treatment of skin injuries induced by sulfur mustard with calmodulin antagonists, using the pig model. J Appl Toxicol. 2000;20(Suppl 1):S133–136. doi: 10.1002/1099-1263(200012)20:1+<::aid-jat668>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Xu ZZ, Wang X, Park JY, Zhuang ZY, Tan PH, Gao YJ, Roy K, Corfas G, Lo EH, Ji RR. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat Med. 2008;14:331–336. doi: 10.1038/nm1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehe K, Balszuweit F, Emmler J, Kreppel H, Jochum M, Thiermann H. Sulfur mustard research–strategies for the development of improved medical therapy. Eplasty. 2008;8:e32. [PMC free article] [PubMed] [Google Scholar]
- Kehe K, Balszuweit F, Steinritz D, Thiermann H. Molecular toxicology of sulfur mustard-induced cutaneous inflammation and blistering. Toxicology. 2009;263:12–19. doi: 10.1016/j.tox.2009.01.019. [DOI] [PubMed] [Google Scholar]
- Kichko TI, Kobal G, Reeh PW. Cigarette smoke has sensory effects through nicotinic and TRPA1 but not TRPV1 receptors on the isolated mouse trachea and larynx. American journal of physiology Lung cellular and molecular physiology. 2015;309:L812–820. doi: 10.1152/ajplung.00164.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YB, Hur GH, Choi DS, Shin S, Han BG, Lee YS, Sok DE. Effects of calmodulin antagonists and anesthetics on the skin lesions induced by 2-chloroethylethyl sulfide. Eur J Pharmacol. 1996;313:107–114. doi: 10.1016/0014-2999(96)00504-3. [DOI] [PubMed] [Google Scholar]
- McGaraughty S, Chu KL, Perner RJ, Didomenico S, Kort ME, Kym PR. TRPA1 modulation of spontaneous and mechanically evoked firing of spinal neurons in uninjured, osteoarthritic, and inflamed rats. Molecular pain. 2010;6:14. doi: 10.1186/1744-8069-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara CR, Mandel-Brehm J, Bautista DM, Siemens J, Deranian KL, Zhao M, Hayward NJ, Chong JA, Julius D, Moran MM, Fanger CM. TRPA1 mediates formalin-induced pain. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:13525–13530. doi: 10.1073/pnas.0705924104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill HC, White CW, Veress LA, Hendry-Hofer TB, Loader JE, Min E, Huang J, Rancourt RC, Day BJ. Treatment with the catalytic metalloporphyrin AEOL 10150 reduces inflammation and oxidative stress due to inhalation of the sulfur mustard analog 2-chloroethyl ethyl sulfide. Free radical biology & medicine. 2010;48:1188–1196. doi: 10.1016/j.freeradbiomed.2010.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opree A, Kress M. Involvement of the proinflammatory cytokines tumor necrosis factor-alpha, IL-1 beta, and IL-6 but not IL-8 in the development of heat hyperalgesia: effects on heat-evoked calcitonin gene-related peptide release from rat skin. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2000;20:6289–6293. doi: 10.1523/JNEUROSCI.20-16-06289.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panahi Y, Sahebkar A, Parvin S, Saadat A. A randomized controlled trial on the anti-inflammatory effects of curcumin in patients with chronic sulphur mustard-induced cutaneous complications. Annals of clinical biochemistry. 2012;49:580–588. doi: 10.1258/acb.2012.012040. [DOI] [PubMed] [Google Scholar]
- Panahi Y, Taherzadeh ES, Davoudi SM, Sahebkar A, Ranjbar R. Investigation of serum substance P status in patients with chronic pruritic skin lesions due to sulfur mustard: a cross-sectional study. Cutaneous and ocular toxicology. 2013;32:4–8. doi: 10.3109/15569527.2012.686077. [DOI] [PubMed] [Google Scholar]
- Petrus M, Peier AM, Bandell M, Hwang SW, Huynh T, Olney N, Jegla T, Patapoutian A. A role of TRPA1 in mechanical hyperalgesia is revealed by pharmacological inhibition. Molecular pain. 2007;3:40. doi: 10.1186/1744-8069-3-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poursaleh Z, Ghanei M, Babamahmoodi F, Izadi M, Harandi AA, Emadi SE, Taghavi NO, Sayad-Nouri SS, Emadi SN. Pathogenesis and treatment of skin lesions caused by sulfur mustard. Cutaneous and ocular toxicology. 2012;31:241–249. doi: 10.3109/15569527.2011.636119. [DOI] [PubMed] [Google Scholar]
- Ren K, Torres R. Role of interleukin-1 beta during pain and inflammation. Brain Res Rev. 2009;60:57–64. doi: 10.1016/j.brainresrev.2008.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Reyes M, Pardi V, Akerman S. A potent and selective calcitonin gene-related peptide (CGRP) receptor antagonist, MK-8825, inhibits responses to nociceptive trigeminal activation: Role of CGRP in orofacial pain. Exp Neurol. 2015;271:95–103. doi: 10.1016/j.expneurol.2015.05.005. [DOI] [PubMed] [Google Scholar]
