

Abstract
Background
Heterotaxy (Htx) syndrome comprises a class of congenital disorders resulting from malformations in left-right body patterning. Approximately 90% of patients with heterotaxy have serious congenital heart diseases; as a result, the survival rate and outcomes of Htx patients are not satisfactory. However, the underlying etiology and mechanisms in the majority of Htx cases remain unknown. The aim of this study was to investigate the function of rare copy number variants (CNVs) in the pathogenesis of Htx.
Methods
We collected 63 sporadic Htx patients with congenital heart defects and identified rare CNVs using an Affymetrix CytoScan HD microarray and real-time polymerase chain reaction. Potential candidate genes associated with the rare CNVs were selected by referring to previous literature related to left-right development. The expression patterns and function of candidate genes were further analyzed by whole mount in situ hybridization, morpholino knockdown, clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9)-mediated mutation, and over-expressing methods with zebrafish models.
Results
Nineteen rare CNVs were identified for the first time in patients with Htx. These CNVs include 5 heterozygous genic deletions, 4 internal genic duplications, and 10 complete duplications of at least one gene. Further analyses of the 19 rare CNVs identified six novel potential candidate genes (NUMB, PACRG, TCTN2, DANH10, RNF115, and TTC40) linked to left-right patterning. These candidate genes exhibited early expression patterns in zebrafish embryos. Functional testing revealed that downregulation and over-expression of five candidate genes (numb, pacrg, tctn2, dnah10, and rnf115) in zebrafish resulted in disruption of cardiac looping and abnormal expression of lefty2 or pitx2, molecular markers of left-right patterning.
Conclusions
Our findings show that Htx with congenital heart defects in some sporadic patients may be attributed to rare CNVs. Furthermore, DNAH10 and RNF115 are Htx candidate genes involved in left-right patterning which have not previously been reported in either humans or animals. Our results also advance understanding of the genetic components of Htx.
Electronic supplementary material
The online version of this article (10.1186/s13073-018-0549-y) contains supplementary material, which is available to authorized users.
Keywords: Copy number variants, Congenital heart defects, Heterotaxy, Zebrafish, Left-right
Background
Heterotaxy (Htx) syndrome is a serious congenital malformation with high mortality and morbidity characterized by the failure to establish normal left-right (LR) body asymmetry. Patients often present with abnormal arrangement of the thoraco-abdominal organs, including ectopia of the heart, lungs, spleen, or liver [1]. The survival rate and outcomes of patients with Htx are unsatisfactory, as approximately 90% of cases are associated with complex congenital heart diseases, including malposition of the great arteries, presence of a single right ventricle, total anomalous pulmonary venous drainage, and double-outlet right ventricle [2, 3].
Researchers have made significant progress in enhancing our understanding of the molecular and cellular mechanisms that determine laterality during early embryogenesis. In the primitive node, asymmetry signaling is activated by leftward “nodal flow” created by the unidirectional rotation of monocilia. The asymmetry signals are then transmitted to the left lateral plate mesoderm, where they upregulate the expression of a series of left determinants, such as Nodal, left-right determination factor 2 (Lefty2), and paired-like homeodomain 2 (Pitx2). Several signaling pathways are involved in establishment of the LR axis, including Notch, Nodal, Hedgehog, Wnt, and transforming growth factor beta (TGF-β) [3–5]. In humans, mutations in several genes have been associated with Htx, including CFC1, NODAL, ACVR2B, LEFTY2, GDF1, ZIC3, CRELD1, and NKX2.5 [6–13]. However, the mutations reported in these genes can explain only 10–20% of Htx cases; the underlying cause in the majority of patients remains unknown [14–16].
Copy number variants (CNVs) are DNA fragments whose copy number varies between individuals in a population due to duplication or deletion events. CNVs can range in size from 1 kilobase (kb) to several megabases (Mb). Numerous studies have demonstrated that a variety of diseases, especially syndrome-related diseases, are associated with CNVs. Several researchers recently reported a relationship between CNVs and Htx. Both Brueckner and Mills identified several novel rare CNVs in congenital heart disease patients with abnormal LR patterning [15–17], suggesting that CNVs may account for a proportion of patients. But the role of CNVs in the occurrence of Htx in patients with complex congenital heart disease should be examined in greater detail.
