

Abstract
A substantial portion of the genomes of most multicellular eukaryotes consists of large arrays of tandemly repeated sequence, collectively called satellite DNA. The processes generating and maintaining different satellite DNA abundances across lineages are important to understand as satellites have been linked to chromosome mis-segregation, disease phenotypes, and reproductive isolation between species. While much theory has been developed to describe satellite evolution, empirical tests of these models have fallen short because of the challenges in assessing satellite repeat regions of the genome. Advances in computational tools and sequencing technologies now enable identification and quantification of satellite sequences genome-wide. Here, we describe some of these tools and how their applications are furthering our knowledge of satellite evolution and function.
Introduction
High copy number tandemly repeated DNA sequences, known as satellites, form a substantial part of many eukaryotic genomes [1–3]. Satellites were discovered in cesium-chloride density gradients as distinct bands of DNA that differ from the rest of the genome due to skewed nucleotide composition [4]. Now, these bands are known to include members of multi-copy gene families (e.g. rDNA, histones), transposable elements (TEs), and non-coding repetitive sequences in large arrays that can span megabases. Modern discussions of satellite DNA generally focus on only the latter, and that is our focus here1. We note that arrays of non-coding satellite repeats may also have other sequences such as TEs interspersed within them [5]. Satellite arrays are generally found in heterochromatin and may form essential chromosome structures such as centromeres and telomeres (reviewed in [6]). Despite their key roles in these critical structures, satellites show astonishing variation in both sequence and copy number among species, even among close relatives [7], suggesting that they evolve rapidly. Various models of satellite evolution have been proposed to explain this variation (Box 1) but genome-wide testing of these models has been lacking due to technological and computational limitations in assessing the repetitive portion of the genome. It is important to understand how and why satellite DNA varies among individuals and species because there are established links between satellites and phenotypes in a wide range of organisms, including humans. For example, satellite derepression is associated with cancer outcomes [8], chromosome mis-segregation and aneuploidy [9], and aging [10]. In addition, variation in satellite copy number has been associated with genetic incompatibilities between species [11] and differences in gene expression [12–14].
Box 1. Satellite DNA mechanisms and evolution1.
Origins
Any process that generates tandemly-arrayed sequence is a potential source of new satellite DNA. Polymerase slippage is likely the major mechanism generating tandemly repeated simple sequences. Other processes such as rolling-circle replication and multiple TE insertions at the same site can generate tandem arrays of longer sequences.
Change in copy number
Once tandemly repeated sequences are formed, recombination with unequal exchange can change the repeat copy number in the array, (reviewed in [55–57]. Recombination can involve any pair of repeats in homologous or sister arrays (i). Thus, ectopic recombination allows expansions and contractions of arrays (ii). Intrastrand exchange may also cause array size contractions and expansions through loop deletions and reinsertion of resulting extrachromosomal circles [58]. Because unequal exchange occurs on tandemly repeated sequences, the contraction of an array to a single repeat unit is a dead end; any neutrally evolving array will eventually reach this state and become extinct [59].
Sequence variation
The same ectopic recombination that leads to copy number change in tandem arrays also permits variant alleles to replace or be replaced by wild-type repeat units (ii). Noncrossover, “gene conversion”, events may also allow variants to spread. Variants may fix in some lineages and be lost in others, resulting in arrays which are homogeneous within populations/species but distinct between them, a process termed concerted evolution [60]. Further, the interplay between unequal exchange and mutation generates more complex higher order repeats (HORs), where variant repeat units reoccur at particular periodicities (e.g. human centromeric repeats [9]).
Spread in the population
For many satellites, the spread of copy number and sequence variants is likely neutral. However some satellites may be functional and selection may shape their population variation. Extremely large arrays are likely deleterious, with selection imposing upper limits on copy number expansions [61,62]. Functional satellite array size may be under constraint [9,63], though the role of sequence identity in satellite function/constraint remains debatable [43,64]. Satellite variation may also reflect selfish processes rather than organismal selection. For example, size variation in centromeric satellite arrays may bias chromosome segregation during meiosis [65,66], thus permitting some satellite alleles to selfishly increase their population frequency [42].
Interplay of recombination and selection yields predictable patterns
Because recombination ultimately results in array loss, satellites are more likely to persist in heterochromatic genomic regions where recombination is suppressed [59]. Here, satellites can persist at low copy number and differentially expand in copy number in daughter lineages, known as the library hypothesis [67]. In addition, since recombination is less efficient at the ends of arrays, divergent satellite repeat units are expected at the boundaries of arrays. However, selection for particular sequence variants could homogenize arrays. The relative contributions of mutational mechanisms (recombination, gene conversion) and estimates of mutational parameters (change in array size, birth/death of sequence variants within an array; [61]) under neutral evolution are open questions.
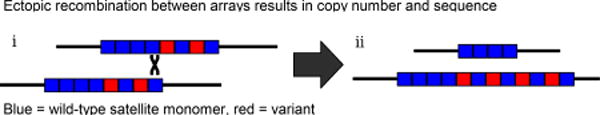
Ectopic recombination between arrays results in copy number and sequence
1Here, we briefly describe major models of satellite evolution. Please see [6] for a recent, detailed review.
Within the past several years there has been an explosion of software resources specifically aimed at improving our ability to assess repeat variation across entire genomes. Combined with improvements in sequencing technology and the growing availability of genomic datasets in public databases, the time is ripe for testing models of satellite DNA evolution and functional impact across a broad array of organisms. This will lend insight into the generality of proposed models of neutral satellite evolution in taxa with diverse life histories and enable detection of adaptive evolution of candidate functional satellites.
