

Abstract
BACKGROUND
The safety and efficacy of a novel cobalt alloy-based coronary stent with a durable elastomeric polymer eluting the antiproliferative agent ridaforolimus for treatment of patients with coronary artery disease is undetermined.
METHODS
A prospective, international 1:1 randomized trial was conducted to evaluate in a noninferiority design the relative safety and efficacy of ridaforolimus-eluting stents (RESs) and slow-release zotarolimus-eluting stents among 1919 patients undergoing percutaneous coronary intervention at 76 centers. Inclusion criteria allowed enrollment of patients with recent myocardial infarction, total occlusions, bifurcations lesions, and other complex conditions.
RESULTS
Baseline clinical and angiographic characteristics were similar between the groups. Overall, mean age was 63.4 years, 32.5% had diabetes mellitus, and 39.7% presented with acute coronary syndromes. At 12 months, the primary end point of target lesion failure (composite of cardiac death, target vessel-related myocardial infarction, and target lesion revascularization) was 5.4% for both devices (upper bound of 1-sided 95% confidence interval 1.8%, Pnoninferiority=0.001). Definite/probable stent thrombosis rates were low in both groups (0.4% RES versus 0.6% zotarolimus-eluting stent, P=0.75); 13-month angiographic in-stent late lumen loss was 0.22±0.41 mm and 0.23±0.39 mm (Pnoninferiority=0.004) for the RES and zotarolimus-eluting stent groups, respectively, and intravascular ultrasound percent neointimal hyperplasia was 8.10±5.81 and 8.85±7.77, respectively (Pnoninferiority=0.01).
CONCLUSIONS
In the present trial, which allowed broad inclusion criteria, the novel RESs met the prespecified criteria for noninferiority compared with zotarolimus-eluting stents for the primary end point of target lesion failure at 12 months and had similar measures of late lumen loss. These findings support the safety and efficacy of RESs in patients who are representative of clinical practice.
CLINICAL TRIAL REGISTRATION
URL: http://www.clinicaltrials.gov. Unique identifier: NCT01995487.
Keywords: coronary intervention, drug-eluting stent, percutaneous, ridaforolimus, stent thrombosis
The benefits of treatment with drug-eluting stents (DESs) to avoid restenosis and repeat revascularization have been consistently demonstrated in clinical trials in both selected and broad populations with varying clinical and angiographic characteristics. Iterative DES development has focused on features that may enhance procedural outcomes and improve clinical safety and efficacy. Large comparative studies involving newer generation DESs demonstrate outcomes superior to conventional bare metal stents and first-generation DESs.1–3
A novel cobalt alloy-based coronary stent has been designed incorporating a unique durable elastomeric polymer eluting the antiproliferative drug ridaforolim-us. Ridaforolimus, a rapamycin derivative, is associated with a higher therapeutic-to-toxicity margin compared with most other sirolimus analogues (Appendix I in the online-only Data Supplement), and the elastomeric properties of the polymer confer resistance to the disruption of polymer integrity during stent expansion that has been observed with contemporary DESs.4,5 The safety and efficacy of the ridaforolimus-eluting stent (RES) has not been established in human patients. Therefore, we conducted a large-scale, randomized, multicenter trial to examine the clinical, angiographic, and intravascular ultrasound (IVUS) outcomes of RESs compared with the contemporary slow-release zotarolimus-eluting stent (ZES) in patients undergoing percutaneous coronary intervention (PCI) representative of routine clinical practice.
METHODS
Trial and Study Population
The BIONICS trial (BioNIR Ridaforolimus-Eluting Coronary Stent System in Coronary Stenosis; clinicaltrials.gov identifier NCT01995487) was a prospective, randomized, single-blinded multicenter trial comparing RESs (BioNIR; Medinol Ltd.) and ZESs (Resolute Integrity, Medtronic) in patients undergoing PCI. The trial was designed with guidance from the US Food and Drug Administration and was intended to support US device approval. Patients ≥18 years of age with ischemic heart disease undergoing planned stent implantation were eligible for enrollment. Enrollment criteria were developed with the US Food and Drug Administration to permit a less restrictive, more complex population than allowed in most prior regulatory approval trials. Angiographic inclusion criteria included a reference vessel diameter between 2.5 and 4.25 mm, with ≤ 2 lesions per vessel in ≤2 major coronary arteries, provided that the total planned stent length was not >100 mm. Calcified lesions requiring atherectomy were included, as were chronic total occlusions, planned 1-stent bifurcations, bypass graft stenoses, and bare metal in-stent restenosis. Patients with recent (<24 hours) ST-segment elevation myocardial infarction, left ventricular ejection fraction <30%, active stent thrombosis, creatinine clearance <30 mL/min, and prior PCI ≤12 months, and those unlikely to adhere to dual antiplatelet therapy, were excluded. The study was approved by the institutional review board or ethics committee at each enrolling site, and eligible patients signed written informed consent before the interventional procedure.
Device Description
The BioNIR stent is constructed of an 87-μm strut thickness cobalt alloy platform with adaptive cells capable of differential lengthening to provide uniform drug distribution in variable vessel anatomy. The 3.0-mm-diameter stent system has a crossing profile of 1.07 mm. The stent is manufactured in an unconventional method, in which a thin sheet of cobalt alloy is laser-cut into the stent design, coated with polymer and drug, rolled into a cylindrical shape, and laser-welded at discrete spots along its axial length. A proprietary copolymer made of thermoplastic silicone polycarbonate polyurethane and poly-butyl methacrylate with elastomeric properties resistant to disruption in integrity is circumferentially coated on the stent and is 7 μm thick. The polymer permits controlled elution of ridaforolimus, an analog of sirolimus, present on the stent at a concentration of 1.1 μg/mm2. In preclinical studies, >95% of the drug is eluted from the stent over a 180-day period (Appendix I in the online-only Data Supplement). Unlike previous DESs, however, an initial peak (burst) concentration followed by diffusion does not occur. Rather, persistently low drug concentrations are measured in the surrounding vascular tissue for 3 months. RESs were available in diameters ranging from 2.5 to 4.0 mm and in lengths from 8 to 33 mm. The comparator ZES (Resolute Integrity or Resolute Onyx, Medtronic, Inc.) was available in diameters between 2.25 and 4.0 mm and in lengths ranging from 8 to 38 mm.
Randomization, Interventional Procedures, and Adjunctive Drug Therapy
Patients were blinded to treatment assignment and ran-domized to RESs or ZESs in a 1:1 fashion. Randomization was stratified according to presence or absence of medically treated diabetes mellitus, acute coronary syndrome versus stable angina presentation, and site. Patients were considered enrolled after guide wire crossing of the target lesions. Treatment of nontarget vessel lesions was permitted provided that procedural success criteria were achieved before ran-domization. Direct stenting without predilation was permitted, and postdilation was recommended but not required.
