

ABSTRACT
Immunotherapies targeting programmed cell death protein 1 (PD-1) or its ligand, programmed cell death ligand 1 (PD-L1), dramatically improve the survival of melanoma patients. However, only ∼40% of treated patients demonstrate a clinical response to single-agent anti-PD-1 therapy. An intact tumor response to type-II interferon (i.e. IFN-γ) correlates with response to anti-PD-1, and patients with de novo or acquired resistance may harbor loss-of-function alterations in the JAK/STAT pathway, which lies downstream of the interferon gamma receptor (IFNGR1/2). In this study, we dissected the specific roles of individual JAK/STAT pathway members on the IFN-γ response, and identified JAK1 as the primary mediator of JAK/STAT signaling associated with IFN-γ-induced expression of antigen-presenting molecules MHC-I and MHC-II, as well as PD-L1 and the cytostatic response to IFN-γ. In contrast to the crucial role of JAK1, JAK2 was largely dispensable in mediating most IFN-γ effects. In a mouse melanoma model, inhibition of JAK1/2 in combination with anti-PD-L1 therapy partially blocked anti-tumor immunologic responses, while selective JAK2 inhibition appeared to augment therapy. Amplification of JAK/STAT signaling in tumor cells through genetic inhibition of the negative regulator PTPN2 potentiated IFN-γ response in vitro and in vivo, and may be a target to enhance immunotherapy efficacy.
KEYWORDS: IFN-γ, JAK/STAT, PTPN2, checkpoint immunotherapy
Introduction
Melanoma is a highly aggressive type of skin cancer and is the 5th most common cancer, approaching 5% of all new cancer cases in the USA. Approximately 50% of melanomas harbor activating BRAF mutations which render constitutive and sustained activation of the MAPK pathway and promote survival and proliferation of melanoma cells.1,2 While targeted inhibition of the Ras/MAPK pathway has offered a precision medicine approach to treating melanoma, the introduction of immunotherapy has dramatically improved survival of melanoma patients, regardless of BRAF status.
Immune checkpoint inhibitors targeting PD-1 (i.e., nivolumab, and pembrolizumab) have been approved for treatment of advanced melanoma.3,4 Treatment with nivolumab, or pembrolizumab strikingly improves the clinical outcome in advanced melanoma patients, but only ∼40% of melanoma patients benefit from anti-PD-1. Therefore, both the search for biomarkers that accurately identify patients who may benefit from immunotherapy and approaches to augment immunotherapy efficacy remain important areas of investigation. In this regard, PD-L1 has been clinically validated in three large phase III studies as a predictive biomarker that enriches for patients who may benefit from anti-PD-1/PD-L1 immunotherapy.5 In addition, IFN-γ, MHC-I, and MHC-II are also predictive of clinical benefit from pembrolizumab.6-8
Interferon gamma (IFN-γ) is a soluble cytokine and the only member of the type-II interferon family. IFN-γ plays an important role in innate and adaptive immunity and also demonstrates anti-viral, immune-regulatory, and anti-tumor activity. IFN-γ is produced primarily by natural killer (NK) cells, natural killer T (NKT) cells, CD4 helper T cells, and CD8 cytotoxic T cells and initiates signaling by binding to a heterodimeric receptor composed of interferon gamma receptor 1 (IFNGR1) and interferon gamma receptor 2 (IFNGR2). After dimerized IFN-γ binds to its receptor, IFN-γ induces trans-phosphorylation and activation of JAK1 and JAK2. Activated JAK1/2 triggers the IFNGR to provide a STAT docking site through the SH2 domain and to recruit STATs to the JAK-IFNGR complex. After recruitment, STATs becomes activated and form dimers, allowing them to translocate to the nucleus to promote the expression of target genes involved in cell proliferation, differentiation and inflammation.9,10
IFN-γ and its regulated genes have been shown in numerous studies to be associated with immunotherapy responsiveness in cancer patients and in mouse models.11-18 In a large-scale CRISPR/Cas9 screen, tumor cell-specific loss of Ifngr1 and Ifngr2, the cognate receptors for IFN-γ, resulted in tumors growing through anti-PD-1 with granulocyte-macrophage colony-stimulating factor (GM-CSF)-secreting, irradiated tumour cell vaccine (GVAX).19 Thus, early preclinical and clinical data support a critical role for IFN-γ in tumor response to T cell-targeting immunotherapies.
IFN-γ secretion by cytotoxic T lymphocytes and Th1 T helper cells induces the expression of MHC-I, MHC-II, and PD-L1 via the JAK/STAT pathway in tumor cells and stroma.20-22 However, the specific roles of individual members of the JAK/STAT pathway in IFN-γ-induced immune response, remain unclear. Interestingly, JAK1 and JAK2 loss-of-function alterations occur in melanoma patients who are de novo-resistant or acquire resistance to anti-CTLA4 or anti-PD-1/PD-L1 immunotherapy.15-17 This finding suggests that signaling via the JAK/STAT pathway in melanoma is critical for inflammation signals that trigger anti-tumor immune responses in patients. As targeted inhibitors become more selective for individual JAK/STAT family members, and as numerous clinical trials are initiated testing combinations of these inhibitors with checkpoint therapy (e.g. NCT03012230), identification of specific members of the JAK/STAT pathway that are required for response to immune checkpoint inhibitor therapy is critical.
In this study, using both pharmacological and genetic approaches, we identify JAK1 as the primary mediator of IFN-γ-induced JAK/STAT pathway activation. JAK1 is required for IFN-γ mediated expression of MHC-I, MHC-II, and PD-L1, while JAK2 has an ancillary or dispensable role in melanoma cells. In addition, we find that combined JAK1/2 inhibition with ruxolitinib suppresses tumor response to immunotherapy using a murine melanoma cell line (YUMM2.1) that typically responds to anti-PD-L1 immunotherapy, while selective JAK2 inhibition appears to improve response. JAK2 inhibition across multiple melanoma cell line models demonstrates selectivity for inhibiting oncogenic STAT3 and STAT5 activation, while sparing STAT1 activation, downstream of the IFN-γ receptor. Conversely, we demonstrate that genetic suppression of PTPN2, a negative regulator of the JAK/STAT pathway, augments JAK/STAT signaling and anti-PD-L1 response using a murine melanoma cell line that is innately resistant to anti-PD-L1 monotherapy (YUMM1.1). Our results indicate that PTPN2 (recently identified in a large in vivo CRISPR screen(19)) serves as a potential target to amplify the anti-PD-L1 efficacy by modulating cellular JAK/STAT responses to IFN-γ. Furthermore, our data suggest that combined JAK1/2 inhibition or selective JAK1 inhibition may thwart immunotherapy responses in combination, and these approaches should be used with caution in patients.
