

Abstract
Spinal bulbar muscular atrophy (SBMA) is a motor neuron disease caused by toxic gain-of-function of the androgen receptor (AR). In a prior study we showed that coregulator binding through the activation function-2 (AF2) domain of AR is essential to pathogenesis, suggesting that AF2 may be a druggable target for selective modulation of toxic AR activity. Here we screened previously identified AF2 modulators for their ability to rescue toxicity in a Drosophila model of SBMA. We identified two compounds, tolfenamic acid (TA) and 1-[2-(4-methylphenoxy)ethyl]-2-[(2-phenoxyethyl)sulfanyl]-1H-benzimidazole (MEPB), as top candidates for rescuing lethality, locomotor function, and neuromuscular junction defects in SBMA flies. Pharmacokinetic analyses in mice showed a more favorable bioavailability and tissue retention of MEPB compared with TA in muscle, brain, and spinal cord. In a preclinical trial in a novel mouse model of SBMA, MEPB treatment yielded a dose-dependent rescue from loss of body weight, rotarod activity, and grip strength. In addition, MEPB ameliorated neuronal loss, neurogenic atrophy, and testicular atrophy, validating AF2 modulation as a potent androgen-sparing strategy for SBMA therapy.
Introduction
Spinal bulbar muscular atrophy (SBMA), or Kennedy’s disease, is a progressive neurodegenerative disease affecting approximately 1 in 40,000 men worldwide1; however, this number is likely underestimated due to common misdiagnoses (e.g., limb girdle muscular dystrophy and amyotrophic lateral sclerosis [ALS])2. Although SBMA is not typically fatal, quality of life is profoundly affected as patients experience bulbar dysfunction, limb weakness, loss of ambulation, and partial androgen insensitivity that often leads to feminization, testicular atrophy and fertility problems. Neuromuscular symptoms generally first appear as muscle spasms and weakness in the extremities, mouth, and throat, which progress to muscle wasting due to loss of motor neurons. There is no cure for SBMA, and treatment is symptomatic, usually entailing physical therapy and rehabilitation. Patients with SBMA frequently become confined to a wheelchair later in life and require assistance with common daily tasks, such as eating2.
SBMA results from a CAG trinucleotide repeat expansion in the androgen receptor (AR) gene, resulting in an expanded polyglutamine (polyQ) tract within the protein3. In healthy, unaffected males, AR typically contains 15 to 37 CAG repeats, whereas the presence of 40 or greater CAG repeats results in manifestation of disease4,5. However, the presence of a polyQ expansion within the AR alone is not sufficient to drive disease pathology, as evidenced by the absence of SBMA-associated symptoms in female carriers6. Specifically, androgen stimulation of the AR, typically by the cognate ligands testosterone or dihydrotestosterone (DHT), has been extensively characterized to be required for induction of polyQ-expanded AR-mediated toxicity7, indicating that activation of the downstream AR signaling pathway via receptor–ligand interaction is a necessary first step to drive neurodegeneration.
In a previous study using a Drosophila model of SBMA, we found that two additional native functions of the AR were required to perpetuate disease: 1) interaction between the AR and its transcriptional target genes through the AR DNA-binding domain (DBD) and 2) interaction between the AR and transcriptional coregulators through the AR activation function-2 (AF2) domain8. Expression of a polyQ-expanded, human AR transgene containing a point mutation rendering the DBD inactive (A574D substitution) abolished DHT-dependent degeneration, suggesting that the SBMA degenerative phenotype is due to a change in polyQ-expanded AR-mediated transcriptional activity. The transcriptional activity of AR is modified by binding of various coregulatory proteins, which can serve as coactivators or corepressors of AR target genes9. The AF2 domain is a regulatory pocket located within the ligand binding domain (LBD) that becomes accessible to binding of coregulator proteins containing LXXLL or FXXLF motifs, following androgen stimulation10. Expression of polyQ-expanded AR containing a point mutation resulting in a K720A substitution, which inhibits binding of LXXLL-bearing coregulators and attenuates binding of FXXLF-bearing coregulators, partially rescued DHT-dependent degeneration in the fly eye8. Expression of polyQ-expanded AR containing an E897K substitution, which inhibits binding of coregulators bearing either LXXLL or FXXLF motifs, fully rescued DHT-induced degeneration, demonstrating a pivotal role for coregulators in SBMA-associated toxicity8.
Studies in mouse models of SBMA have utilized chemical and physical castration as a method for rescuing the SBMA degenerative phenotype11-13. Consequently, androgen antagonists and drugs that promote AR degradation have been suggested for therapeutic treatment of SBMA14,15. In a phase II placebo-controlled clinical trial, patients treated with the anti-androgen leuprorelin for 144 weeks exhibited significantly diminished serum levels of creatine kinase, significantly improved functional scores, and significantly better swallowing parameters than those who received a placebo16. More recently, a larger randomized placebo-controlled, multicentric clinical trial of this drug showed no definite effect on motor functions, although swallowing function improved in a subgroup of patients whose disease duration was less than 10 years14. Although an androgen ablation strategy may be effective at reducing SBMA-associated neuromuscular degeneration in patients, long-term, widespread loss of the AR signaling pathway would likely result in severe endocrine disruption, producing multiple adverse side effects and further antagonizing the loss of QOL in patients with SBMA. Alternatively, modulation of native coregulator–AR interactions with AF2-specific small-molecule compounds such as selective AR modulators (SARMs) holds great promise for a targeted therapeutic approach.
Results
AF2 modulation rescues degeneration in SBMA flies
We compiled a panel of small-molecule compounds that were previously identified by in silico or in vitro screening to modulate coregulator binding to the AR AF2 domain by specifically binding to a proximal regulatory pocket termed the binding function-3 (BF3) domain17-22. Upon binding of such compounds to the BF3 domain, a conformational shift occurs in the AF2 domain that modulates the ability of coregulators bearing FXXLF and LXXLL motifs to bind the AF2 domain (Fig. 1a). To determine whether such compounds may be advantageous for SBMA therapy, we tested their ability to rescue DHT-induced lethality in flies expressing polyQ-expanded AR in pan-neuronal tissues (ELAV>UAS-AR52Q) or specifically in motor neurons (OK371>UAS-AR52Q) (Supplementary Fig. 1 and data not shown). TA and MEPB treatment significantly increased SBMA fly viability in a dose-dependent manner (Fig. 1b,c), similar to the known AR antagonist bicalutamide, whereas an AR-unrelated compound, ibuprofen, did not (Supplementary Fig. 1). Consistent with these results, both TA and MEPB significantly restored locomotor function in flies expressing polyQ-expanded AR in motor neurons (OK371>UAS-AR52Q), measured as increased displacement and velocity of walking in adult flies (Fig.1d–f), to similar levels observed in flies expressing AR variants that modulate coregulator binding to the AF2 domain (OK371>UAS-AR52Q-K720A and OK371>UAS-AR66Q-E897K). Furthermore, TA and MEPB restored DHT-dependent neuromuscular junction defects in SBMA larvae of OK371>UAS-AR52Q flies by reducing the prevalence of satellite boutons and preventing loss of neuromuscular junction branching (Fig. 1g). These results were corroborated in flies expressing polyQ-expanded AR in the eye (GMR>UAS-AR52Q), in which DHT-dependent degeneration was mitigated by TA and MEPB, similar to that of bicalutamide and dimethylcurcumin (Supplementary Fig. 2a), without reducing monomeric or aggregated forms of AR protein levels (Supplementary Fig. 2b–e).
Figure 1. AF2 modulation rescues degeneration in a fruit fly model of SBMA.
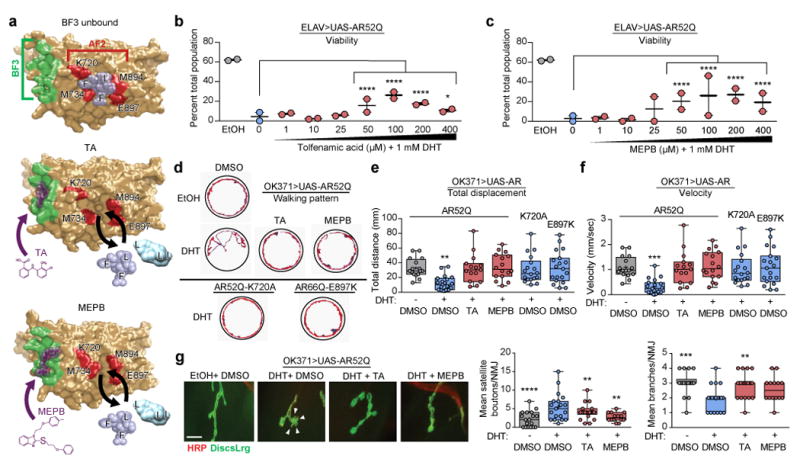
(a) Structural depiction of the AR LBD in complex with TA or MEPB. The BF3 domain is shaded green, the core AF2 domain is shaded red, TA and MEPB are shaded purple, and modeled FXXLF or LXXLL motifs are shaded light blue. (b) Viability of flies expressing human AR52Q in pan-neuronal tissues in the presence of vehicle (gray), DHT (blue), and DHT + TA (red). (c) Viability of flies expressing human AR52Q in pan-neuronal tissues in the presence of vehicle (gray), DHT (blue), and DHT + MEPB (red); Data shown in b–c were evaluated by Chi-square analysis; comparisons between the actual population frequencies of each treatment group and the predicted population frequency were based on the DHT + DMSO group, n = 50 adult flies/treatment group per experiment, two independent experiments. *P = 0.0154, ****P ≤ 0.0001. (d) Representative traces of the walking pattern of adult flies expressing AR52Q, AR52Q-K720A, or AR66Q-E897K in motor neurons for 90 seconds in a well of a 12-well tissue culture plate. Adult flies were reared on food containing 1% ethanol + 0.1% DMSO, 1 mM DHT + 0.1% DMSO, 1 mM DHT + 100 μM TA, or 1 mM DHT + 100 μM MEPB. (e) Quantification of mean distance walked by adult SBMA flies, determined from 15 individual tracing patterns per genotype or treatment group. (f) Quantification of mean velocity of adult SBMA flies, determined from 15 individual tracing patterns per genotype or treatment group. Data shown in e-f were evaluated by one-way ANOVA and Dunnett’s multiple comparison test between each treatment group and the -DHT/+DMSO group. **P = 0.0031 for (e) and ***P = 0.0005 for (f).
(g) Staining and quantification of neuromuscular junctions of larval SBMA flies. Red, HRP (presynaptic marker); green, DiscsLrg (postsynaptic marker). Arrowheads indicate satellite boutons. Scale bar, 10 μm. Mean satellite boutons and neuromuscular junction branches were quantified for 2–3 muscle segments from 12 dissected larval pelts per treatment group. Data shown in g were evaluated by one-way ANOVA and Dunnett’s multiple comparison test between each treatment group and the +DHT/+DMSO group. **P = 0.0047 (+DHT/+TA), **P = 0.0035 (+DHT/+MEPB), ****P ≤ 0.0001 for boutons, **P = 0.0029 (+DHT/+TA), ***P = 0.0009 (-DHT/DMSO) for branches. All graphs represent mean ± s.e.m.
