

Abstract
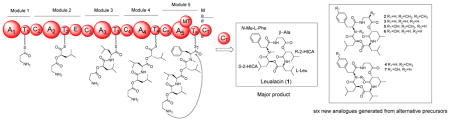
Seven cyclic depsipeptides were isolated from Hapsidospora irregularis and structurally characterized as the calcium channel blocker leualacin and six new analogues based on the NMR and HR-ESI-MS data. These new compounds were named leualacins B-G. The absolute configurations of the amino acids and 2-hydroxyisocaproic acids were determined by recording the optical rotation values. Biological studies showed that calcium influx elicited by leualacin F in primary human lobar bronchial epithelial cells involves the TRPA1 channel. Through genome sequencing and targeted gene disruption, a non-iterative nonribosomal peptide synthetase was found to be involved in the biosynthesis of leualacin. A comparison of the structures of leualacin and its analogues indicated that the A2 and A4 domains of the leualacin synthetase are substrate specific, while A1, A3 and A5 can accept alternative precursors to yield new molecules.
Keywords: Cyclic depsipeptides, Hapsidospora irregularis, Nonribosomal peptide synthetase, Gene disruption, Calcium channel blocker
Graphical abstract
INTRODUCTION
Cyclic depsipeptides are a group of natural products that have shown a wide range of biological activities, and show immunosuppressant,1, 2 antibiotic,3, 4 anti-inflammatory5, 6 and antitumor7–9 properties. They are assembled from proteinogenic/non-proteinogenic amino acids and hydroxy acids through amide and ester linkages. Fungi are an important source of biologically active molecules. Leualacin (1) was previously isolated from the fungus Hapsidospora irregularis FERM BP-2511 as a new cyclic depsipeptide and potent calcium channel blocker, which inhibits the binding of H-nitrendipine to porcine heart membranes in vitro and lowers the blood pressure of spontaneous hypertensive rats.10, 11 Calcium channel blockers block the entry of calcium into cells. These agents are used to treat hypertension, angina pectoris, myocardial infarction, and abnormal heart rhythms.12, 13
Current clinic calcium channel blockers are mainly divided into three groups: phenylalkylamines (e.g., verapamil), benzothiazepines (e.g., diltiazem), and dihydropyridines (e.g., cilnidipine). Compound 1 represents a new type of calcium channel blocker. It is of significant interest to find more analogues of 1 for bioactivity screening. To this end, we reinvestigated the chemical constituents of H. irregularis. Herein we report the isolation and structure elucidation of six new depsipeptides from this fungus. The absolute configurations of amino and α-hydroxy acids were determined by hydrolysis of the parent depsipeptides and measurement of the optical rotation values of the isolated moieties. The leualacin synthetase gene was discovered by genome sequencing. Its involvement in leualacin biosynthesis was confirmed by double crossover gene disruption through Agribacterium-mediated transformation. Production of the six leualacin analogues revealed that this giant fungal noniterative peptide synthetase has relaxed substrate specificity, which presents great potential for precursor-directed biosynthesis of new cyclic depsipeptides.
RESULTS AND DISCUSSION
H. irregularis FERM BP-2511 was grown in K-2 broth at 30 °C for 8 days, and HPLC analysis revealed the production of 1–7, in addition to the previously characterized cephalosporin P1, isocephalosporin P1, Sch210971 and Sch210972 (Figure S1).14, 15 The strain was cultured in K2 broth with and without 0.5% tyrosine (3 L each), from which 1 and six new cyclic depsipeptides were isolated (2–7).
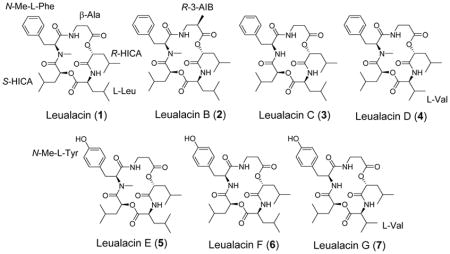
The molecular formula of 1 was determined to be C31H47N3O7 based on HR-ESI-MS analysis, which is the same as that of leualacin. It was further confirmed to be leualacin by a comparison of its NMR data (Figures S2 and S3; Tables 1 and 2) with those previously reported.6 Compound 2 was obtained as an off-white powder. The molecular formula was elucidated as C32H49N3O7 based on HR-ESI-MS analysis as well as its 1H and 13C NMR data (Figures S4 and S5; Tables 1 and 2), which indicated that 2 has an additional CH2 group compared to 1. By comparing the 1H-NMR data of 2 with those of 1, one CH at δH 2.64–2.69 (1H, m) in 2 replaced the CH2 at 2.54–2.61 (2H, m) in 1. Compound 2 has an additional methyl group. Analysis of its HSQC, HMBC, and 1H-1H COSY spectra (Figure 1 and Figures S6–S8) revealed that 2 contains one unit of N-methyl-L-phenylalanine (N-Me-L-Phe), two units of 2-hydroxyisocaproic acid (2-HICA or leucic acid), and one unit of leucine (Leu) that are also present in 1. However, the β-alanine (β-Ala) moiety in 1 was replaced with 3-aminoisobutyric acid (3-AIB) based on the specific COSY signals of the spin system of NH-CH2-CH-CH3 and the HMBC correlations of CH3 at δH 1.21 (3H, d, 7.1) and CH at δH 2.64–2.69 (1H, m) to the carbonyl group at δC 174.9. The connection order of the five moieties was determined by HMBC correlations of α-CH of the N-Me-L-Phe moiety at δH 4.63 (1H, dd, 2.5, 10.5), β-CH2 at δH 3.60–3.71 (2H, m) and NH at δH 7.45 (1H, dd, 5.0, 5.5) of the 3-AIB unit to the carbonyl carbon of the N-Me-L-Phe moiety at δC 170.0, α-CH of the 3-AIB moiety at δH 2.64–2.69 (1H, m) and α-CH of the R-HICA moiety at δH 5.12 (1H, dd, 5.0, 7.6) to the carbonyl group of the 3-AIB moiety at δC 174.9, α-CH of R-HICA, α-CH at δH 4.50 (1H, ddd, 3.0, 9.7, 10.8) and NH at δH 6.32 (1H, d, 9.4) of the Leu moiety to the carbonyl group of the R-HICA unit at δC 171.0, α-CH of the Leu moiety and α-CH of the S-HICA moiety at δH 4.73 (1H, dd, 2.1, 10.5) to the carbonyl group of the Leu moiety at δC 174.1, α-CH of the S-HICA moiety, α-CH of the N-Me-L-Phe moiety and NCH3 of the N-Me-L-Phe moiety at δH 2.87 (3H, s) to the carbonyl group of the S-HICA moiety at δC 170.1. Thus, 2 was identified as a new compound that is identical to 1 except a 3-AIB moiety that replaces β-Ala in 1. It was named as leualacin B.
