

Abstract
The system-wide site-specific analysis of intact glycopeptides is crucial for understanding the exact functional relevance of protein glycosylation. Dedicated workflow with the capability to simultaneously characterize and quantify intact glycopeptides in a site-specific and high-throughput manner is essential to reveal specific glycosylation alteration patterns in complex biological systems. In this study, an enhanced, dedicated, large-scale site-specific quantitative N-glycoproteomics workflow has been established, which includes improved specific extraction of membrane-bound glycoproteins using filter aided sample preparation (FASP) method, enhanced enrichment of N-glycopeptides using sequential hydrophilic interaction liquid chromatography (HILIC) and multi-lectin affinity (MLA) enrichment, site-specific N-glycopeptide characterization enabled by EThcD, relative quantitation utilizing isobaric N, N-dimethyl leucine (DiLeu) tags and automated FDR-based large-scale data analysis by Byonic. For the first time, our study shows that HILIC complements to a very large extent to MLA enrichment with only 20% overlapping in enriching intact N-glycopeptides. When applying the developed workflow to site-specific N-glycoproteome study in PANC1 cells, we were able to identify 1067 intact N-glycopeptides, representing 311 glycosylation sites and 88 glycan compositions from 205 glycoproteins. We further applied this approach to study the glycosylation alterations in PKM2 knockout cells vs. parental breast cancer cells and revealed altered N-glycoprotein/N-glycopeptide patterns and very different glycosylation microheterogeneity for different types of glycans. To provide a more comprehensive map of glycoprotein alterations, N-glycopeptides after treatment with PNGase F were also analyzed. A total of 484 deglycosylated peptides were quantified, among which 81 deglycosylated peptides from 70 glycoproteins showed significant changes. KEGG activation of Akt for their survival. With glycosylation being one of the most important signaling modulators, our results provide additional evidence that signaling pathways is closely regulated by metabolism.
Keywords: glycoproteomics, mass spectrometry, PTM enrichment, PKM2, N-glycosylation, cancer
Table of Contents Entry
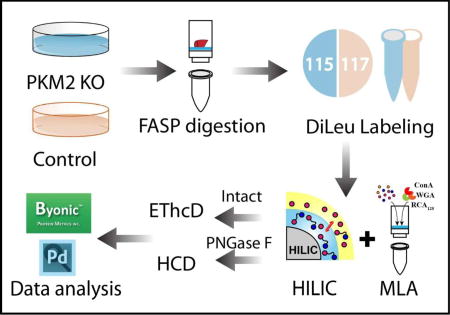
An enhanced large-scale site-specific quantitative N-glycoproteomics workflow was developed, integrating novel isobaric tagging, multiple enrichment strategies and hybrid fragmentation method for intact glycopeptide characterization and quantitation of complex cellular samples.
Introduction
Glycosylation is one of the most prevalent and complex protein post-translational modifications (PTMs). It has been reported to play essential role in many key biological processes including cell adhesion, molecular trafficking and clearance, receptor activation, signal transduction and endocytosis.1–4 Altered glycosylation has been associated with different diseases such as cancer,5–7 neurodegenerative diseases,8 and rheumatoid arthritis.9 Thus, the accurate site-specific characterization and quantitation of N-glycopeptides is of vital importance to provide insights into the molecular mechanisms of diseases and reveal potential biomarkers and therapeutic targets.10
Enrichment of N-glycopeptides is often needed due to their low abundance and signal suppression by more abundant non-glycosylated peptides. Various glycopeptide enrichment strategies have been developed, including hydrophilic interaction chromatography (HILIC),11–14 lectin affinity,15, 16 TiO2 affinity,17 immobilized metal affinity chromatography (IMAC),18, 19 hydrazide 20 and boronic acid chemistry.21, 22 Among them, MLA and HILIC are the most widely used strategies, which not only enables the analysis of deglycosylated peptides after PNGase F treatment but also the analysis of intact N-glycopeptides.23 The former strategy relies on N-glycopeptides’ glycan structure motif recognition, while the latter takes advantage of the differences in physicochemical properties between non-glycosylated peptides and glycopeptides due to glycan attachment. Previous studies 24, 25 have indicated these two methods complement each other in mapping glycosylation sites, and recommended combined strategies should be used for better glycosylation site coverage. Very recently Zhang et al 26 showed that HILIC alone is almost as effective as the combined strategies in mapping glycosylation sites. However, these prior studies targeted at deglycosylated peptides after treatment with PNGase F and none of these studies had investigated performance of HILIC and MLA in enriching intact N-glycopeptides. We propose that an evaluation of the performance of these two most commonly used strategies in enriching intact N-glycopeptides will provide valuable guidance for the site-specific glycoproteomics.
Previous glycoproteomics studies were somewhat biased towards glycosylation sites mapping or released glycan analysis mainly due to a multitude of structural complexity encompassing attached glycans and a lack of enabling analytical technology.23, 27, 28 However, with the rapid development of various glycoproteomics approaches, especially more advanced instrumentation equipped with new features, site-specific characterization of glycoproteins has become increasingly feasible.29 Among various emerging techniques, the electron transfer and higher-energy collision dissociation (EThcD) technique has significantly contributed to the enhanced site-specific glycoproteomics analyses. Commonly used for PTMs analysis, electron-transfer dissociation (ETD) was well known to preserve labile PTM on peptide backbones. It provides peptide backbone fragmentation c/z-ion series with glycan unit attached for confident mapping of glycosylation site and peptide identification; however, it only reveals the glycan as a “blind” modification with an accurate mass. On the other hand, glycan diagnostic oxonium ions and glycan backbone B- and Y-ion series produced by high-energy collision dissociation (HCD) provides confident glycan identification,30, 31 but without glycosylation site information. Since its initial introduction by Heck and coworkers,32 the “hybrid” dissociation method (EThcD) has shown great potential for PTM analysis with improved site localization, as well as generating richer and more informative backbone fragmentation spectra.33, 34 When applied to intact glycopeptide analysis, EThcD can produce rich fragment ion information for glycan, peptide and glycosylation site identification in one spectrum, providing the opportunity for site-specific glycoproteomics.35, 36
Cancer cells display altered patterns of metabolism in order to support their growth and survival.37 These changes in metabolism are often described as the Warburg effect,38, 39 which diverts intermediates away from oxidation respiration in mitochondria into the biosynthesis of carbohydrates, proteins, lipids and nucleic acids, supporting the biosynthetic requirements of uncontrolled proliferation. One of the metabolism pathway alteration is the preferential expression of pyruvate kinase isozymes M2 (PKM2) in the glycolysis pathway. Unlike other pyruvate kinases such as PKL, PKR, and PKM1 that form stable tetramers (the active form of PK), PKM2 exists as both dimers and tetramers.40 The PKM2 dimer has a reduced enzyme activity, which results in a buildup of glycolytic intermediates that enhances anabolic metabolism.41, 42 Pentose phosphate pathway (PPP) and hexosamine biosynthetic pathway (HBP) are two of those affected pathways that will utilize these glycolytic intermediates as substrates to synthesize nucleotide sugars such as uridine diphosphate N-acetylglucosamine (UDP-GlcNAc), which are utilized as donor substrates for protein glycosylation.43 In fact, studies have shown glycosylation alterations in various cancers, including breast cancer, pancreatic cancer and lung cancer.44 With an increased expression of PKM2 in cancer cells at the same time, this leads to the speculation that PKM2 plays a vital role in the glycosylation alterations during cancer progression.37 However, no study has been conducted to explore how PKM2 affects the overall glycosylation patterns in cancer cells.