Transgenic male mice carrying full-length human AR with 121 CAG repeats recapitulate SBMA symptoms and pathology
Previously reported mouse models of SBMA that exhibit disease-relevant phenotypes express the polyQ-AR transgene at levels several-to-many fold higher than endogenous AR. For example, the most frequently used mouse model of SBMA12 expresses exogenous human AR at levels approximately three times higher than endogenous AR levels (Supplementary Fig. 3c,d). Although these prior mouse models of SBMA have been a valuable resource to the research community, we were concerned about the possibility that high levels of mutant AR expression might mask the therapeutic potential of SARMs. Thus, to evaluate the efficacy of SARM therapy in a mammalian system, we developed an animal model that phenocopies the disease-relevant features of SBMA by expressing physiologically relevant levels of mutant AR. Founder SBMA mice were produced by pronuclear injection of human AR cDNA containing 121 CAG/CAA alternating repeats driven by the pCAGGS (CMV-IE enhancer + chick β-Actin) promoter (Supplementary Fig. 3a,b). We identified one line that expressed the AR transgene in spinal cord and muscle at endogenous levels (Supplementary Fig. 3c,d) and this line was chosen for further characterization.
Beginning at approximately 4 weeks of age, male AR121Q mice demonstrated rapid androgen-dependent declines in body weight, rotarod activity, grip strength, and survival (Fig. 2a–d) when compared with nontransgenic FVB/NJ (NTG) control littermates. Immunohistochemistry using anti-AR, anti-polyQ, and anti-ubiquitin revealed the presence of aggregates in the brain, spinal cord, and skeletal muscle but not in the testis or liver of AR121Q mice (Fig. 2e,f and Supplementary Fig. 3e–g). These ubiquitin-positive nuclear inclusions also colocalized with polyQ in the spinal cord and skeletal muscle (Fig. 2g and Supplementary Fig. 3h). Muscle fiber type switching from glycolytic type II fibers to oxidative type I fibers has recently been characterized as a pathologic hallmark of muscle atrophy in mice and patients with SBMA23. Consistent with this characterization, AR121Q mice displayed a marked presence of atrophied skeletal muscle fibers and switching of muscle fibers from glycolytic type II to oxidative type I (Fig. 2h,i). Furthermore, we observed significantly fewer numbers, but no change in area of, choline acetyltransferase (ChAT)–positive motor neurons in the anterior horn of the thoracic spinal cord in AR121Q mice compared to NTG controls (Fig. 2j). Immunofluorescence with antibodies against neuronal cell adhesion molecule (NCAM) and its polysialic acid form (PSA-NCAM), a protein known to be upregulated during muscle reinnervation24-26, revealed marked sarcoplasmic staining of muscle fibers in AR121Q mice but not in NTG controls (Fig. 2k), demonstrating ongoing denervation/reinnervation in AR121Q mice. AR121Q mice also exhibited markedly decreased hindlimb muscle mass and increased angular fibers (data not shown). Although centralized nuclei were present in some muscle fibers of AR121Q mice (Fig. 2f), quantification revealed that both AR121Q and NTG mice had fewer than 3% of fibers with centralized nuclei, with no difference between the two groups (Supplementary Fig. 3i). Together, these data suggest that AR121Q mice exhibit primarily neurogenic rather than myogenic atrophy and weakness.
Figure 2. AR121Q-expressing mice recapitulate SBMA symptoms and pathology.
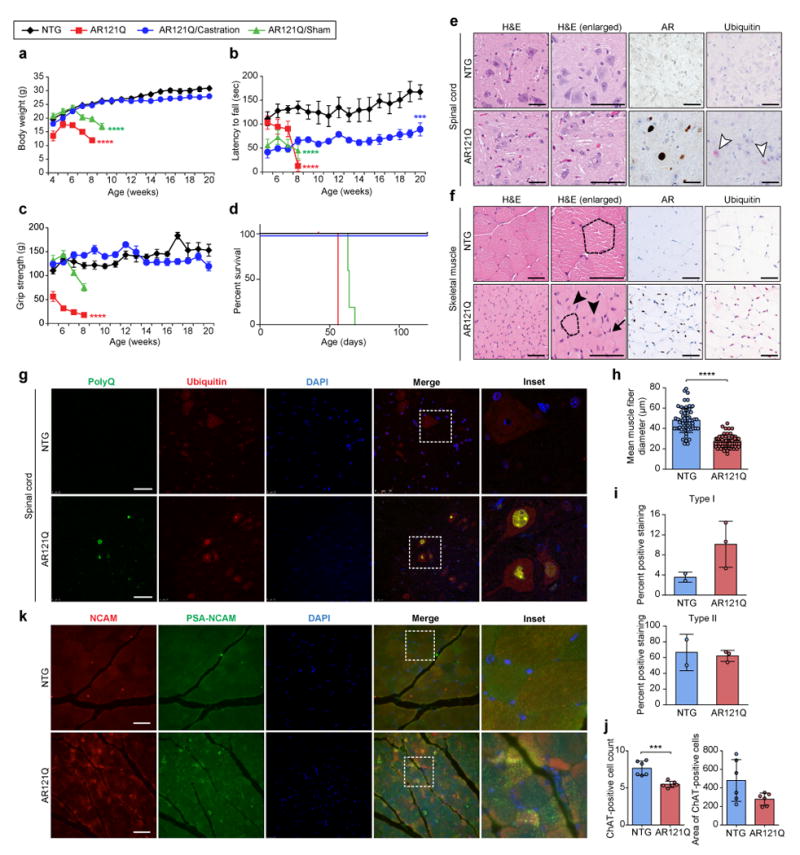
(a-d) Phenotypic measures of SBMA degeneration (mean body weight (a), mean rotarod activity (b), mean grip strength (c), and survival (d) of NTG (black diamonds) and AR121Q mice (red squares), in addition to sham-operated (green triangles) and castrated (blue circles) AR121Q mice. n = 5 mice per group, ****P ≤ 0.0001, ***P =0.0007 by two-way ANOVA and Dunnett’s posthoc test compared to the NTG group (a-c) or Kaplan-Meier estimation (log-rank test) (d). Note that the abrupt reduction in survival reflects animal protocol criteria that mandate euthanasia of animals with loss of 10% body weight or the development of substantial weakness. (e-f) Representative spinal cord (e) and skeletal muscle (f) sections from 7-week-old NTG and AR121Q mice from one independent experiment. Sections were stained with hematoxylin and eosin (H&E) stain for assessment of morphology, in addition to AR (N20) and ubiquitin antibodies. White arrowheads indicate ubiquitin-positive nuclear inclusions in the spinal cord. Dotted lines trace around representative myofibers, demonstrating differences in myofiber size. Arrow indicates atrophied myofibers. Arrowheads indicate myofibers containing centralized nuclei. (g) PolyQ (5TF1-1C2) and ubiquitin costaining in spinal cord of NTG and AR121Q mice. Representative images are shown from one independent experiment. (h-j) Quantification of the mean muscle fiber diameter of gastrocnemius/soleus myofibers (h), mean type I and type II hindlimb muscle fiber staining (i), and ChAT-positive motor neuron count and area in the anterior horn of the thoracic spinal cord (j) of NTG and AR121Q mice. Data shown in h-j were evaluated by unpaired t-test, two-tailed. n = 30 muscle fibers per mouse and 2 mice per genotype, ****P ≤ 0.0001 (h), n=2 and 3 mice for NTG and AR121Q (i), n = 3 slides per mouse (neuron count), n = 6 sections per mouse (neuron area), 2 mice per genotype were counted, ***P =0.0005 (j). (k) Immunofluorescence with antibodies against NCAM and PSA-NCAM in skeletal muscle tissues of NTG and AR121Q mice. Representative images are shown from one independent experiment. Scale bars represent 50 μm. All graphs represent mean ± s.e.m.
Finally, we also observed testicular atrophy in the SBMA mice, similar to that described for an AR113Q knock-in mouse model of SBMA (Supplementary Fig. 3j)27. This feature is consistent with the partial androgen insensitivity experienced by patients with SBMA, which manifests as testicular atrophy, gynecomastia, and reduction in secondary sexual characteristics, and can impair QOL. The partial androgen insensitivity experienced by SBMA patients is not reversed and may be aggravated by conventional, non-selective androgen ablation.
AF2 modulation improves neurodegenerative outcomes in a pilot study in SBMA mice
To ascertain the potential efficacy of AF2 modulation in the SBMA mice, we performed a preclinical pilot assessment of the effects of AF2 modulation on SBMA-associated degeneration in a small cohort of mice. Male SBMA mice were injected intraperitoneally (50 mg/kg body weight, three times per week) with TA, MEPB, or vehicle (1% DMSO in corn oil) from 3 to 8 weeks of age. MEPB treatment significantly improved body weight, rotarod activity, and grip strength (Fig. 3a–c), despite no significant change in survival (Fig. 3d), and improved gait and hindlimb clasping (Fig. 3e,f). In addition, the pathologic appearance of skeletal muscle and spinal cord degeneration was qualitatively recovered by MEPB treatment (Fig. 3g,h). TA treatment did not significantly alter any measurements of SBMA-associated degeneration (Fig. 3), suggesting that MEPB provides superior therapeutic potential in SBMA mice. Indeed, pharmacokinetic analysis of TA and MEPB in NTG mice revealed marked penetration and, most importantly, duration of MEPB but not TA in muscle, spinal cord, and testes (Supplementary Fig. 4), indicating sufficient bioavailability of MEPB in SBMA-affected tissues, but less favorable bioavailability of TA in SBMA mice. Moreover, prior studies indicated that MEPB has greater potency than TA with respect to altering coregulator binding by and AR activity 19, 21. Thus, we selected MEPB for a blinded preclinical trial in SBMA mice.
Figure 3. MEPB improves phenotypic outcomes and pathologic degeneration in a pilot preclinical trial in SBMA mice.