Materials and methods
Cell lines and treatment
Human melanoma cell lines A375, SKMEL5, SKMEL28, CHL-1 and HMCB were obtained from ATCC and grown in DMEM + 10% fetal bovine serum (FBS). WM115 was obtained from ATCC and cultured in EMEM + 10% FBS. MEWO was a gift from the laboratory of Jonathan Irish (Vanderbilt University) and was cultured in DMEM + 10% FBS. Murine melanoma cell lines YUMM1.1 and YUMM2.1, generated by Dr. Marcus Bosenberg (Yale University), were provided by the laboratory of Antoni Ribas (UCLA), with permission from Dr. Bosenberg, and cultured in DMEM + 10% FBS and 2 nmol/L L-glutamine. To determine IFN-γ-induced immune response markers expression, melanoma cells were pre-treated either with 1 μM ruxolitinib (SelleckChem) or 1 μM NVP-BSK805 (kindly provided by Novartis) for 30mins, then treated with 100 ng/ml recombinant (mouse or human, depending on cell line derivation) IFN-γ (Gibco) for 24 hrs. To determine IFN-γ-induced STAT phosphorylation, melanoma cells were pre-treated either with 1 μM ruxolitinib or 1 μM NVP-BSK805 for 30 mins, then stimulated with 100 ng/ml recombinant (appropriate species) IFN-γ for another 30 mins.
siRNA transfection
Cells in 6-well plates were transfected with a siRNA targeting JAK1 (s7647, Ambion), JAK2 (s7649, Ambion), STAT1 (s279, Ambion), STAT3 (s745, Ambion), STAT5A (s13535, Ambion), STAT5B (s13539, Ambion), PTPN2 (s11509, s11510, Ambion) or non-silencing control using Dharmafect-1 (Dharmacon) transfection reagent according to the manufacturer's protocol. Forty-eight hours after transfection, melanoma cells were treated with 100 ng/ml recombinant IFN-γ either for 24hrs or 30mins.
Standard flow cytometry
Cells were washed in phosphate-buffered saline (PBS) and harvested with Accutase (EMD Millipore, #SCR005) for 1 min at 37 °C. Dissociated cells were washed once in flow staining buffer and incubated with respective flow antibodies at 4 °C for 30 min in dark. Flow cytometry was performed using the following antibodies: HLA-A,B,C/Alexa Fluor488 (Biolegend, clone W6/32, 1:200), HLA-DR/PE-Cy7 (Biolegend, clone L243, 1:200), and CD274(PD-L1)/APC (Biolegend, clone 29E.2A3, 1:200). mouse MHC-I (H-2Kd/H-2Dd)/PE (Biolegend, clone 34-1-2 S, 1:200), mouse MHC-II (I-A/I-E)/Alexa Fluor488 (Biolegend, clone M5/114.15.2, 1:200), mouse CD274(PD-L1)/APC (Biolegend, clone 10 F.9 G2, 1:200), and mouse EpCAM/PE-Cy7 (Biolegend, clone G8.8, 1:350). DAPI was used as a viability dye for dead cell exclusion. Samples were analyzed on an Attune NxT flow cytometer (Life Technologies).
Phospho-flow cytometry
Cells were harvested with Accutase for 1 min at 37 °C. Dissociated cells were stimulated with 100 ng/ml IFN-γ at 37 °C for 30 min in a CO2 incubator. Then cells were fixed for 5 min at room temperature with a final concentration of 1.6% paraformaldehyde (Electron Microscopy Services). Cells were pelleted and permeabilized by resuspension in 2 ml of methanol and stored over night at -20 °C. Cells were washed 3 times in flow staining buffer and incubated with respective phospho-flow antibodies at 4 °C for 30 min in the dark. Flow cytometry was performed using the following antibodies: p-STAT1/Alexa Fluor488 pY701 (BD Phosflow, clone 4 a, 1:100), p-STAT3/Alexa Fluor647 pY705 (BD Phosflow, clone 4, 1:100), and p-STAT5/PE-Cy7 pY694 (BD Phosflow, clone 47, 1:10). Samples were analyzed on an Attune NxT system (Life Technologies).
Establishment of YUMM1.1-shPTPN2 stable cell lines
Murine PTPN2 shRNA (shPTPN2) was selected from the pGIPZ lentiviral shRNAmir library (GE Dharmacon). The shRNA sequence utilized was 5’-TGCTGTTGACAGTGAGCGAGCACAAAGAAGTTACATCTTATAGTGAAGCCACAGATGTATAAGATGTAACTTCTTTGTGCCTGCCTACTGCCTCGGA-3’. The shPTPN2 plasmid was transfected using Fugene HD (Promega) into 293FT cells along with psPAX2 and pMD2G helper plasmids (Addgene) in order to produce lentivirus. Lentiviral-rich conditioned media was applied to YUMM1.1 cells in the presence of polybrene for two days, prior to selection with 1 ug/ml puromycin.
Immunoblotting
Immunoblotting was performed as previously described.23 Briefly, cells were washed in cold phosphate-buffered saline, collected and lysed in 1X RIPA buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1.0% NP-40, 0.5% Deoxycholic Acid, 0.1% SDS, 1 mM EDTA, 1 mM EGTA, 5 mM sodium pyrophosphate, 50 mM NaF, 10 mM b-glycerophosphate) with added phosphatase inhibitors (PhosSTOP, Roche) and protease inhibitors (cOmplete, Roche) for 30 min on ice. Lysates were, centrifuged at 13,000 rpm for 15 min at 4 °C. Protein concentrations of the lysates were determined by BCA assay (Thermo). Samples were separated by 8% SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blocked with 5% non-fat dry milk or 5% BSA in tris-buffered saline (TBS) with 0.1% Tween-20 for 1 h at room temperature and then incubated overnight at 4 °C with the appropriate antibody in blocking buffer as indicated. Following incubation with appropriate horseradish peroxidase-conjugated secondary antibodies, proteins were visualized using an enhanced chemiluminescence detection system (Thermo). This study was performed using the following antibodies: JAK1 (#3332), JAK2 (#3230), p-STAT1 (Y701) (#7649), p-STAT3 (Y705) (#9145), p-STAT5 (Y694) (#9359), STAT3 (#9139), PTPN2 (#58935) and PD-L1 (#13684), all of which were purchased from Cell Signaling Technologies. β-actin (sc-47778), calnexin (sc-11397), STAT1 (sc-592), STAT5 A (sc-1081), STAT5B (sc-1656), HLA-A/B/C (sc-52810) and HLA-DR (sc-53319), all of which were purchased from Santa Cruz Biotechnology.
Immunohistochemistry
The following antibodies and conditions were used for IHC: p-STAT1 (Tyr701), Cell Signaling, Catalog# 9167, antigen retrieval HpH Buffer pH9 (Decloaking Chamber), one-hour incubation at room temperature, dilution 1:100, Envision, DAB (Dako); p-STAT3 (Tyr705), Cell Signaling, Catalog# 9145, antigen retrieval HpH Buffer pH9 (Decloaking Chamber), overnight incubation at 4 °C, dilution 1:100, Envision, DAB (Dako); p-STAT5 (Y694), Cell Signaling, Catalog# 9359, antigen retrieval HpH Buffer pH9 (Decloaking Chamber), overnight incubation at 4 °C, dilution 1:100, Envision, DAB (Dako); tGFP, ThermoFisher, Catalog# PA5-22688, antigen retrieval Citrate Buffer pH6 (Decloaking Chamber), overnight incubation at 4 °C, dilution 1:8,000, Envision, DAB (Dako). Stained tumor sections were scored at 40X magnification by a medical research pathologist (MES and PIG-E) and quantified as % tumor cells expressing the indicated marker (nuclear specific staining for p-STAT1/3/5 and cytoplasmic staining for tGFP).