In this study, we identified 19 rare CNVs in 63 patients with Htx by genotyping their DNA using an Affymetrix CytoScan HD microarray. We further identified six potential candidate genes involved in several pathways reported to be related to LR development: ciliary proteome and function, Notch signaling pathway, or ubiquitination (ubiquitin ligase E3 family). Downregulation and over-expression of the Htx candidate genes numb, Parkin co-regulated gene (pacrg), tectonic family member 2 (tctn2), dynein axonemal heavy chain 10 (dnah10), and ring-finger protein 115 (rnf115) in zebrafish resulted in disruption of both heart looping and expression of lefty2 or pitx2. To our knowledge, this study is the first to identify DNAH10 and RNF115 as novel Htx candidate genes. These two genes have not been previously implicated in LR patterning in either humans or animals; numb, pacrg, and tctn2 have been linked to LR development in animals but are previously unreported in patients with Htx.
Methods
Patient ascertainment and study populations
Our study recruited patients with Htx in Xinhua Hospital and Shanghai Children’s Medical Center (SCMC) whose diagnoses were confirmed by echocardiography, cardiac catheterization examinations, computed tomography, abdominal ultrasonography, and other operation recordings. Patients exhibiting abnormal arrangement of the visceral organs and complex congenital heart disease were included, while those with complete situs solitus or other syndromes were excluded.
Affymetrix CytoScan HD microarray analysis
Peripheral blood samples were obtained from each patient, and DNA was extracted using the QIAamp DNA Blood Midi Kit (Qiagen, Duesseldorf, Germany) following the manufacturer’s instructions. The CNVs were detected by CytoScan HD microarray platform (Affymetrix, Santa Clara, CA, USA), which is a high-density chip that contains 2,636,550 probes. In total, 59 samples passed initial quality control. Gains and losses were analyzed using Chromosome Analysis Suite (ChAS) software and the annotations of the Genome Reference Consortium (GRC) human reference genome version GRCh37 (hg19). The data were filtered, and only those regions larger than 50 kb comprising at least 25 contiguous markers were considered. Finally, we distinguished common CNVs from rare CNVs by comparing the results with the known CNVs in the Database of Genomic Variants (DGV, http://dgv.tcag.ca/) and Online Mendelian Inheritance in Man (OMIM, http://omim.org).
Quantitative real-time polymerase chain reaction validation
The selected segments, which are related with Htx, were verified by quantitative real-time polymerase chain reaction (qPCR). The qPCR validation was performed according to SYBR® Premix Ex TaqTM II protocol (Applied TaKaRa). We used 50 ng/μL of genomic DNA in a 20 μL reaction, consisting of 10 μL of 2× SYBR Premix Ex Taq, 0.4 μL of 50× ROX Reference Dye II, 0.3 μL of forward primer, 0.3 μL of reverse primer, 8.0 μL of ddH2O, and 1 μL of DNA. Genomic DNAs extracted from healthy people were mixed, serving as normal controls, and the house-keeping gene GAPDH was used as the control in qPCR. The reactions were performed in triplicate.
Whole-exome sequencing analysis and mutation detection
We performed whole-exome sequencing in patients with Htx and also in 100 healthy control people. The DNA was sequenced using the Illumina HiSeq 2500 platform at a commercial provider (Shanghai Biotechnology Co, Ltd., Shanghai, China). We defined functional mutations to be nonsynonymous mutations, stop-gain mutations, stop-loss mutations, frameshift or non-frameshift deletions or insertions, and splice site mutations.
Zebrafish husbandry
Adult zebrafish (Danio rerio, AB line and Tg [cmlc2:EGFP] line) were raised under standard laboratory conditions with an automatic fish housing system (ESEN, Beijing, China) at 28 °C. All zebrafish experiments were conducted at the Institute of Neuroscience, Chinese Academy of Sciences, according to standard protocols. Embryo stages were determined according to their developmental morphology [18].
Morpholino oligo injection and target gene knockdown
Morpholino oligos (MOs) designed against target genes were purchased from Gene Tools (Philomath, OR, USA) and dissolved in nuclease-free water. According to Gene Tools’ protocol, concentrations of MOs were checked by spectrophotometry (A265 in 0.1 N HCl). MOs were diluted to different working concentrations and 1 nL was pressure-injected into one-cell-stage embryos with a Picospritzer II injector.