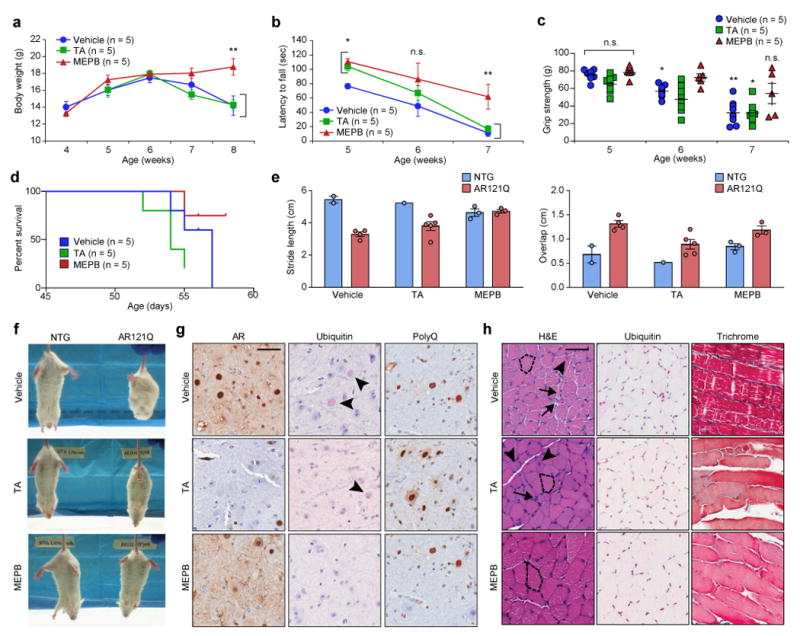
(a) Mean body weight of AR121Q mice from 4 weeks until 8 weeks of age. (b) Mean rotarod performance of AR121Q mice from 5 weeks until 7 weeks of age. (c) Mean grip strength (measured as grams force) of all four paws of AR121Q mice. Data shown in a-c were evaluated by one-way ANOVA with repeated measures followed by Sidak’s posthoc analysis for all pairwise comparisons. **P = 0.007 (a), *P = 0.043 and 0.044 for TA and MEPB at 5 weeks, **P = 0.01 at 7 weeks vs vehicle treated (b), *P = 0.032 at 6 weeks, *P = 0.012, **P = 0.001 at 7 weeks vs 5 weeks (c). (d) Kaplan-Meier survival estimation of AR121Q mice (log-rank test). (e) Quantification of footprint/gait analysis of AR121Q mice. Average stride length and forepaw and hindpaw overlap are depicted. n = 2, 1, and 3 mice for vehicle, TA, and MEPB treated NTG group, n = 4, 5, and 3 mice for vehicle, TA, and MEPB treated AR121Q group. All graphs represent mean ± s.e.m. (f) Video stills of clasping behavior in representative NTG and AR121Q mice from three independent experiments. (g) Representative spinal cord sections from AR121Q mice treated with vehicle, TA, or MEPB from one independent experiment. Sections were stained with AR (N20), ubiquitin, or polyQ (5TF1-1C2) antibodies. Arrowheads indicate the presence of ubiquitin-positive nuclear inclusions. (h) Representative skeletal muscle sections (gastrocnemius/soleus) from AR121Q mice treated with vehicle, TA, or MEPB from one independent experiment. Sections were stained with H&E or Gomori trichrome stain for muscle morphology, in addition to ubiquitin antibody. Dotted lines trace around representative myofibers, demonstrating differences in myofiber size. Arrows indicate atrophied myofibers. Arrowheads indicate myofibers containing centralized nuclei. Vehicle is 1% DMSO in corn oil administered three times per week; TA is 50 mg/kg administered three times per week; and MEPB is 50 mg/kg administered three times per week. Scale bars represent 50 μm.
MEPB demonstrates efficacy in a blinded preclinical trial in SBMA mice
To further evaluate the therapeutic capacity of MEPB, we next performed a blinded, multi-dose preclinical trial of MEPB treatment in male SBMA mice. Using quantitative measures of SBMA-associated degeneration (body weight, rotarod activity, grip strength, and survival) from the previous pilot study, we performed a statistical power analysis to establish cohort numbers required for a full-scale trial (Supplementary Table 1). On the basis of this analysis, we assigned at least 10 male NTG or SBMA mice per treatment group. Mice were injected intra-peritoneally with low-dose (50 mg/kg) MEPB, high-dose (100 mg/kg) MEPB, or vehicle (1% DMSO in corn oil) three times per week starting at 4 weeks of age until completion of the trial when mice were 30 weeks of age. Low-dose and especially high-dose MEPB treatment significantly augmented body weight, rotarod activity, and grip strength (Fig. 4) and also qualitatively improved gait and hindlimb clasping (Supplementary Fig. 5a,b). Consistent with improvement in these behavioral parameters, both low-dose and high-dose MEPB treatment significantly reduced the presence of ubiquitin-positive nuclear inclusions in the spinal cord (Fig. 5a,b), as well as degenerating myofibers in skeletal muscle (Fig. 5c,d). Importantly, both low-dose and high-dose MEPB treatment significantly reduced colocalization of NCAM/PSA-NCAM staining in SBMA mice as compared with vehicle-treated SBMA mice, decreasing these levels similar to those in NTG controls, consistent with amelioration of neurogenic atrophy (Fig. 5e,f). Indeed, high-dose MEPB treatment was found to prevent the loss of ChAT-positive spinal cord motor neurons, with no effect on neuron area (Supplementary Fig. 5c,d). Moreover, both low-dose and high-dose MEPB treatment restored the frequency of type I myofibers to levels observed in NTG control mice, whereas type II myofiber frequency remained unaffected (Supplementary Fig. 5e,f), further demonstrating the efficacy potential of AF2 modulation for attenuation of SBMA-associated muscle degeneration.
Figure 4. MEPB improves phenotypic outcomes and improves quality of life parameters in a preclinical trial in SBMA mice.
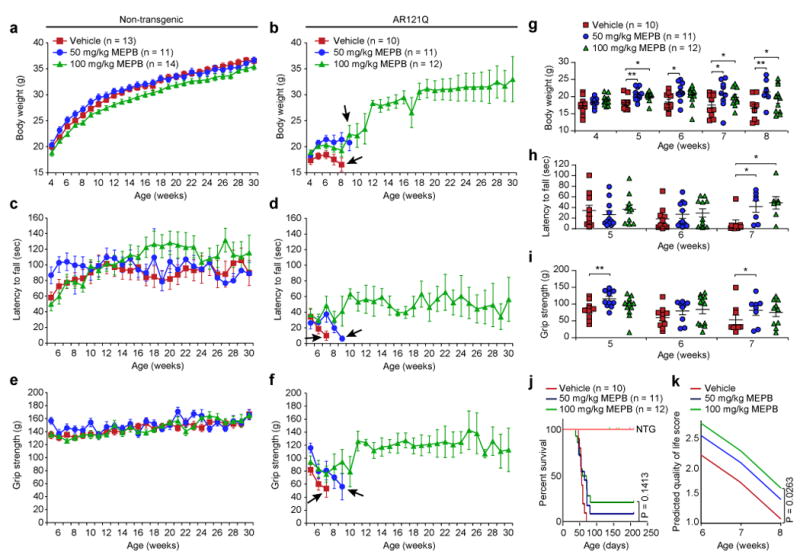
Graphs for each phenotypic assay (mean body weight in (a) and (b), mean rotarod activity in (c) and (d), mean grip strength in (e) and (f) are depicted using identical scales to allow comparison between NTG mice (a), (c), (e) and AR121Q mice (b), (d), (f). Mice were treated with vehicle (red squares; 1% DMSO in corn oil), low-dose MEPB (blue circles; 50 mg/kg, three times per week), or high-dose MEPB (green triangles; 100 mg/kg, three times per week). Arrows indicate last graphed data point for each treatment group due to attrition of animal numbers (< 3 mice/group) from loss of > 10% body weight or limb paralysis/paresis. Data shown in a, c, and e were evaluated by one-way ANOVA with repeated measures followed by Tukey posthoc analysis for all pairwise comparisons. (g to i) Statistical analysis of b, d, and f up to 7 or 8 weeks of age by one-way ANOVA with repeated measures followed by Sidak’s posthoc analysis for all pairwise comparisons. Mean body weight (g), mean rotarod activity (h), and mean grip strength (i) in vehicle (n=10) -, low-dose MEPB (n=11) -, and high-dose MEPB (n=12) -treated AR121Q mice for time points were assayed. (g) 5 weeks: *P = 0.017, **P = 0.005, 6 weeks: *P = 0.028, 7 weeks: *P = 0.042 (50 mg/kg MEPB), *P = 0.049 (100 mg/kg MEPB), 8 weeks: *P = 0.046, **P = 0.008. (h) 7 weeks: *P = 0.0462 (50 mg/kg MEPB), *P = 0.017 (100 mg/kg MEPB). (i) 5 weeks: **P = 0.007, 7 weeks: *P = 0.045. (j) Kaplan-Meier survival estimation in treated (vehicle, low-dose, and high-dose) NTG and AR121Q mice (log-rank test). 95% confidence intervals were used for the analysis. (k) QOL score of AR121Q mice from each treatment group from 6 to 8 weeks of age. In all panels, mice were treated with vehicle (red; 1% DMSO in corn oil), low-dose MEPB (blue; 50 mg/kg, three times per week), or high-dose MEPB (green; 100 mg/kg, three times per week). A mixed-effect model was applied with SAS software to determine statistical significance. 95% confidence intervals were used for the analysis. All graphs represent mean ± s.e.m.
Figure 5. MEPB reduces degeneration in spinal cord and skeletal muscle of SBMA mice.
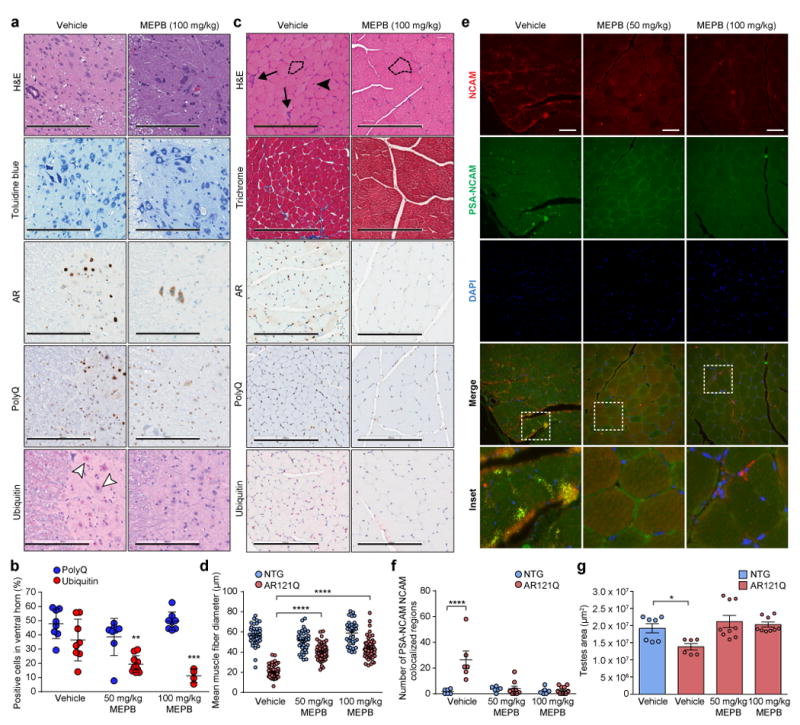
(a) Representative spinal cord sections from AR121Q mice treated with vehicle (1% DMSO in corn oil) or high-dose MEPB (100 mg/kg, three times per week) from one independent experiment. Sections were stained with H&E and toluidine blue to assess spinal cord morphology. Sections were also stained with AR (N20), polyQ (5TF1-1C2), and ubiquitin antibodies. White arrowheads indicate ubiquitin-positive nuclear inclusions. Scale bars represent 200 μm. (b) Quantification of the mean relative number of cells in the spinal cord ventral horn containing positive polyQ (blue) or ubiquitin (red) staining in AR121Q mice. Quantification was performed from total 8 fields for polyQ staining, 8 (vehicle), 10 (50 mg/kg MEPB), and 4 (100 mg/kg MEPB) fields for ubiquitin staining, 2-4 mice per treatment group, **P =0.0022, ***P =0.0005 by two-way ANOVA followed by Dunnett’s posthoc analysis for pairwise comparisons between each MEPB treatment group and vehicle. (c) Representative skeletal muscle (gastrocnemius/soleus) sections from AR121Q mice treated with either vehicle or high-dose MEPB from one independent experiment. Sections were stained with H&E and Gomori trichrome to evaluate morphology, in addition to AR, polyQ, and ubiquitin antibodies. Dotted lines trace around representative myofibers, demonstrating differences in myofiber size. Arrows indicate atrophied myofibers. Arrowhead indicates myofibers containing centralized nuclei. Scale bars represent 200 μm. (d) Quantification of the mean muscle fiber diameter of gastrocnemius/soleus myofibers of NTG (gray) and AR121Q mice (black). Quantification was performed from 20 muscle fibers per mouse, 2 (NTG) and 3 (AR121Q) mice per treatment group, ****P ≤ 0.0001 by two-way ANOVA followed by Dunnett’s posthoc analysis for pairwise comparisons between each MEPB treatment group and vehicle. Quantification was performed from 2–4 fields/mouse, 3 mice/treatment group. (e) Representative images of skeletal muscle stained with antibodies against NCAM and PSA-NCAM in AR121Q mice treated with vehicle (1% DMSO in corn oil), low-dose MEPB (50 mg/kg, three times per week), or high-dose MEPB (100 mg/kg, three times per week). Scale bars represent 50 μm. (f) Quantification of PSA-NCAM/NCAM colocalized regions. n = 6 fields for all NTG groups, 6, 9, and 10 fields for vehicle, 50 mg/kg, and 100 mg/kg MEPB treated AR121Q group, 2 mice for all NTG and vehicle treated AR121Q groups, 3 mice for 50 mg/kg and 100 mg/kg MEPB treated AR121Q groups, ****P ≤ 0.0001 by two-way ANOVA followed by Dunnett’s posthoc analysis for pairwise comparisons between each MEPB treatment group and vehicle. (g) The area of testis were measured in n = 7, 6, 9, and 9 sections from n=2, 2, 3, and 3 mice for vehicle treated NTG, vehicle, 50 mg/kg, and 100 mg/kg MEPB treated AR121Q group, respectively. Data were evaluated by one-way ANOVA followed by Dunnett’s posthoc analysis for comparison between each AR121Q treatment group and NTG vehicle. *P =0.0342. All graphs represent mean ± s.e.m.
