

Abstract
Immunotherapy using immune checkpoint inhibitors and CAR-T cells has revolutionized treatment for patients with malignant tumors. However, measuring tumor cell cytotoxicity mediated by immune effector cells in clinical laboratories has been difficult due to the requirement for radioactive substances. In this study, a series of novel terpyridine derivative proligands were synthesized and a non-radioactive cellular cytotoxicity assay using the newly synthesized compounds was developed for use in preclinical and clinical studies for cancer immunotherapy. Once internalized into target cells, the compounds are hydrolyzed by esterases, resulting in the intracellular accumulation of the negatively-charged terpyridine derivatives. When the labeled target cells are recognized and killed by immune effector cells, the integrity of the cell membrane is disrupted and the terpyridine derivatives are released. Upon combining the culture supernatant with europium (Eu3+), the cytotoxicity of immune effector cells for the target cells can be quantitatively determined by measuring the intensity of the Eu3+/ligand-derived time-resolved fluorescence. Thus, the assay developed in this study would facilitate the development of novel cancer immunotherapies.
Keywords: cellular cytotoxicity assay, cancer immunotherapy, terpyridine proligand, T cell, NK cell
Entry for the Table of Contents
Immune checkpoint inhibitors have revolutionalized treatment of cancer patients. Although many clinical trials have been conducted to develop novel immunotherapies, measuring cellular cytotoxicity has been difficult. In this study, a series of chelate-forming proligands were synthesized to improve non-radioactive cellular cytotoxicity assays. This assay is ideal for measuring the effector functions of immune cells in clinical laboratories.
Introduction
The immune system plays a pivotal role in protecting humans from pathogenic microbes as well as cancers. Recently, immunotherapy has been used to treat patients with a variety of malignancies, including melanoma, non-small cell lung carcinoma, and renal cell carcinoma.[1] We initially focused on the physiological functions of PD-1 immune checkpoints and showed that the PD-1 deficiency leads to autoimmune diseases such as cardiomyopathy in Balb/c mice, indicating that PD-1 delivers a co-inhibitory signal in immune effector cells.[2] Based on these findings, we tested anti-PD-L1 monoclonal antibodies for their effectiveness in treating cancer and demonstrated that the blockade of PD-1 immune checkpoints inhibits tumor cell growth in animal models.[3] The effectiveness of blocking PD-1 interactions was later confirmed in clinical studies using anti-PD-1 and anti-PD-L1 monoclonal antibodies.[1c, 4]
Immune checkpoint therapy is dependent on the number of nonsynonymous mutations[5] in the tumor as well as the number of immune effector cells specific for these mutations.[6] Ex vivo expansion of immune effector cells with or without chimeric antigen receptors followed by their adoptive transfer to patients is a practical means to increase the number of immune effector cells and is likely to be more feasible than in vivo expansion using neoantigens or tumor-specific antigens and interleukin-2 (IL-2). It is thus essential to establish practical methods for the efficient ex vivo expansion of immune effector cells and methods to monitor their effector functions, especially cell-mediated cytotoxicity.
The most commonly used method in research laboratories to assess the cytotoxicity of immune cells is the [51Cr]-sodium chromate release assay.[7] In this assay system, target cells are first labeled with radioactive [51Cr]-sodium chromate. When target cells are incubated with the probe, the radioactive compound enters the cells. Because the probe is present in its ionic form, 51CrO42-, in the cytoplasm, the radioactive ion binds to intracellular proteins. The radioactive 51CrO42-/protein complex can no longer pass through the cell membrane and the cells are labeled. When the labeled target cells are killed by immune effector cells, the radioactive 51CrO42-/protein complexes are released into the culture supernatants and the specific lysis can be determined by counting radioactivity. A major drawback of this assay is the use of [51Cr]-sodium chromate that emits toxic high-energy γ-radiation with the inherent problems of working with radioactivity, disposal of radioactive waste, and the requirement for thick lead shielding. Also, the [51Cr]-release assay takes a significant amount of time and is difficult to automate and setup as a high throughput assay without specialized instrumentation.
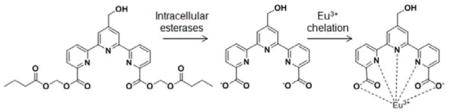
To overcome these drawbacks, numerous attempts have been made to develop non-radioactive cellular cytotoxicity assays, including an enzyme release assay, a dye release assay, and a flow cytometric assay.[8] However, most assay methods using non-radioactive probes have been less sensitive and/or more difficult to perform compared to the [51Cr]-release assay.[8b, 9] The most promising strategy is one that uses europium (III) (Eu3+) and bis(acetoxymethyl) 2,2′:6′,2″-terpyridine-6,6″-dicarboxylate (BATDA), a proligand of a terpyridine derivative (Supporting Information 1).[8b] When target cells are incubated with BATDA, the terpyridine proligand diffuses into the membrane of the cells and is hydrolyzed by intracellular esterases to yield 2,2′:6′,2″-terpyridine-6,6″-dicarboxylic acid (TDA) in a di-anionic form that accumulates in the cytoplasm due to its negative charges. When the labeled target cells are killed by immune effector cells, TDA is rapidly released into the culture media. Upon addition of Eu3+ to the culture supernatant, Eu3+ interacts with TDA. The resulting chelate complex possesses a long fluorescence decay time. When a time-resolved fluorometry detector is used, fluorescence derived from the long-life fluorescent chelate can be specifically measured after the short-lived background fluorescence has decayed. However, the spontaneous release of TDA from the labeled target cells is relatively high and increases rapidly with incubation time and the intensity of the time-resolved fluorescence is not always adequate. This ability of terpyridines to form metal complexes and supramolecular polymeric structures has been extensively studied for drug delivery, catalytic, and optoelectronic applications.[10]
In this study, we have designed and synthesized a series of terpyridine derivative proligands to improve on the BATDA probe. Using the newly synthesized non-radioactive probes, we can measure the functions of immune effector cells even in the clinical laboratory setting. This advance should allow routine monitoring of the effectivenss of present cancer immunotherapies and aid in the development of novel cancer cytotherapies.