Table 1.
1H NMR data of compounds 1–7 (1–6 recorded in CDCl3, 7 recorded in CD3OD, δH in ppm, J in Hz).
| Position | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| L-Phenylalanine | L-Tyrosine | ||||||
|
| |||||||
| α | 4.63–4.67, m | 4.63, dd (2.5, 10.5) | 4.69–4.77, m | 4.65, dd (3.4, 11.4) | 4.61, dd (3.0, 11.0) | 4.61–4.67, m | 4.52, dd (6.2, 8.3) |
| α-NH | - | - | 6.54, d (8.6) | - | - | 6.8, d (8.3) | N/A |
| N-CH3 | 2.87, s | 2.87, s | - | 2.87, s | 2.87, s | - | - |
| β | 2.94 dd (10.5, 14.4) 3.38, dd (2.7, 14.3) |
2.95, dd (10.9, 12.4) 3.30, dd (2.52, 12.2) |
3.12–3.23, m | 2.94, dd (11.7, 14.4) 3.41, dd (3.4, 14.1) |
2.88, dd (11.5, 14.5) 3.31, dd (3.0, 14.5) |
3.05–3.10, m | 2.99 dd (8.6, 14.1) 3.12, dd (6.2, 14.1) |
| o | 7.10, d (7.5) | 7.07, d (6.9) | 7.17, d (7.1) | 7.09, d (6.9) | 6.91, d (8.5) | 6.96, d (7.9) | 6.99, d (8.6) |
| m | 7.26–7.31, m | 7.23–7.29, m | 7.23–7.32, m | 7.22–7.31, m | 6.75, d (8.5) | 6.74, d (7.9) | 6.99, d (8.6) |
| p | 7.23–7.25, m | 7.23, t (6.9) | 7.23, t (6.9) | 7.29, t (7.6) | - | - | - |
|
| |||||||
| β-Alanine | R-3-Aminoisobutyric acid (R-3-AIB) | β-Alanine | |||||
|
| |||||||
| α | 2.54–2.61, m | 2.64–2.69, m | 2.53–2.67, m | 2.57, dd (3.8, 8.3) | 2.54–2.57, m | 2.49–2.52, m | 2.44–2.59, m |
| α-CH3 | - | 1.21, d (7.1) | - | - | - | - | - |
| β | 3.31–3.39, m 3.98–4.09, m |
3.60–3.71, m | 3.82–3.94, m 3.25–3.34, m |
4.04, dt (3.8, 13.7) 3.33–3.42, m |
3.97–4.10, m 3.34–3.42, m |
3.80–3.84, m 3.21–3.25, m |
3.73–3.80, m 3.22–3.30, m |
| β-NH | 7.45–7.48, m | 7.45, dd (5.0, 5.5) | 7.10–7.15, m | 7.44–7.49, m | 7.49, d (6.8) | 7.55–7.57, m | N/A |
|
| |||||||
| (R)-2-hydroxyisocaproic acid (R-HICA) | |||||||
|
| |||||||
| α | 5.13–5.18, m | 5.12, dd (5.0, 7.6) | 5.19, dd (3.8, 9.9) | 5.21, dd (6.2, 7.9) | 5.17, dd (6.4, 7.6) | 5.17–5.21, m | 5.17–5.24, m |
| β | 1.68–1.85, m | 1.68–1.88, m | 1.66–1.90, m | 1.72–1.87, m | 1.71–1.85, m | 1.62–1.83, m | 1.61–1.78, m |
| γ | 1.59–1.71, m | 1.58–1.73, m | 1.60–1.77, m | 1.59–1.70, m | 1.60–1.69, m | 1.54–1.68, m | 1.64–1.74, m |
|
| |||||||
| L-Leucine | L-Valine | L-Leucine | L-Valine | ||||
|
| |||||||
| α | 4.48–4.56, m | 4.50, ddd (3.0, 9.7, 10.8) | 4.08–4.18, m | 4.43, d (4.1) | 4.48–4.55, m | 4.13–4.17, m | 3.90, d (8.9) |
| α-NH | 6.16, d (9.8) | 6.32, d (9.4) | 6.89, d (6.6) | 6.41, d (9.6) | 6.32, d (9.6) | 7.41, d (6.6) | N/A |
| β | 1.57–1.72, m | 1.48–1.73, m | 1.54–1.73, m | 2.35, qqd (4.1, 6.5, 6.9) | 1.50–1.81, m | 1.50–1.70, m | 2.08–2.16, m |
| γ | 1.65–1.86, m | 1.61–1.76, m | 1.60–1.77, m | 1.64–1.82, m | 1.54–1.65, m | ||
|
| |||||||
| (S)-2-Hydroxyisocaproic acid (S-HICA) | |||||||
|
| |||||||
| α | 4.65–4.70, m | 4.73, dd (2.1, 10.5) | 5.07, dd (3.6, 6.1) | 4.66, dd (2.3, 11.3) | 4.72, dd (1.8, 11.0) | 5.15–5.17, m | 5.17–5.24, m |
| β | 1.35–1.59, m −0.38 – −0.47, m |
1.36–1.48, m −0.32 – −0.40, m |
1.35–1.54, m | 1.37–1.52, m −0.38 – −0.47, m |
1.41–1.50, m −0.21 – −0.27, m |
1.45–1.57, m | 1.29–1.54, m |
| γ | 1.35–1.59, m | 1.36–1.48, m | 1.33–1.52, m | 1.23–1.40, m | 1.43–1.60, m | 1.30–1.43, m | 1.43–1.49, m |
|
| |||||||
| R/S-2-Hydroxyisocaproic acid (R/S-HICA) and δ-H of L-leucine (L-Leu)/γ-H of L-valine (L-Val) | |||||||
|
| |||||||
|
R-HICA 0.56, d (6.5) 0.67, d (6.5) S-HICA/L-Leu 0.89, d (6.5) 0.93, d (6.5) |
R-HICA/L-Leu 0.92, d (5.5) 0.87, d (5.9) S-HICA 0.66, d (6.2) 0.56, d (6.4) |
S-HICA 0.84, d (5.9) 0.86, d (6.8) R-HICA/L-Leu 0.92, d (6.2) 0.93, d (6.2) 0.95, d (6.5) 1.00, d (5.9) |
R-HICA 0.91, d (6.5) 0.95, d (6.1) L-Val 0.91, d (6.5) 1.01, d (6.9) S-HICA 0.56, d (6.5) 0.67, d (6.5) |
R-HICA/L-Leu 0.90, d (6.2) 0.9, d (6.4) S-HICA 0.62, d (6.4) 0.73, d (6.2) |
S-HICA 0.91, d (5.9) 0.92, d (6.2) L-Leu 0.91, d (5.9) 0.97, d (5.5) R-HICA 0.83, d (5.8) 0.87, d (5.2) |
R-HICA 0.95, d (6.5) 0.99, d (6.2) L-Val 0.97, d (6.5) 1.00, d (6.5) S-HICA 0.86, d (5.8) 0.89, d (5.8) |
|
Table 2.