Viability assays
Viability was determined by sulfarhodamine B (SRB), as previously described.23-26
In vivo mouse models
All mouse studies were performed with prior protocol approval and in compliance with local and national ethical animal use guidelines and committees. To establish subcutaneous (s.c.) tumors, 1 × 106 YUMM2.1, 1 × 106 YUMM1.1, or 1 × 106 YUMM1.1-shPtpn2 cells per mouse were injected s.c. into the flanks of female C57 BL/6 J mice or athymic nude mice. Mice were randomized for treatment when tumors ≥50 mm3. For the YUMM2.1 mouse model, mice were randomized to 4 groups: 1) IgG (BioXCell, Clone LTF-2) (100 ug, i.p. every 3 days), 2) anti-PD-L1 (BioXCell, Clone 10 F.9 G2) (100 ug i.p. every 3 days), 3) anti-PD-L1 + ruxolitinib (60 mg/kg/day, p.o.), or 4) anti-PD-L1 + BSK805 (60 mg/kg/day, p.o.). For the YUMM1.1/shPtpn2 mouse model, mice were randomized to 2 groups: 1) IgG (100 ug, i.p. every 3 days), or 2) anti-PD-L1 (100 ug, i.p. every 3 days). For nude mouse experiments, no therapy was given, and tumors were tracked 3 times weekly for 30 days. Tumor diameters were measured using calipers twice per week and volume in mm3 calculated with the formula: volume = width2 x length/2.
Statistics
For flow cytometry, geometric mean fluorescence intensity (GMFI) analysis, and IFN-γ-induced inhibition of cell proliferation assays, significant differences were determined by ANOVA with a Tukey's post hoc-test for multiple comparisons. A p-value of <0.05 was considered statistically significant. Bar graphs show mean ± SD, unless otherwise stated in the Fig. legend. For all comparisons, statistical significance is noted by *p<0.05; **p<0.01, and ***p<0.001.
Results
JAK1 is the primary mediator of IFN-γ-induced MHC-I, MHC-II and PD-L1 expression
IFN-γ is a key cytokine in the adaptive immune response. Mutations in the IFN-γ/JAK/STAT pathway render melanoma patients unresponsive to immunotherapy.15-17 In order to determine which components of this pathway are critical to the IFN-γ response, we studied the specific role of JAK1 and JAK2 (JAK3 is not expressed in melanoma tumor cells) on the IFN-γ-regulated proteins MHC-I, MHC-II, and PD-L1. MHC-I and MHC-II are required for antigen presentation to CD8+ and CD4+ T cells, respectively, and are therefore important markers in the adaptive immune response. PD-L1 is robustly regulated by IFN-γ, and all three of these molecules are associated with anti-PD-L1 response in patients.6-8 We first investigated IFN-γ-induced expression of MHC-I, MHC-II, and PD-L1 after treatment with NVP-BSK805 (a specific JAK2 inhibitor, BSK) and ruxolitinib (a JAK1/2 inhibitor, RUX) in BRAF-mutant melanoma cell lines (A375, WM115, SKMEL5, and SKMEL28) and BRAF-wildtype melanoma cell lines (CHL-1, HMCB, and MEWO) by flow cytometry. IFN-γ increased MHC-I and PD-L1 in each cell line, and induced MHC-II in human BRAF-mutant cell lines, but not in BRAF-wildtype cell lines, an observation we previously noted.6 Surprisingly, however, selective JAK2 inhibition with NVP-BSK805 largely spared IFN-γ-induced expression of MHC-I and MHC-II, but significantly decreased IFN-γ-induced expression of PD-L1 (Fig. 1A and Supplementary Fig. 1B). In contrast, combined JAK1/2 inhibition with ruxolitinib completely abrogated IFN-γ-induced expression of MHC-I, MHC-II, and PD-L1. These results indicate that JAK1 is required for IFN-γ-induced expression of MHC-I, MHC-II and PD-L1, whereas JAK2 is dispensable for MHC-I/II induction, but contributes to induction of PD-L1.
Figure 1.

JAK1 shows a greater contribution to IFN-γ-induced expression of MHC-I, MHC-II, and PD-L1 than JAK2. (A) A heatmap illustrates MHC-I, MHC-II, and PD-L1 GMFI in seven human melanoma cell lines under different treatment conditions as determined by flow cytometry. The red color denotes higher expression levels whereas the blue color denotes lower expression levels. The data are the average of at least 4 independent experiments, and the individual plots appear in Supplemental Fig. 1. (B) Western blots show IFN-γ-induced MHC-I, MHC-II, and PD-L1 expression levels in BRAFmutant and BRAF-wildtype melanoma cell lines under conditions of non-targeting control siRNA (NTC), specific JAK1 siRNA (siJAK1) or JAK2 siRNA (siJAK2) transfection. Knockdown efficiency was confirmed by examining JAK1 or JAK2 expression and actin was used as a loading control. (C) Western blots show IFN-γ-induced MHC-I, MHC-II, and PD-L1 expression levels in BRAF-mutant and BRAF-wildtype melanoma cell lines under conditions of non-targeting control siRNA (NTC), specific JAK1 siRNA (siJAK1) and JAK2 siRNA (siJAK2) transfection. Knockdown efficiency was confirmed by examining JAK1 or JAK2 expression and actin was used as a loading control.
To confirm these findings using genetic manipulation, we treated melanoma cell lines with IFN-γ after JAK1 siRNA (siJAK1) or JAK2 siRNA (siJAK2) transfection (Fig. 1B). siJAK1 decreased IFN-γ-induced MHC-I expression, an effect which was primarily observed in BRAF-wildtype cell lines and modestly decreased IFN-γ-induced MHC-II expression in 3 of 4 BRAF-mutant cell lines. siJAK1 also substantially decreased IFN-γ-induced PD-L1 expression in all melanoma cell lines. We also found that JAK2 knockdown did not substantially alter IFN-γ-induced expression of MHC-I, had variable effects on MHC-II expression and broadly repressed IFN-γ-induced PD-L1 expression in all melanoma cell lines. Taken together, the above results suggest that JAK1 is the dominant mediator of IFN-γ-induced expression of MHC-I, MHC-II and PD-L1 whereas JAK2 contributes only to IFN-γ-induced expression of PD-L1.
We observed that the specific knockdown of either JAK1 or JAK2 only modestly impeded IFN-γ-induced MHC-I or MHC-II expression in BRAF-mutant cells, but combined chemical inhibition of both JAK1 and JAK2 completely abrogated cell surface expression of both markers in BRAF-mutant cells. Therefore, we asked whether there are overlapping or redundant roles for JAK1 and JAK2 in IFN-γ-induced MHC-I, MHC-II, or PD-L1 expression in BRAF-mutant cell lines. To answer this question, we used siJAK1 and siJAK2 inhibition, followed by IFN-γ treatment in BRAF-mutant and BRAF-wildtype melanoma cell lines (Fig. 1C). Although knockdown was incomplete, we observed moderately enhanced effects of JAK1/2 knockdown on expression of MHC-I and II, suggesting that BRAF-mutant cell lines may selectively engage both JAK1 and JAK2 downstream of IFN-γ activation, leading to partial redundancy. In contrast, only JAK1 is required to induce PD-L1 expression in both BRAF-wildtype and BRAF-mutant cells. In BRAF-wildtype cells, the effect of a simultaneous knockdown of both JAK1 and JAK2 appeared similar to the effect of a specific knockdown of JAK1 alone. Thus, our data support the concept that JAK1 is the primary mediator of IFN-γ-induced MHC-I, MHC-II, and PD-L1 expression (particularly in BRAF-wildtype cells), while JAK2 plays a limited accessory role. Given that inhibition of JAK1, JAK2, or both JAK1/2 incompletely abrogated both basal and IFN-γ-induced expression of MHC-I and II, these data also point to the possibility that other molecules besides JAKs may play a contributory role in mediating IFN-γ-induced signals.