Results and Discussion
To improve on the currently available non-radioactive cellular cytotoxicity assays based on time-resolved fluorescence emitted from Eu3+/ligand chelate complexes, we sought to synthesize a proligand with a higher maximum release rate and a lower spontaneous release rate when used to label tumor cells. Because the currently available bis(acetoxymethyl)-2,2′:6′,2″-terpyridine-6,6″-dicarbocylate (BATDA) (32) exhibited reasonably good physicochemical and biological properties, we used this as a lead compound, and designed and synthesized a series of BATDA-derivatives.
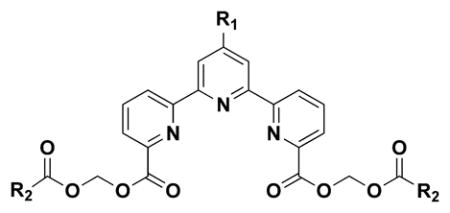
We first synthesized three different kinds of bis(alkanoyloxymethyl) 2,2′:6′,2″-terpyridine-6,6″-dicarboxylates 1–3 (Table 1) and 32. The compounds were synthesized based on the procedure described in Scheme 1. In brief, 2,2′:6′,2″-terpyridine-6,6″-dicarboxylic acid (TDA, 0.05 mmol) was reacted with halomethyl alkanoate (0.13 mmol) in the presence of N, N-diisopropylethylamine (0.23 mmol) in DMF at 50°C for 24 h. The yield was 24%, 28%, and 25% for 1, 2, and 3, respectively.
Table 1.
A list of terpyridine derivatives.
| |||||
|---|---|---|---|---|---|
| Compd. | R1 | R2 | Compd. | R1 | R2 |
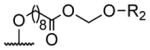
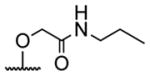
| 1 | H | butyryl | 17 |
|
butyryl |
| 2 | H | isobutyryl | 18 |
|
acetyl |
| 3 | H | pivaloyl | 19 |
|
acetyl |
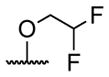
| 4 |
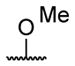
|
butyryl | 20 |
|
acetyl |
| 5 |
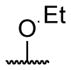
|
butyryl | 21 |
|
acetyl |
| 6 |
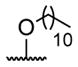
|
acetyl | 22 |
|
butyryl |
| 7 |
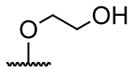
|
acetyl | 23 |
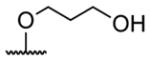
|
butyryl |
| 8 |
|
propionyl | 24 |
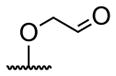
|
butyryl |
| 9 |
|
isobutyryl | 25 |
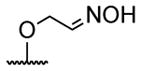
|
butyryl |
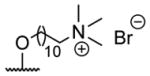
| 10 |
|
butyryl | 26 |
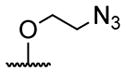
|
butyryl |
| 11 |
|
heptanoyl | 27 |
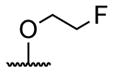
|
butyryl |
| 12 |
|
benzoyl | 28 |
|
butyryl |
| 13 |
|
butyryl | 29 |
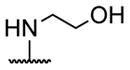
|
butyryl |
| 14 |
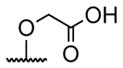
|
butyryl | 30 |
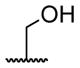
|
acetyl |
| 15 |
|
acetyl | 31 |
|
butyryl |
| 16 |
|
isobutyryl | 32 | H | acetyl |
A series of 4′-substituted bis(alkanoyloxymethyl) 2,2′:6′,2″-terpyridine-6,6″-dicarboxylate derivatives were designed and synthesized as described in Supporting Information. BATDA is shown as compound 32.
Scheme 1.

General procedure for the synthesis of 4′-substituted bis(alkanoyloxymethyl) 2,2′:6′,2″-terpyridine-6,6″-dicarboxylate. 4′-Substituted 2,2′:6′,2″-terpyridine-6,6″-dicarboxylic acid in DMF was reacted with halomethyl alkanoate in the presence of Et3N or i-Pr2EtN at 50°C or 60°C. Purification using SiO2 gel column chromatography afforded 4′-substituted bis(alkanoyloxymethyl) 2,2′:6′,2″-terpyridine-6,6″-dicarboxylate (Step-A)
We next compared the biological properties of the newly synthesized compounds with those of 32. Because the proligands are used for labeling tumor cells, the effects of the compounds on the viability of K562 erythroleukemia cells and U937 histocytoma cells were determined using a luciferase-based assay system. As summarized in Supporting Information 2, the cytotoxic effects of 1, 2, and 3 against tumor cells were essentially the same as those of 32, indicating that the newly synthesized compounds could be used for labeling tumor cells at concentrations below 33.3 μM.
Because the ideal proligand should have a high maximum release and a low spontaneous release rate when labeling tumor cells, we used the index M/[(100 × S)/M], where M represents maximum release, S spontaneous release, and (100 × S)/M the spontaneous release rate as described in the Experimental Section. We then compared the indices of the newly synthesized compounds when U937 histocytoma cells were used as target tumor cells (Fig. 1A). When R2 of the alkanoyloxymethyl group in Table 1 was pivaloyl group (3), the index was greatly lower than those of butyryl (1) and isobutyryl derivatives (2), suggesting that bulky substituent groups failed to be recognized by intracellular esterases. The result demonstrates that less bulky substituent groups at the position of R2 in Table 1 can be used for the improvement of the proligand of the fluorescent chelate.
Figure 1.