13C NMR data of compounds 1–7 (1–6 recorded in CDCl3, 7 recorded in CD3OD, δC in ppm).
| Position | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| L-Phenylalanine | L-Tyrosine | ||||||
|
| |||||||
| C=O | 173.5 (s) | 170.0 (s) | 171.4 (s) | 168.5 (s) | 168.8 (s) | 172.0 (s) | 173.6 (s) |
| α | 63.9 (d) | 63.3 (d) | 54.4 (d) | 63.8 (d) | 64.0 (d) | 54.8 (d) | 56.5 (d) |
| N-CH3 | 30.3 (q) | 30.2 (q) | - | 30.2 (q) | 30.3 (q) | - | - |
| β | 34.4 (t) | 34.4 (t) | 37.8 (t) | 34.3 (t) | 33.3 (t) | 37.2 (t) | 37.6 (t) |
| γ | 138.1 (s) | 137.5 (s) | 136.9 (s) | 138.0 (s) | 128.7 (s) | 127.6 (s) | 128.8 (s) |
| o | 129.8 (2C, d) | 129.7 (2C, d) | 129.5 (2C, d) | 129.7 (2C, d) | 130.7 (2C, d) | 130.4 (2C, d) | 131.5 (2C, d) |
| m | 129.5 (2C, d) | 129.4 (2C, d) | 128.9 (2C, d) | 129.4 (2C, d) | 116.4 (2C, d) | 115.9 (2C, d) | 116.4 (2C, d) |
| p | 127.4 (d) | 127.3 (d) | 127.1 (d) | 127.3 (d) | 155.9 (s) | 155.7 (s) | 157.6 (s) |
|
| |||||||
| β-Alanine | R-3-Aminoisobutyric acid (R-3-AIB) | β-Alanine | |||||
|
| |||||||
| C=O | 174.8 (s) | 174.9 (s) | 171.8 (s) | 173.5 (s) | 173.2 (s) | 171.8 (s) | 172.2 (s) |
| α | 34.7 (t) | 40.4 (d) | 34.8 (t) | 34.8 (t) | 34.5 (t) | 34.8 (t) | 35.6 (t) |
| α-CH3 | - | 13.6 (q) | - | - | - | - | - |
| β | 34.9 (t) | 41.4 (t) | 36.1 (t) | 34.3 (t) | 34.7 (t) | 36.1 (t) | 37.1 (t) |
|
| |||||||
| (R)-2-Hydroxyisocaproic acid (R-HICA) | |||||||
|
| |||||||
| C=O | 170.6 (s) | 171.0 (s) | 171.0 (s) | 170.7 (s) | 171.0 (s) | 171.2 (s) | 173.0 (s) |
| α | 70.9 (d) | 72.8 (d) | 71.7 (d) | 72.9 (d) | 72.9 (d) | 71.9 (d) | 73.9 (d) |
| β | 37.9 (t) | 37.8 (t) | 39.0 (t) | 37.4 (t) | 37.5 (t) | 39.5 (t) | 41.3 (t) |
| γ | 24.4 (d) | 24.4 (d) | 24.6 (d) | 24.3 (d) | 24.3 (d) | 24.6 (d) | 25.9 (d) |
|
| |||||||
| L-Leucine | L-Valine | L-Leucine | L-Valine | ||||
|
| |||||||
| C=O | 171.6 (s) | 174.1 (s) | 171.2 (s) | 174.1 (s) | 174.6 (s) | 170.6 (s) | 171.0 (s) |
| α | 51.5 (d) | 51.6 (d) | 53.2 (d) | 57.8 (d) | 51.6 (d) | 53.2 (d) | 61.9 (d) |
| β | 38.9 (t) | 38.5 (t) | 39.2 (t) | 29.0 (d) | 38.6 (t) | 39.1 (t) | 30.5 (d) |
| γ | 25.4 (d) | 25.2 (d) | 25.2 (d) | 25.2 (d) | 25.1 (d) | ||
|
| |||||||
| (S)-2-Hydroxyisocaproic acid (S-HICA) | |||||||
|
| |||||||
| C=O | 168.6 (s) | 171.7 (s) | 170.1 (s) | 171.2 (s) | 172.1 (s) | 170.8 (s) | 173.3 (s) |
| α | 72.9 (d) | 70.6 (d) | 74.5 (d) | 70.8 (d) | 71.0 (d) | 74.1 (d) | 74.3 (d) |
| β | 37.6 (t) | 37.9 (t) | 40.4 (t) | 37.9 (t) | 37.7 (t) | 40.6 (t) | 42.4 (t) |
| γ | 24.2 (d) | 24.2 (d) | 24.7 (d) | 24.4 (d) | 24.4 (d) | 24.6 (d) | 25.6 (d) |
|
| |||||||
| δ-C of L-Leucine and R/S-HICA, γ-C of L-valine | |||||||
|
| |||||||
|
R-HICA 20.9 (q) 23.1(q) S-HICA/L-Leu 23.2 (q) 23.1 (q) 22.5 (q) 21.3 (q) |
R-HICA 22.2/22.9 (q) L-Leu 20.8/22.9 (q) S-HICA 21.2/22.9 (q) |
S-HICA 21.7 (q) 23.1 (q) R-HICA/L-Leu 23.3 (q) 23.2 (q) 21.8 (2C, q) |
R-HICA 22.4/23.0 (q) L-Val 17.2/19.6 (q) S-HICA 20.8/23.0 (q) |
R-HICA 22.3/23.0 (q) L-Leu 20.8/22.8 (q) S-HICA 21.1/22.9 (q) |
R-HICA 21.7 (q) 22.9 (q) S-HICA/L-Leu 23.1 (2C, q) 21.8 (q) 21.7 (q) |
R-HICA 22.7/23.5 (q) L-Val 20.2/19.6 (q) S-HICA 22.2/23.3 (q) |
|
Figure 1.