JAK1 is the mediator of IFN-γ-stimulated STAT phosphorylation
STATs are the downstream effectors of JAK1 and JAK2 following IFN-γ stimulation. However, the specific JAK members that activate distinct STATs in the IFN-γ signaling pathway remain unresolved. Thus, we next examined the contribution of JAK1 and JAK2 to IFN-γ stimulated phosphorylation of STAT1, STAT3, and STAT5 using NVP-BSK805 or ruxolitinib (Fig. 2A). Our results indicated that selective JAK2 inhibition with NVP-BSK805 largely spared phosphorylation of STAT1 while suppressing STAT3 and STAT5 phosphorylation following IFN-γ stimulation in most cell lines. In contrast, combined JAK1/2 inhibition with ruxolitinib blocked phosphorylation of STAT1, STAT3, and STAT5 upon IFN-γ stimulation.
Figure 2.

JAK1 shows a greater contribution to IFN-γ stimulated expression of p-STAT1, p-STAT3, and p-STAT5 expression than JAK2. (A) Western blots demonstrating the effect of RUX or BSK pretreatment on the IFN-γ stimulated expression levels of p-STAT1, p-STAT3, and p-STAT5 in BRAF-mutant and BRAF-wildtype melanoma cell lines. Actin was used as a loading control. (B) Western blots demonstrating IFN-γ-induced p-STAT1, p-STAT3, and p-STAT5 expression levels in BRAF-mutant and BRAF-wildtype melanoma cell lines under conditions of non-targeting control siRNA (NTC), specific JAK1 siRNA (siJAK1), or JAK2 siRNA (siJAK2) transfection. (C) Western blots demonstrating IFN-γ-induced p-STAT1, p-STAT3, and p-STAT5 expression levels in BRAF-mutant and BRAF-wildtype melanoma cell lines under conditions of non -targeting control siRNA (NTC), specific JAK1 siRNA (siJAK1), and JAK2 siRNA (siJAK2) transfection.
Next, we studied IFN-γ stimulated STAT1, STAT3, and STAT5 phosphorylation after siJAK1 or siJAK2 transfection (Fig. 2B). We found that JAK1 knockdown decreased IFN-γ-induced phosphorylation of STAT1 and STAT3, which was again most pronounced in BRAF-wildtype cell lines upon IFN-γ stimulation. JAK1 knockdown also substantially decreased phosphorylation of STAT5 upon IFN-γ stimulation. In contrast, JAK2 knockdown primarily decreased STAT5 and, to a lesser degree, STAT3 phosphorylation upon IFN-γ stimulation while largely sparing STAT1 phosphorylation. Taken together, these results suggest that JAK1 is required for IFN-γ-stimulated phosphorylation of STAT1, STAT3 and STAT5, whereas JAK2 is again largely dispensable for STAT1 signaling and primarily contributes to IFN-γ stimulated STAT3 and STAT5 phosphorylation. Interestingly, STAT1 is generally known to mediate inflammatory signals in cancer cells, while STAT3 and STAT5 are thought to be oncogenic mediators. Thus, there is a potential role for selective inhibition of JAK2 in cancer to downregulate oncogenic STAT signaling, while preserving inflammatory signals required for anti-tumor immunity.
Since the specific knockdown of either JAK1 or JAK2 did not completely abrogate IFN-γ-induced STAT phosphorylation in BRAF-mutant cells, we asked whether JAK1 and JAK2 demonstrate redundant roles in IFN-γ-induced STAT phosphorylation in these cell lines. Thus, we tested STAT1, STAT3, and STAT5 phosphorylation following IFN-γ stimulation after combined siJAK1 and siJAK2 transfection (Fig. 2C). The effect of simultaneous knockdown of both JAK1 and JAK2 appeared similar to the effect of a specific knockdown of JAK1 alone with regard to IFN-γ-induced STAT1 phosphorylation, although the effect on STAT3 phosphorylation was greater. Nonetheless, these data again point to the possibility that other molecules besides JAKs may play a role in IFN-γ-induced STAT1 phosphorylation in BRAF-mutant cell lines.
STAT1 is the primary downstream molecule of JAKs that mediates IFN-γ-induced expression of MHC-I, MHC-II and PD-L1
Next, we asked which specific STAT molecules downstream of JAK1/2 mediate IFN-γ-induced expression of MHC-I, MHC-II and PD-L1. Thus, we investigated IFN-γ-induced expression of these markers after siSTAT1, siSTAT3, siSTAT5A, or siSTAT5B transfection (Fig. 3A). We found that siSTAT1 modestly decreased IFN-γ-induced expression of MHC-I and MHC-II, but this effect varied substantially among cell lines. Furthermore, siSTAT1 decreased IFN-γ-induced expression of PD-L1 (with the exception of SKMEL28) while STAT3, STAT5A, or STAT5B knockdown did not alter IFN-γ-induced expression of MHC-I, MHC-II, and PD-L1. We assessed the impact of combined STAT5A, and STAT5B knockdown in IFN-γ-induced PD-L1 finding only modest diminution of PD-L1 induction in IFN-γ treated cells, thus suggesting a relatively minor role for STAT5A/B in this context. (Fig. 3B). The results suggest that STAT1 plays a dominant role, whereas STAT3, STAT5A and STAT5B have limited roles in IFN-γ-induced expression of PD-L1.
Figure 3.

STAT1 is the primary molecule downstream of JAKs involved in IFN-γ-induced expression of MHC-I, MHC-II, and PD-L1. (A) Western blots show IFN-γ-induced MHC-I, MHC-II, and PD-L1 expression in BRAF-mutant and BRAF-wildtype melanoma cell lines under conditions of non-targeting control siRNA (NTC), specific siRNA for STAT1 (siSTAT1), STAT3 (siSTAT3), STAT5A (siSTAT5A), and STAT5B (siSTAT5B) transfection. Knockdown efficiency was confirmed by examining STAT1, STAT3, STAT5A, or STAT5B expression and actin was used as a loading control. (B) Western blots show IFN-γ-induced PD-L1 expression in BRAF-mutant and BRAF-wildtype melanoma cell lines under conditions of non-targeting control siRNA (NTC), specific siRNA for STAT5A (siSTAT5A) and STAT5B (siSTAT5B) transfection.