Live cell labeling with novel terpyridine derivative proligands. (A) Selection of proligands with a high maximum release and a low spontaneous release rate (%). U937 histocytoma cells were treated with a series of newly synthesized terpyridine proligands, and the index M/[(100 × S)/M] was determined for each compound. The index of 32 was 5091±96 (mean ± SD) and indicated as a dotted line. Compounds with the values greater than 5091±96 were indicated with asterisk. (B) Comparison of the spontaneous release rates of the compounds with the M/[(100 × S)/M] values being greater than that of 32. U937 cells were treated with compounds with the M/[(100 × S)/M] values greater than 5091±96. The spontaneous release and the maximum release values of each compound are depicted as bar graphs and the spontaneous release rates (%, mean ± SD) are indicated in the graphs. All experiments were performed in triplicate.
Because 32 contains the shortest hydrocarbon chain (R2 is an acetoxymethyl group) and have a higher index than 1 and 2, it is difficult to improve 32 by modifying the alkanoyloxymethyl moiety. We thus attempted to modify the terpyridine backbone structure of 32, and designed a series of compounds. First, 4′-O-substituted derivatives 4–28 were synthesized according to the procedure illustrated in Scheme 2, followed by Scheme 1. In brief, diethyl 1′-hydro-4′-oxo-2,2′:6′,2″-terpyridine-6,6″-dicarboxylate (393 mg, 1.0 mmol) was combined with NaH (60 mg, 60% 1.5 mmol) at room temperature for 30 min and allowed to react with an electrophile (1.5 mmole) in DMF (5.0 mL) for 15 h to yield 4′-O-substituted diethyl 2,2′:6′,2″-terpyridine-6,6″-dicarboxylate derivatives. The products (1.0 mmol) were hydrolyzed by treating with NaOH (3.5 mmol) for 20 h and the pH of the hydrolyzates was adjusted to approximately 4 with 1 N HCl to yield 4′-O-substituted 2,2′:6′,2″-terpyridine-6,6″-dicarboxylic acids. Finally, the resulting products (0.4 mmol) were reacted with halomethyl alkanoate (1.1 mmol) in the presence of Et3N (1.3 mmol) in DMF at 60°C for 18 h to yield 4′-O-substituted bis(alkanoyloxymethyl) 2,2′:6′,2″-terpyridine-6,6″-dicarboxylates. The yield of each compound is described in Supporting Information 3.
Scheme 2.

General procedure for the synthesis of 4′-O-substituted 2,2′:6′,2″-terpyridine-6,6′ ′-dicarboxylic acid. Diethyl 1′-hydro-4′-oxo-2,2′:6′,2″-terpyridine-6,6″-dicarboxylate was reacted with an electrophile in the presence of NaH in DMF for 15 h. Purification using SiO2 gel column chromatography afforded 4′-O-substituted diethyl 2,2′:6′,2″-terpyridine-6,6″-dicarboxylate derivative (Step-B). The product was hydrolyzed using NaOH to yield 4′-O-substituted 2,2′:6′,2″-terpyridine-6,6″-dicarboxylic acid (Step-C).
We then determined the cytotoxicity and the index M/[(100 × S)/M] of each compound 4–28 and compared them with those of 32. As shown in Supporting Information 2, the cytotoxic effects of the newly synthesized compounds against K562 erythroleukemia cells and U937 histocytoma cells were almost the same as those of 32. In fact, all the compounds were not cytotoxic at concentrations below 33.3 μM and considered to be used for live cell labeling. When the index M/[(100 – S)/M] of each compound was determined using U937 cells, compounds 7, 8, 10, 13, 21, 23 and 24 exhibited higher cell labeling efficiencies than 32 (Fig. 1A). The spontaneous release and the maximum release of each compound were summarized in Fig. 1B. Because it is generally accepted that live cell labeling is possible when the spontaneous release rate (100 × S)/M is less than 20% for the determination of cell-mediated cytotoxicity,[11] the newly synthesized compounds 7, 8, 10, 13, 21, 23 and 24 could be used for the assay.
Based on the above results, compounds with a bulky group at the 4′-position of the terpyridine backbone exhibited relatively low maximum release and those with a polar group gave low spontaneous release, suggesting that the ideal compounds should have a polar and less bulky substituent at 4′-position. In addition, it is known that electron-rich atoms like oxygen and nitrogen generally interfere with the emission of fluorescence. Because carbon atom is less electron-rich than oxygen and nitrogen atoms, we hypothesized that 4′-C-substitution was better than 4′-O or -N-substitution. We thus synthesized a 4′-N-substituted derivative, bis(butyryloxymethyl) 4′-(N-2-hydroxyethylamino)-2,2′:6′,2″-terpyridine-6,6″-dicarboxylate (29), and 4′-C-substituted derivates, bis(acetoxymethyl) 4′-(hydroxymethyl)-2,2′:6′,2″-terpyridine-6,6″-dicarboxylate (30) and bis(butyryloxymethyl) 4′-(hydroxymethyl)-2,2′:6′,2″-terpyridine-6,6″-dicarboxylate (31). The synthetic procedures of 29, 30, and 31 are described in Supporting Information 3 and the synthetic route of 31 in Scheme 3. These three compounds did not show cytotoxicity at concentrations below 33.3 μM as shown in Supporting Information 2. Although the index M/[(100 × S)/M] of compound 29 was lower than that of 32, compounds 30 and 31 exhibited higher cell labeling efficiencies than 32 (Fig. 1A). In fact, 31 showed the highest M/[(100 × S)/M] index among the compounds synthesized in this study. In addition, the spontaneous release rates of 30 and 31 were below 20% (Fig. 1B).
Scheme 3.