The MOs for examining heart looping in morphants range from 2 ng to 8 ng in dosage: 8 ng pacrg MO, 8 ng tctn2 MO, 8 ng numb MO, 8 ng dnah10 MO, 2 ng rnf115 MO, 8 ng cfap46/ttc40 MO, and 8 ng galnt11 MO per embryo. As negative controls, we injected 8 ng of standard control MO. A summary of MO doses and sequences is provided in Additional file 1: Table S1. The knockdown efficiencies of splice blocking and translation blocking MOs are illustrated in Additional file 2: Figure S1.
CRISPR/Cas9-mediated mutation of dnah10 and rnf115 in zebrafish embryos
As previously reported, the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) system was applied to introduce dnah10 and rnf115 gene mutation in zebrafish embryos [19]. The sequences of dnah10 guide RNA (gRNA) (5′-GGCTCAGTTCTATGCTTACT -3′) and rnf115 gRNA (5′-GGACAGTCTTGACTCTGAG -3′) were designed to target the sequences of mature dnah10 and rnf115, respectively. We co-injected 600 pg zCas9 messenger RNA (mRNA) and 100 pg dnah10 gRNA or 100 pg rnf115 gRNA into zebrafish embryos at the one-cell stage. The gene mutation and knockout efficiency in F0 embryos was examined by PCR and sequencing analysis with the following primers:
rnf115 forward: 5′-GAGAAGCACTGGTTCCGTCA-3′.
rnf115 reverse: 5′-AACATACCCCTCAACAGCGG-3′.
dnah10 forward: 5′-ATTCATCCAACGTGGAAACCA-3′.
dnah10 reverse: 5′-GTCAGGACCTCGGTTTATTGTC-3′.
The knockout efficiency in F0 embryos of dnah10 is 84.6%; for rnf115 it is 75% (Additional file 2: Figure S2).
mRNA synthesis and injection
Zebrafish total RNA was extracted from 1 to 2 days post fertilization (dpf) wild-type embryos (AB strain) and then was retrotranscribed into coding DNA (cDNA) with PrimeScripttm RT reagent (TaKaRa, Shiga, Japan) according to the manufacturer’s instructions. The full-length coding sequence DNA of tctn2, pacrg, galnt11, numb, and rnf115 was amplified using specific primers with restriction enzyme digestion loci, which were next subcloned into the pCS2+ vector (Additional file 1: Table S2). Positive clones selected by DNA sequencing were applied to generate the corresponding full-length mRNAs using a T7 or SP6 mMessage mMachine kit (Ambion). For the over-expression experiment, pacrg, tctn2, numb, and galnt11 mRNAs were injected into one-cell-stage embryos and green fluorescent protein (GFP) was used as a negative control; for the rescue experiment, 6.25 pg rnf115 mRNA was injected into one-cell-stage embryos mixed with 2 pg rnf115 MO.
Whole mount in situ hybridization
The whole mount in situ hybridization (WMISH) with anti-digoxigenin probes was performed according to the previously described protocol [20]. The anti-digoxigenin RNA probes (Roche) were synthesized with a length of 400–1300 necleotides, complementary to numb, pacrg, tctn2, dnah10, rnf115, pitx2, and lefty2, respectively (Additional file 1: Table S3).
The injected doses of MOs are from 2 ng to 8 ng for scoring of pitx2 and lefty2 expression in morphants: 8 ng pacrg MO, 8 ng tctn2 MO, 8 ng numb MO, 8 ng dnah10 MO, 2 ng rnf115 MO, 8 ng cfap46/ttc40 MO, and 4 ng galnt11 MO per embryo. As negative controls, we injected 8 ng of standard control MO (Additional file 1: Table S1).
Statistical analysis
In all figures, statistical comparisons between groups were analyzed by the chi-squared test (continuity corrected) or Fisher’s exact test. We defined P < 0.05 as statistically significant with *P < 0.05, **P < 0.01, ***P < 0.001.
Results
Clinical data
A total of 63 Chinese children with sporadic Htx were recruited. All of the patients exhibited abnormal arrangement of the visceral organs and complex congenital heart disease, not including complete situs solitus or other syndromes [21]. Among the patients we recruited, no one had central nervous system malformations, vertebral defects, or genitourinary malformations. According to the patients’ medical history, there was no family history of heterotaxy or other malformations. The patients’ ages ranged from 12 days to 113 months; 40 patients were male (63.5%) and 23 were female (36.5%). The detailed cardiac and extracardiac clinical phenotypes are summarized in Table 1. Pulmonary outflow obstruction was discovered in 56 patients, complete or partial atrioventricular canal in 20 patients, and single atrium or single ventricle in 35 patients. Twenty-seven patients had malposed or transposed great arteries, and 21 patients had double outlet of the right ventricle.