Selected 1H-1H COSY and HMBC correlations for 2–7.
Compounds 3 and 4 were obtained as an off-white powder. HR-ESI-MS analysis revealed that 3 and 4 have the same molecular formula of C30H45N3O7. Thus, they have one CH2 less than 1. A comparison of the NMR data of 3 (Figures S9 and S10) and 1 and analysis of the 2D NMR spectra (Figures S11–S13) revealed that 3 does not contain a methyl group at the amino group of the L-Phe moiety. Instead, a NH signal was found at δH 6.54 (1H, d, 8.6), which was confirmed by its 1H-1H COSY correlation to the α-CH proton of the L-Phe moiety at δH 4.69–4.77 (1H, m) as well as its HMBC correlation to the carbonyl group of the S-HICA moiety at δC 170.1. Further analysis of the HSQC, HMBC, and 1H-1H COSY spectra (Figure 1) indicated that 3 is also a cyclic depsipeptide composed of five units, including β-Ala, R-HICA, L-Leu, and S-HICA and L-Phe. Unlike 3, compound 4 was found to have the methyl group at the amino group of the L-Phe moiety (Figures S14–18). Except for the leucine unit, the 1H and 13C NMR data of other four units in compound 4 were nearly identical to those of compound 1. Furthermore, the 1H-1H COSY spectrum revealed a spin system of NH-CH-CH-(CH3)2. The HMBC correlations of the CH at δH 4.43 (1H, d, 4.1) and CH at δH 2.35 (1H, qqd, 4.1, 6.5, 6.9) of this spin system to the carbonyl group at δC 174.1 showed that the L-Leu moiety in 1 was replaced with valine (Val) in this compound. The connection of the five units was confirmed by its HMBC spectrum (Figure 1). Both 3 and 4 are new compounds and were named as leualacins C and D, respectively.
The molecular formula of 5 was determined to be C31H47N3O8 according to the HR-ESI-MS data, suggesting that it has one more oxygen atom than 1. The NMR spectra of 5 (Figures S19–23) were similar to those of 1, except the difference in the N-Me-L-Phe moiety. The aromatic proton signals at δH 6.91 (2H, d, 8.5) and δH 6.75 (2H, d, 8.5) in the 1H NMR spectrum and carbon signals at δC 127.6 (C), 130.4 (2C, CH), 115.9 (2C, CH), and 155.7 (C) in the 13C NMR spectrum (Tables 1 and 2) indicated that there is a 1,4-disubstituted phenyl ring, with a hydroxy as one of the substituents. The HMBC correlations (Figure 1) revealed the presence of an N-Me-L-Tyr moiety. According to the 1D and 2D NMR spectra, 5 was characterized as a new leualacin analogue that has the same structure as 1 except that the N-Me-L-Phe was replaced with N-methyl-L-tyrosine (N-Me-L-Tyr). This compound was named as leualacin E. Compound 6 also has eight oxygen atoms in its molecular formula of C30H45N3O8 based on the HR-ESI-MS, but it has one less CH2 than 5. The NMR spectra (Figures S24–28) also showed that it has a Tyr unit and the detailed 2D NMR signals confirmed it contains the same units as those in 5 with the same connection order, except that there is no methyl group at the amino group of the L-Tyr moiety. This was confirmed by the 1H-1H COSY correlation (Figure 1) between NH at δH 6.83 (1H, d, 8.3) and the α-CH of L-Tyr at δH 4.61–4.67 (1H, m). 6 was named as leualacin F.
The molecular formula of 7 was determined to be C29H43N3O8 by HR-ESI-MS analysis, which is one CH2 unit less than 6. Analysis of the 2D-NMR spectra of 7 (Figure 1 and Figures S29–S33; Tables 1 and 2) revealed that it contains the N-Me-L-Tyr, β-Ala, R-HICA, and S-HICA moieties that are also present in 6. However, 7 has a Val moiety in the same position as the L-Leu moiety in 6. It was named as leualacin G.