JAK1 is the primary mediator IFN-γ-induced inhibition of cell proliferation
Next, we investigated the role of JAK/STAT signaling in IFN-γ-induced inhibition of cell proliferation after JAK1, JAK2, STAT1, STAT3, STAT5A, or STAT5B (or STAT5A and STAT5B) siRNA transfection across both BRAF-wildtype and mutant cell lines (Fig. 4A and 4B, Supplementary Fig. 3). IFN-γ treatment significantly inhibited cell proliferation in all melanoma cell lines. Interestingly, siJAK1, and in some cases, siSTAT1 abrogated IFN-γ-induced inhibition of cell proliferation, while siJAK2 only modestly protected against IFN-γ-induced inhibition of cell proliferation in the SKMEL5 and MEWO cell lines. In contrast, STAT3, STAT5A, or STAT5B knockdown had, little effect on IFN-γ-induced inhibition of cell proliferation. Taken together, these results indicate that JAK1 (and to some degree, STAT1) mediates IFN-γ-induced inhibition of cell proliferation. In contrast, JAK2 has the potential to modestly mediate IFN-γ-induced inhibition of cell proliferation in certain cell lines, but this effect is substantially less than JAK1.
Figure 4.

Knockdown of JAK1 partially recovers IFN-γ-induced inhibition of cell proliferation. (A) Graphs show IFN-γ-induced inhibition of cell proliferation in BRAF-mutant and BRAF-wildtype melanoma cell lines under conditions of nontargeting control (NTC) or specific siRNA (siJAK1, siJAK2, siSTAT1, siSTAT3, siSTAT5A, and siSTAT5B) transfection during a 96 hr period. The box graphs denote an absorbance ratio of IFN-γ treatment to control group under each specific siRNA transfection conditions. The absorbance ratio of control group under NTC transfection condition at 96 hr was used as the baseline. (B) Western blots demonstrating knockdown efficiency of siRNA transfection by examining JAK1, JAK2, STAT1, STAT3, STAT5A or STAT5B expression.
JAK1/2 inhibition partially blocks anti-PD-L1 efficacy
The IFN-γ signaling pathway plays a crucial role in the response to anti-PD-L1 immunotherapy and our in vitro results indicate that JAK1 is the primary mediator for IFN-γ-induced STAT1 phosphorylation and the expression of immune response markers. Thus, we hypothesized that JAK1/2 inhibition, but not JAK2 inhibition could inhibit the response to anti-PD-L1 immunotherapy. We tested this hypothesis using the YUMM2.1 murine melanoma in vivo model (Fig. 5A). YUMM2.1 harbors Braf activation, Pten inactivation, Cdkn2 a inactivation, and β-catenin inactivation.27 YUMM2.1 has been previously shown to be responsive to anti-PD-L1 immunotherapy.13 In this model, we found that the co-administration of ruxolitinib and anti-PD-L1 partially blocked the anti-tumor activity induced by anti-PD-L1 immunotherapy, whereas selective JAK2 inhibition with NVP-BSK805 and anti-PD-L1 did not impact anti-PD-L1 immunotherapy response (Fig. 5B-C). This suggested that JAK1 signaling, but not JAK2 signaling, is critical for response to anti-PD-L1 immunotherapy. Consistent with our findings in cell culture models, JAK1/2 inhibition in melanomas in vivo using ruxolitinib blocked constitutive P-STAT3 and P-STAT5, whereas JAK2-specific inhibition using NVP-BSK805 blocked only P-STAT5. P-STAT1 was below the level of detection in these experiments (Fig. 5D). Importantly, these experiments do not rule out potential immune-independent effects of anti-PD-L1 on tumor cells, as have been described by others.28
Figure 5.
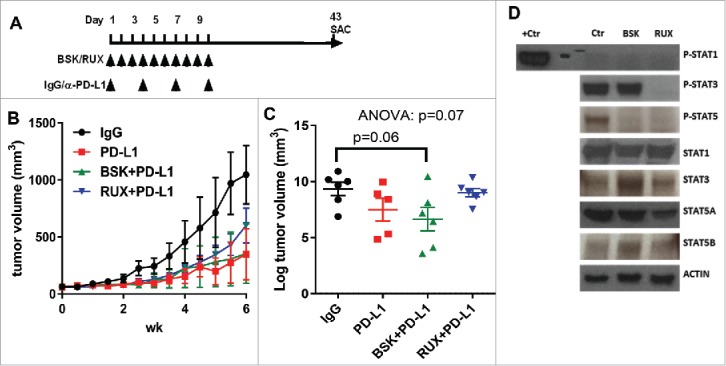
Co-administration of ruxolitinib with anti-PD-L1 antibody partially blocks the immunotherapy efficacy. (A) Schema for the in vivo experiment with results shown in panels B, C and D. (B) Tumor growth curves of YUMM2.1 with at least 5 mice in each group (mean ± SEM) after NVP-BSK805 or ruxolitinib along with anti-PD-L1, or isotype control treatment. (C) The scatter plots illustrate the final tumor volume of YUMM2.1 with at least 5 mice in each group (mean ± SEM) after NVP-BSK805 or ruxolitinib along with anti-PD-L1, or isotype control treatment. (D) Western blots from tumor lysates harvested 1 hr after the last of three consecutive doses demonstrating the effect of NVP-BSK805 or ruxolitinib treatment on STAT1, STAT3 and STAT5 phosphorylation of YUMM2.1 in vivo model tumor samples. Actin was used as a loading control.
PTPN2 suppression amplifies IFN-γ-induced expression of MHC-I, MHC-II, and PD-L1
Since inhibition of JAK1/STAT1 signaling inhibited anti-tumor immunity from anti-PD-L1 therapy, we asked if inhibition of the negative regulator PTPN2 could potentiate immunotherapy responses. PTPN2 is a non-receptor type tyrosine-specific phosphatase that negatively regulates JAK/STAT pathway.29,30 Since PTPN2 is not selectively targetable by chemical means, we utilized siPTPN2 in BRAF-mutant and BRAF-wildtype melanoma cell lines and found that PTPN2 knockdown increased IFN-γ-induced phosphorylation of STAT1, STAT3, and STAT5 (Fig 6A). In addition, PTPN2 knockdown increased IFN-γ-induced expression of MHC-I, MHC-II, and PD-L1 expression in most cell lines, which was most pronounced in BRAF-wildtype cell lines. Of particular note, PTPN2 knockdown prominently augmented IFN-γ-induced expression of MHC-II in CHL-1, HMCB, and MEWO cell lines, which do not induce expression of MHC-II in PTPN2-expressing conditions (Fig. 6B)(6). The results imply that PTPN2 suppression could improve the IFN-γ response in melanoma, which could augment immunotherapy efficacy, and may also enhance antigen presentation on tumor cells.
Figure 6.