The challenge of assessing genome-wide satellites
Satellites have been understudied across taxa due to the limitations of widely used sequencing technology and software tools for assessing variation [15]. For example, while Illumina sequencing provides data with low error rates at low cost, the short read lengths preclude inclusion of large repetitive regions in genome assemblies. In addition, typical short-read library preparation techniques include a PCR step that biases against amplification of sequences with extreme GC content, resulting in underrepresentation of some satellites prior to sequencing. Further, many computational methods are designed to assess repeats only in assembled data (e.g. Tandem Repeats Finder [16], Table I) and/or employ detection strategies that rely on similarity to known repetitive sequences (e.g. RepeatMasker [17]), which can preclude detection of novel repeats. Thus, accurately assessing and quantifying genome-wide satellite sequences across diverse taxonomic groups requires developing alternative approaches. New software and emerging library preparation and sequencing technologies reduce bias through interrogation of repetitive regions via either assembly-free computational methods or improved satellite inclusion in assemblies due to longer read lengths.
Table I. Software for assessing satellite DNA.
This table is not comprehensive. We focus on recently developed assembly-free methods for analysis of large tandem arrays (excluding microsatellites). We also include more widely used methods for assessing tandem repeats in genome assemblies (i) as a comparison to methods designed specifically for assembly-free data, (ii) because some can also be applied to unassembled long reads (as long as the repeat monomers are shorter than the read length), and (iii) because they will continue to prove useful as improved read lengths enable assembly of satellite arrays.
| Name | Purpose* | Approach | Input** | Pros | Cons | Examples | |
|---|---|---|---|---|---|---|---|
| Identification | Assessment | ||||||
| Assembly-free | |||||||
| RepeatExplorer [19,45] | Identify and quantify abundant repeats | Sequence similarity across reads | Graph-based clustering, followed by assembly (CAP3) | <0.5× Illumina (SE, PE) or high-quality long reads | de novo; uses low-coverage data; identifies all repetitive DNA (including TEs, gene families); accessible via Galaxy web server | Does not detect low-abundance repeats; identification of tandem structures must be manually curate; cannot analyze high coverage data (but see [24]) | [24,36,40] |
| Tandem Repeat Analyzer (TAREAN) [20] | Identify and quantify abundant tandem repeats | Sequence similarity across reads | Graph-based clustering followed by kmer-based reconstruction of satellite consensus sequence | <0.5× Illumina (PE) | de novo; specifically assesses tandem repeats; accessible via Galaxy web server | Cannot analyze high coverage data; parameter optimization necessary to tune sensitivity; detection limited by genomic abundance, sequence homogeneity, monomer length, and homology to TE sequences | [46] |
| k-Seek [22] | Identify and quantify abundance of 2–10 bp tandem repeats | Within-read kmer decomposition | kmer counts | Illumina (SE, PE) | de novo; specifically assesses tandem repeats; employs sequencing error correction; PE data can yield monomer interspersion information | Assesses only simple satellites; limited by read length, sensitive to library and batch effects (but see GC correction pipeline in [26]); requires reference genome for absolute copy number estimation | [22,26] |
| ConTExt (McGurk and Barbash, bioRxiv doi: 10.1101/158386) | Structural variant discovery | Align to consensus sequences | Clustering of aligned sequences | Illumina (PE), repeat consensus sequences | Built-in GC-bias correction; can also assess copy number and sequence polymorphism; designed for population-level surveys | Requires a set of consensus sequences and a reference genome (to correct for GC bias) | see Column 1 |
| alpha-CENTAURI [18] | HOR discovery | Similarity to consensus sequences | Clustering of reads by sequence similarity | Long reads | Identifies structural and sequence polymorphism | Requires a training set of known repeat sequences | see Column 1 |
| Assembly-based | |||||||
| RepeatMasker [17] | Identify and mask repetitive regions of genome assemblies | Similarity to a known repeat database | Alignment | Assembly | The most widely used for genome annotation | Repeats must be present in assemblies and similar to known repeats in the database; ignores repeats with monomers less than 20 bp; not satellite-specific | [40] |
| Tandem Repeats Finder (TRF) [16] | Identify and quantify tandem repeat sequence | String matching | Local alignment | Assembly | de novo; focused on satellites | Repeats must be present in assemblies | [47] |
| Global Repeat Map (GRM) [48,49] | HOR discovery | Detects high frequency 8-bp sequences in computationally fragmented assemblies | Alignment | Assembly | de novo; assesses divergence of each monomer from the consensus | Repeats must be present in assemblies | see Column 1 |
| Spectral Repeat Finder [50] | Identify tandem and dispersed repeats | Sequence similarity within computationally-generated fragments detected using Fourier transform | Local alignment | Assembly | de novo; identifies periodicities in data even with divergence; including insertions and deletions | Repeats must be present in assemblies; maximum fragment length of 10 kb | [51] |
| Phobos [52] | Identify tandem repeats | String matching | Local alignment | Assembly | de novo; can detect repeat monomers up to 10,000 bp | Repeats must be present in assemblies; divergence in repeat monomers less than 50 bp tolerated better than divergence in longer repeats | [53] |
| MixTaR [54] | Identify repeats | Sequence similarity of short reads | Graph-based representation and confirmation using long reads | Short + long reads | de novo; internal validation using data from two different sequencing methods | Requires both long and short reads; performs better on more complex sequences | see Column 1 |
HOR: Higher Order Repeat
SE: single-end, PE: paired-end
i. Assembly-free methods for assessing satellites
A variety of methods have been developed to avoid assembly bias by identifying satellites in unassembled read data (Table I). These methods employ different strategies to (i) identify reads derived from repetitive sequences, (ii) assign these reads to discrete repeat families, and (iii) quantify the genomic abundance and/or sequence variation of each repeat. In the first step, repeat-derived reads can be identified by alignment or similarity to known repeat sequences (Figure 1A, e.g ConTExt: McGurk and Barbash, bioRxiv doi: 10.1101/158386, alpha-CENTAURI [18]). However, this biases downstream analysis to known repeats. If comprehensive consensus sequences are not available or the goal is de novo discovery, then other strategies are more appropriate, depending on the repeat type. Reads derived from complex repeats can be identified by sequence similarity to each other in low coverage sequencing (“genome skimming”) data or by down-sampling high coverage data such that only repetitive sequences are likely to be represented by multiple similar reads (e.g. RepeatExplorer [19], TAREAN [20]). One disadvantage to this approach is reduced power to detect low-copy repeats. In contrast, simple repeats can be identified by looking for recurrent motifs within reads, such as by kmer decomposition (Figure 1B, e.g. [21], k-Seek [22]). While this approach is not biased against low abundance repeats, the maximum detectable repeat monomer size is constrained by read length.