Before PCI, all patients received treatment with aspirin (325 mg if no prior therapy, 75–325 mg if chronic therapy) and clopidogrel, ticagrelor, or prasugrel per investigator discretion. For patients not receiving chronic P2Y12 receptor antagonist therapy, a loading dose was administered according to individual product labeling. Dual antiplatelet therapy use was mandatory for ≥6 months after the procedure. Anticoagulation with unfractionated heparin, bivalirudin, or low-molecular-weight heparin with or without a glycoprotein IIb/IIIa inhibitor was prescribed according to local standards. Clinical events were assessed during hospital stay, at 30 days, and at 12 months after the index procedure. Follow-up angio-graphic and IVUS imaging at 13 months was planned in a consecutive cohort of patients from participating sites.
Data Management and Core Laboratories
All data were submitted to a central data coordinating facility (Cardiovascular Research Foundation). An independent clinical events committee (Cardiovascular Research Foundation) adjudicated all primary and secondary clinical end points blinded to stent type. An independent data safety monitoring board (Appendix II in the online-only Data Supplement) was responsible for regular review of the clinical safety data and could recommend study discontinuation or modification. Coronary angiograms performed at baseline and any time during the follow-up period in addition to IVUS images were reviewed by an independent core laboratory (Cardiovascular Research Foundation). Reviewers from the core laboratory were unaware of the type of stent implanted.
Study End Points and Definitions
The primary end point was target lesion failure (TLF) at 12 months, defined as the composite of cardiac death, target vessel-related myocardial infarction (MI), or ischemia-driven target lesion revascularization. Secondary clinical safety and efficacy end points included major adverse cardiac events (cardiac death, MI, or ischemia-driven target lesion revascularization); target vessel failure (all-cause death, target vessel-related myocardial infarction, or ischemia-driven target vessel revascularization); individual components of the composite end points in-hospital, at 30 days, and at 12 months; and definite or probable stent thrombosis according to Academic Research Consortium criteria.6 Device success was defined as achievement of <50% diameter stenosis of the target lesion (determined by the angiographic core laboratory) with the assigned study stent, and procedure success was defined as a final diameter stenosis <50% with the assigned stent and any adjunctive device, and with no in-hospital major adverse cardiac events.
Periprocedural MI was defined according to the Society of Coronary Angiography and Interventions criteria,7 as a creatine kinase myocardial band measured ≤48 hours of the procedure elevated ≥10 times above the upper limit of normal, or ≥5 times the upper limit of normal with development of new pathological Q waves in 2 contiguous electrocardio-graphic leads or new, persistent left bundle-branch block. In the absence of creatine kinase myocardial band measurements, postprocedural MI was defined as a troponin value ≥70 times the upper limit of normal, or ≥35 times the upper limit of normal with new pathological Q waves or new left bundle-branch block. The Society of Coronary Angiography and Interventions criteria to define periprocedural MI were also used for patients with elevated cardiac enzymes at baseline.7 Spontaneous MI was defined according to the Universal Definition of Myocardial Infarction.8 Ischemia-driven revascularization was identified as any repeat revascularization of the target lesion or target vessel associated with either ischemic symptoms or an abnormal functional study and a ≥50% coronary stenosis by quantitative angiography, or any revascularization of a ≥70% diameter stenosis. Cardiovascular death was considered death caused by any proximate cardiac cause, unwitnessed death, or death of unknown etiology.
Secondary angiographic and IVUS efficacy end points at 13-month follow-up included late lumen loss (both in-stent and in-segment), angiographic binary restenosis (in-stent and in-segment), and percent neointimal hyperplasia. Angiographic binary restenosis was defined as a stenosis ≥50% of the lumen diameter of the target lesion (determined by the core angiographic laboratory). Percent diameter steno-sis was defined as (1- [minimum luminal diameter/reference vessel diameter]) × 100, and acute gain was defined as the minimal luminal diameter immediately after the procedure minus the MLD before the procedure. Restenosis patterns were characterized according to established criteria.9
Statistical Methods
This trial was powered to determine the noninferiority of RESs compared with ZESs for TLF at 12 months assuming a primary event rate of 5.8% in each treatment group and a pre-specified margin of 3.3%. With the assumption of 5% loss to follow-up, a sample size of 1906 patients was required for the trial to have 90% statistical power at a 1-sided alpha level of 0.05. For the secondary angiographic end point of in-stent late loss, assuming in-stent late lumen loss for both stents of 0.29 mm (±0.44 mm), a noninferiority margin of 0.20 mm, and a 1-sided α level of 0.05, a sample size of 150 evaluable patients provided 87% power to demonstrate noninferiority between RESs and ZESs. Similarly, for the secondary IVUS end point of percent neointimal hyperplasia, 80 evaluable patients provided 84% statistical power to demonstrate noninferiority at an α level of 0.05, assuming 3.5% neointimal hyperplasia for both the experimental and control groups and a noninfe-riority margin of 3.0%.
All primary and secondary efficacy and safety end points were performed in the intention-to-treat population. Baseline characteristics of study patients were summarized in terms of frequencies and percentages for categorical variables and by means with standard deviations for continuous variables. Categorical variables were compared by χ-square or Fisher exact test. For continuous variables that met the assumption of normality, the 2 treatment groups were compared by the 2-sample t test. If the data failed to meet the assumption for normality per the Shapiro-Wilk test, then the comparisons were made using the Wilcoxon rank sum test. Noninferiority for the primary end point of TLF at 12 months was evaluated by the Farrington and Manning test for binary variables. For the secondary angiographic and IVUS endpoints of in-stent late loss and percent neointimal hyperplasia at 13 months, noninferiority was evaluated by a 1-way linear mixed model, which accounted for the clustering effect of multiple lesions per patient. The model included treatment as a fixed effect and patient as a random effect. The 12-month clinical events were summarized as Kaplan-Meier estimates and compared with the log-rank test. A P value of 0.05 was established as the level of statistical significance for all superiority tests. All analyses were performed with SAS software (version 9.4, SAS Institute).
RESULTS
Patient Enrollment and Characteristics
Between March 2014 and August 2015, 1919 patients were randomized (958 to RESs, 961 to ZESs) at 76 hospitals in 8 countries in North America, Europe, and Is-rael (Figure 1). No significant differences were present in the baseline clinical or demographic characteristics between the groups (Table 1). Approximately 33% of patients had diabetes mellitus. Presentation with acute coronary syndromes was common and was observed in 39.7% of patients.