The absolute configuration of the major product 1 has been previously determined11 to consist of β-Ala, R-HICA, L-Leu, S-HICA and N-Me-L-Phe. 2–7 are new compounds that have one or two new moieties that substitute the β-Ala, L-Leu, or N-Me-L-Phe moiety in 1. In order to determine the absolute configurations of these new moieties, we hydrolyzed the representative compounds 2, 4, and 6 at the ester bonds using 1 N NaOH (Supporting Information) to get two fragments and analyzed the products by LC-MS. The two fragments were separated by HPLC and then treated with 6 N HCl to hydrolyze the amide bonds, from which individual units of these compounds were obtained (Figure S34). The optical rotations of these units were recorded (Table S1). Their absolute configurations were determined by comparing the recorded optical rotations with those of standards or reported. The results showed the moieties in 2–7 are L-Phe11 or L-Tyr,16 β-Ala or R-3-AIB,17 R-HICA, S-HICA,11, 18 L-Leu19 or L-Val.20
Compound 1 blocks H-nitrendipine binding to porcine heart membranes in vitro and can lower the blood pressure of spontaneous hypertensive rats.5 Calcium is a versatile and critical second messenger in all cells. Stimulation and inhibition of changes in intracellular calcium content caused by 1–7 was assessed in lobar bronchial epithelial cells. TRP ion channels in lung epithelial cells are involved in innate immunity and various other processes. Calcium influx was elicited by 3 and 6 at 100 μM, which was correlated with activation of the human TRPA1 and to a lesser extent M8 channels, but not TRPV1, V3, or V4 (Table S2). Activation of TRPA1 is associated with pulmonary irritation, cough, and edema. Elsewhere, TRPA1 signals pain and is associated with allergic sensitization including asthma and dermatitis.21–23
Compounds 1–7 belong to a large group of structurally diverse natural products, nonribosomal peptides (NRPs). They are synthesized by nonribosomal peptide synthetases (NRPSs). These synthetases are modular enzymes that consist of different catalytic domains, such as condensation (C), adenylation (A), thiolation (T), methylation (MT), epimerization (E), and oxidation (Ox). To understand how 1–7 are synthesized, the genome of H. irregularis was sequenced. Because the structure of 1 contains different amino acid/hydroxy acid units, it is proposed that this molecule is assembled by a non-iterative NRPS which is often present in bacteria. Annotation of the sequenced genome revealed the presence of a large non-iterative NRPS (Figure 2, GenBank accession number KU994894) that consists of five A domains, five T domains, and five C domains. Additionally, there is an E domain sitting between the T2 and C3 domains. An N-MT domain is embedded in the A5 domain. The domain organization of this 6,521-aa enzyme (717 kDa, named as leualacin synthetase or LACS) is co-linear with the structure of 1 and thus it represents a possible NRPS involved in leualacin biosynthesis. Although the signature sequences of the five A domains (Table S3) could be extracted from the amino acid sequence of LACS using NRPPSpredictor224 based on the reported bacterial NRPSs, it is hard to conclusively predict the substrate of each A domain due to the lack of the information about genuine signature sequences in fungal NRPSs. In addition, there are several novel precursors used in the biosynthetic process of 1–7, such as HICA that has not been reported as a direct precursor for NRP biosynthesis. Although cryptophycins also have an HICA unit, it was reported that the A4 domain of the dedicated PKS/NRPS selects α-ketoisocaproic acid that is reduced to by a ketoreductase domain in the same module to yield the HICA unit.25 We predicted the substrate of each A domain based on the domain organization of LACS and structure of 1. A1-A5 were proposed to select and activate β-Ala, S-HICA, L-Leu, S-HICA and L-Phe. The E domain in module converts S-HICA to R-HICA, while the MT domain in module 5 methylates the NH2 group of the L-Phe moiety. The C-terminal condensation-like (CT) domain catalyzes the cyclization and concomitant release of the final products.
Figure 2.

Proposed biosynthesis of 1 by a non-iterative NRPS.
To find out whether this NRPS is involved in the biosynthesis of 1, we conducted a gene disruption experiment. Two fragments (3.5 kb and 4.0 kb) of lacs were amplified from the genome of H. irregularis and ligated into the binary donor vector pAG1-H3 as the left and right arms, respectively. The resulting plasmid pAG1-H3-KO (Figure 3A) was introduced into H. irregularis through Agrobacterium tumefaciens-mediated transformation. Correct transformants were then subjected to PCR screening using the genome- and vector-specific primers A-D (Figure 3A). As shown in Figure 3B, a 4,534-bp fragment was amplified from the correct double crossover mutant with primers A and B, and a 5,026-bp fragment with primers C and D. Because primers B and C were specifically from the hygromycin resistance gene (HygR) in the vector, no PCR products were obtained from the wild type. This correct mutant was then grown on the oatmeal agar modified from WSH agar5 for product analysis. As shown in Figure 3C, when grown in the oatmeal medium, the wild type produces 1 and 2 as the major metabolites (traces i-iii). In contrast, the mutant failed to produce 1 or any analogues (trace iv), indicating the essential role of LACS in leualacin biosynthesis.
Figure 3.

Disruption of lacs in H. irregularis. (A) Strategy for targeted disruption of lacs. (B) Verification of the correct mutant by PCR. (C) HPLC analysis of the products of the wild type and mutant on the oatmeal agar plates at 210 nm. (i) wild type; (ii) mutant; (iii) standard of 1; (iv) standard of 2.
H. irregularis is a fungus of the family of Pseudeurotiaceae.26 In this work, six new cyclic depsipeptides were characterized, which significantly expanded the spectrum of natural products from this fungus. These structures consist of two HICA units with different configurations and three amino acid moieties. The main difference among the seven cyclic depsipeptides lies in the amino acid units, while the two HICA units remain the same. This indicated that A2 and A4 of LACS which activate S-HICA are more substrate specific. In contrast, A1, A3 and A5 are more flexible. The structures of 5–7 showed that A5 can use L-Tyr instead of L-Phe in 1. Similarly, A1 was found to take 3-AIB to yield 2, while the discovery of 4 and 7 indicated that A3 activates both L-Val and L-Leu. Thus, LACS is a highly flexible NRPS that synthesizes a variety of leualacin analogues in the presence of different precursors.
EXPERIMENTAL SECTION
General experimental procedures
1D NMR spectra were recorded in CDCl3 or CD3OD at 25 °C on a JEOL ECX-300 instrument (300 MHz for 1H NMR and 75 MHz for 13C NMR). 2D NMR spectra were collected on a JEOL ECX-300 or a Bruker AvanceIII HD Ascend-500 (HMBC spectrum of compound 7) instrument. The chemical shift (δ) values are given in parts per million (ppm). The coupling constants (J values) are reported in Hertz (Hz). LR-ESI-MS were acquired on an Agilent 6130 LC-MS and HR-ESI-MS was measured on an Agilent G6224A TOF mass spectrometer.
Strains, media and culture conditions
Hapsidospora irregularis FERM BP-2511 was purchased from the International Patent Organism Depositary, Japan. It was routinely grown in K2 agar medium consisting of PDA (potato dextrose agar, 3.9%, w/v), sucrose (2%, w/v), casamino acid (1%, w/v), KH2PO4 (0.5%, w/v), and MgSO4 (0.125%, w/v) at 30 °C. E. coli XL-1 Blue (Stratagene) was used for routine cloning and plasmid propagation. It was routinely grown at 37 °C on LB agar plates or in liquid LB medium supplemented with appropriate antibiotics (ampicillin, 50 μg/mL; kanamycin, 50 μg/mL). Agrobacterium tumefaciens LBA4404 was used for the introduction of the disruption plasmid into H. irregularis FERM BP-2511.