In this work, we first evaluated the intact N-glycopeptide enrichment efficiency of HILIC and MLA respectively. The parallel experiments showed that sequential enrichment utilizing both HILIC and MLA largely increased N-glycopeptide coverage by 80% compared to utilizing either one of them alone. With hybrid fragmentation technique EThcD incorporated in the workflow, we confidently achieved glycan, glycosylation site and peptide backbone identification in one spectrum. Besides highly confident intact glycopeptide identification, reporter ions from DiLeu labeled glycopeptides generated in the low mass region enabled accurate quantitation. Utilizing the enhanced workflow, we were able to conduct comparative, quantitative, site-specific glycoproteomics study between PKM2 knockout cells and parental breast cancer cells (Figure 1), which revealed altered N-glycoprotein/N-glycopeptide expression in PKM2 knockout cells with increased fucosylation in several of the examined glycosylation sites. Further deglycoproteomics study confirmed the significantly altered glycosylation pattern after PKM2 knockout and revealed the altered PI3K/Akt signaling pathway, which supports the previous finding that PKM2 knockdown cancer cells rely on activation of Akt for their survival.
Figure 1.

Schematic illustration of the workflow for intact N-glycopeptides and deglycosylated peptide analysis.
Experimental section
Chemicals and materials
Dithiothreitol (DTT), PNGase F and sequencing grade trypsin were from Promega (Madison, WI). Optimal LC/MS grade acetonitrile (ACN), methanol (MeOH) and water were from Fisher Scientific (Pittsburgh, PA). Concanavalin A (ConA), wheat germ agglutinin (WGA), Ricinuscommunis agglutinin (RCA120), iodoacetamide (IAA), acetyl-D18 glucosamine, D-lactose, methyl α-D-mannopyranoside and manganese dichloride were obtained from Sigma-Aldrich (St. Louis, MO). Tris base, urea (UA), sodium chloride, ammonium bicarbonate (ABC) and calcium chloride (CaCl2) were obtained from Fisher Scientific (Pittsburgh, PA). Trifluoroacetic acid (TFA), triethylammonium bicarbonate (TEAB), N, N-dimethylformamide (DMF), 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium tetrafluoroborate (DMTMM), N-methylmorpholine (NMM) and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO). Hydroxylamine solution was purchased from Alfa Aesar (Ward Hill, MA). C18 OMIX tips were obtained from Agilent (Santa Clara, CA). Hydrophilic interaction chromatography material (PolyHYDROXYETHYL A) was obtained from PolyLC (Columbia, MD). Microcon filters YM-30 (30 kDa) was purchased from Merck Millipore (Billerica, MA). PANC-1 pancreatic ductal adenocarcinoma cells were from ATCC (Manassas, VA). Duplex DiLeu tags were custom synthesized in our own lab.45
PANC-1 cells and breast cancer cells
Commercially available PANC1 pancreatic ductal adenocarcinoma cells were routinely maintained in complete media of DMEM/Ham’s F-12 (1:1) (ATCC) supplemented with 10% fetal bovine serum (Hyclone) and 1% antibiotic-antimycotic solution (Cellgro). Cell culture flasks were placed in an incubator containing 5% CO2 and 98% humidity. Cells were used for a maximum of 15 passages and trypsinized using 0.25% trypsin EDTA solution (Gibco) once 80% confluence was achieved. Cell pellets were rapidly washed twice with phosphate-buffered saline, flash frozen in dry ice, and stored at −80 °C. PKM2 knockout cancer cells and parental breast cancer cells were provided by our collaborator Xu’s group.46
Protein extraction and digestion
PANC1 cell pellets were lysed by sonication in a solution containing digest buffer (4% SDS, 100 mM Tris/Base pH 8.0). The bicinchoninic acid assay (BCA assay) was applied to determine the protein concentration. To extract proteins from PKM2 knockout breast cancer cells and parental cells, after removing the cell media, cells were rapidly rinsed with saline, quenched with methanol, and scraped with addition of water. Chloroform was added at certain methanol/water/chloroform ratio (v/v/v). Protein precipitate in the middle layer was collected and dried. The upper and lower layers of supernatant were collected as polar and nonpolar metabolites for a separate study. The proteins were stored at −80°C. The experiments were performed with three biological replicates.
Trypsin digestion was performed based on previously reported filter-aided sample preparation (FASP) protocol47 with some modifications. Briefly, the protein extracts were thawed and centrifuged at 16, 000 ×g for 5 min. Then 200 µg protein was taken out to the vial and 1 M DTT in digest buffer was added to make DTT final concentration 0.1 M. Incubate the sample at 95°C for 3 min to reduce disulfide bonds. 200 µL of urea (UA) buffer (8 M UA in 100 mM Tris/Base) was added into the vial and transferred onto the 30 kDa filter. The filter was centrifuged at 14, 000 ×g for 15 min. Another 200 µL of UA buffer was added to the sample and centrifuge at 14, 000 ×g for 15 min. Add 100 µL of IAA buffer (0.05 M IAA in UA buffer) onto the filter and gently swirl to mix, then incubate in darkness for 20 min, followed by centrifugation at 14, 000 ×g for 10 min. Add 100 µL of UA buffer onto the filter, and centrifuge at 14, 000 ×g for 15 min. Repeat another 2 times. Add 100 µL of ABC buffer (50 mM) onto the filter, and centrifuge at 14, 000 ×g for 15 min. Repeat another 2 times. All the centrifugation was at 20°C. 10 µL of trypsin and 40 µL of ammonium bicarbonate (ABC) buffer was added onto the filter. The filter was incubated at 37°C water bath for 18h. After incubation, the filter was transferred to a fresh collection vial and centrifuged at 14, 000 ×g for 10min. 50 µL 0.5 M NaCl solution was added onto the filter and centrifuged at 14, 000 ×g for 10min. Repeat for one more time. TFA was added into the vial to make TFA final concentration 0.25%. Samples were desalted using a SepPak C18 SPE cartridge (Waters, Milford, MA).
Protein digest labeling with DiLeu tags for quantitation
Digested peptides from PKM2 knockout cells and parental cells were dissolved in 0.5 M TEAB prior to labeling. DiLeu tags were suspended in anhydrous DMF and combined with DMTMM and NMM at 0.6× molar ratios to DiLeu. The mixture was then vortexed at room temperature for 1h. Following centrifugation, the supernatant was immediately mixed with protein digest. Labeling was performed by addition of labeling solution at a 10:1 label to peptide digest ratio by weight and vortexing at room temperature for 2 h. The labeling reaction was quenched by addition of hydroxylamine to a concentration of 0.25%, and the labeled peptide samples were dried in vacuo. The samples labeled with different DiLeu channels were combined and were subject to strong cation exchange (SCX) chromatography to remove excess DiLeu tags.
HILIC enrichment
HILIC enrichment was conducted following a previously reported protocol with minor modification.26 Namely, 5 mg of HILIC beads (PolyLC) were first activated in 100 µL elution buffer (0.1% TFA) by vortexing for 30 min. Then the activated beads were washed with 100 µL binding buffer (0.1% TFA, 19.9% H2O, 80% ACN) for two times. 100 µg tryptic peptides were dissolved in 250 µL of binding buffer and mixed with beads at a 1:50 peptide-to-beads mass ratio. Vortex for 1 h to allow N-glycopeptides to bind to beads. The beads were washed with 50 µL binding buffer for 6 times. N-glycopeptides were eluted by washing the beads with elution buffer for 5 times. The eluted N-glycopeptides were dried down in vacuo. Supernatant during the process were collected for sequential MLA enrichment. The separation between beads and supernatant was achieved by centrifugation.