Finally, testicular atrophy in the SBMA mice was reversed with both low-dose and high-dose MEPB treatment (Fig. 5g), which is not only consistent with overall improvement of the SBMA phenotype, but also underscores the selective nature of AR modulation by MEPB. Consistent with this tissue effect, the testes had the highest concentrations of MEPB in our mouse pharmacokinetic assessments, averaging 11.5 μM over the 48 hour dose interval.
Neither low-dose nor high-dose MEPB treatment significantly altered survival of SBMA mice, although a dose-dependent trend in increased survival was present (Fig. 4j). It should be noted that measurement of mouse “survival” in this trial were confounded by protocol guidelines designed to minimize morbidity in experimental animals. Mice were checked daily by our veterinary staff and recommendations to euthanize individual animals were made based on loss of 10% body weight or subjective signs of hindlimb weakness that might be severe enough to limit an animal’s ability to reach the feeder. However, when other phenotypic measurements (viz., body weight, rotarod activity, and grip strength) were compiled along with survival data to generate a QOL score, high-dose MEPB treatment significantly improved the QOL of SBMA mice (Fig. 4k). Furthermore, MEPB treatment (low-dose or high-dose) did not significantly change blood chemistry of either NTG or SBMA mice (Supplementary Fig. 5g-i), and MEPB treatment (low-dose or high-dose) had no measurable effect on any assays of neuromuscular function in NTG control mice, suggesting minimal or no MEPB-induced toxicity (Fig. 4 and Supplementary Fig. 5b). Together, these findings demonstrate that AF2 modulation by MEPB in SBMA mice improves multiple primary outcomes associated with reduced QOL in patients with SBMA, such as attenuated muscle strength, diminished coordination, and loss of body mass, without apparent adverse effects.
AF2 modulators do not affect normal AR signaling function, but promote corepressor binding
To assess the mechanism by which AF2 modulation attenuates SBMA-associated phenotypes, we performed analyses of polyQ-expanded AR activity in response to TA and MEPB (Fig. 6 and Supplementary Fig. 6). Neither TA nor MEPB treatment reduced steady state levels of polyQ-expanded AR protein in stably transfected motor neuron (MN1) cells or transiently transfected HEK293T cells (Supplementary Fig. 6a–e), indicating that reduced SBMA-associated toxicity as a result of TA and MEPB treatment is not simply due to enhanced degradation of the AR. In addition, the presence of high-molecular weight, multimeric AR complexes and aggregates were unchanged in response to TA and MEPB treatment in HEK293T cells (Supplementary Fig. 6e,f). Consistent with these observations, MEPB treatment did not statistically significantly change the expression level of endogenous mouse AR or transgenic human AR in the muscle and spinal cords of AR121Q mice (Supplementary Fig. 7). Similarly, neither MEPB nor TA treatment changed the expression of transgenic AR in Drosophila (Supplementary Fig. 2d,e), suggesting that TA- and MEPB-mediated attenuation of toxicity occurs independently of AR aggregation. Moreover, neither MEPB nor TA treatment altered DHT-dependent nuclear translocation of polyQ-expanded AR in HEK293T cells and Drosophila (Fig. 6a,b). A dual luciferase reporter assay demonstrated that the transactivational capacity of polyQ-expanded AR was not significantly altered by MEPB treatment, although reduced transcriptional activity was present in cells treated with high concentrations (100 μM) of either TA or Bic (Fig. 6c). Finally, we selected eight AR-responsive genes expressed in motor neurons (Igfbp5, Mt2, Sgk1, Trib1, Camkk2, Tsc22d3, Plk3r3, and AR) and assessed the impact of MEPB on response to ligand-dependent changes in transcription in MN1 cells that stably express human AR (MN1-AR24Q cells). We confirmed statistically significant ligand-dependent changes in transcription for seven of the eight target genes but found no impact for MEPB (Fig. 6d). These results indicate that although the AR signaling pathway remains intact, AF2 modulation by SARMs selectively alters AR activity, reducing the toxicity associated with polyQ-expanded AR but leaving other aspects of AR responsiveness intact. Indeed, the reversal of testicular atrophy in the SBMA mice upon treatment with MEPB is consistent with this conclusion.
Figure 6. AF2 modulation does not inhibit AR functional activity.
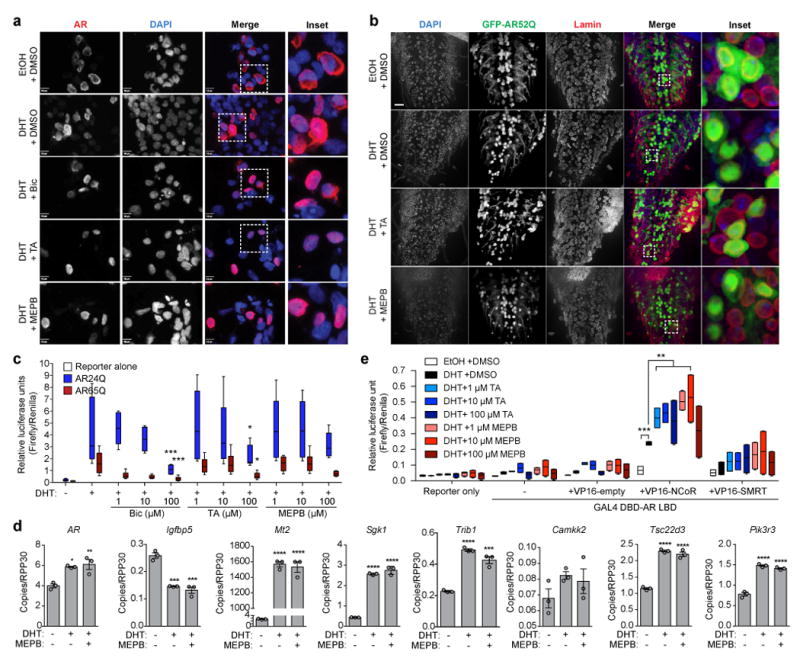
(a) Representative immunofluorescence of HEK293T cells transiently transfected with AR65Q for 48 h in culture media devoid of steroid hormones from three independent experiments. Cells were treated for 24 h with vehicle, 10 nM DHT, or 10 nM DHT + 10 μM bicalutamide (Bic), TA, or MEPB prior to staining with AR (D6F11) and DAPI. Scale bars represent 10 μm. (b) Third instar larvae expressing GFP-AR52Q in motor neurons were dissected and stained for nuclear membrane (lamin, red) and DAPI (blue). Scale bar, 10 μm. Representative images are shown from one independent experiment. (c) AR transcriptional activity reporter assay. HEK293T cells were transiently transfected with AR24Q (blue) or AR65Q (red) in addition to an ARE-luciferase reporter prior to treatment with Bic, TA, or MEPB. Four independent biological replicates were performed on different days with three sample replicates for each treatment group. Data were evaluated by two-way ANOVA followed by Dunnett’s posthoc analysis for pairwise comparisons between each treatment group and the DHT only group. ***P = 0.001 for 100 μM Bic AR24Q and 100 μM Bic AR65Q, *P = 0.017 for 100 μM TA AR24Q and 100 μM TA AR65Q. (d) Digital PCR assay to access the impact of MEPB on AR target gene expression. Data were evaluated by ordinary one-way ANOVA followed by Tukey’s posthoc analysis for all pairwise comparisons. *P = 0.0120 vs. -DHT / -MEPB, **P = 0.0068 for AR vs. -DHT / -MEPB, ***P = 0.0003 (+DHT) vs. -DHT / -MEPB and ***P = 0.0001 (+DHT / +MEPB) vs. -DHT / - MEPB for Igfbp5, ***P = 0.0001 vs. -DHT / -MEPB for Trib1, ****P ≤ 0.0001 vs. -DHT / -MEPB. (e) Mammalian two-hybrid assay to assess binding between the AR LBD and the corepressors NCoR or SMRT in the presence of TA (blue) or MEPB (red). Two independent biological replicates were performed on different days with three sample replicates for each treatment group. Data were evaluated by two-way ANOVA followed by Dunnett’s posthoc analysis for pairwise comparisons between each treatment group and the DHT + DMSO group. ***P = 0.001 for EtOH, 10 μM TA, 1 μM MEPB and 10 μM MEPB, **P = 0.002 for 1 μM TA, ** P = 0.007 for 100 μM TA. All graphs represent mean ± s.e.m.
Because the AR has been well characterized to repress as well as activate transcription of its target genes and these repressor/activator activities are dependent on binding of a specific complement of coactivators and corepressors to the AF2 domain10, we hypothesized that SARM treatment may specifically promote recruitment of corepressors to the AF2 domain rather than block coactivator binding. This would potentially lead to repression of selected target genes driving SBMA-associated toxicity, while allowing transcriptional activation of other target genes. To determine whether association between the AR LBD and steroid receptor corepressors is augmented in response to AF2 modulation by TA or MEPB, we performed a mammalian two-hybrid assay, whereby increased association between the AR LBD and a given coregulator drives luciferase reporter activity. Consistent with our hypothesis, binding between the AR LBD and nuclear receptor corepressor-1 (NCoR) was specifically increased by TA and MEPB treatment, whereas binding of silencing mediator for retinoic acid and thyroid hormone receptors (SMRT) was unchanged (Fig. 6e), suggesting that modulation of the AF2 domain by TA and MEPB occurs by a selective and precisely controlled mechanism.