Knockdown of PTPN2 promotes IFN-γ-induced expression of p-STAT1, p-STAT3, and p-STAT5 along with MHC-I, MHC-II, and PD-L1 and potentiates anti-PD-L1 response. (A) Western blots show IFN-γ-induced p-STAT1, p-STAT3, and p-STAT5 expression in BRAF-mutant and BRAF-wildtype melanoma cell lines under conditions of nontargeting control siRNA (NTC) or specific PTPN2 siRNA (siPTPN2) transfection. Knockdown efficiency was confirmed by examining PTPN2 expression and actin was used as a loading control. (B) Western blots show IFN-γ-induced MHC-I, MHC-II, and PD-L1 expression in BRAF-mutant and BRAF-wildtype melanoma cell lines under conditions of nontargeting control siRNA (NTC) or specific PTPN2 siRNA (siPTPN2) transfection. (C) Schema for the in vivo experiment with results shown in panels D and E. (D) Tumor growth curves of YUMM1.1 with 5 mice in each group (mean ± SEM) with anti-PD-L1, or isotype control treatment under shPTPN2 conditions. (E) Waterfall plot of YUMM1.1 tumors with anti-PD-L1, or isotype control treatment under shPTPN2 conditions. The data are expressed as the percentage of tumor volume change from baseline.
To test this hypothesis, we studied the effect of the knockdown of murine Ptpn2 using stable transduction of shRNA on response to anti-PD-L1 immunotherapy in the YUMM1.1 murine melanoma in vivo model (Fig. 6D, Supplementary Fig. 5). YUMM1.1 harbors Braf activation, Pten inactivation, and Cdkn2a inactivation.27 and is resistant to anti-PD-L1 immunotherapy in vivo.13 Knockdown of Ptpn2 inhibited tumor growth in syngeneic mice and promoted tumor regression, an effect which was not observed when tumor cells were grown in the flanks of athymic nude mice (Supplementary Fig. 6A-B). This observation suggests that Ptpn2 deficiency increases the sensitivity of the tumor cells to anti-tumor immunity (e.g. by IFN-γ-mediated STAT activation). In one published model, melanoma tumors with CRISPR/Cas9-mediated Ptpn2 loss were recently shown to be completely rejected versus control sgRNA counterparts.19 Importantly, in our model a reduced effect (40% tumor rejection) of Ptpn2 shRNA alone may be due to incomplete target knockdown, subclonal selection of proficient clones, or inter-model differences. Consistent with this concept, examination of terminal tumor masses demonstrated clonal selection against shPtpn2-expressing cells (determined by loss of turboGFP [tGFP] expression) and also demonstrated low pSTAT1 expression. Interestingly, shPtpn2 tumors maintained higher expression of pSTAT3 and pSTAT5. These effects were presumably immunologically-mediated as little pSTAT1/3/5 was observed in shPtpn2 tumors grown in athymic nude mice (Supplementary Fig. 6C-F). In addition, knockdown of Ptpn2 further inhibited tumor growth and induced tumor regression when tumor-bearing mice were treated with anti-PD-L1 immunotherapy (Fig. 6D and Fig. 6E). Complete responses/regressions (CR) were observed in 2/5 mice with shPtpn2 tumors, and 4/5 mice with shPtpn2 tumors treated with anti-PD-L1 (χ2 test p<0.01).
Discussion
The advent of immunotherapy has dramatically extended the survival of melanoma patients over the past 5 years. However, not all patients benefit from immunotherapy and the objective response rate even with combined PD-1 and CTLA-4-targeting immunotherapy (a substantially toxic combination) is approximately 60%.31-33 Immunotherapeutic treatment failures may include either intrinsic (de novo) or acquired resistance.34,35 In patients with de novo resistance, most features of anti-tumor immunity and T-cell activation appear to be absent. In acquired resistance, patients initially respond, but eventually relapse due to immunologic evolution induced by the immunotherapy. As such, a large number of clinical trials are now focused on immuno-molecular combinations that target both cell signaling pathways and immunologic checkpoints, to target these refractory patients. However, due to the complexity of the immune system and the suboptimal preclinical models that exist, some therapeutic combinations are being employed in the absence of strong preclinical supportive data. Furthermore, when targeting molecular pathways for the purposes of improving immunotherapy responses, the effects of target inhibition on both the tumor and the immune system must be considered in tandem.
IFN-γ is a critical cytokine for the host immune response that demonstrates antiviral, antitumor, and immune-regulatory activities. IFN-γ regulates tumor antigen loading on tumor cells, antigen presentation by MHCs, and ligands for immune checkpoint receptors, all of which support anti-tumor immunity and possibly response to immunotherapy. In addition, IFN-γ regulates the release of secondary chemokines, cytokines, and interleukins that also participate in the immune response.36 However, controversies concerning the exact role of IFN-γ in immunotherapy exist. Benci et al. reported that chronic IFN-γ treatment promotes epigenetic changes and maintains immune resistance to immunotherapy .37 In contrast, Gao et al. reported that genomic defects in IFN-γ pathway genes results in melanoma tumors that are resistant to ipilimumab immunotherapy.38 In general, however, the majority of literature support a positive role for the IFN-γ response signature as a predictor and functional effector of immunotherapy outcome. Despite the general consensus that IFN-γ is required for immunotherapy responses, the individual signaling components contributing to this effect that are necessary for the immunomodulatory effect are unclear.
In this study, we investigated the specific role of JAK/STAT pathway members on IFN-γ mediated cell and immunologic/immunogenic signaling. We found that JAK1 is the primary and essential mediator for IFN-γ mediated immune activity in regards to: a) downstream STAT1, STAT3, and STAT5 phosphorylation, b) downstream MHC-I, MHC-II, and PD-L1 expression, c) inhibition of cell proliferation, and d) anti-tumor activity of anti-PD-L1 immunotherapy. We also found that JAK2 contributes to IFN-γ-induced STAT5 phosphorylation and PD-L1 expression, but is not required for MHC-I and MHC-II induction. Although JAK2 mainly regulated STAT5 phosphorylation upon IFN-γ stimulation, a double knockout of STAT5A and STAT5B demonstrated that STAT5 is not involved in IFN-γ-induced PD-L1 expression. This finding concurs with a previous report by Garcia-Diaz et al. suggesting that IFN-γ-JAK1/JAK2-STAT1/STAT2/STAT3 drives PD-L1 expression, although this analysis was done largely at the genetic/gene expression level.22 Given the majority of literature that focuses on the pro-tumorigenic and anti-inflammatory role of STAT3 and STAT5 [reviewed in.39], including a suppressive role on STAT1 signaling,40 our data suggest that selective inhibition of JAK2 may be a viable immunotherapy combination strategy without impairing JAK1/STAT1 activity. In contrast, however, inhibition of JAK1 or both JAK1 and JAK2 may be detrimental to IFN-γ mediated tumor responses to activated T cells, and thus immunotherapy response. These hypothesis is supported by the observations that loss-of-function mutations in JAK1 or JAK2 were associated with intrinsic and acquired immunologic resistance to anti-PD-1 immunotherapy in melanoma patients.16-41 Finally, in vivo data support this concept, as JAK1/2, but not JAK2 inhibition thwarted immunotherapeutic responses in a mouse model of melanoma. Importantly, these effects were observed in the presence of short-term (transient) inhibition of the JAK pathway, which we performed in this manner to determine if JAK inhibition during the adaptive immunity phase (first 7–14 days) would circumvent or undermine T cell responses. The long-term effects of JAK/STAT inhibition in the tumor microenvironment remains to be determined.