Figure 1. BIONICS trial (BioNIR Ridaforolimus Eluting Coronary Stent System in Coronary Steno-sis) flow chart.
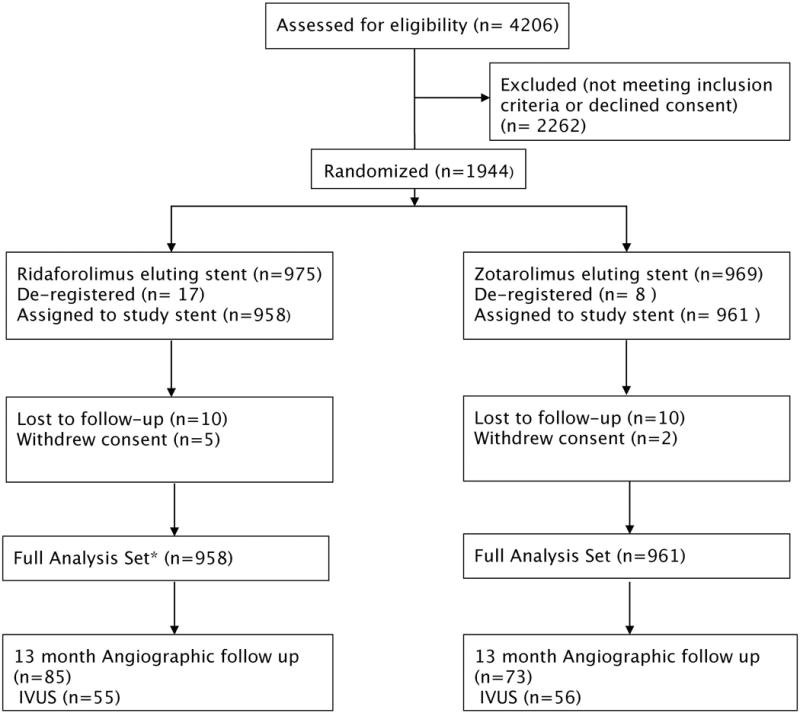
Full analysis set included all randomized subjects assigned to treatment per randomization regardless of whether they received the study stent. Subjects were included for analysis once the study stent had been advanced beyond the guide catheter. IVUS indicates intravascular ultrasound.
Table 1.
Baseline Clinical and Angiographic Characteristics
| Ridaforolimus-Eluting Stent (N=958, 1276 Lesions) | Zotarolimus-Eluting Stent (N=961, 1277 Lesions) | |
|---|---|---|
| Age, y | 63.7±10.2 | 63.1±10.3 |
| Male sex | 78.3 (750/958) | 81.9 (787/961) |
| Diabetes mellitus | 32.8 (314/958) | 32.3 (310/961) |
| Hypertension | 72.4 (687/949) | 74.0 (704/951) |
| Hyperlipidemia | 80.4 (759/944) | 78.1 (744/953) |
| Previous myocardial infarction | 31.1 (298/958) | 30.5 (293/961) |
| Previous PCI | 38.8 (372/958) | 38.2 (367/961) |
| Previous coronary bypass surgery | 8.8 (84/958) | 9.6 (92/961) |
| Current smoker | 23.4 (224/958) | 19.4 (186/961) |
| LV ejection fraction | 55.5±9.8 | 55.8±9.3 |
| Clinical presentation | ||
| Stable coronary disease | 59.3 (568/958) | 61.3 (589/961) |
| ACS | 40.7 (390/958) | 38.7 (372/961) |
| Elevated cardiac biomarkers* | 27.0 (206/763) | 28.2 (216/766) |
| Target lesion vessel | ||
| Left main | 1.1 (14/1276) | 0.4 (5/1277) |
| Left anterior descending | 40.7 (519/1276) | 39.7 (507/1277) |
| Right coronary | 32.0 (408/1276) | 32.2 (411/1277) |
| Left circumflex | 24.4 (311/1276) | 25.1 (320/1277) |
| Bypass graft | 1.9 (24/1276) | 2.7 (34/1277) |
| Multivessel disease | 29.4 (282/958) | 29.3 (282/961) |
| Angiographic complexity | ||
| No. lesions treated per patient | 1.3±0.6 | 1.3±0.6 |
| Reference vessel diameter (mm) | 2.73±0.49 | 2.74±0.49 |
| Lesion length (mm) | 17.7±10.8 | 17.9±10.7 |
| SYNTAX score | 11.2±7.4 | 11.0±7.2 |
| TIMI 0 flow in target vessel | 9.7 (92/953) | 7.7 (74/957) |
| Bifurcation lesion | 28.6 (365/1276) | 29.1 (371/1277) |
| Ostial lesion | 6.0 (77/1276) | 6.1 (78/1277) |
| Chronic total occlusion | 7.2 (92/1272) | 5.8 (74/1276) |
| Overlapping stents | 24.7 (237/958) | 23.1 (222/961) |
| Use of atherectomy device | 0.9 (9/958) | 1.1 (11/961) |
| Thrombus present | 3.4 (43/1270) | 3.0 (38/1275) |
| Severe calcification | 13.3 (169/1272) | 10.5 (134/1274) |
| Severe tortuosity | 3.9 (50/1271) | 2.8 (35/1272) |
| ACC lesion class B2/C | 57.5 (733/1275) | 58.9 (752/1277) |
ACC indicates American College of Cardiology; ACS, acute coronary syndrome; LV, left ventricle; PCI, percutaneous coronary intervention; SYNTAX, synergy between percutaneous coronary intervention with taxus and cardiac surgery; and TIMI, thrombolysis in myocardial infarction. Values are % (n/N) or mean±SD.
Troponin I or T ≥3X upper limit of normal.
Baseline angiographic characteristics were also similar between groups, except for a slightly higher prevalence of left main (1.1% versus 0.4%, P=0.04) and severely calcified (13.3% versus 10.5%, P=0.03) target lesions in the ridaforolimus cohort. The left anterior descending artery was the most commonly treated vessel (40.2%), and complex lesions were frequent, including bifurcation disease (29%), ostial lesions (6%), and overlapping stents (24%). The mean lesion length and reference vessel diameter were 17.8 (±10.8) mm and 2.74 (±0.49) mm, respectively.
Procedural and Clinical Outcomes
The number, length, and diameter of stents implanted were similar in the 2 treatment groups (Table 2). The average number of lesions treated per patient was 1.3±0.6, and the average stent length was 24 mm. Procedural success was similar in both groups, although device success was slightly lower with RESs than ZESs (98.0% versus 99.4%, P=0.001). The reason for device failures in both cohorts was most commonly inability to deliver the assigned study stent to the target lesion.