Table 1.
Cardiac and extracardiac abnormalities in the patients with Htx
| Number of patients (%) | |
|---|---|
| Sex | |
| Male | 40 (63.5%) |
| Female | 23 (36.5%) |
| Cardiac position | |
| Levocardia | 15 (23.8%) |
| Dextrocardia | 35 (55.6%) |
| Mesocardia | 13 (20.6%) |
| Atrial arrangement | |
| Atrial situs inversus | 20 (31.7%) |
| Isomerism of right atrial appendages | 33 (52.4%) |
| Isomerism of left atrial appendages | 7 (11.1%) |
| Ventricular arrangement | |
| Ventricular situs solitus | 14 (22.2%) |
| Ventricular situs inversus | 16 (25.4%) |
| Single ventricle (morphologic right) | 23 (36.5%) |
| Single ventricle (morphologic left) | 3 (4.8%) |
| Single ventricle (morphologic indeterminate) | 5 (7.9%) |
| Other abnormal ventricle arrangement | 2 (3.2%) |
| Bronchi | |
| Bilateral right bronchi (short) | 34 (54.0%) |
| Bilateral left bronchi (long) | 7 (11.1%) |
| Bronchial inversus | 22 (34.9%) |
| Spleen | |
| Polysplenia | 6 (9.5%) |
| Asplenia | 29 (46.0%) |
| Single right spleen | 21 (33.3%) |
| Single left spleen | 7 (11.1%) |
| Stomach | |
| Right-sided stomach | 38 (60.3%) |
| Left-sided stomach | 19 (30.2%) |
| Stomach centrally situated | 4 (6.3%) |
| Unknown | 2 (3.2%) |
| Liver | |
| Left-sided liver | 23 (36.5%) |
| Liver centrally situated | 31 (49.2%) |
| Aortic arch | |
| Left aortic arch | 17 (27.0%) |
| Right aortic arch | 45 (71.4%) |
| Aortic arch centrally descending | 1 (1.6%) |
| SVC | |
| Right SVC | 8 (12.7%) |
| Left SVC | 33 (52.4%) |
| Bilateral SVC | 22 (34.9%) |
| IVC | |
| Interrupted IVC, hemiazygos vein continuation | 1 (1.6%) |
| Interrupted IVC, azygos vein continuation | 6 (9.5%) |
| Relationship of IVC and descending aorta | |
| IVC right of spine and descending aorta left of spine | 4 (6.3%) |
| IVC left of spine and descending aorta right of spine | 17 (27.0%) |
| IVC and descending aorta same side | 32 (50.8%) |
| IVC left of spine and descending aorta anterior of spine | 2 (3.2%) |
| IVC anterior of spine and descending aorta left of spine | 1 (1.6%) |
SVC superior vena cava, IVC inferior vena cava
CNVs in patients with Htx and identification of candidate genes
To identify the molecular causes of Htx, an Affymetrix CytoScan HD microarray was used to identify possible pathogenic CNVs. A total of 59 samples passed initial quality control. Rare CNV segments were identified based on the following criteria: (1) CNV > 50 kb in size; (2) > 25 markers in each segment; (3) present at < 1‰ frequency or have < 50% overlap with published common CNVs or not found in the DGV (http://dgv.tcag.ca/); (4) not identified in either a dataset with microarray results of 216 normal Chinese individuals or another dataset of 720 Chinese non-heterotaxy patients with developmental delay/intellectual disability (DD/ID) (Additional file 1: Table S4). Finally, we identified 19 rare CNVs in 14 patients with Htx (Table 2). The percentage of subjects with rare CNVs was 23.7% (14 of 59 Htx subjects). The selected CNVs ranged in size from 57 to 1009 kb. These CNVs included 5 heterozygous genic deletions, 4 internal genic duplications, and 10 complete duplications of at least one gene [15].
Table 2.