MLA enrichment
MLA enrichment was performed based on a previously reported protocol with some modifications48–51 Tryptic peptides were dissolved in 80 µL 1×binding solution (1 mM CaCl2, 1 mM MnCl2, 0.5 M NaCl in 20 mM Tris/Base, pH 7.3) and transferred to the 30 kDa filter. Adding 36 µL lectin mixtures (90 µg ConA, 90 µg WGA and 90 µg RCA120 in 2×biniding buffer) onto the filter. Incubate at room temperature for 1 h and unbound peptides were eluted by centrifuging at 14, 000 ×g for 10 min at 18°C. Wash with 200 µL binding solution for four times. Transfer the filter to a new collection vial. Add 100 µL sugar mixtures (300 mM N-acetyl-D-glucosamine, D-lactose, methyl α-D-mannopyranoside in 1×binding buffer) and incubate at room temperature for 30 min. This step was repeated once. The N-glycopeptides were eluted by centrifugation. Then the samples were acidified to 0.25% TFA and desalted by C18 OMIX tip. To prepare deglycosylated N-glycopeptides, after adding lectin mixtures and incubation for 1 h, wash with 200 µL binding solution for four times and 50 µl digest buffer (50 mM NH4HCO3) for two times. 8 µl PNGase F in 72 µl digest buffer was added onto the filter and incubated in 37°C water-bath for 3 h. The deglycosylated N-glycopeptides were Eluted with 2×50 µl digest buffer by centrifugation at 14, 000 ×g for 10 min at 18°C.
LC-MS/MS analysis
Samples were dissolved in 0.1% FA and analyzed on the Orbitrap Fusion™ Lumos™ Tribrid™ Mass Spectrometer (Thermo Fisher Scientific, San Jose, CA) coupled to a Dionex UPLC system. A binary solvent system composed of H2O containing 0.1% formic acid (A) and MeCN containing 0.1% formic acid (B) was used for all analyses. Peptides were loaded and separated on a 75 µm × 15 cm homemade column packed with 1.7 µm, 150 Å, BEH C18 material obtained from a Waters UPLC column (part no. 186004661). The LC gradient for intact N-glycopeptides was set as follows, 3%-30% B (18–98min), 30%-75% B (100–108 min) and 75%-95% B (108–118min). The mass spectrometer was operated in data dependent mode to automatically switch between MS and MS/MS acquisition. For intact N-glycopeptide analysis, an MS1 scan was acquired from 400–1800 (120,000 resolution, 4e5 AGC, 100 ms injection time) followed by EThcD MS/MS acquisition of the precursors with the highest charge states in an order of intensity and detection in the Orbitrap (60,000 resolution, 3e5 AGC, 100 ms injection time). EThcD was performed with optimized user defined charge dependent reaction time (2+ 50 ms; 3+ 20 ms; 4+ 20 ms; 5+ 20ms; 6 + 9 ms; 7+; 9 ms; 8+ 9ms) supplemented by 33% HCD activation. The LC gradient for deglycosylated N-glycopeptides was set as follows: 3%-30% B (18–100min), 30%-75% B (100–110 min) and 75%-95% B (110–111min). For deglycosylated N-glycopeptide analysis, an MS1 scan was acquired from 300–1500 (60,000 resolution, 2e5 AGC, 100 ms injection time) followed by MS/MS data-dependent acquisition of the 15 most intense ions with HCD and detection in the Orbitrap (15,000 resolution, 1e4 AGC, 30 NCE, 100 ms injection time).
Data analysis
All raw data files were searched against UniProt Homo sapiens reviewed database (08.10.2016, 20, 152 sequences). Deglycosylated peptides data were searched using Sequest HT algorithm incorporated in Proteome Discoverer (version 2.1.1.21, Thermo Scientific). Trypsin was selected as the enzyme and two maximum missed cleavages were allowed. Searches were performed with a precursor mass tolerance of 20 ppm and a fragment mass tolerance of 0.03 Da. Static modifications consisted of DiLeu labels on peptide N-termini and lysine residue (+145.1240 Da), carbamidomethylation of cysteine residues (+57.02146 Da). Dynamic modifications consisted of oxidation of methionine residues (+15.99492 Da), deamidation of asparagine (+0.98402 Da) and oxidation of methionine (+15.9949 Da). Three maximal dynamic modifications were allowed. Peptide spectral matches (PSMs) were validated based on q-values to 1% FDR (false discovery rate) using percolator. Quantitation was performed in Proteome Discoverer with a reporter ion integration tolerance of 20 ppm for the most confident centroid.
For intact N-glycopeptide analysis, data were searched using PTM-centric search engine Byonic (version 2.9.38, Protein Metrics, San Carlos, CA) incorporated in Proteome Discoverer (Figure 1). All the parameters were set the same as deglycosylated N-glycopeptide analysis except the following settings. N-glycosylation on asparagine and deamidation on asparagine and glutamine were set as common modifications. Oxidation of methionine (+15.9949 Da) was set as rare modification. Two common modification and one rare modification were allowed. Human N-glycan database embedded in Byonic, which included 182 glycan entities, was used. As for glycopeptide FDR control, Byonic default settings was applied that cut the protein list after the 20th decoy protein or at the point in the list at which the protein FDR first reached 1%, whichever cut resulted in more proteins. After that, Byonic estimated the spectrum-level FDR of the remaining PSMs to the reported proteins which typically were in the range 0–5%. Only those N-glycopeptides with PSMs FDR ≤ 1% were reported.