Table 2.
Procedural Results and 30-Day Clinical Events
| Ridaforolimus-Eluting Stent (N=958 patients, 1326 Lesions) | Zotarolimus-Eluting Stent (N=961 patients, 1305 Lesions) | P Value | |
|---|---|---|---|
| No. of stents | |||
| Per patient | 1.6±0.9 | 1.6±0.8 | 0.17 |
| Per lesion | 1.2±0.5 | 1.1±0.5 | 0.49 |
| Per target vessel | 1.4±0.7 | 1.3±0.6 | 0.33 |
| Total stent length (mm) | |||
| Per patient | 33±20.9 | 31.8±19.7 | 0.48 |
| Per lesion | 24.3±14 | 23.7±12 | 0.33 |
| Stent diameter (mm) | 3.04±0.44 | 3.02±0.45 | 0.42 |
| >1 stent implanted | 16.7 (221/1326) | 14.9 (194/1305) | 0.21 |
| Minimal luminal diameter (mm) | |||
| Preprocedure in-lesion | 0.78±0.40 | 0.81±0.40 | 0.07 |
| Postprocedure in-stent | 2.50±0.45 | 2.54±0.47 | 0.002 |
| Postprocedure in-segment | 2.29±0.47 | 2.31±0.49 | 0.13 |
| Diameter stenosis (%) | |||
| Preprocedure in-lesion | 71.5±13.4 | 70.7±12.8 | 0.15 |
| Postprocedure in-stent | 11.7±7.7 | 10.9±8.3 | 0.002 |
| Postprocedure in-segment | 16.4±9.2 | 16.3±9.6 | 0.40 |
| Acute gain (mm) | |||
| In stent | 1.7±0.5 | 1.7±0.5 | 0.19 |
| In segment | 1.5±0.5 | 1.5±0.5 | 0.96 |
| Index PCI procedure characteristics (mm) | |||
| Balloon predilation | 75.4 (1000/1326) | 72.0 (940/1305) | 0.05 |
| Postdilation | 60.3 (800/1326) | 56.7 (740/1305) | 0.06 |
| FFR performed | 4.9 (65/1326) | 4.8 (62/1305) | 0.86 |
| Device success* | 98.0 (1243/1268) | 99.4 (1261/1268) | 0.001 |
| Lesion success† | 99.9 (1257/1258) | 99.8 (1262/1264) | 0.99 |
| Procedure success‡ | 97.6 (929/952) | 97.3 (928/954) | 0.67 |
| 30-day outcomes (mm) | |||
| Death | 0.5 (5/955) | 0.2 (2/958) | 0.29 |
| Myocardial infarction, any | 2.8 (27/953) | 3.2 (31/956) | 0.60 |
| Q-wave myocardial infarction | 0.5 (5/953) | 0.4 (4/956) | 0.75 |
| Non-Q-wave myocardial infarction | 2.4 (23/952) | 2.8 (27/956) | 0.58 |
| Stent thrombosis | 0.4 (4/952) | 0.4 (4/957) | 0.99 |
| TLF§ | 2.6 (25/954) | 3.2 (31/957) | 0.42 |
| TVF¶ | 2.9 (28/955) | 3.4 (33/958) | 0.52 |
| MACEǁ | 3.0 (29/954) | 3.6 (34/957) | 0.53 |
FFR indicates fractional flow reserve; MACE, major adverse cardiac events; PCI, percutaneous coronary intervention; TLF, target lesion failure; TVF, target vessel failure; QCA, quantitative coronary angiography. Values are % (n/N) or mean±SD.
Device success: final in-stent residual QCA diameter stenosis of <50% using the assigned device only and without a device malfunction.
Lesion success: final in-stent residual QCA diameter stenosis of <50% using any percutaneous method.
Procedure success: final in-stent QCA diameter stenosis of <50% using the assigned device or with any adjunctive devices, without the occurrence of cardiac death, Q wave or non-Q wave MI, or repeat revascularization of the target lesion during the hospital stay.
Target lesion failure defined as the composite rate of cardiac death, target vessel myocardial infarction, or clinically driven target lesion revascularization.
Target vessel failure defined as the composite rate of all-cause death, target vessel-related myocardial infarction, or clinically driven target vessel revascularization.
Major adverse cardiac events defined as the composite rate of cardiac death, any myocardial infarction, or clinically driven target lesion revascularization.
Adverse events within 30 days were low in both treatment groups (Table 2). At 12 months, the primary end point of TLF occurred in 5.4% of patients in both groups (relative risk, 1.00; 95% confidence interval, 0.69‒1.47; Pnoninferiority=0.001, Psuperiority=0.98) (Table 3, Figure 2). No significant interactions were observed in the relative rates of TLF in prespecified subgroups (Figure 3). Individual components of TLF were also similar between groups (Table 3). Definite or probable stent thrombosis occurred in 0.4% of RES-treated patients and 0.8% of ZES-treated patients (P=0.53). No stent thrombosis events were observed in the RES group beyond 30 days of the index revascularization procedure. At 12 months, adherence to dual antiplatelet therapy was reported in ≈75% of patients and was similar in both groups.
Table 3.
One-Year Clinical Results
| Ridaforolimus-Eluting Stent (n=958) | Zotarolimus-Eluting Stent (n=961) | P value | |
|---|---|---|---|
| Target lesion failure* | 5.4 (50) | 5.4 (50) | 0.91, 0.001 for noninferiority |
| Death | |||
| All cause | 1.2 (11) | 1.0 (10) | 0.82 |
| Cardiac | 0.5 (5) | 0.2 (2) | 0.27 |
| Target vessel Myocardial infarction Any | 3.2 (30) | 3.2 (31) | 0.91 |
| Q-wave | 0.5 (5) | 0.5 (5) | 0.99 |
| Non-Q-wave | 2.8 (26) | 2.7 (26) | 0.30 |
| Clinically driven target lesion revascularization | |||
| Any | 3.2 (28) | 2.3 (22) | 0.38 |
| PCI | 2.5 (22) | 2.1 (20) | 0.74 |
| CABG | 0.8 (7) | 0.2 (2) | 0.09 |
| Myocardial infarction† | |||
| Periprocedural | 2.4 (23) | 2.8 (27) | 0.58 |
| Nonperiprocedural | 2.6 (24) | 2.5 (23) | 0.87 |
| Target vessel failure‡ | 7.1 (66) | 6.3 (60) | 0.58 |
| MACE§ (%) | 6.8 (63) | 6.6 (63) | 0.99 |
| Stent thrombosis | |||
| Definite/probable | 0.4 (4) | 0.6 (6) | 0.53 |
| Definite | 0.4 (4) | 0.5 (5) | 0.74 |
| Probable | 0.0 (0) | 0.1 (1) | 0.32 |
| Acute (24 h) | 0.1 (1) | 0.1 (1) | 0.99 |
| Subacute (1–30 days) | 0.3 (3) | 0.3 (3) | 0.99 |
| Late (31 days – 1 y) | 0.0 (0) | 0.2 (2) | 0.16 |
CABG indicates coronary artery bypass graft; MACE, major adverse cardiac events; and PCI, percutaneous coronary intervention. Values are % (n).