General procedure for the synthesis of bis(butyryloxymethyl) 4′-(hydroxymethyl)-2,2′:6′,2″-terpyridine-6,6″-dicarboxylate (31)
To further characterize 31, the photophysical properties of the ligand form of 31 were analyzed and compared with those of 10 and 32. After combining with Eu3+, the excitation wavelength (λex), the molar absorptivity at the excitation wavelength (ε), the emission wavelength (λem) and the relative quantum yield (Φ) were 330 nm, 7040 M−1cm−1, 592/615 nm and 0.34 for the ligand form of 32 (2,2′:6′,2″-terpyridine-6,6″-dicarboxylic acid (TDA), 323 nm, 13075 M−1cm−1, 592/615 nm and 0.58 for the ligand form of 10 (4′-(2-hydroxyethoxy)-2,2′:6′,2″-terpyridine-6,6″-dicarboxylic acid) and 333 nm, 12240 M−1cm−1, 592/615 nm and 1.02 for the ligand form of 31 (4′-(hydroxymethyl)-2,2′:6′,2″-terpyridine-6,6″-dicarboxylic acid). The measurement methods and emission spectra for each compound are shown in Supporting Information 3. Based on the above results, compound 31 seems to be the best proligand for live cell labeling to determine cell-mediated cytotoxicity. It is worth noting that not only Eu3+ but also Tb3+ may be used for this assay system (Supporting Information 3).
Because a variety of tumor cell lines are used to assess cell-mediated cytotoxicity by immune effector cells, compound 31 was tested for its ability to label a number of human cell lines including K562 (erythroleukemia), Raji (Burkitt’s lymphoma), P31/FUJ (monocytic leukemia), RAMOS-RAI (Burkitt’s lymphoma), RPMI8226 (plasmacytoma), EJ-1 (bladder carcinoma), and HCT-1, -4, and -5 (HTLV-1-infected, immortalized T cell lines). In all of the tumor cell lines, the maximum release exceeded the arbitrary fluorescence units of 100,000. Even in T cell lines immortalized with HTLV-1, the maximum arbitrary fluorescence units were greater than 4,000. Moreover, the spontaneous release rates were below 20% in all the cell lines used in this study, clearly demonstrating that compound 31 shows promise as a probe for live cell labeling to determine cell-mediated cytotoxicity.
To directly test compound 31 in a cytotoxicity assay, we used it to measure NK cell killing of K562 erythroleukemia cells. The principle of the assay is illustrated in Scheme 4. When K562 cells are treated with 31, the compound permeates into the cells and is hydrolyzed by intracellular esterases labeling the target cells. When NK cells kill the labeled target cells, the chelate-forming ligand is released. Because the negatively charged ligand does not interact avidly with intracellular proteins, it is rapidly released from the cells when the integrity of the cell membrane is disrupted by immune effector cells. Upon addition of Eu3+ to the culture supernatant, the ligand binds to Eu3+, resulting in the formation of a complex that emits long-life fluorescence upon laser excitation. Note that europium is not radioactive and is relatively non-toxic, similar to other rare earth heavy metals, requiring oral doses of 5 g/kg or greater for LD50 in rodents.[12]
Scheme 4.

Schematic representation of an improved non-radioactive cellular cytotoxicity assay method using a novel terpyridine derivative proligand. When target tumor cells are treated with bis(butyryloxymethyl) 4′-hydroxymethyl-2,2′:6′,2″-terpyridine-6,6″-dicarboxylate, a newly designed and synthesized terpyridine derivative proligand, it permeates into the target cells, where it is hydrolyzed by intracellular esterases to yield 4′-hydroxymethyl-2,2′:6′,2″-terpyridine-6,6″-dicarboxylic acid. Because the nascent hydrolyzates are negatively-charged, the ions accumulate in the target cells. When the labeled tumor cells are killed by immune effector cells, the ions are released into the culture medium. Upon addition of Eu3+ to the supernatant, the ion and Eu3+ forms a complex that emits long-life fluorescence under excitation with laser pulses.
NK cell lines with a purity of 98% or greater were established from PMBC from two healthy donors by removing CD3+ T cells and incubating the CD3− cells with IL-2 for 10 days (Figs. 3A and 3B). K562 cells were then labeled with 31 and incubated with the NK cell lines. Both NK cell lines efficiently killed K562 target cells in an effector to target ratio-dependent manner (Fig. 3C, D), clearly demonstrating that the novel terpyridine derivative proligand 31 is an effective reagent for use in cellular cytotoxicity assays.
Figure 3.

Non-radioactive cellular cytotoxicity assay using the compound 31. (A,B) Expansion of NK cells. Peripheral blood mononuclear cells from two healthy donors were each incubated with anti-CD3 monoclonal antibody-conjugated beads and the CD3+ T cells removed. The CD3− cells were incubated with IL-2 and the expanded NK cells were analyzed for CD56 expression by flow cytometry. (C,D) Determination of the specific lysis of K562 cells by NK cell lines. K562 cells labeled with 31 were incubated with the NK cell line from panel A for panel C or NK cells from panel B for panel D at effector to target ratios of 0, 0.625, 1.25, 2.5, 5, 10, 20 and 40. The specific lysis (%) was calculated as described in the Materials and Methods. Mean ± SD is shown for triplicate values.
Conclusions
A series of chelate-forming proligands were designed and synthesized to improve non-radioactive cellular cytotoxicity assays. One of the novel proligands, bis(butanoyloxymethyl) 4′-(hydroxymethyl)-2,2′:6′,2″-terpyridine-6,6″-dicarboxylate (31), efficiently labels target cells. Once the compound is internalized into target cells, it is hydrolyzed by intracellular esterases labeling the target cells. Upon killing, the nascent ligand is released from the cytoplasm into the supernatant where it forms complexes with Eu3+, a long-lived fluorescent species. The new 31 reagent exhibited higher fluorescence labeling of cells coupled with similar or lower toxicity compared to the currently used BATDA compound. Using this improved reagent, the cytotoxic activity of NK cells against K562 erythroleukemia cells was efficiently and reproducibly determined. The assay was also more rapid that the conventional method using radioactive [51Cr]-sodium chromate. Since the discovery that PD-1 immune checkpoint inhibitors have clinical benefits in cancer patients, immunotherapy has become a major focus for research in oncology. To develop new cancer immunotherapy approaches, it is essential to monitor and quantitate the activity of immune effector cells. The non-radioactive assay described in this study is a significant improvement over existing cytotoxicity assays and is ideal for use in clinical laboratories.