Figure 1. Assembly-free strategies for identifying and analyzing repetitive sequences.

A) Alignment based approaches generally collapse repeat-derived reads onto known repeat consensus sequences (ConTExt, McGurk and Barbash, bioRxiv doi: 10.1101/158386). Copy number can be inferred from the alignment depth, while sequence polymorphism can be assessed from the collapsed reads. The large gray rectangle represents consensus sequence, while smaller black rectangles represent individual sequencing reads. Red positions within reads indicate sequence polymorphism. B) Kmer-based approaches decompose sequencing reads into overlapping subsequences of length k. Reads derived from simple satellites will be enriched for a small set of kmers. These kmers can be quantified (k-Seek [22]) or used in more abstract representations of reads [23]. C) Graph-based methods construct representations of repeats using sequence similarity. The nodes in such graphs are sequences: kmers (De Bruijin graphs), sequencing reads (e.g. RepeatExplorer [19]), or entire repeats [44]. The edges reflect neighboring relationships (sequence composition similarity). Because of their tandem nature, satellites often present as as circular graphs. Distinct repeat families can be identified as clusters of nodes. Here, two distinct satellites (the separate circular graphs, left and center) as well as a non-tandem sequence (right) are depicted.
Once repeat-derived reads are identified, several strategies are available for assigning reads to discrete repeats and quantifying variation in those repeats, depending on the question of interest. The abundance of simple satellites can be quantified by their kmer counts, but this approach provides little information about higher order structures. In contrast, graph-based representations provide information on both structure and abundance. These methods work by constructing graphs based on the sequence similarity of reads, which can then be partitioned with clustering algorithms to identify distinct families/subfamiles of repeats (e.g. RepeatExplorer [19], TAREAN [20], [23]). Because of their repeated nature, tandem repeats generally appear as circular structures in the graph (Figure 1C). Subsequently, structural variation, copy number, and sequence polymorphism can be inferred and estimated using the clustered reads. If repeat consensus sequences are available, alignment-based strategies also yield copy-number information as well as sequence and structural variation. Any of these identification and assessment approaches can be combined to comprehensively assess repeats genome-wide (e.g. satMiner [24]) or to perform analysis of a specific repeat family (e.g. [18]).
ii. New sequencing technologies decrease bias and enable assembly
As mentioned above, typical Illumina sequencing library preparation workflows include PCR amplification of fragments prior to sequencing. New PCR-free library preparation techniques can mitigate this bias, although their regular adoption into “standard” sequencing protocols is not yet widespread. In the absence of PCR-free libraries, GC-bias can be accounted for by inferring correction factors from the relationship between coverage and GC content of single copy sequence [25,26] or by including GC composition as a covariate in regression analyses. However, such corrections are unlikely to completely account for bias at the extremes of GC-composition. Spike-in of calibration sequences into sequencing libraries may ultimately be required for accurate correction.
Read lengths have significantly improved for both widely-used (short read: Illumina) and relatively new (long read: Pacific Biosciences, PacBio; Oxford Nanopore) sequencing platforms (Table II). This increases the size of repeat monomers that kmer-based methods can interrogate and can be especially useful when employing targeted strategies, such as sequencing PCR amplicons, to assess copy number or sequence variation in a particular satellite [27]. The very long reads offered by PacBio and Nanopore can even yield assemblies of large satellite arrays using improved algorithms [28]. However, assessing variation in satellite sequence and abundance using these methods requires stringent filtering and validation methods to account for their high error rates.
Table II.
Technological strategies for assessing satellite DNA
| Platform | Read length; method |
Pros | Cons | Example |
|---|---|---|---|---|
| Illumina | Up to 300 bp; clustered amplicon | Inexpensive, low error rate | PCR bias in library prep*; short reads | [40] |
| Ion torrent | Up to 400 bp; on-bead amplicon | Fast, inexpensive | Lower yield; high error rate in homopolymer tracts | [27] |
| Pacific Biosciences | Up to 50 kb; single molecule | Long reads; can assemble complex satellite regions | Expensive; high error rate** | [5] |
| Oxford Nanopore | Up to 300 kb; single molecule | Longest reads | High error rate; extracting high molecular weight DNA is limiting | Jain et al., bioRxiv 10.1101/170373 |
| Optical mapping (nanochannel) | Up to 220 kb; single molecule | Long-range positional information; orthogonal method to sequencing | Requires a reference genome; large nicking intervals preclude mapping simple sequences | [41] |
PCR-free libraries reduce bias.