Target lesion failure defined as the composite rate of cardiac death, target vessel myocardial infarction, or clinically driven target lesion revascularization.
Myocardial infarction defined on the basis of the Society of Coronary Angiography and Interventions definition.7
Target vessel failure defined as the composite rate of all-cause death, target vessel-related myocardial infarction, or clinically driven target vessel revascularization.
Major adverse cardiac events defined as the composite rate of cardiac death, any myocardial infarction, or clinically driven target lesion revascularization.
Figure 2. One-year target lesion failure.
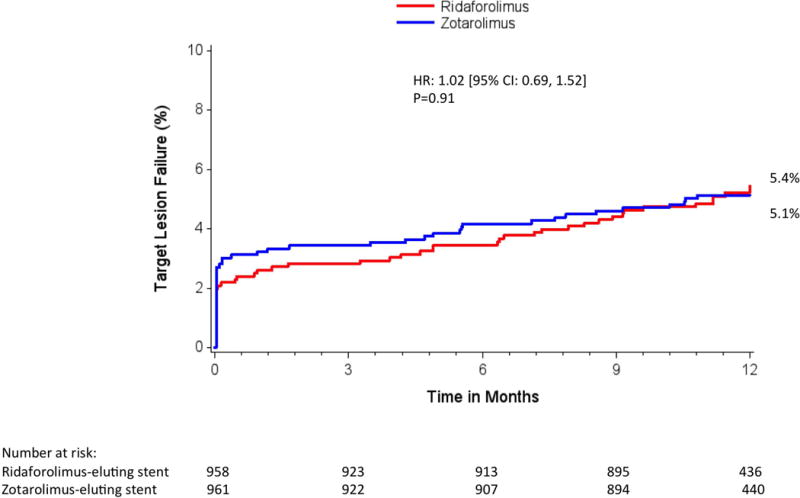
Kaplan-Meier estimates of the primary outcome target lesion failure by 1 year. CI indicates confidence interval, and HR, hazard ratio.
Figure 3. Subgroup analysis for the primary end point of target lesion failure at 1-year follow-up.
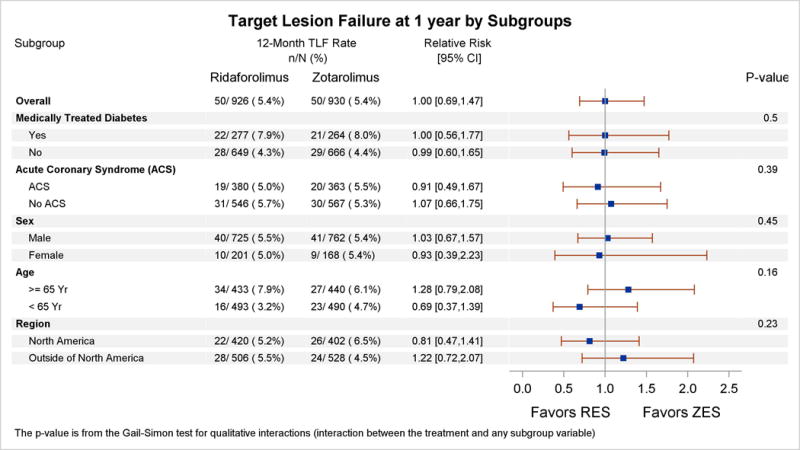
Subgroup analysis for prespecified baseline characteristics is shown. The P value for qualitative interaction (treatment by subgroup) is obtained from the Gail-Simon test, which assesses the heterogeneity of the treatment effect across subgroups. ACS indicates acute coronary syndrome; CI, confidence interval; RES, ridaforolimus-eluting stent; TLF, target lesion failure; and ZES, zotarolimus-eluting stent.
Angiographic and IVUS Outcomes
Follow-up angiography at 13 months was performed in 158 patients, including 85 patients (106 lesions) treated with RESs and 73 patients (97 lesions) treated with ZESs. In-stent late lumen loss and binary restenosis were similar among ridaforolimus and zotarolimus patients (Table 4). In-stent late lumen loss was 0.22±0.41 mm (mean, SD) for RESs and 0.23±0.39 mm for ZESs (Pnoninferiority=0.004). No differences in restenosis occurred within the proximal or distal margins of the stents comparing ridaforolimus versus zotarolimus patients (Table 4).
Table 4.
Thirteen-Month Angiographic and Intravascular Ultrasound Results
| Ridaforolimus-Eluting Stent (n=85 Patients, 105 Lesions) | Zotarolimus-Eluting Stent (n=73 Patients, 96 Lesions) | P Value | |
|---|---|---|---|
| Quantitative coronary angiography | |||
| Late lumen loss (mm) | |||
| In-stent | 0.22±0.41 | 0.23±0.39 | 0.85, 0.004 for noninferiority |
| In-segment | 0.17±0.42 | 0.15±0.38 | 0.58 |
| Minimal luminal diameter (mm) | |||
| In-stent | 2.23±0.61 | 2.32±0.58 | 0.40 |
| In-segment | 2.05±0.61 | 2.14±0.54 | 0.26 |
| Diameter stenosis | |||
| Postprocedure in- stent | 20.1±16.5 | 18.7±15.0 | 0.39 |
| Postprocedure in- segment | 25.0±17.4 | 22.8±14.3 | 0.28 |
| Binary restenosis | |||
| In-stent | 8.9 (9/101) | 7.5 (7/93) | 0.73 |
| In-segment | 10.7 (11/103) | 7.5 (7/93) | 0.43 |
| Proximal edge stent restenosis | 1 (1/96) | 1.1 (1/90) | 0.99 |
| Distal edge stent restenosis | 0 (0/99) | 0 (0/93) | NA |
| IVUS results* | |||
| Minimal lumen area (mm2) | |||
| In-stent | 5.8±1.8 | 6.3±2.2 | 0.17 |
| In-segment | 5.0±2.2 | 5.3±2.1 | 0.33 |
| Neointimal hyperplasia (%) | 8.1±5.8 | 8.9±7.8 | 0.95 |
| Neointimal volume (mm3) | 17.4±21.8 | 17.2±17.3 | 0.33 |
| New stent malapposition (%) | 3.7 (2/54) | 0 (0/51) | 0.50 |
IVUS indicates intravascular ultrasound; and NA, not applicable. Values are % (n/N) or mean±SD.