Nineteen rare copy number variants identified in patients with heterotaxy
| ID | Chromosome | Genomic coordinates | Type | Size (kbp) | Genes altered |
|---|---|---|---|---|---|
| 5 | 4q24 | 104,554,264–105,123,728 | Internal dup | 569.464 | TACR3 |
| 5 | 6p22.2 | 26,019,198–26,227,973 | Genic dup | 208.775 | HIST1H3A, HIST1H4A, HIST1H4B, HIST1H3B, HIST1H2AB, HIST1H2BB, HIST1H3C, HIST1H1C, HFE, HIST1H4C, HIST1H1T, HIST1H2BC, HIST1H2AC, HIST1H1E, HIST1H2BD, HIST1H2BE, HIST1H4D, HIST1H3D, HIST1H2AD, HIST1H2BF, HIST1H4E, HIST1H2BG, HIST1H2AE, HIST1H3E |
| 7 | 1q21.1 | 145,625,128–145,927,662 | Genic del | 302.534 | RNF115 , CD160, PDZK1, GPR89A, GPR89C, PDZK1P1 |
| 10 | 5q23.1 | 115,247,380–115,683,172 | Genic dup | 435.792 | AP3S1, AQPEP, LOC644100, COMMD10 |
| 16 | 6p12.1 | 54,138,106–54,277,341 | Genic dup | 139.235 | TINAG |
| 18 | 12p13.33 | 173,786–356,461 | Genic dup | 182.675 | IQSEC3, LOC574538, SLC6A12, SLC6A13 |
| 20 | 12q24.31 | 123,357,010–124,310,519 | Genic dup | 953.509 | VPS37B, ABCB9, OGFOD2, ARL6IP4, PITPNM2, MIR4304, LOC100507091, MPHOSPH9, C12orf65, CDK2AP1, SBNO1, SETD8, RILPL2, SNRNP35, RILPL1, MIR3908, TMED2, DDX55, EIF2B1, GTF2H3, TCTN2 , ATP6V0A2, DNAH10 |
| 20 | 19q13.32 | 47,308,130–47,418,258 | Genic dup | 110.128 | SNAR-E, AP2S1 |
| 26 | 11q12.2 | 60,408,411–60,465,698 | Genic del | 57.287 | LINC00301 |
| 31 | 10p15.1 | 6,254,055–6,374,584 | Internal dup | 120.529 | PFKFB3, LOC399715 |
| 34 | 4q22.2 | 93,875,432–93,988,049 | Genic del | 112.617 | GRID2 |
| 39 | 8q11.1q11.21 | 47,398,661–48,407,568 | Genic dup | 1008.907 | LINC00293, LOC100287846, KIAA0146 |
| 40 | 14q24.2 | 73,620,299–73,786,493 | Genic dup | 166.194 | PSEN1, PAPLN, NUMB |
| 43 | 4q24 | 101,476,709–101,668,938 | Genic del | 192.229 | EMCN-IT3 |
| 59 | 2q24.1 | 157,170,397–157,315,649 | Internal dup | 145.252 | NR4A2, GPD2 |
| 59 | 6q26 | 163,549,870–163,842,358 | Genic dup | 292.488 | PACRG , PACRG-AS1, DKFZp451B082, CAHM, QKI |
| 59 | 9p22.2 | 16,826,417–16,931,236 | Internal dup | 104.819 | BNC2 |
| 63 | 3q25.32 | 158,198,274–158,256,949 | Genic del | 58.675 | RSRC1 |
| 63 | 10q26.3 | 134,358,785–134,921,135 | Genic dup | 562.35 | INPP5A, NKX6–2, TTC40 , LOC399829, GPR123 |
Bold items are candidate genes we identified from rare CNVs
Genic del deletion of at least one coding exon, Genic dup full duplication of at least one gene, Internal dup duplication of internal exons
Among the 19 rare CNVs, none of the common known Htx-related genes, including ZIC3, CFC1, NKX2.5, GDF1, NODAL, LEFTY1, LEFTY2, ACVR2B, DANH5, DNAH11, DNAI1, FOXH1, CRELD1, or GALNT11, were identified. In order to correlate the phenotypes of the Htx syndrome patients to specific pathologic genes, we first examined the function of the genes associated with the 19 rare CNV segments (Additional file 1: Table S5). Candidate genes were identified based on the following criteria: (1) participation in ciliary proteome and function; (2) relation to signaling pathways Notch, Nodal, Hedgehog, Wnt, and TGF-β; (3) member of ubiquitin ligase E3 family. E3 ubiquitin ligases were reported to play important roles in the Nodal signaling pathway, cilia formation, and cilia assembly [22–24]. We finally found six potential candidate genes in five CNV segments in five subjects: NUMB [MIM: 603728], PACRG [MIM: 608427], TCTN2 [MIM: 613846], DANH10 [MIM: 605884], RNF115, and cilia and flagella associated protein 46 [CFAP46/TTC40].