Production and purification of cyclic depsipeptides
Single colonies were obtained by streaking the fungus on K2 agar medium at 30 °C for 8 days. A single colony was picked and pre-cultured in 4 mL of K2 broth, then cultured in 50 mL of K2 broth consisting of PDB (potato dextrose broth, 2.4%, w/v), sucrose (2%, w/v), casamino acid (1%, w/v), KH2PO4 (0.5%, w/v), and MgSO4 (0.125%, w/v) at 250 rpm and 30 °C for 7 days. The culture was centrifuged at 3,500 rpm for 5 minutes. The supernatant was extracted with 50 mL of ethyl acetate three times and the cells were extracted three times with 50 mL of methanol. The ethyl acetate and methanol extracts were combined and dried under reduced pressure. The residue was re-dissolved in 1 mL of methanol for analysis on an Agilent 1200 HPLC (HPLC conditions are showed in Figure S1).
The strain was also grown in 3 L of K2 broth and 3 L of K2 plus 0.5% L-tyrosine (to enhance the production of 5–7) for the isolation of 1–7. After 8 days, the cultures were combined and centrifuged at 3,500 rpm for 5 minutes. The supernatant was combined and loaded to an HP-20 column (10 cm × 50 cm) and the column was eluted with 4 L of water. The cells were extracted with 500 mL of methanol three times with sonication for 40 minutes. After centrifugation at 3,500 rpm for 5 minutes, the methanol extract was diluted with 3 volumes of water and the solution was loaded to the HP-20 column as the 25% eluant. The column was then successively eluted with 4 L of 50% aqueous methanol, 75% aqueous methanol, and 100% methanol. These fractions were analyzed by HPLC, and the target compounds were found in the 75% and 100% methanol fractions. These two fractions were separated on the Agilent 1220 HPLC with an Agilent ZORBAX SB-C18 column (5 μm, 21.2 mm × 150 mm), eluted with a gradient of methanol-water (0–15 min: 70%, 15–18 min: 70–80%, 18–25 min: 80%, 25–27 min: 80–90%, 27–35 min: 90%, v/v) with 0.1% formic acid at a flow rate of 3 mL min−1 to get six fractions A-F. LC-MS analysis indicated that fraction A contained 7, fraction B contained 5 and 6, fraction D contained 3 and 4, fraction E contained 1, and fraction F contained 1 and 2. Then fractions A-F were repurified by HPLC with an Agilent ZORBAX SB-C18 column (5 μm, 4.6 mm × 250 mm) at 1 mL min−1 and detected at 210 nm. Fraction A was eluted with 40% acetonitrile-water to get pure 7 (18 mg). Fraction B was eluted with 55% acetonitrile-water to get pure 5 (33 mg) and 6 (59 mg). Fraction D was eluted with 60% acetonitrile-water to get pure 3 (35 mg) and 4 (35 mg). Fraction E was eluted with 70% methanol-water to get pure 1 (450 mg). Fraction F was eluted with 85% methanol-water to get pure 2 (39 mg). The purified compounds were subjected to spectral analyses for structure determination (Tables 1 and 2).
Compound 1: off-white powder; −97.8 (c 1.0, MeOH); 1H and 13C NMR data, see Tables 1 and 2; HR-ESI-MS m/z 574.3493 [M + H]+ (calcd for C31H48N3O7, 574.3487).
Compound 2: off-white powder; −99.0 (c 0.3, MeOH); 1H and 13C NMR data, see Tables 1 and 2; HR-ESI-MS m/z 588.3648 [M + H]+ (calcd for C32H50N3O7, 588.3643).
Compound 3: off-white powder; −48.2 (c 1.17, MeOH); 1H and 13C NMR data, see Tables 1 and 2; HR-ESI-MS m/z 560.3332 [M + H]+ (calcd for C30H46N3O7, 560.3330).
Compound 4: off-white powder; −68.0 (c 0.25, MeOH); 1H and 13C NMR data, see Tables 1 and 2; HR-ESI-MS m/z 560.3333 [M + H]+ (calcd for C30H46N3O7, 560.3330).
Compound 5: off-white powder; −96.1 (c 0.62, MeOH); 1H and 13C NMR data, see Tables 1 and 2; HR-ESI-MS m/z 590.3440 [M + H]+ (calcd for C31H48N3O8, 590.3436).
Compound 6: off-white powder; −46.0 (c 0.50, MeOH); 1H and 13C NMR data, see Tables 1 and 2; HR-ESI-MS m/z 576.3284 [M + H]+ (calcd for C30H46N3O8, 576.3279).
Compound 7: off-white powder; −52.9 (c 0.34, MeOH); 1H and 13C NMR data, see Tables 1 and 2; HR-ESI-MS m/z 562.3119 [M + H]+ (calcd for C29H44N3O8, 562.3123).
Determination of absolute configurations of the five units in 1–7 (Table S1 and Figure S34)
25 mg of 6 was dissolved in 600 μL of 30% aqueous acetonitrile and 100 μL of 1 N NaOH in water was added under stirring. The compound was hydrolyzed at 40 °C for 1 hour. The reaction mixture was neutralized with 1 N HCl. The hydrolysis was confirmed by LC-MS and two fragments (with a MW of 366 and 245, respectively) were purified on an Agilent 1220 HPLC with an Agilent ZORBAX SB-C18 (5 μm, 4.6 mm × 250 mm) at 1 mL min−1, eluted with 35% acetonitrile-water and monitored at 210 nm. The two fragments were further hydrolyzed with 6 N HCl at 115 °C for 12 hours. After neutralizing with NaOH, the products were analyzed by LC-MS and compared with the standards. Then the target products were purified on the same HPLC eluted with a gradient of acetonitrile-water from 0% to 100% in 30 minutes. The optical rotations of the purified amino acids and hydroxy acids were recorded on a Rudolph Autopol IV polarimeter with a 10-cm cell at 19 °C. Compounds 2 and 4 were hydrolyzed and purified according to the same procedure for 6. The absolute configurations were determined by comparing the optical data with those reported in the literature. The optical rotations of compounds 1–7 were also recorded on the same equipment (Table S1).
Transient receptor potential channel assay
Fluorometric cell-based Ca2+ flux assays were performed using a BMG Labtech NOVOStar fluorescence plate reader equipped with a plate-to-plate reagent delivery system. Human embryonic kidney-293 (HEK-293) cells that stably overexpress human TRPA1, M8, TRPV3 or V4 have been previously described27 and were grown to confluence in 2% (w/v) gelatin-coated 96-well plates in DMEM: F12 media supplemented with 5% fetal bovine serum, 1× penicillin/streptomycin (Invitrogen) and geneticin (300 μg/mL). Assays using TRPV1 over-expressing and normal BEAS-2B cells have also been described.1 Lobar Bronchial Epithelial cells (Lifeline Cell Technology, Donor Lot # 1344) were grown to 80–90% confluence in BronchiaLife B/T medium (Lifeline Cell Technology).