Experimental Section
General chemistry
All reactions were conducted under an air atmosphere unless otherwise stated. All NMR spectra were recorded on Varian Gemini300, JEOL AL 400 and Varian 500PS spectrometers at 24°C. 1H and 13C NMR spectra are reported as chemical shifts (δ) in parts per million (ppm) relative to the solvent peak using tetramethylsilane, 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt (1H and 13C) and trichlorofluoromethane (19F) as an internal standard. Chemical shifts (δ) are quoted in parts per million (ppm) and coupling constants (J) are measured in hertz (Hz). The following abbreviations are used to describe multiplicities s=singlet, d=doublet, t=triplet, q=quartet, quint.=quintet, sext.=sextet, sept.=septet br=broad, m=multiplet. NMR spectra were processed in ACD/SpecManager. High resolution mass spectra (HRMS, m/z) were obtained on JEOL JMS-700N for FAB using m-nitrobenzylalcohol as a matrix or on JEOL JMS-T100TD for electrospray ionization (ESI+). All reactions were performed in apparatuses with magnetic stirring under an inert atmosphere. Flash column chromatography was performed over Fuji Silysia Chemical Ltd. silica gel C60 (50–200 μm) and CHROMATOREX DIOL (MB 100-40/75) using an eluent system as described for each experiment. Thin-layer chromatography was performed using TLC Silica gel 60 F254 aluminum sheets (Merck) and silica gel F254 glass plates (Merck). NMR spectral information and physical data are shown in Supporting Information (Chemistry and NMR spectral information).
General procedure for the synthesis of 4′-substituted bis(alkanoyloxymethyl) 2,2′:6′,2″-terpyridine-6,6″-dicarboxylate (Scheme 1. Step-A)
To a solution of 4′-substituted 2,2′:6′,2″-terpyridine-6,6″-dicarboxylic acid (0.4 mmol) in DMF (10 mL) was added Et3N (1.3 mmol) and chloromethyl alkanoate (1.1 mmol). The reaction mixture was heated at 60°C. After stirring for 18 h, the solution was cooled to room temperature and the mixture was washed with H2O (20 mL), and the aqueous phase was extracted with AcOEt (2 × 30 mL). The combined organic phase fraction was washed with brine, then dried over MgSO4, filtered and concentrated in vacuo. Purification using SiO2 gel column chromatography afforded 4′-substituted bis(alkanoyloxymethyl) 2,2′:6′,2″-terpyridine-6,6″-dicarboxylate.
General procedure for 4′-O-substitution of diethyl 1′-hydro-4′-oxo-2,2′:6′,2″-terpyridine-6,6″-dicarboxylate (Step-B)
To a suspension of NaH (60 mg, 60% 1.5 mmol) in DMF (5.0 mL) was added diethyl 1′-hydro-4′-oxo-2,2′:6′,2″-terpyridine-6,6″-dicarboxylate (393 mg, 1.0 mmol) and the reaction mixture was allowed to stir for 30 min at room temperature. An electrophile (1.5 mmol) was added dropwise and the reaction mixture was stirred for 15 h. The reaction was quenched by the addition of the saturated solution of NH4Cl. The aqueous phase was extracted with AcOEt (2 × 50 mL). The combined organic phase fraction was washed with water (50 mL) and brine (50 mL), dried over MgSO4, filtered and then concentrated in vacuo. Purification using SiO2 gel column chromatography afforded 4′-O-substituted diethyl-2,2′:6′,2″-terpyridine-6,6″-dicarboxylate derivative.
General procedure for the hydrolysis of 4′-O-substituted diethyl 2,2′:6′,2″-terpyridine-6,6″-dicarboxylate derivative (Step-C)
A mixture of 4′-O-substituted diethyl 2,2′:6′,2″-terpyridine-6,6″-dicarboxylate derivative (1.0 mmol) and NaOH (3.5 mmol) was stirred for 20 h at room temperature. The pH was adjusted to approximately 4 with 1 N HCl. The precipitate was collected by filtration, dried under vacuo affording 4′-O-substituted 2,2′:6′,2″-terpyridine-6,6″-dicarboxylic acid.
Biological assays
Screening of terpyridine derivative proligands
The maximum release (M) was calculated as [maximum release value – background value] and the spontaneous release (S) as [spontaneous release value – background value]. Then, the spontaneous release rate (%) was calculated as [(100 × S)/M]. Because the ideal proligand should be the one with a high maximum release and with a low spontaneous release rate, the proligand with the highest M/[(100 × S)/M] value should be the best compound for use in live cell labeling. In this study, the M/[(100 × S)/M] value was used as an index for the selection of the best proligand.