PacBio also offers a Circular Consensus Sequencing (CSS) approach, where single circular molecules are read multiple times, thus generating a high quality consensus for each molecule.
Combined with whole-genome sequencing approaches, optical mapping [29] provides an orthogonal way to assemble satellite arrays with large-monomers (~ 1 kb) without library preparation bias. This technique uses detection of fluorescently-labeled sequence motifs on single, stretched DNA fragments to construct a physical map of each fragment, thus providing long-range information to improve assemblies. However, short satellite monomer sequences cannot be mapped using this technology because labeled motifs cannot be detected distinctly if they are too close together.
Satellite expression or association with chromatin state/chromosome structures can also be assessed, using RNA sequencing (RNA-seq) or chromatin immunoprecipitation sequencing (ChIP-seq [30]). Newly developed protocols both enhance the sensitivity of these techniques and reduce bias due to PCR-duplicates arising from library preparation [31].
New approaches advance our knowledge of satellite evolution and function
i. Evolutionary patterns across taxa
Satellites can now be assessed from multiple lineages of nonmodel taxa at relatively low cost because (i) methods exist to identify and quantify satellite sequences de novo, (ii) short-read sequencing is relatively inexpensive, and (iii) low coverage sequencing schemes can be applied to even the largest genomes. As a result, there has been an explosion of studies assessing satellite diversity across a wide array of organisms including mammals: canids [32]; fish: Characidae [33], sterlet [34]; insects: cactophilic Drosophila [35], fireflies [36], locust [24], cricket [37]; and plants: Populus [38], bread wheat [39]. Comparison of sequences from males and females has uncovered satellites that differ in abundance between the sexes, enabling identification of sex chromosome satellites in systems with otherwise homomorphic (identical) sex chromosomes [34,40] with implications for the study of sex chromosome evolution. Predicted evolutionary patterns and hypotheses are beginning to be interrogated across taxonomic groups including the library hypothesis [22], age stratification of arrays [27], and concerted evolution [5,35] (Box 1). The taxonomic breadth of available short-read data sets the stage for rigorous comparative analyses of satellite evolution across life histories, mating strategies, polyploidization status, and divergence levels, though careful consideration of bias, including appropriate correction for GC-bias and batch effects, is paramount. Population-level studies remain rare (but see [22]), but will be valuable in investigating satellite evolutionary dynamics given the amount of variation in abundance and sequence even at small timescales.
ii. Neutral mutation and detecting selection
Extensive variation in specific satellite abundances across lineages suggests that they have high rates of copy number change. However, estimates of neutral rates of copy number change and sequence variation, including the relative contributions of gene conversion versus unequal exchange to these parameters, remain unknown. Estimation of these parameters is important in order to detect selection – for example, a satellite under stabilizing selection will have a narrower copy number distribution than expected given neutral processes, and, as the eventual fate of arrays is extinction (Box 1), remain in genomes longer than expected under neutrality. One approach for neutral rate estimation is assessment of satellites in whole-genome sequencing of mutation accumulation studies. Recently, Flynn and colleagues used k-Seek [22] to compare the satellite composition of Daphnia pulex lines derived from a single progenitor either evolving under neutrality (bottlenecked to N = 1 each generation) or under selection (maintained as a large population) [26]. They found very high rates of satellite copy number change in the lines evolving under neutrality, on the order of 10-3 changes in copy number per repeat unit per generation. Further, variation in copy number across individuals from the large population was 33% lower than the variation across the mutation accumulation lines, suggesting that total satellite abundance is under stabilizing selection. Interestingly, some satellites were much more constrained than others.
iii. Including satellites in assemblies
Technologies that generate long-range data, such as long reads from single molecules or optical mapping, are now enabling assembly into or even across satellite regions [5]. Recently, Jain and colleagues (bioRxiv doi: 10.1101/170373) assembled the entire centromere of the human Y-chromosome using Oxford Nanopore long reads. Once assembled, patterns predicted by theoretical models can be tested (Box 1). Combining PacBio long reads and optical mapping, Weissensteiner and colleagues confirmed the inverse relationship of repeat array length and recombination rate in the Eurasian crow [41]. Depending on array size and homogeneity, complete assemblies may still remain out of reach for some regions of the genome.
iv. Functional consequences of satellite variation
Assembly-free methods of quantifying satellite variants applied to ChIP-seq data have revealed some of the selfish and functional roles of satellite sequences. For example, Iwata-Otsubo and colleagues identified differences in array length and sequence diversity associated with selfishly-transmitted centromeres in mice by assessing satellite sequences immunoprecipitated with the centromere protein CenP-A [42]. While, in this study, specific satellite DNA sequences were associated with centromeres, studies in other organisms have suggested that sequence identity may not be as important. In a comparative ChIP-seq approach across races of maize, satellite sequence identity was not as closely associated with centromere function [43]. More work is necessary to further distinguish possible functional roles of different satellites and the relationship of function to copy number and sequence variation across the genome.