IVUS was performed in 55 patients (61 lesions) in the ridaforolimus group and in 56 patients (60 lesions) in the zotarolimus group.
Follow-up IVUS was performed at 13 months in 111 patients, including 55 and 56 patients in the RES and ZES groups, respectively. No significant difference occurred in percent neointimal hyperplasia between RES and ZES (8.1±5.8% versus 8.6±7.8%, respectively, Pnoninferiority=0.01). Acquired late stent malapposition (because of positive vessel remodeling) occurred in only 2 patients (3.7%) treated with RES and in 0 patients treated with ZES (P=0.49).
DISCUSSION
In this large-scale, randomized clinical trial enrolling patients with less restricted clinical indications and expanded lesion complexity than in most US Food and Drug Administration–approval trials, PCI with a novel RES was noninferior to slow-release ZES for the 1-year outcome of TLF. Indeed, TLF was numerically identical in each treatment group, and revascularization with RES resulted in low rates of MI, target lesion revascularization, and stent thrombosis. Results were consistent in predefined patient and lesion subgroups. These results support the safety and efficacy of RESs in patients representative of everyday clinical practice.
Contemporary DESs vary in numerous ways that may affect device performance and clinical outcomes. Selection, dose, and the pharmacokinetic elution profile of the antiproliferative agent may yield variable results. Differences in polymer formulation and physical properties also regulate both the temporal course and uniformity of antiproliferative drug distribution. The metal alloy and underlying stent construction affects device delivery, expansion characteristics, recoil, side branch access, and uniformity of vessel coverage. The RES was designed to optimize each of these components and overcome limitations of existing DES. Ridaforolimus is associated with a higher therapeutic-to-toxicity margin than most other sirolimus analogues (Appendix I in the online-only Data Supplement), and the elastomeric properties of the polymer confer resistance to disruption in polymer integrity (bonding, webbing, cracking), which may be observed with contemporary DESs.4,5 The stent is constructed of a thin-strut cobalt alloy platform with a variable strut width and an adaptive cell design capable of differential lengthening to accommodate uniform drug distribution in variable vessel anatomy. Unlike other contemporary DESs, persistently low drug concentrations are measurable in the surrounding vascular tissue rather than the more typical initial peak (burst) concentration followed by prolonged diffusion. Finally, the stent is manufactured in an unconventional method in which a thin sheet of cobalt alloy is laser-cut into the stent design, coated with polymer and drug, rolled into a cylindrical shape, and laser-welded at discrete points along its axial length. Compared with more traditional manufacturing methods, this process permits greater efficiency and reproducibility that may translate into cost savings.
Mechanistic insight to the safety and efficacy of RESs is supported by the angiographic and IVUS sub-study from the present study. In addition, simultaneous to BIONICS, patients in Europe were enrolled in the NIREUS trial (BioNIR Ridaforolimus-Eluting Cornary Stent System European Angiography Study), a smaller randomized study (n=302) that also compared ridaforolimus- and zotarolimus-eluting stents.10 This latter study was conducted simultaneously with the BIONICS trial, and without predicate human clinical experience, together the 2 trials represented an ambitious first in-human and key registration clinical program with planned interim analyses to ensure patient safety. Angiographic measures of restenosis and late lumen loss measured at 6 months in NIREUS and at 13 months in BIONICS were similar with both RESs and ZESs, translating into low rates of repeat revascularization. Between 6 and 13 months in these 2 studies, in-stent late loss increased from 0.04±0.31 mm to 0.22±0.41 mm with RESs and from 0.03±0.31 mm to 0.23±0.39 mm with ZESs. Although caution should be applied given different enrollment criteria and sites (even though the same angiographic core laboratory was used), these data suggest some increase in late loss between 6 and 13 months with both stent types, consistent with that reported with other rapamycin analogue-based DESs.11,12 Nevertheless, these results are consistent with both intermediate and late angio-graphic measures for ZESs from prior studies,13–15 and the low degree of late loss at 13 months is a favorable finding that resulted in low rates of angiographic restenosis and ischemia-driven repeat target lesion revascularization. Finally, follow-up IVUS imaging in the current study at 13 months reaffirms minimal neo- intimal hyperplasia, with a low rate of late acquired malapposition consistent with vessel healing without toxicity.
Despite these favorable findings and notwithstanding enrollment of a broader, less restricted study population compared with predicate DES-approval trials,16 the low rates of adverse clinical events in the BIONICS study demonstrate the difficulty in eliciting subtle differences between devices. Despite the relative complexity of the patient population, the 1-year rate of repeat revascularization with RESs in the present study was only 3.2%, and stent thrombosis occurred in only 0.4% of patients, rates not signifi-cantly different than with ZESs. Larger trials perhaps restricted to higher-risk patients and more complex lesions would be required to test whether meaningful differences indeed exist between contemporary DESs. However, even trials randomizing all-comers without restriction enroll a lower-risk population than those patients not randomized but instead followed in a registry.17,18
The present study was underpowered for components of the primary end point and stent thrombosis and indeed was not designed as a superiority trial. Despite the more-comers design, the sample size specific to high-risk patient and lesion subgroups was variable, and interaction testing is inherently underpowered. Thus, caution should be applied when considering the comparative rates of low-frequency events and outcomes in subgroups. Finally, device success was slightly lower with RESs than ZESs, a finding that may be attributable to the first-generation delivery system that has since been addressed by modifying the stent delivery balloon catheter in the current generation device.
Conclusions and Clinical Implications
The present large-scale randomized trial intended to include many patients treated for more complex, off-label clinical indications and lesion anatomy. Clinical, angiographic, and IVUS outcomes with a novel RES were comparable to the slow-release ZES, supporting the safety and effectiveness of RESs in the treatment of a broad population of patients with symptomatic coronary artery disease.
Supplementary Material
Clinical Perspective.
What Is New?
A novel thin-strut, cobalt alloy-based coronary stent incorporates a durable elastomeric polymer eluting the antiproliferative drug ridaforolimus.