Results and Discussion
Comparison between HILIC and MLA
In a previous study, ion paring HILIC has been reported to increase the difference in hydrophilicity between glycosylated and non-glycosylated peptides, which largely decreased the non-specific binding and increased the coverage of N-glycosylation sites.52 Thus, ion pairing HILIC (IP-HILIC) method was used in our study by adding TFA as the ion pairing reagent. Compared with single lectin method, multi-lectin affinity (MLA) method can increase N-glycopeptide coverage by binding to different glycan moiety.50 In our study, lectin mixtures containing ConA, WGA and RCA 120 were selected to simultaneously capture mannose, sialic acid, N-acetylglucosamine and galactose to improve N-glycopeptide enrichment ability. Starting from 200 µg proteins from PANC1, a total of 433 intact N-glycopeptides were identified using HILIC method and 582 intact N-glycopeptides were identified using MLA method (Table S1). The Venn diagram analysis in Figure 2 shows that only 103 N-glycopeptides were overlapped. HILIC and MLA enrichment analyses were also carried out in a separate cell aliquot to demonstrate the reproducibility of the results (Figure S2). A closer look at the composition of the 433 N-glycopeptides enriched by HILIC shows that sialylated N-glycopeptides comprise 71% of the identified N-glycopeptides. This is probably because sialic acid increases the hydrophilic interactions between the N-glycopeptides and HILIC beads, resulting in preferential enrichment of sialylated N-glycopeptides. It has been reported that sialylated N-glycopeptides bind HILIC sorbent stronger than non-sialylated N-glycopeptides under commonly used acidic conditions.53 While for MLA method, the sialylated N-glycopeptides enriched only comprise 12%, even though WGA in the lectin mixtures binds to sialic acid. One possible reason is that WGA also binds to N-acetylglucosamine, which may cause other high-mannose and complex glycans to compete for the binding. Our results showed that HILIC preferentially enriches sialylated N-glycopeptides, while MLA preferentially enriches non-sialylated N-glycopeptides. For example, N-glycopeptide [R].EAGNHTSGAGLVQINK.[S] from cation-dependent mannose-6-phosphate receptor, which was N-glycosylated at the 4th asparagine residue, was identified using both of these two methods. With HILIC method, eight out of nine N-glycans identified at this site were sialylated glycans. In sharp contrast, three out of eight were sialylated glycans using MLA method. Another interesting observation between these two methods is the ability to enrich paucimannosidic N-glycopeptides, which consist of two N-acetylglucosamine, 1–3 mannose residues and an optional fucose residue. Out of 433 glycopeptides identified by HILIC, only 3 of them were paucimannosidic N-glycopepitdes. For MLA enrichment method, 80 out of 582 were paucimannosidic N-glycopeptides. This is because, in order for glycopeptides to be captured by HILIC beads, a minimum degree of local hydrophilicity is needed, which does not appear to be provided by the paucimannosidic N-glycopepitdes.54 This indicates that the nature of the individual glycan significantly influences the HILIC retention behavior of these lowly hydrophilic truncated N-glycans attached to peptides. In comparison, lectin captures N-glycans through recognizing a specific glycan moiety, which is not influenced by the hydrophilicity of glycans. Therefore, in order to achieve more comprehensive enrichment of different N-glycopeptides, our results indicate that there is a need for sequential enrichment. With no salt used in HILIC method, MLA enrichment could be directly performed with the flow-through after HILIC enrichment. After combining these two enrichment methods, a total of 1067 intact N-glycopeptides were identified, representing 311 glycosylation sites and 88 N-glycan compositions from 205 glycoproteins as shown in Figure 3A. As a consequence of sequential enrichment, there are about 80% increase in the number of intact N-glycopeptides, 40% increase in glycosylation sites and 35% increase in glycoproteins compared to individual enrichment method. Among the 88 N-glycans identified along with the 1067 N-glycopeptides, we found that almost all categories of N-glycans were identified, including high-mannose, hybrid and complex types along with the N-glycan synthesis pathways (Figure 3B). Besides 79 of N-glycans identified in the classical synthesis pathway, 9 paucimannosidic N-glycans from 84 N-glycopeptides and 38 glycoproteins were also identified in the unconventional truncation pathway. Gene ontology analysis of these glycoproteins revealed that they were most enriched in lysosome, which was in accordance with the previous finding.55
Figure 2.

The Venn diagram analysis of intact N-glycopeptides (A) and N-glycosylation sites (B) enriched by HILIC and MLA methods. (C) The comparison of the percentage of sialylated N-glycopeptides enriched by HILIC and MLA respectively.
Figure 3.

(A) The comparison of the number of intact N-glycopeptides, N-glycosylation sites and N-glycoproteins enriched by HILIC, MLA and sequential HILIC and MLA enrichment. (B) Different types of N-glycans along the N-glycan synthesis enriched by sequential HILIC and MLA enrichment. EThcD was used for N-glycopeptide identification.
Intact N-glycopeptide quantitation enabled by EThcD
Intact N-glycopeptide analysis was conducted by utilizing EThcD as fragmentation method. As shown in Figure 4A, a series of fragment ions including c/z, b/y ions, glycan fragment ions and glycopeptides with one or more loss of monosaccharides were detected. In the low mass region, the glycan signature oxonium ions including m/z 138.06 (HexNAc-2H2O-CH2O), 168.06 (HexNAc-2H2O), 186.08 (HexNAc-H2O) and 204.09 (HexNAc), were detected, confirming that the spectrum belonged to a glycopeptide. For relative quantitation, intact N-glycopeptides from different samples were labeled by isobaric duplex DiLeu tags, which were custom synthesized by incorporating stable isotopes (13C, 2H, 18O and 15N) into the reporter group and mass-balanced group, creating reporter ions at m/z 115.12476 and 117.13731. When designing the DiLeu tags, we have intentionally grouped the deuterium atoms surrounding the tertiary amine group in the DiLeu tag and also only used a small number of deuterium atoms in DiLeu tag to minimize the isotope effect. As a result, the increased polarity of the amine group has successfully offset the small deuterium number difference in the tags with a negligible difference in retention time using RP chromatography.45 The reliable and robust quantitation ability of DiLeu tags up to 12-plex for quantitative proteomics studies have been demonstrated.56–61 Recently, our group has successfully applied DiLeu tags for quantitation of labile PTM enabled by EThcD.62 Even though ETD has been well-known to preserve labile PTMs, the intensity of reporter ions produced is often quite low so that the quantitation accuracy is compromised. While HCD provides high reporter ion intensity, labile PTMs are poorly preserved. Utilizing the novel EThcD hybrid fragmentation method, where a supplemental energy is applied to all fragment ions formed by ETD, we were able to successfully apply DiLeu tags for reliable quantitative phosphoproteomics study with increased phosphorylation site confidence.62 In this regard, we propose quantitation of intact glycopeptides could also benefit from EThcD for site-specific characterization as well as accurate quantitation. As shown in Figure 4A, reporter ions with decent intensities next to the base peak (HexNAc oxonium ion) were produced in the low mass region, which facilitated the accurate and reliable quantitation of intact glycopeptides. As oxonium ions are easily generated due to the labile glycosidic bonds, the oxonium ion would always be the base peak, which was also observed for isobaric tags for relative and absolute quantitation (iTRAQ) and tandem mass tags (TMT) labeled intact N-glycopeptides.63–65
Figure 4.

(A) The EThcD mass spectrum of duplex DiLeu labeled intact N-glycopeptide. (B) The HCD mass spectrum of duplex DiLeu labeled deglycosylated peptide.
To delineate to what extent the labile glycans affect the overall reporter ion yield, we compared the reporter ion yield efficiency between the labeled N-glycopeptides and labeled nonglycosylated peptides. After HILIC and MLA enrichment, there were still a few nonglycosylated peptides residuals, because of overlapping hydrophobicity between N-glycopeptides and non-glycosylated peptides and non-specific binding of nonglycosylated peptides to lectin. Thus, the reporter ions generated from the labeled N-glycopeptides and labeled nonglycosylated peptides in the same run would serve as perfect examples for their reporter ion yield comparison with a minimized instrument and sample variations. As shown in Figure S1, compared to nonglycosylated peptides, the average reporter ion intensity generated by labeled N-glycopeptides was 6× times lower and the average precursor intensity was 3× times lower, which is largely due to the lower abundance and poorer ionization efficiency of N-glycopeptides. In addition, the labile glycosidic bonds could negatively impact the yield of reporter ions, which leads to a 2-fold lower reporter ion yield for N-glycopeptides. To this end, compared to EThcD, HCD mode may generate a higher reporter ion yield of intact N-glycopeptides given sufficient collision energy being used. However, it should be noted that if a higher HCD collision energy is used to increase the reporter ion yield, glycan fragments may dominate and compromise the peptide backbone fragmentation. Unlike the longer cycle time needed for ETD reaction, HCD also affords a faster scan speed that allows more spectra to be collected in one cycle. However, the EThcD fragmentation technique could produce many more information-rich fragments including c/z ions and peptide ions with partial or intact glycan preserved, resulting in improved spectral quality. To conclude, the reporter ion yield, the number of total scans, and spectral quality should be considered when choosing the most effective fragmentation technique for quantitative analysis of intact glycopeptides.