Verification by qPCR showed that these five rare CNVs comprise four duplications and one deletion (Fig. 1 and Additional file 2: Figure S3). The qPCR results and clinical diagnosis of the five patients are summarized in Table 3.
Fig. 1.

Chromosomal view of rare CNVs in candidate Htx patients and the verified results of qPCR. a CytoScan HD array presents a 302.5-kb deletion of 1q21.1 involving RNF115. b A 953.5-kb duplication at 12q24.31 affecting both TCTN2 and DNAH10. In data (a, b), the upper panel depicts log2 ratio data, the middle panel depicts the copy number duplications or deletions, and the lower panel depicts smooth signals of indicated segments. Locations of genes implicated in Htx are shown in the top. Results of qPCR are denoted by yellow stars
Table 3.
Clinical phenotypes of heterotaxy patients with CNVs carrying candidate genes
| ID | Segments | Sizes (kb) | Copy numbers | Genes | Patients’ cardiac abnormalities | Extracardiac abnormalities |
|---|---|---|---|---|---|---|
| 7 | 1q21.1 (145,625,128–145,927,662) | 302.534 | 1 del | RNF115 | D, SA, PAVC, LSVC | RAA, BI, right spleen, RSS, LSL |
| 20 | 12q24.31 (123,357,010–124,310,519) | 953.509 | 3 dup | TCTN2, DNAH10 | D, SA, TA, TGA, PA, VSD, PDA, LSVC | RAA, BI, right spleen, RSS, LSL |
| 40 | 14q24.2 (73,620,299–73,786,493) | 166.194 | 3 dup | NUMB | D, DORV, PS, VSD, LSVC, SIV | RAA, BI, right spleen, RSS, LSL |
| 59 | 6q26 (163,549,870–163,842,358) | 292.488 | 3 dup | PACRG | D, SA, SV, MGA, PS, CAVC, LSVC, IRAA | LAA, BRB, asplenia, LSS, LCS |
| 63 | 10q26.3 (134,358,785–134,921,135) | 562.35 | 4 dup | TTC40 | D, DORV, PS, VSD, PDA, LSVC, SIV | RAA, BI, right spleen, RSS, LSL |
dup duplication, del deletion, PDA patent ductus arteriosus, PS pulmonary stenosis, VSD ventricle septum defect, CAVC complete atrioventricular canal, PAVC partial atrioventricular canal, D dextrocardia, SA single atrium, SV single ventricle, TA tricuspid atresia, PA pulmonary atresia, TGA/MGA translocation of great arteries/malposition of great arteries, DORV double outlet right ventricle, IRAA isomerism of right atrial appendages, SIV superior-inferior ventricle, LSVC left superior vena cava, RAA right aortic arch, LAA left aortic arch, BI bronchial inversus, BRB bilateral right bronchi (short), RSS right-sided stomach, LSS left-sided stomach, LSL left-sided liver, LCS liver centrally situated
To determine whether the patients identified as carrying CNVs of the candidate genes had mutations in other known laterality-related genes (e.g., ZIC3, CFC1, NKX2.5, GDF1, NODAL, LEFTY1, LEFTY2, ACVR2B, DANH5, DNAH11, DNAI1, FOXH1, CRELD1, and GALNT11), we screened the coding sequences of these genes using whole-exome sequencing analysis. Aside from a nonsynonymous heterozygous mutation (c. 841A > G, p.Trp281Arg) in LEFTY1 (Additional file 2: Figure S4 and Additional file 1: Table S6) in one patient with a CNV of CFAP46, the results revealed no functional mutations in these laterality-related genes.
Expression patterns of candidate genes in zebrafish
Zebrafish were used as a model organism to further elucidate the roles of the candidate genes in regulating organ laterality, as all six of the candidate genes have orthologs in zebrafish. We examined the gene expression patterns at two developmental stages using WMISH with digoxigenin-labeled RNA as probes: the 8–10 somite stage (13–15 h post fertilization [hpf]; symmetry was first broken at Kupffer’s vesicle [KV]) and the primordium 5 stage (24 hpf; heart begins to beat). WMISH analysis of the tctn2 gene was unsuccessful. The pacrg gene was expressed in the KV in the 8–10 somite stage, and was expressed in the pronephric duct and floor plate in the primordium 5 stage (Fig. 2a, b). In the 8–10 somite stage, numb, rnf115, dnah10, and cfap46 exhibited nearly ubiquitous expression patterns (Fig. 2c, e, g, i). In the primordium 5 stage, however, these four genes exhibited more localized expression patterns: numb and rnf115 were both expressed in the brain (Fig. 2d, f); dnah10 was restricted to the notochord (Fig. 2h); cfap46 was expressed in the pronephric duct and floor plate (Fig. 2j).