We found that additional effectors besides JAK1 and JAK2 may also play a role in IFN-γ-induced MHC-I, MHC-II, and PD-L1 expression along with STAT1 phosphorylation in BRAF-mutant cell lines, as dual knockdown of both of these kinases was insufficient to completely block IFN-γ mediated upregulation of response markers. However, JAK1 and JAK2 appear to both contribute to IFN-γ-induced MHC-I, MHC-II, and PD-L1 expression along with STAT1, STAT3, and STAT5 phosphorylation, particularly in BRAF-wildtype cell lines. It is currently unclear if BRAF mutational status is a surrogate or a causative feature for these phenotypic differences. Previous reports have indicated that other alternative pathways including the MAPK, PI3K, or NF-κB pathways either cooperate or operate in parallel with the JAK/STAT pathway in order to regulate IFN-γ functions.42 Thus, an investigation into the contribution of these alternative pathways to IFN-γ-induced MHC-I, MHC-II, and PD-L1 expression in BRAF-mutant melanoma cells may lead to the development of new strategies for immuno-molecular combinations.
Finally, we demonstrated that PTPN2 may serve as a potential target to augment immunotherapy efficacy through enhancement of IFN-γ mediated JAK/STAT signaling. This finding supports a previous report by Manguso et al.19using in vivo CRISPR screening which identified that knockout of Ptpn2 in melanoma tumors boosted immunotherapy efficacy by enhancing IFN-γ-induced antigen presentation and growth suppression. Interestingly, in this screen, loss of Jak1 was also associated with immunotherapy resistance, whereas the effect of Jak2 deletion was not significant.19
There are several limitations to the present study. For one, because systemic JAK inhibition was used, it is unclear whether the effects on tumor progression are related to tumor –intrinsic effects, immune-cell-intrinsic effects, or a combination of both. Extended studies using CRISPR or RNAi-mediated genetic ablation of JAK/STAT components in tumor cells or using parallel studies in genetic knock-out mice will be key to identifying which of these mechanisms is most crucial for immunotherapy activity. Certainly translational studies have already demonstrated that genetic loss-of-function (in the tumor cells) of some of these components may be associated with intrinsic or acquired resistance to immunotherapy(15,16). However, key compartments of the tumor microenvironment, including both myeloid and T cells, require various JAK signals for differentiation into inflammatory (M1, Th1, Th17), wound healing (Th2, M2), and regulatory phenotypes (Treg, myeloid-derived suppressor cells). Thus, future studies should be beneficial in evaluating the effect of systemic JAK/STAT inhibition on the differentiation of these compartments and their impact on the anti-tumor immune microenvironment.
Collectively, our findings underscore the importance of careful selection of therapeutic strategies to modulate tumor signaling to favor anti-tumor immunity, and suggest that enhancement of JAK1/STAT1 signaling in the tumor microenvironment should be conserved, while JAK2 appears to play a dispensable role and may actually be therapeutically targetable in combination with anti-PD-1/L1. Thus, current clinical trials combining JAK1 inhibitors (alone or in combination with JAK2 inhibition) with anti-PD-1/L1 therapy are not advisable, and could thwart anti-tumor immunity. In contrast, selective inhibition of JAK2 may actually improve response due to selective inhibition on STAT3 and STAT5 activation.
Supplementary Material
Funding Statement
This work was supported by the HHS | NIH | National Cancer Institute (NCI)(K23CA204726), HHS | NIH | National Cancer Institute (NCI) (R00CA181491).
Conflicts of interest
The authors have no conflicts to disclose.
Acknowledgments
Funding for this work was provided by the Bready Foundation for Melanoma Research (JMB and DBJ), China Scholarship Council 201406205050 (NL), NIH/NCI R00CA181491 (JMB), K23CA204726 (DBJ), James C. Bradford Jr. Melanoma Fund (DBJ), and the Vanderbilt-Ingram Cancer Center Support Grant P30 CA68485.
References
- 1.Ascierto PA, Kirkwood JM, Grob JJ, Simeone E, Grimaldi AM, Maio M, Palmieri G, Testori A, Marincola FM, Mozzillo N. The role of BRAF V600 mutation in melanoma. J Transl Med. 2012;10:85. doi: 10.1186/1479-5876-10-85. PMID:22554099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hall RD, Kudchadkar RR. BRAF mutations: Signaling, epidemiology, and clinical experience in multiple malignancies. Cancer Control. 2014;21(3):221–30. doi: 10.1177/107327481402100307. PMID:24955706. [DOI] [PubMed] [Google Scholar]
- 3.Alexander W. The Checkpoint Immunotherapy Revolution: What Started as a Trickle Has Become a Flood, Despite Some Daunting Adverse Effects; New Drugs, Indications, and Combinations Continue to Emerge. P & T: A peer-reviewed journal for formulary management. 2016;41(3):185–91. [PMC free article] [PubMed] [Google Scholar]
- 4.Redman JM, Gibney GT, Atkins MB. Advances in immunotherapy for melanoma. BMC medicine. 2016;14:20. doi: 10.1186/s12916-016-0571-0. PMID:26850630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Masucci GV, Cesano A, Hawtin R, Janetzki S, Zhang J, Kirsch I, Dobbin KK, Alvarez J, Robbins PB, Selvan SR, et al. Validation of biomarkers to predict response to immunotherapy in cancer: Volume I – pre-analytical and analytical validation. J Immunother Cancer. 2016;4:76. doi: 10.1186/s40425-016-0178-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson DB, Estrada MV, Salgado R, Sanchez V, Doxie DB, Opalenik SR, Vilgelm AE, Feld E, Johnson AS, Greenplate AR, et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat Commun. 2016;7:10582. doi: 10.1038/ncomms10582. PMID:26822383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karachaliou N, Crespo G, Aldeguer E, Drozdowskyj A, Gimenez Capitan A, Teixido C. Interferon-gamma (INFG), an important marker of response to immune checkpoint blockade (ICB) in non-small cell lung cancer (NSCLC) and melanoma patients. Journal of Clinical Oncology. 2017;35(15_suppl):11504–04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Albright A. Relationship between immune gene signatures and clinical response to PD-1 blockade with pembrolizumab (MK-3475) in patients with advanced solid tumors. Journal for immunotherapy of cancer. 2015;3(2):P80. doi: 10.1186/2051-1426-3-S2-P80. [DOI] [Google Scholar]
- 9.Hu X, Ivashkiv LB. Cross-regulation of Signaling and Immune Responses by IFN-γ and STAT1. Immunity. 2009;31(4):539–50. doi: 10.1016/j.immuni.2009.09.002. PMID:19833085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nature reviews Immunology. 2005;5(5):375–86. doi: 10.1038/nri1604. PMID:15864272. [DOI] [PubMed] [Google Scholar]