Cell lines
Human tumor cell lines used for this study were U937 histocytoma, K562 erythroleukemia, Raji Burkitt’s lymphoma, P31/FUJ monocytic leukemia, RAMOS-RAI Burkitt’s lymphoma, RPMI8226 plasmacytoma, EJ-1 bladder carcinoma, HCT-1 HTLV-1, -4 and -5 HTLV-1-infected T cell lines. The tumor cell lines except for HCT-1, -4 and -5 were obtained from the National Institutes of Biomedical Innovation, Health and Nutirition JCRB Cell Bank (Ibaraki, Osaka, Japan). HCT-1, -4 and -5 were established in our laboratory after approval of the institutional review board of Nagasaki University along with written informed consent. If brief, peripheral blood mononuclear cells were infected with HTLV-1 virus and immortalized cells were maintained in complete RPMI1640 media (RPMI1640 media (Merck & Co., Inc.) supplemented with 10% fetal serum albumin (Merck & Co., Inc.), 10−5 M 2-mercaptoethanol (Wako Pure Chemical Industries, Ltd., Chuo-ku, Osaka, Japan), 100 U/ml of penicillin (Meiji Seika Pharma Co., Ltd., Chuo-ku, Tokyo, Japan), 100 μg/ml of streptomycin(Meiji Seika Pharma Co., Ltd.) and 100 U/ml of IL-2 (Shionogi & Co. Ltd., Chuo-ku, Osaka, Japan) at 37°C with 5% CO2 in 75 cm2 culture flasks (Thermo Fisher Scientific, Waltham, MA).
Cytotoxicity assay
K562 erythroleukemia cells and U937 histocytoma cells were grown in 30 ml of complete RPMI1640 media in 75 cm2 culture flasks at 37°C with 5% CO2. After centrifugation at 600 × g for 5 min at 4°C in a refrigerated centrifuge (Tomy Seiko Co., Ltd., Nerima-ku, Tokyo, Japan), the cells were resuspended in complete RPMI1640 media to give a cell concentration of 1 × 105 cells/ml, respectively. The cell suspensions were dispensed into a 96-well flat bottom plate (5 × 103 cells/50 μl/well) (Merck & Co., Inc.) and were treated with 50 μl of a 3-fold serial dilution of synthesized compounds ranging from 100 μM to 5.081 nM at 37°C with 5% CO2. After 2 h, 100 μl of CellTiterGlo® Luminescent Cell Viability Assay reagent (Promega Corp., Madison, WI) were added to each well and the cell lyzates were transferred to OptiPlates-96® (PerkinElmer, Waltham, MA). Then, luminescence was determined through an ARVO multiplate reader (PerkinElmer). All experiments were performed in triplicate.
Live cell labeling
Tumor cells were grown in 30 ml of complete RPMI1640 media in 75 cm2 culture flasks at 37°C with 5% CO2. The cell suspensions were transferred into 50 ml conical tubes (Thermo Fisher Scientific) and centrifuged at 600 × g for 5 min at 4°C. After the supernatants were removed, the cell pellets were resuspended in complete RPMI1640 to give a cell concentration of 1 × 106 cells/ml. The cells (1 × 106 cells in 1 ml) were treated with 25 μM of synthesized compounds at 37°C for 15 min, washed three times with 5 ml of complete RPMI1640 media and resuspended in 1 ml of complete RPMI1640 media. The cell suspensions (100 μl each) were placed in 6 wells of a 96-well round bottom plate (Merck & Co., Inc.), to three wells of which were added 100 μl of complete RPMI1640 media for the determination of spontaneous release and to the remaining three wells of which were added 90 μl of complete RPMI1640 media for the determination of maximum release. The plate was centrifuged at 500 rpm at room temperature for 2 min in a centrifuge (Tomy Seiko Co., Ltd.) and incubated at 37°C with 5% CO2 for 30 min. Then, 10 μl of 0.125% digitonin (Wako Pure Chemical Industries Ltd.) in 19% dimethyl sulfoxide (Wako Pure Chemical Industries Ltd.) were added to three wells for the determination of maximum release. The plates were incubated at 37°C with 5% CO2 for 30 more min and centrifuged at 600 × g for 2 min at 4°C in a refrigerated centrifuge (Tomy Seiko Co., Ltd.). The supernatants (25 μl each) were carefully removed into wells of 96-well round bottom plates (Merck & Co., Inc.) containing 250 μl each of europium solution (50 μM Eu3+ (Merck & Co., Inc.) in 0.3 M sodium acetate buffer, pH 4.0 (Wako Pure Chemical Industries Ltd.)). The samples were mixed well and 200 μl each of them were removed into a 96-well optical bottom plate with polymer base (Thermo Fisher Scientific). Then, luminescence was determined through an ARVO multiplate reader (PerkinElmer).
Derivation of human NK cells
Peripheral blood samples were obtained from two adult volunteers. Heparin (100 U/100 μl, Mochida Pharmaceutical Co., Ltd., Shinjuku-ku, Tokyo, Japan) was added to 10 ml of blood, to which were added 10 ml of phosphate buffered saline (PBS, Nissui Pharmaceutical Co., Ltd., Taito-ku, Tokyo, Japan). The diluted blood samples were loaded onto 20 ml of Ficoll-Paque Plus (GE Healthcare Bio-Sciences, Pittsburgh, PA) in 50 ml conical tubes (Thermo Fisher Scientific). The conical tubes were centrifuged at 600 × g for 30 min at room temperature in a centrifuge (Tomy Seiko Co., Ltd.) and the fluffy layer containing peripheral blood mononuclear cells (PBMC) was removed into new 50 ml conical tubes, to which were added 35 ml of PBS. The tubes were centrifuged at 900 × g for 10 min at 4°C and the supernatants were removed. The cell pellets were washed with 10 ml of PBS resuspended in 80 μl of PBS containing 0.5% bovine serum albumin (BSA, Merck & Co., Inc.) and 2 mM ethylenediaminetetraacetic acid (EDTA, Dojindo Molecular Technologies, Inc., Mashiki, Kumamoto, Japan) and transferred into 15 ml conical tubes, to which were added 20 μl of anti-human CD3 MACS Beads (Miltenyi Biotec Inc., Auburn, CA). After incubation at 4°C for 15 min, the cell/beads suspensions were diluted with 1 ml of PBS/0.5% BSA/2 mM EDTA and centrifuged at 300 × g for 10 min at 4°C. The supernatants were removed and the cell/beads mixtures were resuspended in 1 ml of PBS/0.5% BSA/2 mM EDTA and loaded on an LD column (Miltenyi Biotec Inc.) that had been equilibrated with 2 ml of PBS/0.5% BSA/2 mM EDTA equipped with a magnet (Miltenyi Biotec Inc.). The flow-through fractions were placed in 15 ml conical tubes and the LD column was washed 2 times with 1 ml of PBS/0.5% BSA/2 mM EDTA. The combined flow-through fractions containing CD3− PBMC were centrifuged at 600 × g for 5 min at 4°C and resuspended in 3 ml of Yssel’s media containing 10% human AB serum (Cosmo Bio Co., Ltd., Koto-ku, Tokyo, Japan). The CD3− PBMC were placed in 2 wells of a 24-well plate (Thermo Fisher Scientific), to which were added 100 U/ml of IL-2 every day. After 10 days, the expanded NK cells were harvested and the expression of CD3 and CD56 was analyzed on a FACSCalibur flow cytometer using phycoerythrin (PE)-conjugated mouse anti-human CD56 monoclonal antibody (Becton Dickinson & Co., Franklin Lakes, NJ) and fluorescein isothiocyanate (FITC)-conjugated mouse anti-human CD3 monoclonal antibody (Becton Dickinson & Co.).