Cells were prepared for assay by replacing the growth media with a 1:1 solution of LHC-9 and Fluo 4-Direct (Invitrogen) reagent containing Fluo 4-AM, pluronic F-127, probenecid, and a proprietary quencher dye. Cells were then incubated at 37 °C for 1 h in a cell culture incubator, or at room temperature (BEAS-2B cells). Cells were subsequently washed by replacing the loading solution with LHC-9 containing 1 mM water-soluble probenecid (Invitrogen), 750 μM Trypan Red (ATT Bioquest). All cell types were incubated for an additional 30 minutes at 37 °C to allow for Fluo 4-AM cleavage and activation as well as equilibration to both the test compounds and Fluo 4.
Treatment solutions were prepared in LHC9 (TRPV1, M8, and V4) or calcium assay buffer (TRPA1, V3, LOBAR) at 3× concentration and 25 μL was added to 50 μL of media on the cells in 96 well plates. Changes in cellular fluorescence were monitored for 1 minute at 37 °C. Data were quantified as the maximum rate of change in fluorescence intensity (max ΔF/s), vs. media only treatment (negative control) and in HEK-293 or normal BEAS-2B cells were represented as the percentage of response relative to the specific TRP agonist control (TRPA1-2,4-ditert butylphenol at 250 μM; M8-icilin at 25 μM; V1-capsaicin at 10 μM, V3-carvacrol at 300 μM, and V4-GSK1016790A at 30 nM). A minimum of three replicates were used for all treatments.
Knock-out of lacs in H. irregularis FERM BP-2511 through Agrobacterium-mediated transformation
H. irregularis FERM BP-2511 was cultured in 50 mL of K2 broth at 30 °C and 250 rpm for 6 days. The mycelia were collected by filtration and stored at −80 °C for 2 hours. Then 200 mg of the mycelia were ground in liquid nitrogen. 3 mL of the lysis buffer consisting of 0.5 M NaCl, 10 mM Tris-HCl (pH=7.5), 10 mM EDTA, 1% SDS was added into the mycelia at 65 °C. The extraction of genomic DNA was done with cetyl trimethyl ammonium bromide (CTAB) extraction buffer28 followed by purification through phenol/chloroform extraction and precipitation with isopropanol.29 RNA was removed by adding RNase.
Two DNA fragments were amplified by PCR from the lacs gene using the genomic DNA of H. irregularis FERM BP-2511 as the template. A 3,600-bp fragment upstream of the disruption region of the lacs gene was amplified using the primers BP2511-128-NRPS-L-AvrII-3 (5′-aaCCTAGGacagcctgggtttcgagaac-3′, AvrII site in bold) and BP2511-128-NRPS-L-KpnI-5 (5′-aaGGTACCagagcctgtgacttccagac-3′, KpnI site in bold), and cloned into pJET1.2 for sequencing. After sequence validation, this left-arm gene fragment was excised with AvrII and KpnI and inserted into pAG1-H3 between the same sites. A 3,976-bp fragment downstream of the disruption region of the lacs gene was amplified using the primers BP2511-128-NRPS-R-SbfI-3 (5′-aaCCTGCAGGgtgtcgctctttcggtcgtg-3′, SbfI site in bold) and BP2511-128-NRPS-R-SpeI-5 (5′-aaACTAGTcgagcccgacgttggctgca-3′, SpeI site in bold), and cloned into pJET1.2 for sequencing. After sequence validation, the right-arm gene fragment was excised with SbfI and SpeI, and inserted into pAG1-H3-right arm between the same sites. This disruption plasmid was designed to replace a portion of the lacs gene with the hygromycin phosphotransferase gene of pAg1-H3 upon double homologous recombination with the H. irregularis genome.
Agrobacterium-mediated transformation was performed according to the previously reported procedure.30 We used the modified WSH agar (1.0% crushed oatmeal, 0.1% KH2PO4, 0.1% NaNO3, 0.1% MgSO4, 0.5% glycerol, 0.1% MES, 2.0% agar) plates supplemented with 200 μg/mL hygromycin and 500 μg/mL kanamycin (final concentrations) to select the correct transformants at 30 °C. The growing fungal spores were transferred onto new oatmeal plate with 500 μg/mL carbenicillin and 200 μg/mL hygromycin for the second-round selection.
The spores of the selected mutant were cultured in YM broth (yeast extract, 4 g/L, malt extract, 10 g/L, glucose, 4 g/L) for 7 days to extract DNA according to the extract protocol for the wild type. Four genome- and vector-specific primers (A–D) were designed to confirm the correct mutant. A 4,534-bp fragment was amplified from the correct double crossover mutant with primers A (5′-ATGTATGACAATGGCCATCATAC-3′) and B (5′-CGGAGACGCTGTCGAACTTT-3′), and a 5,026-bp fragment was amplified with primers C (5′-AGCTTGACTATGAAAATTCCGTCAC-3′) and D (5′-CACCAGTCATGTAGACCCTTCC-3′). The genomic DNA of wild type H. irregularis FERM BP-2511 was used as the template to amplify the same fragments by the same primers, but no target bands were obtained.
The mutant and wild type strains were cultured on the same oatmeal plate without any antibiotics at 30 °C for 7 days. The cultures were harvested and extracted three times with methanol. After drying, the extracts were re-dissolved in 1 mL of methanol for analysis on an Agilent 1200 HPLC with an Agilent XDB-C18 column (5 μm, 4.6 mm × 250 mm), eluted with a gradient of acetonitrile-water (0–10 min: 50%, 10–50 min: 50–90%) with 0.1% formic acid at a flow rate of 1 mL/min.