Intact N-glycopeptide quantitation between PKM2 knockout cells vs. parental breast cancer cells
PKM2 knockout breast cancer cell lines were recently generated by CRISPR/cas9 method46 and used in this glycoproteomic study. In total, 45 intact N-glycopeptides (39 up-regulated and 6 down-regulated) from 22 glycoproteins were significantly regulated (p≤ 0.05 and ±1.5-fold) in PKM2 knockout cells vs. parental breast cancer cells (Figure 5B, Table S2). Using the developed approach, we were able to quantify different glycoforms on a single glycosylation site and reveal different glycosylation pattern. One example is lysosome-associated membrane glycoprotein 1 (LAMP-1), which has been shown heavily N-glycosylated previously.66, 67 A total of 20 N-glycopeptides from six glycosylation sites were quantified and N-glycopeptides from four representing glycosylation sites were shown in Figure 7. Five glycoforms were quantified on the 7th asparagine residue on glycopeptide [R].LLNINPNK.[T], including one high-mannose N-glycan and four fucosylated N-glycans. The results show that the high-mannose glycoform was up-regulated, while the four fucosylated glycoforms remained unaltered after PKM2 knockout, which indicates that different kinds of glycoforms respond differently even on the same glycosylation site. On the third asparagine residue of N-glycopeptide [R].KDNTTVTR.[L], 4 out of 5 fucosylated glycoforms detected showed an up-regulation after PKM2 knockout. Overall, there was an up-regulation of fucosylation on glycoprotein LAMP-1, which was also observed for LAMP-2. LAMP-1 and LAMP-2 were mainly found in the lysosomes and late endosomes and were known to maintain the lysosomal acidification and lysosomal membrane integrity.68 In addition to their role in lysosomal biogenesis, it was also reported that LAMP-1 and LAMP-2 are associated with autophagosome accumulation and biogenesis.69, 70 A lack of fucosylation may alter the integrity of LAMP-1 and LAMP-2, which in turn may affect the dynamic distribution of lysosomes. In addition, the fucosylated LAMP-1 and LAMP-2 forms are responsible for regulating autophagy biogenesis, which may influence tumor development and progression.71 Besides LAMP-1 and LAMP-2, an increased fucosylation was also observed for other glycoproteins including prosaposin, palmitoyl-protein thioesterase 1, galectin-3-binding protein and dipeptidyl peptidase 2. In fact, increased fucosylation has been reported to have unique biological significance in cancer.72 For example, in breast cancer, increased core fucosylation of epidermal growth factor receptor (EGFR) has been found to result in increased EGFR-mediated signaling associated with tumor cell growth and malignancy.73, 74
Figure 5.

(A) Volcano plot of the quantitation results of deglycosylated peptides in PKM2 knockout (PKM2 KO) and parental breast cancer cells. (B) Volcano plot of the quantitation results of intact N-glycopeptides in PPKM2 KO and parental breast cancer cells.
Figure 7.

Quantitative analysis of intact N-glycopeptides from Lysosomal-associated membrane protein 1 (LAMP-1). The letter “N” in red denotes the N-glycosylation site.
On the other hand, PKM2 was found to be preferentially expressed in cancer cells75 and it significantly affected the levels of substrates used for protein glycosylation by regulating the last step of glycolysis.76 Thus, it has been speculated that PKM2 attributes to glycosylation alteration in cancer by affecting the availability and abundance of the sugar nucleotide donors.75 However, whether PKM2 alters the glycosylation pattern remains unknown. Our comparative glycoproteomics study using paired parental and PKM2 knockout breast cancer cells revealed alteration in glycosylation patterns after PKM2 knockout, providing evidence that PKM2 contributed to the altered glycosylation in cancer. In particular, fucosylation, one of the most widely reported glycosylation often altered in cancer cells, was notably affected by knocking out PKM2.
Deglycosylated peptide quantitation enabled by HCD
Due to glycosylation micro-heterogeneity, various glycoforms can exist on the same glycosylation site. By enzymatically removing these glycoforms with PNGase F, deglycosylated peptides could be obtained with the marker of asparagine (Asn) being deamidated indicating the exact glycosite.77 After this step, the abundance of glycopeptides with different glycoforms will be merged into one glycosylation site-containing peptide, increasing the chance to be detected. Also the ionization efficiency will be largely increased by removing the hydrophilic glycan and signal suppression was greatly reduced by other more abundant nonglycosylated peptide, increasing the coverage of glycosylation site. Despite losing the site-specific glycoform information, deglycoproteomics approach could provide complementary information about alterations of glycoproteins. Thus, deglycosylated peptides were analyzed using HCD after PNGase F treatment. Figure 4B shows the spectrum of a duplex DiLeu labeled deglycosylated peptide, with a wealth of peptide backbone fragments and reporter ion fragments being detected. The N-glycosylation site was identified by the signature asparagine deamidation after PNGase F treatment. To avoid any false-positive assignment of N-glycosites brought by spontaneous deamidation,77 aliquots of intact N-glycopeptides were used as negative control samples and analyzed directly by LC-MS/MS without PNGase F treatment. Furthermore, all the deglycosylated glycopeptides identified in the study contain NXS/T (where X≠proline) consensus motifs to ensure confident identification. A total of 484 deglycosylated peptides were quantified, among which 81 glycosylations sites from 70 glycoproteins, showed significant changes with more than 1.5-fold (p≤0.05) alterations (Figure 5A, Table S3). KEGG pathway analysis revealed that these altered glycoproteins are highly enriched in ECM-receptor interaction, arrhythmogenic right ventricular cardiomyopathy (ARVC), PI3K/Akt signaling pathway, cell adhesion molecules (CAMs) and focal adhesion pathways (Figure 6A, Table S4).
Figure 6.

KEGG pathway analysis (A), gene ontology cellular component (B) and gene ontology cellular component (C) analysis of the significantly altered glycoproteins.
One interesting observation was the alteration of the PI3K/Akt signaling pathway that was highly enriched. In our study, 10 glycoproteins along the PI3K/Akt pathway were found to be significantly altered in PKM2 knockout cells, including insulin like growth factor 1 receptor (IGF1R), insulin receptor (INSR), integrin subunit alpha V (ITGAV), integrin subunit beta 1 (ITGB1), integrin subunit beta 6 (ITGB6), ephrin A4 (EFNA4), laminin subunit alpha 5 (LAMA5), laminin subunit beta 1(LAMB1), laminin subunit beta 2 (LAMB2) and thrombospondin 1 (THBS1) (Table 1). PKM 2 plays an important role for anabolic glycolysis to support rapid proliferation of cancer cells.76 Previous studies have shown that PKM2 knockdown only leads to a modest impairment of cancer cell survival but the mechanism remains elusive. One study showed that knockdown of PKM2 leads to activation of Akt and inhibition of PI3K/Akt signaling pathway, resulting in significant growth inhibition and apoptosis in PKM2 knockdown cells, which indicated Akt activation was necessary for the survival of PKM2 knockdown cells.78 In fact, activated Akt is a well-established survival factor and activated Akt modulates the function of many substrates involved in the regulation of cell survival, cell cycle progression and cellular growth.79 Thus, our results offer additional evidence to support that PKM2 knockout cells may rely on regulation of the PI3K/Akt pathway to survive. Gene ontology biological process analysis revealed that cell adhesion, positive regulation of cell migration, cell adhesion mediated by integrin, viral entry into host cell, extracellular matrix organization, leukocyte migration and integrin-mediate signaling pathways are the main biological processes that have been affected (Figure 6C, Table S4). According to our observation, there was a decrease in cell migration after PKM2 knockout, which agreed well with the alteration of positive regulation of cell migration pathway. Gene ontology cellular component analysis showed that both intracellular and extracellular glycoproteins were affected, including those localized in extracellular exosome, cell surface, and endoplasmic reticulum, etc. (Figure 6B, Table S4).
Table 1.