Discussion
Recent advances in prostate cancer research have led to the discovery of many novel SARM compounds that specifically bind to and modulate coregulator binding to the AR AF2 domain17-22. Interestingly, a reciprocal relationship between prostate cancer and SBMA has been suggested to be mediated by polyQ length, as prostate cancer risk appears to be elevated in individuals whose AR gene contains fewer than ten CAG repeats28. Increased transactivational activity of the AR is also inversely correlated with CAG repeat length29,30, and polyQ expansion has been suggested to augment intrinsic disorder within the N-terminal domain, reducing coactivator binding to the AR LBD through an unknown mechanism31,32. To avoid the proliferative effects of increased AR transactivation, compounds that reduce coactivator binding or recruit corepressor binding to the AF2 domain have been sought for prostate cancer therapy. The seminal discovery of the BF3 regulatory pocket as an allosteric regulator of the AF2 domain by Estebanez-Perpina et al. (2007) was an unexpected revelation in the search for such drugs19. The development of several compounds that specifically target the BF3 pocket have since been reported20-22.
A recent report by Jehle et al. (2014) demonstrated the importance of the BF3 domain in prostate cancer and the role that BF3 modulation may play in therapeutic treatment33. Specifically, ectopic expression of the co-chaperone Bag-1L in the prostate secretory epithelium may be associated with tumorigenesis by stimulating AR activation via interaction between a duplicated GARRPR hexapeptide motif within Bag-1L and the AR BF3 binding pocket. Interestingly, the binding of LXXLL-bearing coactivators to the AF2 domain was inhibited by Bag-1L binding to the BF3 pocket, further demonstrating the allosteric regulation that BF3 imparts upon the AF2 domain. Moreover, BF3 Bag-1L binding was inhibited by the MEPB analog 2-((2-(2,6-dimethylphenoxy)ethyl)thio)-1H-benzimidazole (compound 49 or CPD49)20,33, suggesting that modulation of the interaction between the AR and Bag-1L by CPD49 may provide a promising novel approach to mitigate the oncogenic program initiated by androgen signaling in prostate cancer.
We previously showed that coactivator binding to the AF2 domain is required for polyQ-expanded AR toxicity in Drosophila8. Despite the apparent reciprocal relationship between prostate cancer and SBMA, we reasoned that targeting the BH3 interaction surface to modulate coregulator binding to the AF2 domain may represent a common strategy for treatment of both SBMA and prostate cancer. Therefore, we initiated a Drosophila-based screen to determine whether any previously described BF3-binding compounds may be advantageous for the treatment of SBMA. As expected, several compounds either had no effect on, or even exacerbated, SBMA toxicity (data not shown), suggesting that some compounds may be more effective in the treatment of prostate cancer whereas others may be more effective for SBMA. Nevertheless, two promising compounds (TA and MEPB) emerged that ameliorated polyQ-expanded AR toxicity in flies and became candidate therapeutics for a preclinical trial in a mammalian model of SBMA.
TA is a well-known and well-studied compound belonging to the nonsteroidal anti-inflammatory drug family of small molecules34-37. Although it has been approved for treatment of migraines in the United Kingdom by the National Health Service and is available as a general analgesic for humans and animals in several countries in Europe, Latin America, and Asia, it has not been approved for any use in the United States. This compound was found to have an unfavorable murine pharmacokinetic profile, i.e. low bioavailability in mouse brain, spinal cord, and muscle, and no significant effect in a pilot trial in AR121Q mice. Thus, TA was not subsequently pursued in this detailed proof of concept study.
MEPB was first described by Lack et al. (2011) through virtual screening for BF3-binding compounds21. X-ray crystallography revealed MEPB to specifically bind the BF3 pocket of the AR, where the benzimidazole moiety of MEPB is oriented toward the interior of the BF3 pocket and is stabilized by strong hydrophobic interactions with Pro723, Phe673, and Tyr834 and an arene–arene conjugation between a benzene ring of MEPB and Phe826. MEPB was shown unequivocally to bind to the BF3 domain and as a consequence modulate AF2 binding to coregulators. MEPB was found to comparatively favorable bioavailability in mouse brain, spinal cord and muscle and, after showing a beneficial effect in a pilot trial in SBMA mice, was selected for detailed evaluation. In a blinded, multi-dose preclinical trial in male SBMA mice, MEPB treatment was found to significantly augment body weight, reduce hindlimb clasping, and improve rotarod activity, grip strength, gait, and QOL score. Whereas MEPB did not significantly extend AR121Q survival, this aspect of the pre-clinical trial was confounded by protocol guidelines that were designed to minimize morbidity in experimental animals. There were no spontaneous deaths; rather all mouse deaths represented euthanasia due to loss of 10 % body weight or the development of substantial weakness. Nevertheless, we do not believe that lifespan extension should be used as the primary criterion for judging the efficacy of a pre-clinical therapeutic for SBMA. Men with SBMA do not exhibit a shortened lifespan. Rather, they experience substantial progressive morbidity due to weakness that impairs bulbar functions and ambulation, and feminization (testicular atrophy and gynecomastia). In our mouse pre-clinical trial, MEPB treatment resulted in significant improvement in phenotypic measurements (viz., body weight, rotarod activity and grip strength). These data, compiled with survival data, generate a QOL score that was significantly improved in MEPB-treated SBMA mice. Consistent with improvement in the behavioral phenotype, MEPB treatment rescued motor neuron loss and neurogenic atrophy. Finally, MEPB treatment was found to reverse testicular atrophy in the SBMA mice, a finding that underscores the selective activity of MEPB. Prior in vitro analyses found that MEPB binding to BF3 enhances AF2 interaction with coregulators bearing an extended LXXLL motif (termed the “corepressor nuclear receptor box”), such as that found in the corepressor NCoR38,39. This observation is consistent with our observation that NCoR was recruited to the AR LBD by MEPB, and suggests that MEPB may relieve polyQ-expanded AR-mediated toxicity by promoting the binding of corepressors to the AF2 domain.
Altogether, these results provide considerable evidence for the utility of AR AF2 domain modulation by BF3-binding compounds as a novel paradigm for SBMA therapy. Subtle modulation of coregulator binding, and thus AR functional activity, rather than ablation of the entire androgen signaling pathway, may provide therapeutic relief of neurodegenerative symptoms in patients with SBMA. Such a targeted approach may reduce or potentially even reverse adverse effects of androgen insensitivity and improve the QOL of patients with SBMA.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Online Methods
Study design
The objective of this study was to determine whether modulation of the AR AF2 domain by small molecules that specifically bind to the BF3 regulatory pocket would be a practicable therapy for improving the QOL of patients with SBMA. To this end, we screened several BF3-specific compounds in Drosophila; performed a pilot preclinical study of the top two candidate compounds; performed an extensive, multidose preclinical study of the lead compound; and determined the mechanism of action of these compounds in cell culture studies. Drosophila NMJ staining and analysis was performed in a blinded manner, whereby treated larvae were coded by an independent investigator. Treatment groups were not uncoded until analyses were completed. Both preclinical trials were performed in a blinded manner, whereby drugs (i.e., vehicle, TA, and/or MEPB) were assigned a specific code by an independent investigator. Treatment groups remained blinded until data collection and analysis were complete. Drug dose and animal numbers were determined empirically for the pilot preclinical study. The data acquired from this study were used in a power analysis to estimate the number of animals required for the preclinical study. Animal endpoints were reached when a mouse exhibited hindlimb paralysis/paresis or greater than 10% loss of bodyweight, at which point the animal was humanely euthanized. Because the yield of AR121Q-positive male mice was generally low throughout our studies, randomization was not possible. Mice were entered into treatment groups as they became available and were followed longitudinally. Replicate number and statistical tests for each dataset are provided in the figure legends.
Reagents
For fly and cell culture experiments, TA (Sigma Aldrich), MEPB (Specs), bicalutamide (3B Scientific Corporation), ibuprofen (Sigma Aldrich), or dimethylcurcumin (Cayman Chemical) were resuspended in DMSO (Sigma Aldrich), whereas DHT (Steraloids) was dissolved in ethanol, prior to addition to fly food or culture media. All cell culture experiments were performed in media containing 10% charcoal–dextran stripped serum (Hyclone) to remove exogenous steroid hormones. For preclinical and pharmacokinetic mouse studies, TA and MEPB were dissolved in either a 10 mg/mL (low dose) or 20 mg/mL (high dose) solution of corn oil containing 1% DMSO at 37°C for approximately 12 h, filtered (0.22 μm filter), and stored at 4°C until injection.
Molecular modeling
BF3-unbound AR LBD (pdb1T7R) in complex with an FXXLF motif was modeled using PyMOL software. BF3 residues (I690, F691, P723, G724, N727, F826, E829, N833, E837, and R840) were highlighted green, charge clamp residues of the AF2 domain (K720, M734, M894, and E897) were highlighted red, and a synthetic FXXLF (FESLF) motif was highlighted blue. MEPB modeling was performed in the same manner using pdb2YLO. TA modeling was performed in a similar manner using pdb2PIX, whereby flufenamic acid was removed and TA was added according the lowest predicted energy conformation using SwissDock software.
Fly stocks and phenotypic characterization
UAS-AR52Q, UAS-AR52Q-K720A, and UAS-AR66Q-E897K fly stocks were generated as previously described8. Fly stocks were crossed to various GAL4 driver lines (ELAV-GAL4 for pan-neuronal, OK371-GAL4 for motor neuron, GMR-GAL4 for eye) to induce expression of the AR transgene in a tissue-specific manner in fly vials containing fly food with either vehicle (1% ethanol +/- 0.1% DMSO) or drugs. Upon the presence of progeny, parental flies were removed, and F1 flies were scored for phenotypic analysis. Viability was determined as previously described40. Briefly, the population frequency of adult F1 SBMA flies (ELAV>UAS-AR52Q or OK371>UAS-AR52Q) and control flies (GAL4-ELAV; CyO-GFP) were determined by the presence or absence of the CyO phenotypic marker. At least three independent biological replicates (F0 crosses) were performed with new drug/food preparations for each treatment group, and a total of at least 50 flies were scored for each treatment group. To determine locomotor activity of SBMA flies, adult F1 flies (OK371>UAS-AR52Q) reared on food containing vehicle or drug were allowed to walk for 90 seconds in one well of a 12-well tissue culture plate while video was recorded using a Leica M205C stereomicroscope and Leica DFC320 digital camera. Videos were recorded for at least 15 flies per treatment group. Tracing of fly movement and analysis of displacement and velocity were performed using ImageJ software. Preparation and staining of larval neuromuscular junctions and ventral ganglions were performed as previously described8.