Supplementary Material
Acknowledgments
This research was supported by a Utah State University SPARC grant, a National Science Foundation Award CHE-1429195, and NIH grants ES017431 and GM107557. The authors thank Dr. Stephen T. Lee at the USDA Poisonous Plant Research Lab for helping measure the optical rotation data, and Dr. Ronald Shin at the University of Alabama at Birmingham for collecting the HMBC spectrum of 2.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI:
The optical rotation values of the parent depsipeptides and hydrolyzed units (Table S1), Ca2+ flux data for 1–7 (Table S2), extracted signature sequences of the A domains of LACS (Table S3), HPLC analysis of the metabolites of Hapsidospora irregularis FERM BP-2511 (Figure S1), NMR spectra of compounds 1–7 (Figures S2–S33), and the hydrolysis of compounds 2, 4, and 6 (Figure S34) (PDF)
References
- 1.Chakraborty TK, Ghosh S, Dutta S. Tetrahedron Lett. 2001;42:5085–5088. [Google Scholar]
- 2.Thell K, Hellinger R, Schabbauer G, Gruber CW. Drug Discov Today. 2014;19:645–653. doi: 10.1016/j.drudis.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boros C, Smith CJ, Vasina Y, Che Y, Dix AB, Darveaux B, Pearce C. J Antibiot. 2006;59:486–494. doi: 10.1038/ja.2006.68. [DOI] [PubMed] [Google Scholar]
- 4.Lang G, Kalvelage T, Peters A, Wiese J, Imhoff JF. J Nat Prod. 2008;71:1074–1077. doi: 10.1021/np800053n. [DOI] [PubMed] [Google Scholar]
- 5.Chung YM, El-Shazly M, Chuang DW, Hwang TL, Asai T, Oshima Y, Ashour ML, Wu YC, Chang FR. J Nat Prod. 2013;76:1260–1266. doi: 10.1021/np400143j. [DOI] [PubMed] [Google Scholar]
- 6.Randazzo A, Bifulco G, Giannini C, Bucci M, Debitus C, Cirino G, Gomez-Paloma L. J Am Chem Soc. 2001;123:10870–10876. doi: 10.1021/ja010015c. [DOI] [PubMed] [Google Scholar]
- 7.Kitagaki J, Shi G, Miyauchi S, Murakami S, Yang Y. Anticancer Drugs. 2015;26:259–271. doi: 10.1097/CAD.0000000000000183. [DOI] [PubMed] [Google Scholar]
- 8.Stoianov AM, Robson DL, Hetherington AM, Sawyez CG, Borradaile NM. PLoS One. 2015;10:e0131269. doi: 10.1371/journal.pone.0131269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang G, Liu S, Liu Y, Wang F, Ren J, Gu J, Zhou K, Shan B. Oncol Lett. 2014;8:248–252. doi: 10.3892/ol.2014.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamano K, Kinoshita M, Furuya K, Miyamoto M, Takamatsu Y, Hemmi A, Tanzawa K. J Antibiot. 1992;45:899–905. doi: 10.7164/antibiotics.45.899. [DOI] [PubMed] [Google Scholar]
- 11.Hamano K, Kinoshita M, Tanzawa K, Yoda K, Ohki Y, Nakamura T, Kinoshita T. J Antibiot. 1992;45:906–913. doi: 10.7164/antibiotics.45.906. [DOI] [PubMed] [Google Scholar]
- 12.Abernethy DR, Schwartz JB. N Engl J Med. 1999;341:1447–1457. doi: 10.1056/NEJM199911043411907. [DOI] [PubMed] [Google Scholar]
- 13.Grossman E, Messerli FH. Prog Cardiovasc Dis. 2004;47:34–57. doi: 10.1016/j.pcad.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 14.Kakule TB, Zhang S, Zhan J, Schmidt EW. Org Lett. 2015;17:2295–2297. doi: 10.1021/acs.orglett.5b00715. [DOI] [PubMed] [Google Scholar]
- 15.Zhang S, Wang S, Zhang Q, Chang CWT, Zhan J. Bioorg Med Chem Lett. 2015;25:1920–1924. doi: 10.1016/j.bmcl.2015.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Katchalski E, Sela M. J Am Chem Soc. 1953;75:5284–5289. [Google Scholar]
- 17.Beddow JE, Davies SG, Ling KB, Roberts PM, Russell AJ, Smith AD, Thomson JE. Org Biomol Chem. 2007;5:2812–2825. doi: 10.1039/b707689d. [DOI] [PubMed] [Google Scholar]
- 18.Bauer T, Gajewiak J. Tetrahedron. 2004;60:9163–9170. [Google Scholar]
- 19.Plaisance GP. J Am Chem Soc. 1917;39:2087–2088. [Google Scholar]
- 20.MacDonald JC. Can J Chem. 1969;47:2739–2746. [Google Scholar]
- 21.Caceres AI, Brackmann M, Elia MD, Bessac BF, del Camino D, D’Amours M, Witek JS, Fanger CM, Chong JA, Hayward NJ, Homer RJ, Cohn L, Huang X, Moran MM, Jordt SE. Proc Natl Acad Sci USA. 2009;106:9099–9104. doi: 10.1073/pnas.0900591106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu B, Escalera J, Balakrishna S, Fan L, Caceres AI, Robinson E, Sui A, McKay MC, McAlexander MA, Herrick CA, Jordt SE. FASEB J. 2013;27:3549–3563. doi: 10.1096/fj.13-229948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bessac BF, Jordt SE. Physiology. 2008;23:360–370. doi: 10.1152/physiol.00026.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rausch C, Weber T, Kohlbacher O, Wohlleben W, Huson DH. Nucleic Acids Res. 2005;33:5799–5808. doi: 10.1093/nar/gki885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Magarvey NA, Beck ZQ, Golakoti T, Ding Y, Huber U, Hemscheidt TK, Abelson D, Moore RR, Sherman DH. ACS Chem Biol. 2006;1:766–779. doi: 10.1021/cb6004307. [DOI] [PubMed] [Google Scholar]
- 26.Malloch D, Cain RF. Can J Bot. 1970;48:1815–1825. [Google Scholar]
- 27.Deering-Rice CE, Romero EG, Shapiro D, Hughen RW, Light AR, Yost GS, Veranth JM, Reilly CA. Chem Res Toxicol. 2011;24:950–959. doi: 10.1021/tx200123z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doyle JJ, Doyle JL. Phytochem Bull. 1987;19:11–15. [Google Scholar]
- 29.Ashktorab H, Cohen RJ. Biotechniques. 1992;13:198–200. [PubMed] [Google Scholar]
- 30.Zhang A, Lu P, Dahl-Roshak AM, Paress PS, Kennedy S, Tkacz JS, An Z. Mol Genet Genomics. 2003;268:645–655. doi: 10.1007/s00438-002-0780-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.