Conclusions
Recent developments in computational methods and sequencing technology are advancing our understanding of satellite evolution and function. Best practices should be established to mitigate biases introduced by library preparation and sequencing technologies. For short read data, incorporation of PCR-free library preparation techniques into short-read genome sequencing workflows should become routine where possible, i.e. when the amount of DNA is not limiting. When PCR-based libraries are necessary, biases in GC composition can be explored and corrected computationally. To control for batch effects, co-preparation and pooling of libraries across multiple lanes of sequences rather than sequential preparation and sequencing of samples is essential [22,26]. Spike-ins of known repeats can, in principle, serve as additional control. Finally, with improved read lengths, it will be important to continue adapting or developing new methods for satellite assessment in long-read, error prone datasets.
To achieve the ultimate goal of identifying satellites evolving under selection, future work should establish the parameters of neutral satellite evolution and develop methods to detect departures from neutrality. Questions include: Do the empirical genome-wide distributions of satellite copy number and sequence variation across lineages fit with our expectations given neutral processes of replication slippage, unequal exchange, point mutation, and gene conversion? What is the respective contribution of each of these mechanisms? How and why do these vary across different satellites genome-wide? How prevalent is selection and what is the source? With this knowledge we will finally be able to identify the dominant mechanistic and evolutionary forces shaping variation in this “black box” of the genome.
Acknowledgments
The authors would like to thank Arvid Ågren, Emily Brown, Dean Castillo, Anne-Marie Dion-Côté, and Jullien Flynn for valuable comments on the manuscript.
Funding: This work was supported by the National Institutes of Health (NIGMS R01GM119125 to DAB and AGC; Ruth L. Kirschstein National Research Service Award F32GM126736 to SSL).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The term “satellite” has also been used for shorter stretches of tandem DNA that exist in euchromatic regions of the genome (e.g micro- and minisatellites; 10s–100s of bp). Here, we focus on large tandem repetitive arrays because (i) microsatellites are generally short and can be fully assembled in sequencing projects, while large satellites require novel methods to analyze and (ii) large satellite arrays likely have unique effects on chromosome structure and function.
References
- 1.John B, Miklos GL. Functional aspects of satellite DNA and heterochromatin. Int Rev Cytol. 1979;58:1–114. doi: 10.1016/s0074-7696(08)61473-4. [DOI] [PubMed] [Google Scholar]
- 2.Choo KH, Vissel B, Earle E. Evolution of α-satellite DNA on human acrocentric chromosomes. Genomics. 5:332–344. doi: 10.1016/0888-7543(89)90066-9. 1989/8. [DOI] [PubMed] [Google Scholar]
- 3.Camacho JPM, Ruiz-Ruano FJ, Martín-Blázquez R, López-León MD, Cabrero J, Lorite P, Cabral-de-Mello DC, Bakkali M. A step to the gigantic genome of the desert locust: chromosome sizes and repeated DNAs. Chromosoma. 2015;124:263–275. doi: 10.1007/s00412-014-0499-0. [DOI] [PubMed] [Google Scholar]
- 4.Kit S. Equilibrium sedimentation in density gradients of DNA preparations from animal tissues. J Mol Biol. 1961;3:711–IN2. doi: 10.1016/s0022-2836(61)80075-2. [DOI] [PubMed] [Google Scholar]
- 5.Khost DE, Eickbush DG, Larracuente AM. Single-molecule sequencing resolves the detailed structure of complex satellite DNA loci in Drosophila melanogaster. Genome Res. 2017;27:709–721. doi: 10.1101/gr.213512.116. The authors employ PacBio sequencing of Drosophila melanogaster to achieve detailed assemblies of the major autosomal complex satellite DNA loci, particularly Responder and 1.688 gm/cm3. They show that PacBio assembly with error correction can significantly extend contigs into repetitive pericentromeric regions. They urge careful examination of assemblies across multiple parameters, using orthogonal molecular and genomic evidence to validate assemblies to ensure accuracy. Their data support models of TE gain at the ends of arrays and are consistent with the homogenizing effect of concerted evolution. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garrido-Ramos MA. Satellite DNA: An Evolving Topic. Genes. 2017;8 doi: 10.3390/genes8090230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jagannathan M, Warsinger-Pepe N, Watase GJ, Yamashita YM. Comparative Analysis of Satellite DNA in the Drosophila melanogaster Species Complex. G3. 2017;7:693–704. doi: 10.1534/g3.116.035352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bersani F, Lee E, Kharchenko PV, Xu AW, Liu M, Xega K, MacKenzie OC, Brannigan BW, Wittner BS, Jung H, et al. Pericentromeric satellite repeat expansions through RNA-derived DNA intermediates in cancer. Proc Natl Acad Sci U S A. 2015;112:15148–15153. doi: 10.1073/pnas.1518008112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aldrup-MacDonald ME, Kuo ME, Sullivan LL, Chew K, Sullivan BA. Genomic variation within alpha satellite DNA influences centromere location on human chromosomes with metastable epialleles. Genome Res. 2016;26:1301–1311. doi: 10.1101/gr.206706.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang W, Li J, Suzuki K, Qu J, Wang P, Zhou J, Liu X, Ren R, Xu X, Ocampo A, et al. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science. 2015;348:1160–1163. doi: 10.1126/science.aaa1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferree PM, Barbash DA. Species-specific heterochromatin prevents mitotic chromosome segregation to cause hybrid lethality in Drosophila. PLoS Biol. 2009;7:e1000234. doi: 10.1371/journal.pbio.1000234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lemos B, Branco AT, Hartl DL. Epigenetic effects of polymorphic Y chromosomes modulate chromatin components, immune response, and sexual conflict. Proc Natl Acad Sci U S A. 2010;107:15826–15831. doi: 10.1073/pnas.1010383107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feliciello I, Akrap I, Ugarković Đ. Satellite DNA Modulates Gene Expression in the Beetle Tribolium castaneum after Heat Stress. PLoS Genet. 2015;11:e1005466. doi: 10.1371/journal.pgen.1005466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joshi SS, Meller VH. Satellite Repeats Identify X Chromatin for Dosage Compensation in Drosophila melanogaster Males. Curr Biol. 2017;27:1393–1402.e2. doi: 10.1016/j.cub.2017.03.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet. 2011;13:36–46. doi: 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smit A, Hubley R, Green P. RepeatMasker Open-4.0. 2013--2015. Institute for Systems Biology. 2015 http://repeatmaskerorg.