Generation of AR121Q transgenic mice and surgical castration
Human AR cDNA containing 121 CAG/CAA alternating repeats was subcloned into the pCAGGS vector. An 11.5 kb fragment containing the pCAGGS promoter and AR cDNA was released by ApaL1 digestion, purified by QIAEXII (Qiagen), and injected into FVB pronuclei, which were transferred into female FVB recipients. Founders were screened by DNA genotyping of tail biopsies. Fluorescence in situ hybridization analysis was performed on lung tissue to confirm transgene insertion. Out of 15 founders, one line was viable, demonstrated germline transmission, and expressed levels of hAR protein comparable with that of endogenous mouse AR. Therefore, the phenotype of this line was extensively characterized. Mice were maintained on a purebred FVB background for all studies.
Castration was performed according to USDA guidelines for aseptic technique in animal survival surgery and oversight was provided by veterinary staff at St. Jude Children’s Research Hospital. Briefly, mice were anesthetized by isoflurane exposure, and the surgical site was prepared by shaving and disinfection. A 10-mm incision was made along the scrotal midline, and the testes were removed using forceps. The connection between the testes and vas deferens, as well the associated blood vessels, were cauterized by hot forceps. Incisions were closed using tissue glue, and preemptive analgesia (0.2 mg/kg meloxicam) was administered. Mice were bred and maintained in accordance with the guidelines set forth by the National Institutes of Health Guide for the Care and Use of Laboratory Animals, published by the U.S. Public Health Service. All experimental protocols were approved by the Institutional Animal Care and Use Committee at St. Jude Children’s Research Hospital.
Preclinical trial design in AR121Q mice
To determine the efficacy of TA and/or MEPB in a mouse model of SBMA, we first performed a pilot study. Five mice were assigned to each drug treatment group: 50 mg/kg TA, 50 mg/kg MEPB, or vehicle (1% DMSO in corn oil). Drug identities were coded to ensure investigator blinding, and drugs were administered three times per week (Monday, Wednesday, and Friday) by intraperitoneal injection from 3 to 8 weeks of age. At 8 weeks, all mice were sacrificed and tissues were collected for pathologic analysis. To fully characterize the efficacy of MEPB in SBMA mice, a multi-dose preclinical trial was performed. A power analysis (Supplementary Table 1) was performed based on the pilot study data to determine the adequate number of mice required for 80% power. At least 10 mice were assigned to each drug treatment group: 50 mg/kg MEPB, 100 mg/kg MEPB, and vehicle (1% DMSO in corn oil). Drug identities were coded to ensure investigator blinding, and drugs were administered three times per week (Monday, Wednesday, and Friday) by intraperitoneal injections from 4 weeks until 30 weeks of age.
AR121Q mouse phenotypic characterization
Body weight, rotarod activity, and grip strength data were collected weekly. Footprint analysis and clasping phenotype were assessed at 7 and 8 weeks of age. Body weight was measured using a standard laboratory scale (Fisher Scientific). Rotarod activity analysis was performed on an accelerating rotarod apparatus (IITC Life Science) using a two-day, weekly protocol. Mice were trained on the first day with one session set at 4 rpm for 5 min. The following day, mice were placed on the apparatus, rotation speed was set to accelerate from 4–40 rpm at a rate of 0.1 rpm/s, and the latency to fall was recorded for four separate trials per mouse. Mice were given a 15-min rest period between each trial. Grip strength was measured using a grip strength meter (Bioseb). Grip strength was measured as grams of force in six repeated measurements for forepaws and hindpaws of each animal. To perform gait/footprint analysis, the forepaws and hindpaws of each animal were dipped in red and blue, respectively, water-soluble, non-toxic paint. The animal was then placed in a 70-cm long tunnel lined on the bottom with Whatman filter paper, the entrance was sealed, and the animal was allowed to walk through one time. Footprints were scanned and analyzed with Image J for stride length and forepaw/hindpaw overlap41. To determine clasping phenotype, mice were recorded using an Apple iPod camera (iOS version 6.1.6) for approximately 60 seconds and still frames were extracted using ImageJ.
AR121Q mouse pathologic assessment
A separate cohort of five mice per treatment group was generated for pathologic and biochemical analysis. Mice were anesthetized by isoflurane inhalation and transcardially perfused with either PBS for frozen tissue samples or 10% formalin for fixed tissue samples. Selected tissues (brain, spinal cord, gastrocnemius/soleus muscle, testes, and liver) were dissected and either snap-frozen in liquid nitrogen or processed for paraffin embedding/sectioning. Tissue sections were stained with hematoxylin and eosin, toluidine blue, or Gomori trichrome to ascertain overall morphology and pathologic changes. To determine AR staining and colocalization patterns, tissue sections were stained with anti-AR (EPR1535, Abcam), anti-polyQ (5TF1-1C2, EMD Millipore), or anti-ubiquitin (Dako) primary antibodies. To quantify type I and type II myofibers, hindlimb tissue sections were stained with myosin heavy chain–slow twitch and myosin heavy chain–fast twitch antibodies (Leica Biosystems), respectively. For the purposes of counting ChAT-positive neurons, three stepped sections separated by 50 μm were analyzed for the cervical, thoracic, and lumbosacral areas of spinal cords. The slides contained three to six tissue sections at each level and were labeled with an anti-ChAT antibody (EMD Millipore). Following primary antibody staining, slides were incubated with rabbit anti-IgG1 + IgG2a + IgG3 secondary antibody (M204-3, Abcam) if needed, and staining visualized with the OmniMap rabbit detection system (Roche). All slides were digitally scanned at scalable magnifications up to 20× (objective lens) using an Aperio XT Slide Scanner (Leica Biosystems). Whole slide images were imported into the Halo software program (Indica Labs) and positive staining was quantified by an area quantification algorithm. For spinal cord motor neuron quantification, static images were made at a 2× magnification with ImageScope (Leica Biosystems) and the number of ChAT-positive neurons located within the anterior horn were counted with FIJI software42.
Pharmacokinetics
MEPB and TA concentrations in plasma, liver, muscle, testes, brain, and spinal cord of male FVB/NJ mice were measured at multiple time points (5 min to 48 h) after a single intraperitoneal injection of either 100 mg/kg body weight MEPB or 50 mg/kg body weight TA. A 25 μL aliquot of MEPB spiked with 100 ng/mL internal standard SLV320 (Tocris Biosciences) were extracted from plasma and tissue homogenate samples with 600 μL methyl tert-butyl ether. After 5 min of vortexing, samples were centrifuged at 10,000 rpm for 5 min, the organic layer was then transferred to a glass vial and dried in a CentriVap Console (Labconco) at 35°C for 25 min. The dried extracts were then reconstituted with 400 μL methanol, and an aliquot of 3 μL was injected onto the chromatographic system. For TA, a 25 μL aliquot of sample was spiked with 1000 ng/mL internal standard diclofenac (Alfa Aesar), and extracted from plasma and tissue homogenate samples with 100 μL acetonitrile. After vortexing for 5 min, samples underwent centrifugation at 10,000 rpm for 5 min, and an aliquot of 5 μL supernatant was injected into the chromatographic system. All chemicals were HPLC grade or higher and were obtained from Fisher Scientific (Fair Lawn) unless otherwise specified. Quantitation of MEPB and TA was performed using an API 4000 mass spectrometer (SCIEX) equipped with a Prominence Ultra-Fast Liquid Chromatograph (UFLCXR) system (Shimadzu). Chromatographic separation was achieved with a Phenomenex Luna C18 column (3 μm, 100 A 50 × 2.00 mm) by using mobile phases consisting of 0.1% formic acid in water (A) and acetonitrile (B). Mass spectrometric analysis was performed with the turbo ion spray in positive ionization mode. All MS data were acquired using Analyst 1.5.2 software and processed using MultiQuant 2.1.1 software (SCIEX).
For all matrices (plasma, liver, muscle, testes, brain, and spinal cord), the lower limit of quantification (LLOQ) of MEPB was 6.18 nM with a calibration range of 6.18 ng/mL to 247 nM. However, due to dilution during homogenization, the effective LLOQ was 37.1 nM for solid tissues. The LLOQ of TA was 19.1 nM for plasma and 229 nM for tissues. Assays were found to be linear and reproducible with a correlation coefficient (R) > 0.99. The MEPB and TA concentration-time (Ct) data for plasma, liver, muscle, testes, brain, and spinal cord were grouped by mouse and analyzed using a two-stage, semi-physiologic, nonlinear mixed effects approach with maximum likelihood expectation maximization in ADAPT 543. For data below the LLOQ, Beal’s M3 method was applied44. First, compartmental models were fit to plasma data – one- and two-compartment models were fit to MEPB and TA plasma Ct data respectively, with first order absorption. Population mean PK parameters with inter-mouse variability and post-hoc individual mouse parameters were estimated. The resulting individual mouse plasma parameters were inputted into the semi-physiological tissue PK model. Briefly, the tissue Ct data were well described with a rapid equilibrium, perfusion-limited model, using specific mouse tissue blood flows fixed to literature values45-49. The population mean and inter-mouse variability of the tissue to plasma partition coefficients (Kp,tissue) were estimated, along with proportional residual error.
Monte Carlo simulations (n=1000 subjects) using the population mean PK parameters and variances for MEPB and TA were conducted using ADAPT 5, with a 100-step 48-hour Ct profile simulated for each compound, tissue, and theoretical subject. Each profile was subjected to noncompartmental analysis (NCA) using standard formulae50 with the package ‘PKNCA’51 in R52. Estimated parameters from the simulated data included maximum concentration (Cmax), time of Cmax (Tmax), and the area under the Ct curve from 0 to the last time point of 48 hours (AUClast). The terminal phase was defined as at least three time points at the end of the Ct profile and an elimination rate constant (Kel) was also estimated using an unweighted log-linear regression of the terminal phase. The terminal elimination half-life (T1/2) was estimated as ln(2)/Kel, and the AUC from time 0 to infinity (AUCinf) was estimated as the AUC to the last time point (AUClast) + predicted last concentration/Kel. Each NCA parameter was summarized using the median and 90% prediction interval, and tabulated for presentation.
Immunoblotting and immunofluorescence
Lysates from mouse tissues were prepared by grinding snap-frozen tissues on dry ice with a pestle until pulverized into a fine powder, resuspending in ice-cold RIPA (1% Triton-X, 0.1% sodium deoxycholate, and 0.1% SDS in Tris/NaCl) buffer, briefly sonicating, and centrifuging at 14,000 g for 20 min. Drosophila lysates were prepared as previously described8. Cell lysates were prepared by scraping transfected (mycoplasma-free) HEK293T cells (ATCC) or MN1 (first described by Brooks et al. (1997)53, gift from Kurt Fischbeck) cells into room temperature PBS, centrifuging at 400 g for 5 min, and resuspending pellets in ice-cold RIPA buffer, briefly sonicating, and centrifuging at 14,000 g for 20 min. Supernatants were then collected, and protein levels were measured and adjusted using Bradford analysis. Proteins were separated on 4% to 20% or 8% Tris-glycine gels and transferred overnight at 4°C onto PVDF membranes. Membranes were stained with REVERT total protein stain (Li-Cor) and immunoblotted using anti-AR (H280 or N20 [Santa Cruz], D6F11 [Cell Signaling Technology], or EPR1535(2) [Abcam]), anti-tubulin (Sigma Aldrich), anti-GAPDH (Cell Signaling Technology), or anti-β-actin (Santa Cruz) primary antibodies, followed by anti-mouse or anti-rabbit HRP-linked secondary antibodies (Cell Signaling Technology) and ECL detection (ThermoFisher Scientific).