Altered glycoproteins involved in PI3K–Akt signaling pathway
| Uniprot ID |
Gene Name |
Glycosylation position |
Fold change |
Up or down |
|---|---|---|---|---|
| P52798 | EFNA4 | P52798 [28–38] | 0.63 | ↓ |
| P08069 | IGF1R | P08069 [223–252] | 0.56 | ↓ |
| P08069 [741–753] | 0.47 | ↓ | ||
| P06213 | INSR | P06213 [697–714] | 1.67 | ↑ |
| P06756 | ITGAV | P06756 [73–88] | 1.84 | ↑ |
| P06756 [865–883] | 2.07 | ↑ | ||
| P05556 | ITGB1 | P05556 [403–414] | 1.89 | ↑ |
| P18564 | ITGB6 | P18564 [459–466] | 3.17 | ↑ |
| P18564 [567–594] | 3.21 | ↑ | ||
| O15230 | LAMA5 | O15230 [3106–3123] | 0.61 | ↓ |
| P07942 | LAMB1 | P07942 [1271–1283] | 3.58 | ↑ |
| P55268 | LAMB2 | P55268 [1347–1365] | 0.57 | ↓ |
| P07996 | THBS1 | P07996 [1065–1077] | 0.50 | ↓ |
Both altered glycosylation and metabolic reprogramming have been extensively reported in cancer cells.80, 81 Glycosylation is generated from metabolites and is also a nutrient-sensitive post-translational modification of proteins. At the same time, as one of the key cues for cellular signals, glycosylation also regulates metabolism through various receptors and signal transduction pathways that regulate metabolism.37 This implies that these two most common features, glycosylation signaling and metabolism, of cancers cells might be intervened.82 PKM2, a limiting glycolytic enzyme that catalyzes the final step in glycolysis, plays essential role in metabolic changes in cancer cells. Many studies83–85 have shown PKM2 was preferentially expressed in cancer cells and it contributes to the accumulation of metabolites intermediates, which are the sources of substrates for protein glycosylation. Thus, PKM2 has been implicated to be involved in the glycosylation alteration in cancer.37 To directly address the question whether and how PKM2 knockout alters the overall glycosylation pattern in cancer cells, we employed PKM2 knockout cell lines for quantitative glycosylation studies. Indeed, altered glycosylation was detected by knocking out PKM2, which confirms the previous speculation as well as provides direct evidence that metabolism and signaling pathway were tightly regulated. On the other hand, previous study showed that activation of Akt is necessary for the survival of PKM2 knockdown cells, suggesting that combination of PKM2 knockdown with PI3K or Akt inhibitors may be therapeutically effective. Our study identified 10 N-glycoproteins with altered glycosylation in the PI3K/Akt pathway upon loss of PKM2, which could provide the potential targets for follow-up studies.
Conclusions
In the present study, a dedicated large-scale site-specific quantitative N-glycoproteomics workflow has been established. By employing FASP method for trypsin digestion, SDS could be used to enhance the extraction efficacy of membrane glycoproteins. Intact N-glycopeptide enrichment efficiencies with HILIC and MLA were compared for the first time. The results show that HILIC preferentially enriched sialylated N-glycopeptides while MLA preferentially enriched nonsialylated glycopeptides, implying that a sequential enrichment procedure would improve intact N-glycopeptide enrichment efficiency. Benefiting from EThcD, site-specific intact glycopeptide structure characterization and quantitation can be achieved in one spectrum, which also provides an opportunity for FDR control at the intact glycopeptide level. Using PTM-centric search engine Byonic, FDR-based large-scale glycoproteomics was achieved. Furthermore, we successfully applied this developed workflow to study the glycosylation alteration in PKM2 knockout breast cancer cells vs. parental cells. Upon loss of PKM2, the abundance ratios of different glycoforms on the same glycosylation vary differently and an increased fucosylation was observed in several of the examined glycosylation sites. Further deglycoproteomics study revealed that the 10 glycoproteins in PI3K/Akt signaling pathway were altered, which supported the previous finding that PKM2 knockdown cancer cells rely on the activation of Akt for their survival.
Supplementary Material
Figure S1. The reporter ions intensity (A), precursor ions intensity (B) and reporter ion yield (C) of glycopeptides and nonglycosylated peptides in the same run.
Figure S2. The Venn diagram analysis of (A) intact N-glycopeptides (A) and (B) N-glycosylation sites identified with HILIC and MLA enrichment methods. (C) The comparison of the percentage of sialylated N-glycopeptides enriched between two methods.
The identified N-glycopeptides enriched by HILIC alone, lectin alone and sequential HILIC and lectin enrichment.
Glycoproteomics results between PKM2 knockout cells vs. parental breast cancer cells.
Deglycoproteomics results between PKM2 knockout cells vs. parental breast cancer cells.
Gene ontology analysis results.
Acknowledgments
This research was supported in part by the National Institutes of Health (NIH) grants R21AG055377, R01AG052324, R01 DK071801, R01 CA213293, and P41GM108538. The Orbitrap instruments were purchased through the support of an NIH shared instrument grant (NIH-NCRR S10RR029531) and Office of the Vice Chancellor for Research and Graduate Education at the University of Wisconsin-Madison. LL acknowledges a Vilas Distinguished Achievement Professorship and Janis Apinis Professorship with funding provided by the Wisconsin Alumni Research Foundation and University of Wisconsin-Madison School of Pharmacy. We thank Dr. Marshall Bern from Protein Metrics for providing access to Byonic software package.
Footnotes
Conflicts of interest
There are no conflicts of interest to declare.
Supplementary information is available for the online version of this article through publisher’s web site.