- 18.Sevim V, Bashir A, Chin C-S, Miga KH. Alpha-CENTAURI: assessing novel centromeric repeat sequence variation with long read sequencing. Bioinformatics. 2016;32:1921–1924. doi: 10.1093/bioinformatics/btw101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Novák P, Neumann P, Pech J, Steinhaisl J, Macas J. RepeatExplorer: a Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics. 2013;29:792–793. doi: 10.1093/bioinformatics/btt054. [DOI] [PubMed] [Google Scholar]
- 20.Novák P, Ávila Robledillo L, Koblížková A, Vrbová I, Neumann P, Macas J. TAREAN: a computational tool for identification and characterization of satellite DNA from unassembled short reads. Nucleic Acids Res. 2017;45:e111. doi: 10.1093/nar/gkx257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krassovsky K, Henikoff S. Distinct chromatin features characterize different classes of repeat sequences in Drosophila melanogaster. BMC Genomics. 2014;15:105. doi: 10.1186/1471-2164-15-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei KH-C, Grenier JK, Barbash DA, Clark AG. Correlated variation and population differentiation in satellite DNA abundance among lines of Drosophila melanogaster. Proc Natl Acad Sci U S A. 2014;111:18793–18798. doi: 10.1073/pnas.1421951112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Altemose N, Miga KH, Maggioni M, Willard HF. Genomic characterization of large heterochromatic gaps in the human genome assembly. PLoS Comput Biol. 2014;10:e1003628. doi: 10.1371/journal.pcbi.1003628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ruiz-Ruano FJ, López-León MD, Cabrero J, Camacho JPM. High-throughput analysis of the satellitome illuminates satellite DNA evolution. Sci Rep. 2016;6:28333. doi: 10.1038/srep28333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benjamini Y, Speed TP. Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res. 2012;40:e72. doi: 10.1093/nar/gks001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flynn JM, Caldas I, Cristescu ME, Clark AG. Selection Constrains High Rates of Tandem Repetitive DNA Mutation in Daphnia pulex. Genetics. 2017;207:697–710. doi: 10.1534/genetics.117.300146. The extent to which satellite DNA is neutrally evolving versus under selection is an open question. Here, the authors use the k-Seek pipeline to characterize tandem simple sequence repeats across the genomes of Daphnia pulex mutation accumulation lines originating from a single progenitor strain and that were maintained over generations either neutrally (bottlenecked each generation) or under selection (kept in a large population). They found less variance in copy number across lines kept under selection conditions versus lines kept under neutral evolution conditions, suggesting that tandem repeat content is constrained by selection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cacheux L, Ponger L, Gerbault-Seureau M, Richard FA, Escudé C. Diversity and distribution of alpha satellite DNA in the genome of an Old World monkey: Cercopithecus solatus. BMC Genomics. 2016;17:916. doi: 10.1186/s12864-016-3246-5. Alpha satellites are the major component of primate centromeres. Here, the authors use targeted Ion-torrent sequencing of digested genomic DNA to identify all members of the alpha-satellite family in an Old World monkey. Stringent filtering of sequences was required due to a high error rate, resulting in a greatly reduced dataset. Nevertheless, the authors found an interesting pattern of age-stratification, where old sequences are displaced from the centromere. Fluorescence in situ hybridization showed that satellite families are not homogeneously distributed among chromosomes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chakraborty M, Baldwin-Brown JG, Long AD, Emerson JJ. Contiguous and accurate de novo assembly of metazoan genomes with modest long read coverage. Nucleic Acids Res. 2016;44:e147–e147. doi: 10.1093/nar/gkw654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lam ET, Hastie A, Lin C, Ehrlich D, Das SK, Austin MD, Deshpande P, Cao H, Nagarajan N, Xiao M, et al. Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat Biotechnol. 2012;30:771–776. doi: 10.1038/nbt.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 31.Cerutti F, Gamba R, Mazzagatti A, Piras FM, Cappelletti E, Belloni E, Nergadze SG, Raimondi E, Giulotto E. The major horse satellite DNA family is associated with centromere competence. Mol Cytogenet. 2016;9:35. doi: 10.1186/s13039-016-0242-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vozdova M, Kubickova S, Cernohorska H, Fröhlich J, Rubes J. Satellite DNA Sequences in Canidae and Their Chromosome Distribution in Dog and Red Fox. Cytogenet Genome Res. 2016;150:118–127. doi: 10.1159/000455081. [DOI] [PubMed] [Google Scholar]
- 33.Utsunomia R, Ruiz-Ruano FJ, Silva DMZA, Serrano ÉA, Rosa IF, Scudeler PES, Hashimoto DT, Oliveira C, Camacho JPM, Foresti F. A Glimpse into the Satellite DNA Library in Characidae Fish (Teleostei, Characiformes) Front Genet. 2017;8:103. doi: 10.3389/fgene.2017.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Biltueva LS, Prokopov DY, Makunin AI, Komissarov AS, Kudryavtseva AV, Lemskaya NA, Vorobieva NV, Serdyukova NA, Romanenko SA, Gladkikh OL, et al. Genomic Organization and Physical Mapping of Tandemly Arranged Repetitive DNAs in Sterlet (Acipenser ruthenus) Cytogenet Genome Res. 2017;152:148–157. doi: 10.1159/000479472. [DOI] [PubMed] [Google Scholar]