Fig. 2.
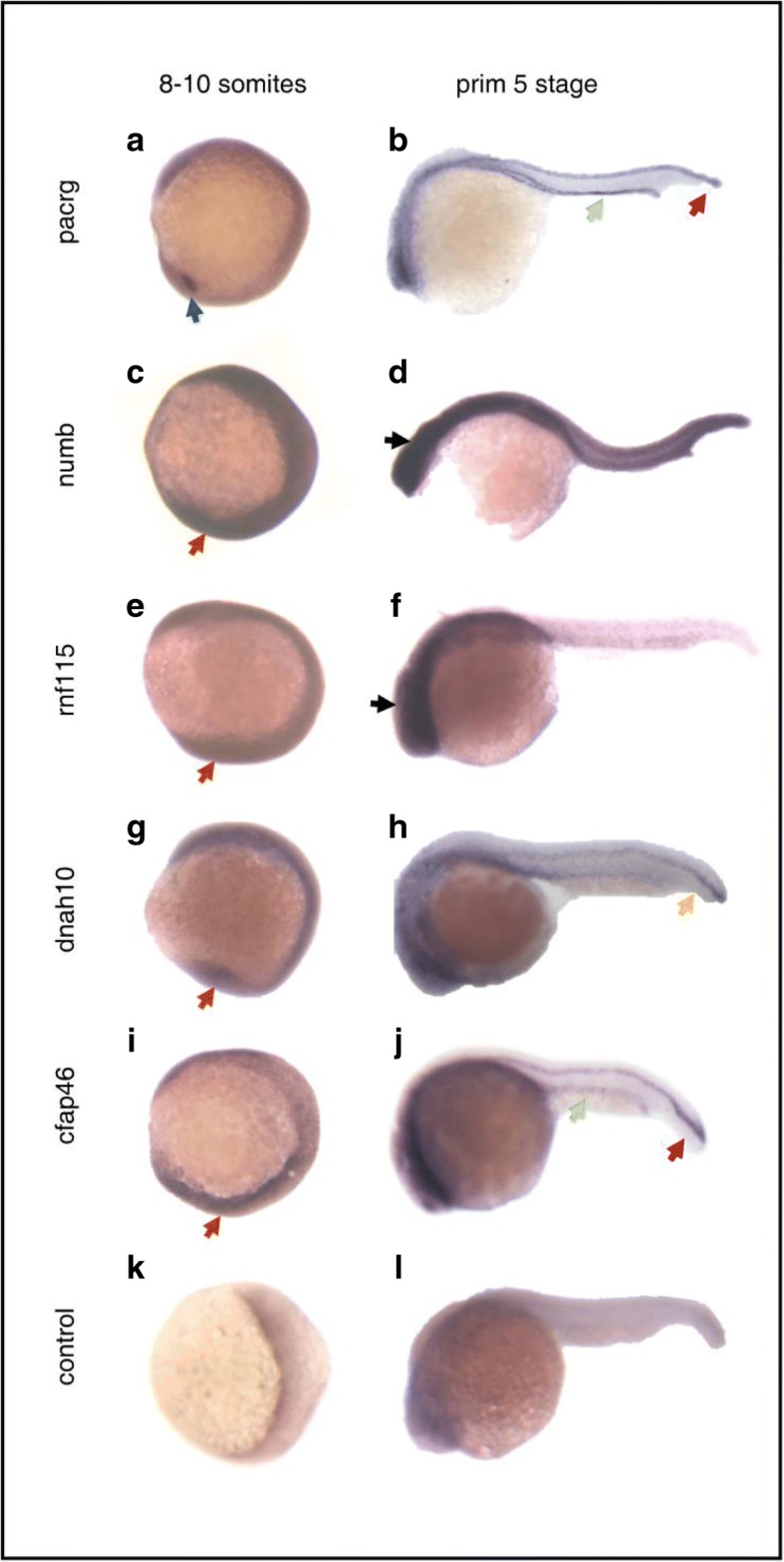
Whole mount in situ hybridization analysis of candidate genes at two stages: 8–10 somites and primordium 5 stage. a, c, e, g, i, k Results of in situ hybridization of candidate genes and standard control at 13–15 hpf (8–10 somites). Embryos are viewed laterally with anterior to the top to examine KV expression. b, d, f, h, j, l Results of in situ hybridization of candidate genes and standard control at 24 hpf (primordium 5 stage). Lateral view of embryos with anterior to the left. KV (blue arrow), floor plate (red arrows), pronephric duct (green arrows), notochord (yellow arrow), head (black arrows), ubiquitous expression (orange arrows)
Knockdown and mutation of the candidate genes disturbs cardiac looping
Expression of the candidate genes in zebrafish was downregulated using MO knockdown to examine the effect on cardiac looping, which depends on normal LR patterning. Three heart tube morphologies occur in zebrafish: dextral loop (d-loop), sinistral loop (s-loop), or no loop (Fig. 3a) [25]. In our study, galnt11 was used as a positive control. GALNT11, encoding the polypeptide N-acetylgalactosaminyltransferase 11, was previously identified as a candidate Htx gene in a patient by CNV analysis. The gene plays an important role in the development of the LR axis by activating the Notch signaling pathway and modulating the balance between motile and nonmotile cilia [15, 26]. A standard control provided by Gene Tools was injected as a negative control.