References
- 1.Gewinner C, Hart G, Zachara N, Cole R, Beisenherz-Huss C, Groner B. J. Biol. Chem. 2004;279:3563–3572. doi: 10.1074/jbc.M306449200. [DOI] [PubMed] [Google Scholar]
- 2.Alikhani Z, Alikhani M, Boyd CM, Nagao K, Trackman PC, Graves DT. J. Biol. Chem. 2005;280:12087–12095. doi: 10.1074/jbc.M406313200. [DOI] [PubMed] [Google Scholar]
- 3.Phan UT, Waldron TT, Springer TA. Nat. Immunol. 2006;7:883–889. doi: 10.1038/ni1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Partridge EA, Le Roy C, Di Guglielmo GM, Pawling J, Cheung P, Granovsky M, Nabi IR, Wrana JL, Dennis JW. Science. 2004;306:120–124. doi: 10.1126/science.1102109. [DOI] [PubMed] [Google Scholar]
- 5.Hakomori S-i. Cancer Res. 1996;56:5309–5318. [PubMed] [Google Scholar]
- 6.Couldrey C, Green JE. Breast Cancer Res. 2000;2:321. doi: 10.1186/bcr75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drake PM, Cho W, Li B, Prakobphol A, Johansen E, Anderson NL, Regnier FE, Gibson BW, Fisher SJ. Clin. Chem. 2010;56:223–236. doi: 10.1373/clinchem.2009.136333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hwang H, Zhang J, Chung KA, Leverenz JB, Zabetian CP, Peskind ER, Jankovic J, Su Z, Hancock AM, Pan C. Mass spectrometry reviews. 2010;29:79–125. doi: 10.1002/mas.20221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rademacher TW, Parekh RB, Dwek RA, Isenberg D, Rook G, Axford JS, Roitt I. Springer seminars in immunopathology. 1988 doi: 10.1007/BF01857227. [DOI] [PubMed] [Google Scholar]
- 10.Thaysen-Andersen M, Packer NH. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics. 2014;1844:1437–1452. doi: 10.1016/j.bbapap.2014.05.002. [DOI] [PubMed] [Google Scholar]
- 11.Hägglund P, Bunkenborg J, Elortza F, Jensen ON, Roepstorff P. J. Proteome Res. 2004;3:556–566. doi: 10.1021/pr034112b. [DOI] [PubMed] [Google Scholar]
- 12.Wuhrer M, Koeleman CA, Hokke CH, Deelder AM. Anal. Chem. 2005;77:886–894. doi: 10.1021/ac048619x. [DOI] [PubMed] [Google Scholar]
- 13.Thaysen-Andersen M, Thøgersen IB, Lademann U, Offenberg H, Giessing AM, Enghild JJ, Nielsen HJ, Brünner N, Højrup P. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics. 2008;1784:455–463. doi: 10.1016/j.bbapap.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 14.Chen Z, Zhong X, Tie C, Chen B, Zhang X, Li L. Anal. Bioanal. Chem. 2017;409:4437–4447. doi: 10.1007/s00216-017-0387-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirabayashi J. Glycoconjugate J. 2004;21:35–40. doi: 10.1023/B:GLYC.0000043745.18988.a1. [DOI] [PubMed] [Google Scholar]
- 16.Bunkenborg J, Pilch BJ, Podtelejnikov AV, Wiśniewski JR. Proteomics. 2004;4:454–465. doi: 10.1002/pmic.200300556. [DOI] [PubMed] [Google Scholar]
- 17.Larsen MR, Jensen SS, Jakobsen LA, Heegaard NH. Mol. Cell. Proteomics. 2007;6:1778–1787. doi: 10.1074/mcp.M700086-MCP200. [DOI] [PubMed] [Google Scholar]
- 18.Glover MS, Yu Q, Chen Z, Shi X, Kent KC, Li L. Int. J. Mass spectrom. 2017 doi: 10.1007/s13361-017-1701-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu J, Wang F, Cheng K, Dong J, Sun D, Chen R, Wang L, Ye M, Zou H. Proteomics. 2013;13:1306–1313. doi: 10.1002/pmic.201200367. [DOI] [PubMed] [Google Scholar]
- 20.Zhang H, Li X-j, Martin DB, Aebersold R. Nat. Biotechnol. 2003;21:660–666. doi: 10.1038/nbt827. [DOI] [PubMed] [Google Scholar]
- 21.Chen W, Smeekens JM, Wu R. Mol. Cell. Proteomics. 2014;13:1563–1572. doi: 10.1074/mcp.M113.036251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li X, Liu H, Qing G, Wang S, Liang X. Journal of Materials Chemistry B. 2014;2:2276–2281. doi: 10.1039/c4tb00130c. [DOI] [PubMed] [Google Scholar]
- 23.Thaysen-Andersen M, Packer NH, Schulz BL. Mol. Cell. Proteomics. 2016;15:1773–1790. doi: 10.1074/mcp.O115.057638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Calvano CD, Zambonin CG, Jensen ON. J. Proteomics. 2008;71:304–317. doi: 10.1016/j.jprot.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 25.Ma C, Zhao X, Han H, Tong W, Zhang Q, Qin P, Chang C, Peng B, Ying W, Qian X. Electrophoresis. 2013;34:2440–2450. doi: 10.1002/elps.201200662. [DOI] [PubMed] [Google Scholar]
- 26.Zhang C, Ye Z, Xue P, Shu Q, Zhou Y, Ji Y, Fu Y, Wang J, Yang F. J. Proteome Res. 2016;15:2960–2968. doi: 10.1021/acs.jproteome.6b00098. [DOI] [PubMed] [Google Scholar]
- 27.Chen Z, Glover MS, Li L. Curr. Opin. Chem. Biol. 2018;42:1–8. doi: 10.1016/j.cbpa.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhong X, Chen Z, Snovida S, Liu Y, Rogers JC, Li L. Anal. Chem. 2015;87:6527–6534. doi: 10.1021/acs.analchem.5b01835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thaysen-Andersen M, Packer NH, Schulz BL. Molecular & Cellular Proteomics. 2016;15:1773–1790. doi: 10.1074/mcp.O115.057638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Domon B, Costello CE. Glycoconjugate J. 1988;5:397–409. [Google Scholar]
- 31.Biemann K. Methods Enzymol. 1990;193:886. doi: 10.1016/0076-6879(90)93460-3. [DOI] [PubMed] [Google Scholar]
- 32.Frese CK, Altelaar AM, van den Toorn H, Nolting D, Griep-Raming J, Heck AJ, Mohammed S. Anal. Chem. 2012;84:9668–9673. doi: 10.1021/ac3025366. [DOI] [PubMed] [Google Scholar]
- 33.Frese CK, Zhou H, Taus T, Altelaar AM, Mechtler K, Heck AJ, Mohammed S. J. Proteome Res. 2013;12:1520–1525. doi: 10.1021/pr301130k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liao R, Zheng D, Nie A, Zhou S, Deng H, Gao Y, Yang P, Yu Y, Tan L, Qi W. J. Proteome Res. 2017;16:780–787. doi: 10.1021/acs.jproteome.6b00788. [DOI] [PubMed] [Google Scholar]
- 35.Yu Q, Wang B, Chen Z, Urabe G, Glover MS, Shi X, Guo L-W, Kent KC, Li L. J. Am. Soc. Mass Spectrom. 2017;28:1751–1764. doi: 10.1007/s13361-017-1701-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marino F, Bern M, Mommen GP, Leney AC, van Gaans-van den Brink JA, Bonvin AM, Becker C, van Els CA, Heck AJ. J. Am. Chem. Soc. 2015;137:10922–10925. doi: 10.1021/jacs.5b06586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cairns RA, Harris IS, Mak TW. Nature Reviews Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 38.Warburg O, Wind F, Negelein E. The Journal of general physiology. 1927;8:519. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Warburg O. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 40.Mazurek S. The international journal of biochemistry & cell biology. 2011;43:969–980. doi: 10.1016/j.biocel.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 41.Vander Heiden MG, Locasale JW, Swanson KD, Sharfi H, Heffron GJ, Amador-Noguez D, Christofk HR, Wagner G, Rabinowitz JD, Asara JM. Science. 2010;329:1492–1499. doi: 10.1126/science.1188015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ye J, Mancuso A, Tong X, Ward PS, Fan J, Rabinowitz JD, Thompson CB. Proceedings of the National Academy of Sciences. 2012;109:6904–6909. doi: 10.1073/pnas.1204176109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shirato K, Nakajima K, Korekane H, Takamatsu S, Gao C, Angata T, Ohtsubo K, Taniguchi N. J. Clin. Biochem. Nutr. 2010;48:20–25. doi: 10.3164/jcbn.11-015FR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pinho SS, Reis CA. Nature Reviews Cancer. 2015;15:540–555. doi: 10.1038/nrc3982. [DOI] [PubMed] [Google Scholar]