Ridaforolimus is associated with a higher therapeutic-to-toxicity margin compared with most other sirolimus analogues; the elastomeric properties confer resistance to disruption of polymer integrity during stent expansion.
The stent is manufactured by laser-cutting a thin cobalt alloy sheet into the stent design, coating it with polymer and drug, and then rolling it into a cylindrical shape.
The BIONICS trial (BioNIR Ridaforolimus-Eluting Coronary Stent System in Coronary Steno-sis) was a prospective, randomized, multicenter study comparing ridaforolimus-eluting stents and zotarolimus-eluting stents in patients undergoing percutaneous coronary intervention.
What Are the Clinical Implications?
In a large-scale, international, randomized trial enrolling patients treated for more complex, off-label clinical indications and lesion anatomy, per-cutaneous coronary intervention with a novel ridaforolimus-eluting stent was noninferior to zotarolimus-eluting stents for the 1-year outcome of target lesion failure.
Treatment with ridaforolimus-eluting stents resulted in low rates of myocardial infarction, repeat revascularization, and stent thrombosis. Results were consistent in predefined patient and lesion subgroups.
Angiographic and intravascular ultrasound measures of restenosis, late lumen loss, and neointimal hyperplasia measured at 13 months were similar with both ridaforolimus-eluting stents and zotarolimus-eluting stents.
Acknowledgments
SOURCES OF FUNDING
This trial was funded by Medinol Ltd, Tel Aviv, Israel.
Dr Kandzari reports receiving consulting fees from Medtronic, Boston Scientific, Micell, and St Jude Medical and receiving research support from Medtronic, Abbott Vascular, Boston Scientific, Biotronik, St Jude Medical, and Medinol. Dr Smits reports receiving consulting fees from Medinol, Abbott Vascular, and St Jude Medical and receiving research support from Abbott Vascular, St Jude Medical, and Terumo. Dr Feldman reports receiving consulting fees from Boston Scientific. Dr Jonas reports receiving consulting fees from Medinol. Dr Chandna reports owning stock options of Gilead Pharma and being on speaker boards for Pfizer, Jennsen, and Novartis Pharma. Dr Leon reports having an equity relationship with Medinol and serving on the medical advisory board of Medtronic. Dr Edelman reports being the founder of and holding equity in BioRest. Dr Ben-Yehuda and Ms Ozan are employees of the Cardiovascular Research Foundation, which received funding from Medi-nol Ltd for the conduct of the trial. Dr Perlman reports being an employee of Medinol.
Footnotes
DISCLOSURES
The other authors report no conflicts.
Contributor Information
David E. Kandzari, Piedmont Heart Institute, Atlanta, GA.
Pieter C. Smits, Maasstad Hospital, Rotterdam, The Netherlands.
Michael P. Love, Queen Elizabeth II Health Sciences Center, Halifax, Nova Scotia.
Ori Ben-Yehuda, Columbia University Medical Center and the Cardiovascular Research Foundation, New York.
Shmuel Banai, Tel Aviv Medical Center, Israel.
Simon D. Rob-inson, Victoria Heart Institute Foundation, British Columbia, Canada.
Michael Jonas, Kaplan Medical Center, Rehovot, Israel.
Ran Kornowski, Rabin Medical Center, Petach Tikva, Israel.
Rodrigo Bagur, Centre Hospitalier Universitaire de Quebec, Canada.
Andres Iniguez, Hospital Meixoeiro, Vigo, Spain.
Haim Danenberg, Hadassah Hebrew University Medical Center, Jerusalem, Israel.
Robert Feldman, MediQuest Research Group, Ocala, FL.
Rajiv Jauhar, North Shore University Hospital, New York.
Harish Chandna, Victoria Heart and Vascular Center, TX.
Manish Parikh, Columbia University Medical Center and the Cardiovascular Research Foundation, New York.
Gidon Y. Perlman, Hadassah Hebrew University Medical Center, Jerusalem, Israel; Medinol Ltd, Tel Aviv, Israel.
Mer-cedes Balcells, Massachusetts Institute of Technology, Cambridge.
Peter Markham, CBSET Inc, Lexington, MA.
Melek Ozgu Ozan, Columbia University Medical Center and the Cardiovascular Research Foundation, New York.
Philippe Genereux, Columbia University Medical Center and the Cardiovascular Research Foundation, New York.
Elazer R. Edelman, Massachusetts Institute of Technology, Cambridge.
Martin B. Leon, Columbia University Medical Center and the Cardiovascular Research Foundation, New York.
Gregg W. Stone, Columbia University Medical Center and the Cardiovascular Research Foundation, New York.
References
- 1.Palmerini T, Biondi-Zoccai G, Della Riva D, Mariani A, Sabaté M, Smits PC, Kaiser C, D’Ascenzo F, Frati G, Mancone M, Genereux P, Stone GW. Clinical outcomes with bioabsorbable polymer- versus durable polymer-based drug-eluting and bare-metal stents: evidence from a comprehensive network meta-analysis. J Am Coll Cardiol. 2014;63:299–307. doi: 10.1016/j.jacc.2013.09.061. [DOI] [PubMed] [Google Scholar]
- 2.Bangalore S, Kumar S, Fusaro M, Amoroso N, Attubato MJ, Feit F, Bhatt DL, Slater J. Short- and long-term outcomes with drug-eluting and bare-metal coronary stents: a mixed-treatment comparison analysis of 117 762 patient-years of follow-up from randomized trials. Circulation. 2012;125:2873–2891. doi: 10.1161/CIRCULA-TIONAHA.112.097014. [DOI] [PubMed] [Google Scholar]
- 3.Palmerini T, Biondi-Zoccai G, Della Riva D, Stettler C, Sangiorgi D, D’Ascenzo F, Kimura T, Briguori C, Sabatè M, Kim HS, De Waha A, Kedhi E, Smits PC, Kaiser C, Sardella G, Marullo A, Kirtane AJ, Leon MB, Stone GW. Stent thrombosis with drug-eluting and bare-metal stents: evidence from a comprehensive network meta-analysis. Lancet. 2012;379:1393–1402. doi: 10.1016/S0140-6736(12)60324-9. [DOI] [PubMed] [Google Scholar]