- Rothenberg C, Achanta S, Svendsen ER, Jordt SE. Tear gas: an epidemiological and mechanistic reassessment. Ann N Y Acad Sci. 2016 doi: 10.1111/nyas.13141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabourin CL, Danne MM, Buxton KL, Casillas RP, Schlager JJ. Cytokine, chemokine, and matrix metalloproteinase response after sulfur mustard injury to weanling pig skin. J Biochem Mol Toxicol. 2002;16:263–272. doi: 10.1002/jbt.10050. [DOI] [PubMed] [Google Scholar]
- Sabourin CL, Petrali JP, Casillas RP. Alterations in inflammatory cytokine gene expression in sulfur mustard-exposed mouse skin. J Biochem Mol Toxicol. 2000;14:291–302. doi: 10.1002/1099-0461(2000)14:6<291::AID-JBT1>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Sabourin CLK, Danne MM, Buxton KL, Rogers JV, Niemuth NA, Blank JA, Babin MC, Casillas RP. Modulation of sulfur mustard-induced inflammation and gene expression by olvanil in the hairless mouse vesicant model. Journal of Toxicology-Cutaneous and Ocular Toxicology. 2003;22:125–136. [Google Scholar]
- Schuster NM, Rapoport AM. New strategies for the treatment and prevention of primary headache disorders. Nat Rev Neurol. 2016;12:635–650. doi: 10.1038/nrneurol.2016.143. [DOI] [PubMed] [Google Scholar]
- Shakarjian MP, Bhatt P, Gordon MK, Chang YC, Casbohm SL, Rudge TL, Kiser RC, Sabourin CL, Casillas RP, Ohman-Strickland P, Riley DJ, Gerecke DR. Preferential expression of matrix metalloproteinase-9 in mouse skin after sulfur mustard exposure. J Appl Toxicol. 2006;26:239–246. doi: 10.1002/jat.1134. [DOI] [PubMed] [Google Scholar]
- Shakarjian MP, Heck DE, Gray JP, Sinko PJ, Gordon MK, Casillas RP, Heindel ND, Gerecke DR, Laskin DL, Laskin JD. Mechanisms mediating the vesicant actions of sulfur mustard after cutaneous exposure. Toxicological sciences: an official journal of the Society of Toxicology. 2010;114:5–19. doi: 10.1093/toxsci/kfp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silny W, Czarnecka-Operacz M, Silny P. The new scoring system for evaluation of skin inflammation extent and severity in patients with atopic dermatitis. Acta Dermatovenerol Croat. 2005;13:219–224. [PubMed] [Google Scholar]
- Stenger B, Popp T, John H, Siegert M, Tsoutsoulopoulos A, Schmidt A, Muckter H, Gudermann T, Thiermann H, Steinritz D. N-Acetyl-L-cysteine inhibits sulfur mustard-induced and TRPA1-dependent calcium influx. Archives of toxicology. 2017;91:2179–2189. doi: 10.1007/s00204-016-1873-x. [DOI] [PubMed] [Google Scholar]
- Stenger B, Zehfuss F, Muckter H, Schmidt A, Balszuweit F, Schafer E, Buch T, Gudermann T, Thiermann H, Steinritz D. Activation of the chemosensing transient receptor potential channel A1 (TRPA1) by alkylating agents. Archives of toxicology. 2015;89:1631–1643. doi: 10.1007/s00204-014-1414-4. [DOI] [PubMed] [Google Scholar]
- Tewari-Singh N, Jain AK, Inturi S, Agarwal C, White CW, Agarwal R. Silibinin attenuates sulfur mustard analog-induced skin injury by targeting multiple pathways connecting oxidative stress and inflammation. PloS one. 2012;7:e46149. doi: 10.1371/journal.pone.0046149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevisani M, Siemens J, Materazzi S, Bautista DM, Nassini R, Campi B, Imamachi N, Andre E, Patacchini R, Cottrell GS, Gatti R, Basbaum AI, Bunnett NW, Julius D, Geppetti P. 4-Hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:13519–13524. doi: 10.1073/pnas.0705923104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayaraghavan R, Sugendran K, Pant SC, Husain K, Malhotra RC. Dermal intoxication of mice with bis(2-chloroethyl)sulphide and the protective effect of flavonoids. Toxicology. 1991;69:35–42. doi: 10.1016/0300-483x(91)90151-p. [DOI] [PubMed] [Google Scholar]
- Voss T, Lipton RB, Dodick DW, Dupre N, Ge JY, Bachman R, Assaid C, Aurora SK, Michelson D. A phase IIb randomized, double-blind, placebo-controlled trial of ubrogepant for the acute treatment of migraine. Cephalalgia. 2016;36:887–898. doi: 10.1177/0333102416653233. [DOI] [PubMed] [Google Scholar]
- Wattana M, Bey T. Mustard gas or sulfur mustard: an old chemical agent as a new terrorist threat. Prehospital and disaster medicine. 2009;24:19–29. doi: 10.1017/s1049023x0000649x. discussion 30-11. [DOI] [PubMed] [Google Scholar]
- Wilson MR, Takata M. Inflammatory mechanisms of ventilator-induced lung injury: a time to stop and think? Anaesthesia. 2013;68:175–178. doi: 10.1111/anae.12085. [DOI] [PubMed] [Google Scholar]
- Wilson SR, Gerhold KA, Bifolck-Fisher A, Liu Q, Patel KN, Dong X, Bautista DM. TRPA1 is required for histamine-independent, Mas-related G protein-coupled receptor-mediated itch. Nature neuroscience. 2011;14:595–602. doi: 10.1038/nn.2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wormser U, Brodsky B, Green BS, Arad-Yellin R, Nyska A. Protective effect of povidone-iodine ointment against skin lesions induced by sulphur and nitrogen mustards and by non-mustard vesicants. Archives of toxicology. 1997;71:165–170. doi: 10.1007/s002040050371. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.