- 45.Xiang F, Ye H, Chen R, Fu Q, Li L. Anal. Chem. 2010;82:2817–2825. doi: 10.1021/ac902778d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu F, Ma F, Wang Y, Hao L, Zeng H, Jia C, Wang Y, Liu P, Ong IM, Li B. Nat. Cell Biol. 2017;19:1358. doi: 10.1038/ncb3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Nat. Methods. 2009;6:359. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 48.Zielinska DF, Gnad F, Schropp K, Wiśniewski JR, Mann M. Mol. Cell. 2012;46:542–548. doi: 10.1016/j.molcel.2012.04.031. [DOI] [PubMed] [Google Scholar]
- 49.Deeb SJ, Cox J, Schmidt-Supprian M, Mann M. Mol. Cell. Proteomics. 2014;13:240–251. doi: 10.1074/mcp.M113.033977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zielinska DF, Gnad F, Wiśniewski JR, Mann M. Cell. 2010;141:897–907. doi: 10.1016/j.cell.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 51.Pan Y, Bai H, Ma C, Deng Y, Qin W, Qian X. Talanta. 2013;115:842–848. doi: 10.1016/j.talanta.2013.06.031. [DOI] [PubMed] [Google Scholar]
- 52.Mysling S, Palmisano G, Højrup P, Thaysen-Andersen M. Analytical chemistry. 2010;82:5598–5609. doi: 10.1021/ac100530w. [DOI] [PubMed] [Google Scholar]
- 53.Thaysen-Andersen M, Larsen MR, Packer NH, Palmisano G. Rsc Advances. 2013;3:22683–22705. [Google Scholar]
- 54.Loke I, Packer NH, Thaysen-Andersen M. Biomolecules. 2015;5:1832–1854. doi: 10.3390/biom5031832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Medzihradszky KF, Kaasik K, Chalkley RJ. Mol. Cell. Proteomics. 2015;14:2103–2110. doi: 10.1074/mcp.M115.050393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Greer T, Hao L, Nechyporenko A, Lee S, Vezina CM, Ricke WA, Marker PC, Bjorling DE, Bushman W, Li L. PLoS One. 2015;10:e0135415. doi: 10.1371/journal.pone.0135415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hao L, Johnson J, Lietz CB, Buchberger A, Frost D, Kao WJ, Li L. Anal. Chem. 2017;89:1138–1146. doi: 10.1021/acs.analchem.6b03482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Frost DC, Greer T, Li L. Anal. Chem. 2014;87:1646–1654. doi: 10.1021/ac503276z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yu Q, Shi X, Greer T, Lietz CB, Kent KC, Li L. J. Proteome Res. 2016;15:3420–3431. doi: 10.1021/acs.jproteome.6b00641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Frost DC, Greer T, Xiang F, Liang Z, Li L. Rapid Commun. Mass Spectrom. 2015;29:1115–1124. doi: 10.1002/rcm.7201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xiang F, Ye H, Chen R, Fu Q, Li L. Anal. Chem. 2010;82:2817–2825. doi: 10.1021/ac902778d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu Q, Shi X, Feng Y, Kent KC, Li L. Anal. Chim. Acta. 2017;968:40–49. doi: 10.1016/j.aca.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shah P, Wang X, Yang W, Eshghi ST, Sun S, Hoti N, Chen L, Yang S, Pasay J, Rubin A. Mol. Cell. Proteomics. 2015;14:2753–2763. doi: 10.1074/mcp.M115.047928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ye H, Boyne MT, Buhse LF, Hill J. Anal. Chem. 2013;85:1531–1539. doi: 10.1021/ac3026465. [DOI] [PubMed] [Google Scholar]
- 65.Lee H-J, Cha H-J, Lim J-S, Lee SH, Song SY, Kim H, Hancock WS, Yoo JS, Paik Y-K. J. Proteome Res. 2014;13:2328–2338. doi: 10.1021/pr4011519. [DOI] [PubMed] [Google Scholar]
- 66.Carlsson S, Fukuda M. J. Biol. Chem. 1990;265:20488–20495. [PubMed] [Google Scholar]
- 67.Carlsson SR, Lycksell P-O, Fukuda M. Arch. Biochem. Biophys. 1993;304:65–73. doi: 10.1006/abbi.1993.1322. [DOI] [PubMed] [Google Scholar]
- 68.Huynh KK, Eskelinen EL, Scott CC, Malevanets A, Saftig P, Grinstein S. The EMBO journal. 2007;26:313–324. doi: 10.1038/sj.emboj.7601511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Eskelinen E-L, Illert AL, Tanaka Y, Schwarzmann G, Blanz J, von Figura K, Saftig P. Mol. Biol. Cell. 2002;13:3355–3368. doi: 10.1091/mbc.E02-02-0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tanaka Y, Guhde G, Suter A, Eskelinen E-L, Hartmann D, Lüllmann-Rauch R, Janssen PM, Blanz J, von Figura K, Saftig P. Nature. 2000;406:902–906. doi: 10.1038/35022595. [DOI] [PubMed] [Google Scholar]
- 71.Tan K-P, Ho M-Y, Cho H-C, Yu J, Hung J-T, Yu AL-T. Cell Death Dis. 2016;7:e2347. doi: 10.1038/cddis.2016.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Miyoshi E, Moriwaki K, Nakagawa T. J. Biochem. 2008;143:725–729. doi: 10.1093/jb/mvn011. [DOI] [PubMed] [Google Scholar]
- 73.Takahashi M, Kuroki Y, Ohtsubo K, Taniguchi N. Carbohydr. Res. 2009;344:1387–1390. doi: 10.1016/j.carres.2009.04.031. [DOI] [PubMed] [Google Scholar]
- 74.Liu Y-C, Yen H-Y, Chen C-Y, Chen C-H, Cheng P-F, Juan Y-H, Chen C-H, Khoo K-H, Yu C-J, Yang P-C. Proceedings of the National Academy of Sciences. 2011;108:11332–11337. doi: 10.1073/pnas.1107385108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cairns RA, Harris IS, Mak TW. Nature Reviews Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 76.Wong N, De Melo J, Tang D. Int. J. Cell Biol. 2013:2013. doi: 10.1155/2013/242513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Palmisano G, Melo-Braga MN, Engholm-Keller K, Parker BL, Larsen MR. J. Proteome Res. 2012;11:1949–1957. doi: 10.1021/pr2011268. [DOI] [PubMed] [Google Scholar]
- 78.Qin X, Du Y, Chen X, Li W, Zhang J, Yang J. Cell & bioscience. 2014;4:20. doi: 10.1186/2045-3701-4-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Di Cristofano A, Pandolfi PP. Cell. 2000;100:387–390. doi: 10.1016/s0092-8674(00)80674-1. [DOI] [PubMed] [Google Scholar]
- 80.Hsu PP, Sabatini DM. Cell. 2008;134:703–707. doi: 10.1016/j.cell.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 81.Peracaula R, Barrabés S, Sarrats A, Rudd PM, de Llorens R. Dis. Markers. 2008;25:207–218. doi: 10.1155/2008/797629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wellen KE, Thompson CB. Nature reviews Molecular cell biology. 2012;13:270–276. doi: 10.1038/nrm3305. [DOI] [PubMed] [Google Scholar]
- 83.Schneider J, Neu K, Grimm H, Velcovsky H-G, Weisse G, Eigenbrodt E. Anticancer Res. 2001;22:311–318. [PubMed] [Google Scholar]
- 84.Brinck U, Fischer G, Eigenbrodt E, Oehmke M, Mazurek S. Virchows Archiv. 1994;424:177–185. doi: 10.1007/BF00193498. [DOI] [PubMed] [Google Scholar]
- 85.Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. Nature. 2008;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The reporter ions intensity (A), precursor ions intensity (B) and reporter ion yield (C) of glycopeptides and nonglycosylated peptides in the same run.
Figure S2. The Venn diagram analysis of (A) intact N-glycopeptides (A) and (B) N-glycosylation sites identified with HILIC and MLA enrichment methods. (C) The comparison of the percentage of sialylated N-glycopeptides enriched between two methods.
The identified N-glycopeptides enriched by HILIC alone, lectin alone and sequential HILIC and lectin enrichment.
Glycoproteomics results between PKM2 knockout cells vs. parental breast cancer cells.
Deglycoproteomics results between PKM2 knockout cells vs. parental breast cancer cells.
Gene ontology analysis results.