Filter trap assay for aggregation was performed by resuspending cell lysates in a 10% SDS buffer to achieve a final 2% SDS concentration, heating for 5 min at 95°C, and applying to a 0.22 μm cellulose acetate membrane (GE Healthcare Life Sciences) with a vacuum dot blot apparatus (Schleicher and Schuell). Membranes were washed three times with 0.1% SDS and blotted with anti-FLAG (M2, Sigma Aldrich), followed by anti-mouse HRP-lined secondary antibody (Cell Signaling Technology) and ECL detection (ThermoFisher Scientific). For immunofluorescence experiments in Drosophila, UAS-AR flies were crossed to OK371-GAL4 flies at 25°C on food containing either 1 mM DHT (Steraloids) or 1% ethanol together with indicated drug. Third instar larvae were heat killed, dissected in PBS, and fixed with 4% PFA for 20 minutes. Primary antibody staining (anti-AR D6F11, Cell Signaling Technolofy) was performed at 4°C overnight followed by staining waswith the appropriate Alexa Fluor-conjugated secondary antibody (ThermoFisher Scientific) at room temperature for 4 hours. After staining, pelts were mounted in Fluoromount-G (SouthernBiotech). For immunofluorescence experiments in HEK293T cells (ATCC) or MN1 cells, HEK293T cells were transiently transfected with AR65Q for 48 h in culture media devoid of steroid hormones and treated for 24 h with vehicle, 10 nM DHT, or 10 nM DHT + 10 μM bicalutamide (Bic), TA, or MEPB before being fixed in 4 % formaldehyde in PBS for 10 min at room temperature. Thereafter, cells were permeabilized with 0.5 % Triton-X 100 and incubated with an anti-AR antibody (D6F11, Cell Signaling Technology) for 1 h, followed by incubation for 2 h at room temperature with the appropriate Alexa Fluor-conjugated secondary antibody (ThermoFisher Scientific). Cells were then washed, stained with DAPI, and visualized on an inverted widefield fluorescence microscope (Leica DMI3000b) using a 63X objective.
For immunofluorescence experiments in mice, deparaffinized and rehydrated cross-sections of spinal cord and hindlimb muscle were permeabilized in 2% Triton-X 100, treated with TrueBlack autofluorescence quencher (Biotium), and blocked in PBS with 4% BSA and 2% NGS. Sections were incubated overnight at 4°C with anti-NCAM (Proteintech), anti-PSA-NCAM (EMD Millipore), anti-ubiquitin (Abcam), or anti-polyQ (EMD Millipore) antibodies, followed by incubation for 2 h at room temperature with the appropriate Alexa Fluor-conjugated secondary antibodies (ThermoFisher Scientific). Sections were mounted with ProLong Gold Antifade Mountant with DAPI (ThermoFisher Scientific) and allowed to dry for at least 24 h at room temperature before imaging on a Leica TCS SP8 STED 3X confocal microscope (Leica Biosystems for ubiquitin-polyQ colocalization or Leica DMI8 Widefield microscope for NCAM) with 40× objective and LASX software (Leica).
NCAM imaging and analysis
One image was acquired in a region of three to four slices where bright punctate NCAM and PSA-NCAM could be seen within the muscle fibers. If no punctate regions greater than that of autofluorescence intensity in either channel could be seen, a representative field without punctate signal was taken for that muscle slice. Imaging and analysis was performed for two mice from each of the following conditions: untreated NTG, untreated SBMA, NTG treated with 50 mg/kg MEPB, and NTG treated with 100 mg/kg MEPB. Three mice each were analyzed from each of the following conditions: SBMA treated with 50 mg/kg MEPB and SBMA treated with 100 mg/kg MEPB. The same images were subjected to colocalization analysis with LASX software. For each image, the background out-of-focus light was subtracted out and the remaining white detail was enhanced to make the punctate regions more easily recognizable by the program.
Following processing, an intensity-based mask was created for each channel, recognizing the regions where the signal was above that of tissue autofluorescence. These masks were then dilated and smoothed to combine nearby punctate regions. Using a binary “AND” operand, the number of overlapping regions larger than 5 pixels that had signal in both masks was calculated. Two-way ANOVA with Dunnett’s multiple comparisons test was performed using GraphPad Prism version 6. P < 0.05 was considered significant.
Luciferase reporter assays
To determine AR-dependent transcriptional activity, HEK293T cells were transiently transfected in culture media containing 10% charcoal-dextran stripped serum with ARE-firefly luciferase and CMV-Renilla luciferase (Cignal ARE Reporter Assay Kit, Qiagen), in addition to either FLAG-AR24Q or FLAG-AR65Q (gift from Maria Pennuto). Following 24 h of transfection, cells were washed and treated with vehicle, bicalutamide, TA, or MEPB for 24 h. Firefly and Renilla luciferase substrates (Dual-Luciferase Reporter Assay, Promega) were added, and luciferase activity was measured using a microplate spectrophotometer (BioTek). Mammalian two-hybrid assays were performed by transiently transfecting HEK293T cells with pG5Luc firefly luciferase reporter (Checkmate Mammalian Two-Hybrid kit, Promega), CMV-Renilla luciferase (Cignal ARE Reporter Assay Kit, Qiagen), and GAL4 DBD-AR LBD (gift from Elizabeth Wilson), in addition to either VP16 empty vector, VP16-NCoR, or VP16-SMRT (gifts from Vivian Bardwell) for 24 h. Cells were then washed and treated with vehicle, TA, or MEPB for 24 h. Renilla and firefly luciferase were quantified using Dual-Luciferase Reporter Assay (Promega) and a microplate spectrophotometer (BioTek).
Droplet digital PCR
The QX200 droplet digital PCR (ddPCR) system (Bio-Rad) was used to measure gene expression levels in 20 μl emulsion PCR reactions that contain 20,000 droplets. Total RNAs were firstly treated with DNase (ThermoFisher Scientific, AM1907) to remove genomic DNA, and 5 ng of treated RNA was used in each assay. ddPCR assay consisted of the following components: 1x one-step RT-ddPCR mix for probes (Bio-Rad, 1864021), forward primer (900 nM), reverse primer (900 nM), probe (FAM or VIC, 250 nM), nuclease-free water, and 5 ng RNA. All primers and probes were purchased from ThermoFisher Scientific. Droplets were generated in the droplet generator (Bio-Rad) and PCR was performed in a C1000 Touch thermal cycler (Bio-Rad) according to the manufacturer’s recommendation. After PCR, read-out of positive versus negative droplets was performed using the QX200 droplet reader (Bio-Rad) and calculated by QuantaSoft software version 1.7.4.0917 (Bio-Rad).
Statistical analyses
Significant changes in Drosophila population frequencies (i.e., viability) were determined by Chi square analysis. Survival of SBMA mice was determined by Kaplan-Meier estimation, and comparisons between survival curves were made with the log-rank (i.e., Mantel-Cox) test. All other data, except power analysis and QOL score, were analyzed by one-way or two-way ANOVA and either Tukey’s or Dunnett’s post-hoc analysis, where appropriate, with Prism software (version 6.0, GraphPad). Power analysis was performed to determine sample size requirements to achieve 80% power using SAS software (SAS Institute). QOL scores were determined by averaging the change from baseline measurements for each behavioral phenotype (body weight, grip strength, and rotarod activity). A score of zero was applied at each time point for any animal that could not complete the task due to hindlimb paralysis or that had been euthanized. A mixed effect model was applied using SAS software to determine statistical significance.
Life sciences reporting summary
Further information on experimental design is available in the Life Sciences Reporting Summary.
Supplementary Material
Acknowledgments
We thank N. Nedelsky for editorial assistance. We thank the transgenic core facility at St. Jude for production of AR121Q transgenic mice, and ARC Technical Services at St. Jude for surgical and animal care assistance. We also thank W. Denney for guidance in use of the ‘PKNCA’ R package. This work was supported by the Howard Hughes Medical Institute (JPT), the American-Lebanese-Syrian Associated Charities (JPT, HT), NIH R01 NS053825 (JPT), NIH R01 NS100023 (ARL), the Muscular Dystrophy Association (ARL), and the Kennedy’s Disease Association (HCM).
Footnotes
Author contributions
N.M.B., A.K., H.J.K., A.R.L., and J.P.T. designed the fruit fly, mouse, and cell-based experiments. N.M.B., A.K., and H.C.M. performed the experiments and data analyses. P.K.V., Y.W., and B.B.F. performed the pharmacokinetics analyses. R.B.S., E.R.Q., and N.C.K. provided assistance in the fruit fly experiments. J.D., J.Messing, B.D.F., and P.-C.C. provided assistance in the mouse experiments. B.J.W. performed in silico docking of TA into the AR LBD and generated the structural depictions of the AR LBD. B.L.S. and A.P.K. performed the cloning and preparation of the constructs used to generate AR121Q transgenic mice. J.Moore and B.F. provided assistance in cell-based experiments for digital PCR. J.Moore provided assistance in mouse tissue and plasma collection for pharmacokinetics analysis. H.T. performed histopathologic analysis of AR121Q transgenic mice. Y.L. performed the sample size estimation (power analysis) and statistical QOL analysis. N.M.B., A.K., H.J.K., and J.P.T. wrote the manuscript. All authors provided scientific input, read, and approved the manuscript.
Competing interests
The authors declare no competing interests.