- 11.Taube JM, Young GD, McMiller TL, Chen S, Salas JT, Pritchard TS, Xu H, Meeker AK, Fan J, Cheadle C, et al. Differential Expression of Immune-Regulatory Genes Associated with PD-L1 Display in Melanoma: Implications for PD-1 Pathway Blockade. Clin Cancer Res. 2015;21(17):3969–76. doi: 10.1158/1078-0432.CCR-15-0244. PMID:25944800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. Loss of IFN-gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell. 2016;167(2):397–404 e9. doi: 10.1016/j.cell.2016.08.069. PMID:27667683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Homet Moreno B, Zaretsky JM, Garcia-Diaz A, Tsoi J, Parisi G, Robert L, Meeth K, Ndoye A, Bosenberg M, Weeraratna AT, et al. Response to Programmed Cell Death-1 Blockade in a Murine Melanoma Syngeneic Model Requires Costimulation, CD4, and CD8 T Cells. Cancer Immunol Res. 2016;4(10):845–57. doi: 10.1158/2326-6066.CIR-16-0060. PMID:27589875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McNamara MJ, Hilgart-Martiszus I, Barragan Echenique DM, Linch SN, Kasiewicz MJ, Redmond WL. Interferon-gamma Production by Peripheral Lymphocytes Predicts Survival of Tumor-Bearing Mice Receiving Dual PD-1/CTLA-4 Blockade. Cancer Immunol Res. 2016;4(8):650–7. doi: 10.1158/2326-6066.CIR-16-0022. PMID:27262113. [DOI] [PubMed] [Google Scholar]
- 15.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. Cancer Immunol Res. 2016;375(9):819–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, Grasso CS, Hugo W, Sandoval S, Torrejon DY, et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017;7(2):188–201. doi: 10.1158/2159-8290.CD-16-1223. PMID:27903500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165(1):35–44. doi: 10.1016/j.cell.2016.02.065. PMID:26997480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–71. doi: 10.1038/nature13954. PMID:25428505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, Collins NB, Bi K, LaFleur MW, Juneja VR, et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature. 2017;547(7664):413–18. doi: 10.1038/nature23270. PMID:28723893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou F. Molecular mechanisms of IFN-gamma to up-regulate MHC class I antigen processing and presentation. Int Rev Immunol. 2009;28(3-4):239–60. doi: 10.1080/08830180902978120. PMID:19811323. [DOI] [PubMed] [Google Scholar]
- 21.Steimle V, Siegrist CA, Mottet A, Lisowska-Grospierre B, Mach B. Regulation of MHC class II expression by interferon-gamma mediated by the transactivator gene CIITA. Science. 1994;265(5168):106–9. doi: 10.1126/science.8016643. PMID:8016643. [DOI] [PubMed] [Google Scholar]
- 22.Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X, et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Reports. 2017;19(6):1189–201. doi: 10.1016/j.celrep.2017.04.031. PMID:28494868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balko JM, Jones BR, Coakley VL, Black EP. MEK and EGFR inhibition demonstrate synergistic activity in EGFR-dependent NSCLC. Cancer Biol Ther. 2009;8(6):522–30. doi: 10.4161/cbt.8.6.7690. PMID:19305165. [DOI] [PubMed] [Google Scholar]
- 24.Balko JM, Giltnane JM, Wang K, Schwarz LJ, Young CD, Cook RS, Owens P, Sanders ME, Kuba MG, Sánchez V, et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014;4(2):232–45. doi: 10.1158/2159-8290.CD-13-0286. PMID:24356096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Balko JM, Schwarz LJ, Bhola NE, Kurupi R, Owens P, Miller TW, Gómez H, Cook RS, Arteaga CL. Activation of MAPK pathways due to DUSP4 loss promotes cancer stem cell-like phenotypes in basal-like breast cancer. Cancer research. 2013;73(20):6346–58. doi: 10.1158/0008-5472.CAN-13-1385. PMID:23966295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balko JM, Cook RS, Vaught DB, Kuba MG, Miller TW, Bhola NE, Sanders ME, Granja-Ingram NM, Smith JJ, Meszoely IM, et al. Profiling of residual breast cancers after neoadjuvant chemotherapy identifies DUSP4 deficiency as a mechanism of drug resistance. Nature medicine. 2012;18(7):1052–59. doi: 10.1038/nm.2795. PMID:22683778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meeth K, Wang JX, Micevic G, Damsky W, Bosenberg MW. The YUMM lines: A series of congenic mouse melanoma cell lines with defined genetic alterations. Pigment Cell Melanoma Res. 2016;29(5):590–7. doi: 10.1111/pcmr.12498. PMID:27287723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clark CA, Gupta HB, Sareddy G, Pandeswara S, Lao S, Yuan B, Drerup JM, Padron A, Conejo-Garcia J, Murthy K, et al. Tumor-Intrinsic PD-L1 Signals Regulate Cell Growth, Pathogenesis, and Autophagy in Ovarian Cancer and Melanoma. Cancer research. 2016;76(23):6964–74. doi: 10.1158/0008-5472.CAN-16-0258. PMID:27671674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pike KA, Tremblay ML. TC-PTP and PTP1B: Regulating JAK-STAT signaling, controlling lymphoid malignancies. Cytokine. 2016;82:52–7. doi: 10.1016/j.cyto.2015.12.025. PMID:26817397. [DOI] [PubMed] [Google Scholar]
- 30.Vainchenker W, Constantinescu SN. JAK/STAT signaling in hematological malignancies. Oncogene. 2013;32(21):2601–13. doi: 10.1038/onc.2012.347. PMID:22869151. [DOI] [PubMed] [Google Scholar]
- 31.Ott PA, Hodi FS, Robert C. CTLA-4 and PD-1/PD-L1 blockade: New immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin Cancer Res. 2013;19(19):5300–9. doi: 10.1158/1078-0432.CCR-13-0143. PMID:24089443. [DOI] [PubMed] [Google Scholar]
- 32.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373(1):23–34. doi: 10.1056/NEJMoa1504030. PMID:26027431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372(21):2006–17. doi: 10.1056/NEJMoa1414428. PMID:25891304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kelderman S, Schumacher TN, Haanen JB. Acquired and intrinsic resistance in cancer immunotherapy. Molecular oncology. 2014;8(6):1132–9. doi: 10.1016/j.molonc.2014.07.011. PMID:25106088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Restifo NP, Smyth MJ, Snyder A. Acquired resistance to immunotherapy and future challenges. Nature reviews Cancer. 2016;16(2):121–6. doi: 10.1038/nrc.2016.2. PMID:26822578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nature reviews Cancer. 2016;16(3):131–44. doi: 10.1038/nrc.2016.14. PMID:26911188. [DOI] [PubMed] [Google Scholar]
- 37.Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint\sVictor C, Cucolo L, Lee DSM, Pauken KE, Huang AC, et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell. 2016;167(6):1540–54 e12. doi: 10.1016/j.cell.2016.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell. 2016;167(2):397–404.e9. doi: 10.1016/j.cell.2016.08.069. PMID:27667683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798–809. doi: 10.1038/nrc2734. PMID:19851315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wellbrock C, Weisser C, Hassel JC, Fischer P, Becker J, Vetter CS, Behrmann I, Kortylewski M, Heinrich PC, Schartl M. STAT5 contributes to interferon resistance of melanoma cells. Curr Biol. 2005;15(18):1629–39. doi: 10.1016/j.cub.2005.08.036. PMID:16169484. [DOI] [PubMed] [Google Scholar]
- 41.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med. 2016;375(9):819–29. doi: 10.1056/NEJMoa1604958. PMID:27433843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gough DJ, Levy DE, Johnstone RW, Clarke CJ. IFNγ signaling—Does it mean JAK–STAT? Cytokine Growth Factor Rev. 2008;19(5):383–94. doi: 10.1016/j.cytogfr.2008.08.004. PMID:18929502. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.