Cellular cytotoxicity assay
K562 erythroleukemia cells were grown in 30 ml of complete RPMI1640 media in 75 cm2 culture flasks at 37°C with 5% CO2. The cell suspensions were centrifuged at 600 × g for 5 min at 4°C, the cell pellets were resuspended in complete RPMI1640 to give a cell concentration of 1 × 106 cells/ml. The cells (1 × 106 cells in 1 ml) were treated with 25 μM of compound 31 at 37°C for 15 min, washed three times with 5 ml of complete RPMI1640 media and resuspended in 1 ml of complete RPMI1640 media. The cell suspensions (100 μl each) were placed in a 96-well round bottom plate (Merck & Co., Inc.), to which were added 100 μl of a 2-fold serial dilution of human NK cells at an effector to target ratio of 40 : 1 through 0.625 : 1. The plate was centrifuged at 500 rpm at room temperature for 2 min in a centrifuge (Tomy Seiko Co., Ltd.) and incubated at 37°C with 5% CO2 for 60 min. Then, the plate was centrifuged at 600 × g for 2 min at 4°C in a refrigerated centrifuge (Tomy Seiko Co., Ltd.). The supernatants (25 μl each) were carefully removed into wells of 96-well round bottom plates (Merck & Co., Inc.) containing 250 μl each of europium solution (50 μM Eu3+ (Merck & Co., Inc.) in 0.3 M sodium acetate buffer, pH 4.0 (Wako Pure Chemical Industries Ltd.)). The samples were mixed well and 200 μl each of them were removed into a 96-well optical bottom plate with polymer base (Thermo Fisher Scientific). Then, luminescence was determined through an ARVO multiplate reader (PerkinElmer). All experiments were performed in triplicate.
Supplementary Material
Figure 2.

Live cell labeling with the compound 31. Tumor cell lines and HTLV-1-transformed cell lines were treated with 31. The spontaneous release and the maximum release values for the cell lines are depicted as bar graphs and the spontaneous release rates (%, mean ± SD) are indicated in the graphs. All experiments were performed in triplicate.
Acknowledgments
This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Science, Culture, Sports, and Technology of Japan 16K08844 (MEXT)(to Y.T.), by the Japan Agency for Medical Research and Development 12723070 and DNW-17004 (to Y.T.), by the Department of Veterans Affairs (Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development) Grant 1 I01 BX000972-01A1 (to C.T.M.), National Cancer Institute Grants CA097274 (University of Iowa/Mayo Clinic Lymphoma Specialized Program of Research Excellence) and P30CA086862 (Core Support) (to C.T.M.). C.T.M. is the Kelting Family Scholar in Rheumatology.
Footnotes
Supporting information and the ORCID identification numbers for the authors of this article can be found under https://doi.org/10.1002/cmdc.201700626.
References
- 1.a) Topalian SL, Drake CG, Pardoll DM. Cancer Cell. 2015;27:450. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Iwai Y, Hamanishi J, Chamoto K, Honjo T. J Biomed Sci. 2017;24:26. doi: 10.1186/s12929-017-0329-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM. N Engl J Med. 2012;366:2455. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, Larkin J, Lorigan P, Neyns B, Blank CU, Hamid O, Mateus C, Shapira-Frommer R, Kosh M, Zhou H, Ibrahim N, Ebbinghaus S, Ribas A. N Engl J Med. 2015;372:2521. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]; e) Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E, Savage KJ, Hernberg MM, Lebbe C, Charles J, Mihalcioiu C, Chiarion-Sileni V, Mauch C, Cognetti F, Arance A, Schmidt H, Schadendorf D, Gogas H, Lundgren-Eriksson L, Horak C, Sharkey B, Waxman IM, Atkinson V, Ascierto PA. N Engl J Med. 2015;372:320. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]; f) Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, Waterhouse D, Ready N, Gainor J, Aren Frontera O, Havel L, Steins M, Garassino MC, Aerts JG, Domine M, Paz-Ares L, Reck M, Baudelet C, Harbison CT, Lestini B, Spigel DR. N Engl J Med. 2015;373:123. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, Barlesi F, Kohlhaufl M, Arrieta O, Burgio MA, Fayette J, Lena H, Poddubskaya E, Gerber ED, Gettinger SN, Rudin CM, Rizvi N, Crino L, Blumenschein GR, Jr, Antonia SJ, Dorange C, Harbison CT, Graf Finckenstein F, Brahmer JR. N Engl J Med. 2015;373:1627. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, Carcereny E, Ahn MJ, Felip E, Lee JS, Hellmann MD, Hamid O, Goldman JW, Soria JC, Dolled-Filhart M, Rutledge RZ, Zhang J, Lunceford JK, Rangwala R, Lubiniecki GM, Roach C, Emancipator K, Gandhi L. N Engl J Med. 2015;372:2018. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]; i) McDermott DF, Drake CG, Sznol M, Choueiri TK, Powderly JD, Smith DC, Brahmer JR, Carvajal RD, Hammers HJ, Puzanov I, Hodi FS, Kluger HM, Topalian SL, Pardoll DM, Wigginton JM, Kollia GD, Gupta A, McDonald D, Sankar V, Sosman JA, Atkins MB. J Clin Oncol. 2015;33:2013. doi: 10.1200/JCO.2014.58.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, Mizoguchi A, Hiai H, Minato N, Honjo T. Science. 2001;291:319. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- 3.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Proc Natl Acad Sci USA. 2002;99:12293. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M. N Engl J Med. 2012;366:2443. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, Ibrahim F, Bruggeman C, Gasmi B, Zappasodi R, Maeda Y, Sander C, Garon EB, Merghoub T, Wolchok JD, Schumacher TN, Chan TA. Science. 2015;348:124. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, Stephens PJ, Daniels GA, Kurzrock R. Mol Cancer Ther. 2017 doi: 10.1158/1535-7163.MCT-17-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini KS, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, Watkins TB, Shafi S, Murugaesu N, Mitter R, Akarca AU, Linares J, Marafioti T, Henry JY, Van Allen EM, Miao D, Schilling B, Schadendorf D, Garraway LA, Makarov V, Rizvi NA, Snyder A, Hellmann MD, Merghoub T, Wolchok JD, Shukla SA, Wu CJ, Peggs KS, Chan TA, Hadrup SR, Quezada SA, Swanton C. Science. 2016;351:1463. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brunner KT, Mauel J, Cerottini JC, Chapuis B. Immunology. 1968;14:181. [PMC free article] [PubMed] [Google Scholar]
- 8.a) Korzeniewski C, Callewaert DM. J Immunol Methods. 1983;64:313. doi: 10.1016/0022-1759(83)90438-6. [DOI] [PubMed] [Google Scholar]; b) Blomberg K, Hautala R, Lovgren J, Mukkala VM, Lindqvist C, Akerman K. J Immunol Methods. 1996;193:199. doi: 10.1016/0022-1759(96)00063-4. [DOI] [PubMed] [Google Scholar]; c) Sheehy ME, McDermott AB, Furlan SN, Klenerman P, Nixon DF. J Immunol Methods. 2001;249:99. doi: 10.1016/s0022-1759(00)00329-x. [DOI] [PubMed] [Google Scholar]; d) Zaritskaya L, Shurin MR, Sayers TJ, Malyguine AM. Expert Rev Vaccines. 2010;9:601. doi: 10.1586/erv.10.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Granberg C, Blomberg K, Hemmila I, Lovgren T. J Immunol Methods. 1988;114:191. doi: 10.1016/0022-1759(88)90173-1. [DOI] [PubMed] [Google Scholar]; b) Blomberg K, Ulfstedt AC. J Immunol Methods. 1993;160:27. doi: 10.1016/0022-1759(93)90005-r. [DOI] [PubMed] [Google Scholar]; c) Cui J, Bystryn JC. J Immunol Methods. 1992;147:13. doi: 10.1016/s0022-1759(12)80023-8. [DOI] [PubMed] [Google Scholar]; d) He L, Hakimi J, Salha D, Miron I, Dunn P, Radvanyi L. J Immunol Methods. 2005;304:43. doi: 10.1016/j.jim.2005.06.005. [DOI] [PubMed] [Google Scholar]; e) Kolber MA, Quinones RR, Gress RE, Henkart PA. J Immunol Methods. 1988;108:255. doi: 10.1016/0022-1759(88)90427-9. [DOI] [PubMed] [Google Scholar]; f) Lovgren J, Blomberg K. J Immunol Methods. 1994;173:119. doi: 10.1016/0022-1759(94)90289-5. [DOI] [PubMed] [Google Scholar]; g) Maley DT, Simon P. J Immunol Methods. 1990;134:61. doi: 10.1016/0022-1759(90)90112-9. [DOI] [PubMed] [Google Scholar]; h) Pacifici R, Di Carlo S, Bacosi A, Altieri I, Pichini S, Zuccaro P. J Immunol Methods. 1993;161:135. doi: 10.1016/0022-1759(93)90205-l. [DOI] [PubMed] [Google Scholar]; i) Volgmann T, Klein-Struckmeier A, Mohr H. J Immunol Methods. 1989;119:45. doi: 10.1016/0022-1759(89)90379-7. [DOI] [PubMed] [Google Scholar]; j) von Zons P, Crowley-Nowick P, Friberg D, Bell M, Koldovsky U, Whiteside TL. Clin Diagn Lab Immunol. 1997;4:202. doi: 10.1128/cdli.4.2.202-207.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hofmeier H, Schubert US. Chem Soc Rev. 2004;33:373. doi: 10.1039/b400653b. [DOI] [PubMed] [Google Scholar]
- 11.Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. Nature. 1998;393:480. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- 12.a) Haley TJ, Komesu N, Colvin G, Koste L, Upham HC. J Pharm Sci. 1965;54:643. doi: 10.1002/jps.2600540435. [DOI] [PubMed] [Google Scholar]; b) Bruce DW, Hietbrink BE, Dubois KP. Toxicol Appl Pharmacol. 1963;5:750. doi: 10.1016/0041-008x(63)90067-x. [DOI] [PubMed] [Google Scholar]; c) Ogawa Y, Suzuki S, Naito K, Saito M, Kamata E, Hirose A, Ono A, Kaneko T, Chiba M, Inaba Y, et al. J Environ Pathol Toxicol Oncol. 1995;14:1. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.