References
- 1.Kennedy WR, Alter M, Sung JH. Progressive proximal spinal and bulbar muscular atrophy of late onset. A sex-linked recessive trait. Neurology. 1968;18:671–680. doi: 10.1212/wnl.18.7.671. [DOI] [PubMed] [Google Scholar]
- 2.Chahin N, Klein C, Mandrekar J, Sorenson E. Natural history of spinal-bulbar muscular atrophy. Neurology. 2008;70:1967–1971. doi: 10.1212/01.wnl.0000312510.49768.eb. [DOI] [PubMed] [Google Scholar]
- 3.Poletti A, Negri-Cesi P, Martini L. Reflections on the diseases linked to mutations of the androgen receptor. Endocrine. 2005;28:243–262. doi: 10.1385/ENDO:28:3:243. [DOI] [PubMed] [Google Scholar]
- 4.Fratta P, et al. Correlation of clinical and molecular features in spinal bulbar muscular atrophy. Neurology. 2014;82:2077–2084. doi: 10.1212/WNL.0000000000000507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- 6.Soraru G, et al. Spinal and bulbar muscular atrophy: skeletal muscle pathology in male patients and heterozygous females. J Neurol Sci. 2008;264:100–105. doi: 10.1016/j.jns.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 7.Finsterer J. Perspectives of Kennedy’s disease. J Neurol Sci. 2010;298:1–10. doi: 10.1016/j.jns.2010.08.025. [DOI] [PubMed] [Google Scholar]
- 8.Nedelsky NB, et al. Native functions of the androgen receptor are essential to pathogenesis in a Drosophila model of spinobulbar muscular atrophy. Neuron. 2010;67:936–952. doi: 10.1016/j.neuron.2010.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith CL, O’Malley BW. Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocr Rev. 2004;25:45–71. doi: 10.1210/er.2003-0023. [DOI] [PubMed] [Google Scholar]
- 10.Askew EB, Gampe RT, Jr, Stanley TB, Faggart JL, Wilson EM. Modulation of androgen receptor activation function 2 by testosterone and dihydrotestosterone. J Biol Chem. 2007;282:25801–25816. doi: 10.1074/jbc.M703268200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chevalier-Larsen ES, et al. Castration restores function and neurofilament alterations of aged symptomatic males in a transgenic mouse model of spinal and bulbar muscular atrophy. J Neurosci. 2004;24:4778–4786. doi: 10.1523/JNEUROSCI.0808-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katsuno M, et al. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron. 2002;35:843–854. doi: 10.1016/s0896-6273(02)00834-6. [DOI] [PubMed] [Google Scholar]
- 13.Katsuno M, et al. Leuprorelin rescues polyglutamine-dependent phenotypes in a transgenic mouse model of spinal and bulbar muscular atrophy. Nat Med. 2003;9:768–773. doi: 10.1038/nm878. [DOI] [PubMed] [Google Scholar]
- 14.Katsuno M, et al. Efficacy and safety of leuprorelin in patients with spinal and bulbar muscular atrophy (JASMITT study): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2010;9:875–884. doi: 10.1016/S1474-4422(10)70182-4. [DOI] [PubMed] [Google Scholar]
- 15.Yang Z, et al. ASC-J9 ameliorates spinal and bulbar muscular atrophy phenotype via degradation of androgen receptor. Nat Med. 2007;13:348–353. doi: 10.1038/nm1547. [DOI] [PubMed] [Google Scholar]
- 16.Banno H, et al. Phase 2 trial of leuprorelin in patients with spinal and bulbar muscular atrophy. Ann Neurol. 2009;65:140–150. doi: 10.1002/ana.21540. [DOI] [PubMed] [Google Scholar]
- 17.Axerio-Cilies P, et al. Inhibitors of androgen receptor activation function-2 (AF2) site identified through virtual screening. J Med Chem. 2011;54:6197–6205. doi: 10.1021/jm200532b. [DOI] [PubMed] [Google Scholar]
- 18.Ban F, et al. Discovery of 1H-indole-2-carboxamides as novel inhibitors of the androgen receptor binding function 3 (BF3) J Med Chem. 2014;57:6867–6872. doi: 10.1021/jm500684r. [DOI] [PubMed] [Google Scholar]
- 19.Estebanez-Perpina E, et al. A surface on the androgen receptor that allosterically regulates coactivator binding. Proc Natl Acad Sci U S A. 2007;104:16074–16079. doi: 10.1073/pnas.0708036104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Munuganti RS, et al. Targeting the binding function 3 (BF3) site of the androgen receptor through virtual screening. 2. development of 2-((2-phenoxyethyl) thio)-1H-benzimidazole derivatives. J Med Chem. 2013;56:1136–1148. doi: 10.1021/jm3015712. [DOI] [PubMed] [Google Scholar]
- 21.Lack NA, et al. Targeting the binding function 3 (BF3) site of the human androgen receptor through virtual screening. J Med Chem. 2011;54:8563–8573. doi: 10.1021/jm201098n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Munuganti RS, et al. Identification of a potent antiandrogen that targets the BF3 site of the androgen receptor and inhibits enzalutamide-resistant prostate cancer. Chem Biol. 2014;21:1476–1485. doi: 10.1016/j.chembiol.2014.09.012. [DOI] [PubMed] [Google Scholar]
- 23.Rocchi A, et al. Glycolytic-to-oxidative fiber-type switch and mTOR signaling activation are early-onset features of SBMA muscle modified by high-fat diet. Acta Neuropathol. 2016;132:127–144. doi: 10.1007/s00401-016-1550-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cashman NR, Covault J, Wollman RL, Sanes JR. Neural cell adhesion molecule in normal, denervated, and myopathic human muscle. Ann Neurol. 1987;21:481–489. doi: 10.1002/ana.410210512. [DOI] [PubMed] [Google Scholar]
- 25.Gosztonyi G, Naschold U, Grozdanovic Z, Stoltenburg-Didinger G, Gossrau R. Expression of Leu-19 (CD56, N-CAM) and nitric oxide synthase (NOS) I in denervated and reinnervated human skeletal muscle. Microsc Res Tech. 2001;55:187–197. doi: 10.1002/jemt.1170. [DOI] [PubMed] [Google Scholar]
- 26.Marbini A, et al. Immunohistochemical study of muscle biopsy in children with cerebral palsy. Brain Dev. 2002;24:63–66. doi: 10.1016/s0387-7604(01)00394-1. [DOI] [PubMed] [Google Scholar]
- 27.Yu Z, et al. Abnormalities of germ cell maturation and sertoli cell cytoskeleton in androgen receptor 113 CAG knock-in mice reveal toxic effects of the mutant protein. Am J Pathol. 2006;168:195–204. doi: 10.2353/ajpath.2006.050619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun JH, Lee SA. Association between CAG repeat polymorphisms and the risk of prostate cancer: a meta-analysis by race, study design and the number of (CAG)n repeat polymorphisms. Int J Mol Med. 2013;32:1195–1203. doi: 10.3892/ijmm.2013.1474. [DOI] [PubMed] [Google Scholar]
- 29.Albertelli MA, et al. Glutamine tract length of human androgen receptors affects hormone-dependent and -independent prostate cancer in mice. Hum Mol Genet. 2008;17:98–110. doi: 10.1093/hmg/ddm287. [DOI] [PubMed] [Google Scholar]
- 30.Robins DM, Albertelli MA, O’Mahony OA. Androgen receptor variants and prostate cancer in humanized AR mice. J Steroid Biochem Mol Biol. 2008;108:230–236. doi: 10.1016/j.jsbmb.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buchanan G, et al. Structural and functional consequences of glutamine tract variation in the androgen receptor. Hum Mol Genet. 2004;13:1677–1692. doi: 10.1093/hmg/ddh181. [DOI] [PubMed] [Google Scholar]
- 32.Buchanan G, et al. Corepressor effect on androgen receptor activity varies with the length of the CAG encoded polyglutamine repeat and is dependent on receptor/corepressor ratio in prostate cancer cells. Mol Cell Endocrinol. 2011;342:20–31. doi: 10.1016/j.mce.2011.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jehle K, et al. Coregulator control of androgen receptor action by a novel nuclear receptor-binding motif. J Biol Chem. 2014;289:8839–8851. doi: 10.1074/jbc.M113.534859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corell T. Pharmacology of tolfenamic acid. Pharmacol Toxicol. 1994;75(Suppl 2):14–21. doi: 10.1111/j.1600-0773.1994.tb01991.x. [DOI] [PubMed] [Google Scholar]
- 35.Hendel J. The overall safety of tolfenamic acid. Pharmacol Toxicol. 1994;75(Suppl 2):53–55. doi: 10.1111/j.1600-0773.1994.tb01998.x. [DOI] [PubMed] [Google Scholar]
- 36.Hendel L, Larsen E, Bonnevie O. A comparative study of the influence of tolfenamic acid (Clotam) and diclofenac sodium (Voltaren) on the gastrointestinal mucosa in patients with a history of NSAID-related dyspeptic symptoms. Pharmacol Toxicol. 1994;75(Suppl 2):49–50. doi: 10.1111/j.1600-0773.1994.tb01996.x. [DOI] [PubMed] [Google Scholar]
- 37.Pedersen SB. Biopharmaceutical aspects of tolfenamic acid. Pharmacol Toxicol. 1994;75(Suppl 2):22–32. doi: 10.1111/j.1600-0773.1994.tb01992.x. [DOI] [PubMed] [Google Scholar]
- 38.Yuan H, et al. Suppression of the androgen receptor function by quercetin through protein-protein interactions of Sp1, c-Jun, and the androgen receptor in human prostate cancer cells. Mol Cell Biochem. 2010;339:253–262. doi: 10.1007/s11010-010-0388-7. [DOI] [PubMed] [Google Scholar]
- 39.Hodgson MC, Shen HC, Hollenberg AN, Balk SP. Structural basis for nuclear receptor corepressor recruitment by antagonist-liganded androgen receptor. Mol Cancer Ther. 2008;7:3187–3194. doi: 10.1158/1535-7163.MCT-08-0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bott LC, et al. A small-molecule Nrf1 and Nrf2 activator mitigates polyglutamine toxicity in spinal and bulbar muscular atrophy. Hum Mol Genet. 2016;25:1979–1989. doi: 10.1093/hmg/ddw073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Glynn MW, Glover TW. Incomplete processing of mutant lamin A in Hutchinson-Gilford progeria leads to nuclear abnormalities, which are reversed by farnesyltransferase inhibition. Hum Mol Genet. 2005;14:2959–2969. doi: 10.1093/hmg/ddi326. [DOI] [PubMed] [Google Scholar]
- 42.Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.D’Argenio DZ, Schumitzky A, Wang X. ADAPT 5 User’s Guide: Pharmacokinetic/Pharmacodynamic Systems Analysis Software. Biomedical Simulations Resource; Los Angeles: 2009. [Google Scholar]
- 44.Beal SL. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn. 2001;28:481–504. doi: 10.1023/a:1012299115260. [DOI] [PubMed] [Google Scholar]
- 45.Brown RP, Delp MD, Lindstedt SL, Rhomberg LR, Beliles RP. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol Ind Health. 1997;13:407–484. doi: 10.1177/074823379701300401. [DOI] [PubMed] [Google Scholar]
- 46.Gargas ML, et al. Physiologically based pharmacokinetic modeling of chloroethane disposition in mice, rats, and women. Toxicol Sci. 2008;104:54–66. doi: 10.1093/toxsci/kfn064. [DOI] [PubMed] [Google Scholar]
- 47.Kaliss N, Pressman D. Plasma and blood volumes of mouse organs, as determined with radioactive iodoproteins. Proc Soc Exp Biol Med. 1950;75:16–20. doi: 10.3181/00379727-75-18083. [DOI] [PubMed] [Google Scholar]
- 48.McIlwain DL, Hoke VB, Kopchick JJ, Fuller CR, Lund PK. Differential inhibition of postnatal brain, spinal cord and body growth by a growth hormone antagonist. BMC Neurosci. 2004;5:6. doi: 10.1186/1471-2202-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Duhamel G, Callot V, Cozzone PJ, Kober F. Spinal cord blood flow measurement by arterial spin labeling. Magn Reson Med. 2008;59:846–854. doi: 10.1002/mrm.21567. [DOI] [PubMed] [Google Scholar]
- 50.Gibaldi M, Perrier D. Pharmacokinetics. CRC Press; New York: 1982. [Google Scholar]
- 51.Denney W, Duvvuri S, Buckeridge C. Simple, Automatic Noncompartmental Analysis: The PKNCA R Package. J Pharmacokinet Pharmacodyn. 2015;42:11–107. [Google Scholar]
- 52.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2016. [Google Scholar]
- 53.Brooks BP, et al. Characterization of an expanded glutamine repeat androgen receptor in a neuronal cell culture system. Neurobiol Dis. 1997;3:313–323. doi: 10.1006/nbdi.1997.0126. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.