- 35.de Lima LG, Svartman M, Kuhn GCS. Dissecting the Satellite DNA Landscape in Three Cactophilic Drosophila Sequenced Genomes. G3. 2017;7:2831–2843. doi: 10.1534/g3.117.042093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lower SS, Johnston JS, Stanger-Hall KF, Hjelmen CE, Hanrahan SJ, Korunes K, Hall D. Genome Size in North American Fireflies: Substantial Variation Likely Driven by Neutral Processes. Genome Biol Evol. 2017;9:1499–1512. doi: 10.1093/gbe/evx097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Palacios-Gimenez OM, Dias GB, de Lima LG, Kuhn GCES, Ramos É, Martins C, Cabral-de-Mello DC. High-throughput analysis of the satellitome revealed enormous diversity of satellite DNAs in the neo-Y chromosome of the cricket Eneoptera surinamensis. Sci Rep. 2017;7:6422. doi: 10.1038/s41598-017-06822-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Usai G, Mascagni F, Natali L, Giordani T, Cavallini A. Comparative genome-wide analysis of repetitive DNA in the genus Populus L. Tree Genet Genomes. 2017;13:96. [Google Scholar]
- 39.Tiwari VK, Wang S, Danilova T, Koo DH, Vrána J, Kubaláková M, Hribova E, Rawat N, Kalia B, Singh N, et al. Exploring the tertiary gene pool of bread wheat: sequence assembly and analysis of chromosome 5M(g) of Aegilops geniculata. Plant J. 2015;84:733–746. doi: 10.1111/tpj.13036. [DOI] [PubMed] [Google Scholar]
- 40.Puterova J, Razumova O, Martinek T, Alexandrov O, Divashuk M, Kubat Z, Hobza R, Karlov G, Kejnovsky E. Satellite DNA and Transposable Elements in Seabuckthorn (Hippophae rhamnoides), a Dioecious Plant with Small Y and Large X Chromosomes. Genome Biol Evol. 2017;9:197–212. doi: 10.1093/gbe/evw303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weissensteiner MH, Pang AWC, Bunikis I, Höijer I, Vinnere-Petterson O, Suh A, Wolf JBW. Combination of short-read, long-read, and optical mapping assemblies reveals large-scale tandem repeat arrays with population genetic implications. Genome Res. 2017;27:697–708. doi: 10.1101/gr.215095.116. Genome assemblies are generally fragmented by heterochromatic repetitive arrays. Here, the authors compare genome assemblies using short-read (Illumina), long-read (PacBio), and optical-mapping techniques (and combinations thereof). Long-read techniques clearly improve the length and orientation of assemblies, while optical mapping allows extension of scaffolds into previously unassembled repetitive sequence. Combined with genome scans of population genetic parameters, this allows testing of molecular correlates, such as recombination rate, with repetitive array location, size, and/or sequence variation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iwata-Otsubo A, Dawicki-McKenna JM, Akera T, Falk SJ, Chmátal L, Yang K, Sullivan BA, Schultz RM, Lampson MA, Black BE. Expanded Satellite Repeats Amplify a Discrete CENP-A Nucleosome Assembly Site on Chromosomes that Drive in Female Meiosis. Curr Biol. 2017;27:2365–2373.e8. doi: 10.1016/j.cub.2017.06.069. Despite their essential function in proper chromosome segregation, centromeric sequences show high sequence diversity across closely-related taxa. Here, the authors investigate one potential hypothesis for this diversity - centromere drive, where, during asymmetric female meiosis, centromeres can bias their transmission to the egg cell, rather than the polar bodies. Using ChIP sequencing followed by mapping to satellite consensus sequences, they find differences in satellite repeat array length across mouse strains that differ in centromere strength. Using immunostaining, they find that stronger centromeres more frequently orient toward the pole of the cell destined to become the egg. This study is an intriguing look into the mechanism of centromere drive and demonstrates how combining sequencing with other approaches can begin to elucidate satellite function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gent JI, Wang N, Dawe RK. Stable centromere positioning in diverse sequence contexts of complex and satellite centromeres of maize and wild relatives. Genome Biol. 2017;18:121. doi: 10.1186/s13059-017-1249-4. The degree to which centromeres are determined by sequence identity is an open question. Here the authors use ChIP-seq to identify centromeric regions across maize and its wild relatives. They find that centromere size is variable across lineages, but is not heritable in hybrid crosses. Further, they use RepeatExplorer on the ChIP-seq reads to discover that the satellite CentC is associated with centromeres in most lineages, though it has high sequence polymorphism, and has even been lost in two lineages. This study is an excellent example of combining experimental and bioinformatic analyses to assess centromeric satellite sequence evolution. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miga KH, Newton Y, Jain M, Altemose N, Willard HF, Kent WJ. Centromere reference models for human chromosomes X and Y satellite arrays. Genome Res. 2014;24:697–707. doi: 10.1101/gr.159624.