- 4.Basalus MW, Ankone MJ, van Houwelingen GK, de Man FH, von Birgelen C. Coating irregularities of durable polymer-based drug-eluting stents as assessed by scanning electron microscopy. EuroIntervention. 2009;5:157–165. doi: 10.4244/eijv5i1a24. [DOI] [PubMed] [Google Scholar]
- 5.Farooq V, Gogas BD, Serruys PW. Restenosis: delineating the numerous causes of drug-eluting stent restenosis. Circ Cardiovasc Interv. 2011;4:195–205. doi: 10.1161/CIRCINTERVENTIONS.110.959882. [DOI] [PubMed] [Google Scholar]
- 6.Cutlip DE, Windecker S, Mehran R, Boam A, Cohen DJ, van Es GA, Steg PG, Morel MA, Mauri L, Vranckx P, McFadden E, Lansky A, Hamon M, Krucoff MW, Serruys PW, Academic Research Consortium Clinical end points in coronary stent trials: a case for standard-ized definitions. Circulation. 2007;115:2344–2351. doi: 10.1161/CIRCULATIONAHA.106.685313. [DOI] [PubMed] [Google Scholar]
- 7.Moussa ID, Klein LW, Shah B, Mehran R, Mack MJ, Brilakis ES, Reilly JP, Zoghbi G, Holper E, Stone GW. Consideration of a new definition of clinically relevant myocardial infarction after coronary revascularization: an expert consensus document from the Society for Cardiovascular Angiog-raphy and Interventions (SCAI) J Am Coll Cardiol. 2013;62:1563–1570. doi: 10.1016/j.jacc.2013.08.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thygesen K, Alpert JS, Jaffe AS, Simoons ML, Chaitman BR, White HD, the Writing Group on behalf of the Joint ESC/ACCF/AHA/WHF Task Force for the Universal Definition of Myocardial Infarction Third universal definition of myocardial infarction. Circulation. 2012;126:2020–2035. doi: 10.1161/CIR.0b013e31826e1058. [DOI] [PubMed] [Google Scholar]
- 9.Mehran R, Dangas G, Abizaid AS, Mintz GS, Lansky AJ, Satler LF, Pich-ard AD, Kent KM, Stone GW, Leon MB. Angiographic patterns of in-stent restenosis: classification and implications for long-term outcome. Circulation. 1999;100:1872–1878. doi: 10.1161/01.cir.100.18.1872. [DOI] [PubMed] [Google Scholar]
- 10.Smits P. Primary endpoint results from the NIREUS trial: a prospective randomised trial comparing the novel ridaforolimus-eluting BioNIR stent to the zotarolimus-eluting resolute stent. Presented at Transcatheter Therapeutics 2016; Washington, DC. October 31, 2016. [Google Scholar]
- 11.Serruys PW, Ruygrok P, Neuzner J, Piek JJ, Seth A, Schofer JJ, Richardt G, Wiemer M, Carrié D, Thuesen L, Boone E, Miquel-Herbert K, Daemen J. A randomised comparison of an everolimus-eluting coronary stent with a paclitaxel-eluting coronary stent: the SPIRIT II trial. EuroIntervention. 2006;2:286–294. [PubMed] [Google Scholar]
- 12.Stone GW, Midei M, Newman W, Sanz M, Hermiller JB, Williams J, Farhat N, Mahaffey KW, Cutlip DE, Fitzgerald PJ, Sood P, Su X, Lansky AJ, SPIRIT III Investigators Comparison of an everolimus-eluting stent and a paclitaxel-eluting stent in patients with coronary artery disease: a randomized trial. JAMA. 2008;299:1903–1913. doi: 10.1001/jama.299.16.1903. [DOI] [PubMed] [Google Scholar]
- 13.Meredith IT, Worthley S, Whitbourn R, Walters D, Popma J, Cutlip D, Fitzgerald P. The next-generation Endeavor Resolute stent: 4-month clinical and angiographic results from the Endeavor Resolute first-in-man trial. EuroIntervention. 2007;3:50–53. [PubMed] [Google Scholar]
- 14.Meredith IT, Worthley S, Whitbourn R, Walters DL, McClean D, Hor-rigan M, Popma JJ, Cutlip DE, DePaoli A, Negoita M, Fitzgerald PJ, RESOLUTE Investigators Clinical and angiographic results with the next-generation resolute stent system: a prospective, multicenter, first-inhuman trial. JACC Cardiovasc Interv. 2009;2:977–985. doi: 10.1016/j.jcin.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 15.Stefanini GG, Serruys PW, Silber S, Khattab AA, van Geuns RJ, Richardt G, Buszman PE, Kelbæk H, van Boven AJ, Hofma SH, Linke A, Klauss V, Wijns W, Macaya C, Garot P, Di Mario C, Manoharan G, Kornowski R, Ischinger T, Bartorelli AL, Gobbens P, Windecker S. The impact of patient and lesion complexity on clinical and angiographic outcomes after revascularization with zotarolimus- and everolimus-eluting stents: a substudy of the RESOLUTE All Comers Trial (a randomized comparison of a zotarolimus-eluting stent with an everolimus-eluting stent for percutaneous coronary intervention) J Am Coll Cardiol. 2011;57:2221–2232. doi: 10.1016/j.jacc.2011.01.036. [DOI] [PubMed] [Google Scholar]
- 16.Kereiakes DJ, Meredith IT, Windecker S, Lee Jobe R, Mehta SR, Sarembock IJ, Feldman RL, Stein B, Dubois C, Grady T, Saito S, Kimura T, Christen T, Allocco DJ, Dawkins KD. Efficacy and safety of a novel bioabsorbable polymer-coated, everolimus-eluting coronary stent: the EVOLVE II Randomized Trial. Circ Cardiovasc Interv. 2015;8:e002372. doi: 10.1161/CIRCINTERVENTIONS.114.002372. [DOI] [PubMed] [Google Scholar]
- 17.Serruys PW, Silber S, Garg S, van Geuns RJ, Richardt G, Buszman PE, Kel-baek H, van Boven AJ, Hofma SH, Linke A, Klauss V, Wijns W, Macaya C, Garot P, DiMario C, Manoharan G, Kornowski R, Ischinger T, Bartorelli A, Ronden J, Bressers M, Gobbens P, Negoita M, van Leeuwen F, Windecker S. Comparison of zotarolimus-eluting and everolimus-eluting coronary stents. N Engl J Med. 2010;363:136–146. doi: 10.1056/NEJMoa1004130. [DOI] [PubMed] [Google Scholar]
- 18.Sen H, Tandjung K, Basalus MW, Löwik MM, van Houwelingen GK, Stoel MG, Louwerenburg HW, de Man FH, Linssen GC, Nijhuis R, Nienhuis MB, Verhorst PM, van der Palen J, von Birgelen C. Comparison of eligible non-enrolled patients and the randomised TWENTE trial population treated with Resolute and Xience V drug-eluting stents. EuroIntervention. 2012;8:664–671. doi: 10.4244/EIJV8I6A104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.