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Novák P, Neumann P, Macas J. Graph-based clustering and characterization of repetitive sequences in next-generation sequencing data. BMC Bioinformatics. 2010;11:378. doi: 10.1186/1471-2105-11-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cook DE, Zdraljevic S, Tanny RE, Seo B, Riccardi DD, Noble LM, Rockman MV, Alkema MJ, Braendle C, Kammenga JE, et al. The genetic basis of natural variation in Caenorhabditis elegans telomere length. Genetics. 2016;204:371–383. doi: 10.1534/genetics.116.191148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Melters DP, Bradnam KR, Young HA, Telis N, May MR, Ruby JG, Sebra R, Peluso P, Eid J, Rank D, et al. Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution. Genome Biol. 2013;14:R10. doi: 10.1186/gb-2013-14-1-r10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vlahovic I, Gluncic M, Rosandic M, Ugarkovic Ð, Paar V. Regular Higher Order Repeat Structures in Beetle Tribolium castaneum Genome. Genome Biol Evol. 2017;9:2668–2680. doi: 10.1093/gbe/evw174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Glunčić M, Paar V. Direct mapping of symbolic DNA sequence into frequency domain in global repeat map algorithm. Nucleic Acids Res. 2013;41:e17. doi: 10.1093/nar/gks721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sharma D, Issac B, Raghava GPS, Ramaswamy R. Spectral Repeat Finder (SRF): identification of repetitive sequences using Fourier transformation. Bioinformatics. 2004;20:1405–1412. doi: 10.1093/bioinformatics/bth103. [DOI] [PubMed] [Google Scholar]
- 51.Mu X, Wang X, Liu Y, Song H, Liu C, Gu D, Wei H, Luo J, Hu Y. An unusual mitochondrial genome structure of the tonguefish, Cynoglossus trigrammus: Control region translocation and a long additional non-coding region inversion. Gene. 2015;573:216–224. doi: 10.1016/j.gene.2015.07.041. [DOI] [PubMed] [Google Scholar]
- 52.Mayer C. Phobos, a tandem repeat search tool for complete genomes. Version. 2008;3:12. [Google Scholar]
- 53.Mayer C, Leese F, Tollrian R. Genome-wide analysis of tandem repeats in Daphnia pulex--a comparative approach. BMC Genomics. 2010;11:277. doi: 10.1186/1471-2164-11-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fertin G, Jean G, Radulescu A, Rusu I. Hybrid de novo tandem repeat detection using short and long reads. BMC Med Genomics. 2015;8(Suppl 3):S5. doi: 10.1186/1755-8794-8-S3-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Charlesworth B, Sniegowski P, Stephan W. The evolutionary dynamics of repetitive DNA in eukaryotes. Nature. 1994;371:215–220. doi: 10.1038/371215a0. [DOI] [PubMed] [Google Scholar]
- 56.Ugarković Ð, Plohl M. Variation in satellite DNA profiles—causes and effects. EMBO J. 2002;21:5955–5959. doi: 10.1093/emboj/cdf612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Plohl M, Luchetti A, Mestrović N, Mantovani B. Satellite DNAs between selfishness and functionality: structure, genomics and evolution of tandem repeats in centromeric (hetero)chromatin. Gene. 2008;409:72–82. doi: 10.1016/j.gene.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 58.Walsh JB. Persistence of tandem arrays: implications for satellite and simple-sequence DNAs. Genetics. 1987;115:553–567. doi: 10.1093/genetics/115.3.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Charlesworth B, Langley CH, Stephan W. The evolution of restricted recombination and the accumulation of repeated DNA sequences. Genetics. 1986;112:947–962. doi: 10.1093/genetics/112.4.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dover G. Molecular drive: a cohesive mode of species evolution. Nature. 1982;299:111–117. doi: 10.1038/299111a0. [DOI] [PubMed] [Google Scholar]
- 61.Stephan W. Quantitative variation and chromosomal location of satellite DNAs. Genet Res. 1987;50:41–52. doi: 10.1017/s0016672300023326. [DOI] [PubMed] [Google Scholar]
- 62.Lyckegaard EM, Clark AG. Ribosomal DNA and Stellate gene copy number variation on the Y chromosome of Drosophila melanogaster. Proc Natl Acad Sci U S A. 1989;86:1944–1948. doi: 10.1073/pnas.86.6.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zeng W, de Greef JC, Chen Y-Y, Chien R, Kong X, Gregson HC, Winokur ST, Pyle A, Robertson KD, Schmiesing JA, et al. Specific Loss of Histone H3 Lysine 9 Trimethylation and HP1γ/Cohesin Binding at D4Z4 Repeats Is Associated with Facioscapulohumeral Dystrophy (FSHD) PLoS Genet. 2009;5:e1000559. doi: 10.1371/journal.pgen.1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maheshwari S, Ishii T, Brown CT, Houben A, Comai L. Centromere location in Arabidopsis is unaltered by extreme divergence in CENH3 protein sequence. Genome Res. 2017;27:471–478. doi: 10.1101/gr.214619.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Henikoff S, Malik HS. Centromeres: selfish drivers. Nature. 2002;417:227. doi: 10.1038/417227a. [DOI] [PubMed] [Google Scholar]
- 66.Walker PM. Origin of satellite DNA. Nature. 1971;229:306–308. doi: 10.1038/229306a0. [DOI] [PubMed] [Google Scholar]
- 67.Fry K, Salser W. Nucleotide sequences of HS-alpha satellite DNA from kangaroo rat Dipodomys ordii and characterization of similar sequences in other rodents. Cell. 1977;12:1069–1084. doi: 10.1016/0092-8674(77)90170-2. [DOI] [PubMed] [Google Scholar]