Fig. 3.

Loss of function of candidate genes in zebrafish disturbed cardiac looping. a Zebrafish heart shows normal dextral loop, abnormal sinistral loop, and no-loop types in cmlc2:eGFP morphants in ventral view. b The percentage of abnormal heart looping with MO injected. The experiments were repeated 3 times, and at each time > 70 embryos were examined for each group. c Summary of the abnormal heart looping of dnah10 and rnf115 mutations generated by co-injection of zebrafish Cas9 mRNA 600 pg and dnah10 gRNA 100 pg or rnf115 gRNA 100 pg. The experiments were repeated 3 times, and at each time > 71 embryos were examined for each group. d Percentage of embryos that exhibit abnormal cardiac looping with mRNA over-expressed. e The rnf115 mRNA can rescue LR randomization. The abnormal heart looping phenotype which is induced by rnf115 MO can be rescued by 6.25 pg rnf115 mRNA. Heart looping direction was assayed in zebrafish at stage 2 dpf. Bars show the total percent of abnormally looped heart including two types: no-loop and sinistral loop heart. Standard control MO (StdCtrl) is negative control. galnt11 is used as positive control. Error bars represent the standard error of the mean (SEM). *P < 0.05, **P < 0.01, ***P < 0.001, respectively vs. StdCtrl. WT, wild type
Zebrafish embryos were injected with MOs at the one-cell stage, and the direction of heart looping was assessed at 48 hpf. We found that MO knockdown of five of the six candidate genes led to altered cardiac looping phenotypes. Compared with the negative control (2.35% abnormality), the phenotypes of five morphants (numb, pacrg, tctn2, dnah10, and rnf115) differed significantly, with 13.16–26.2% of embryos exhibiting either an L-loop or no loop (P < 0.001). However, knockdown of cfap46 had no significant effect on the direction of heart looping (P > 0.05) (Fig. 3b).
Among the five candidate genes, DNAH10 and RNF115 were found to be novel candidate Htx genes in both humans and animals. dnah10 and rnf115 mutations induced by co-injecting zebrafish codon-optimized Cas9 mRNA and dnah10 or rnf115 gRNA also led to a significant increase of abnormal heart looping in F0 embryos (Fig. 3c; P < 0.001), which demonstrates the roles that dnah10 and rnf115 play in LR patterning.
Over-expression of the candidate genes disturbs cardiac looping
The five candidate genes with phenotypes in MO-knockdown embryos were then divided into two groups based on the clinical copy number in the Htx patients: four duplicated genes (numb, pacrg, tctn2, dnah10), and one deleted gene (rnf115). The functions of the duplicated genes were examined using mRNA over-expression analyses. Rescue of the deleted gene, rnf115, was conducted using an rnf115-pCS2+ plasmid.
Compared with negative control (1% abnormality), over-expression of the candidate genes had a significant effect on cardiac looping: numb (10.0% abnormality, P < 0.001); tctn2 (10.2% abnormality, P < 0.001); pacrg (4.95% abnormality, P < 0.05) (Fig. 3d). Over-expression of the duplicated gene dnah10 could not be carried out because its mRNA was too long (14,062 bases). Remarkably, rnf115 MO knockdown led to 14.3% of abnormal cardiac looping, and injection of zebrafish with rnf115 mRNA rescued the normal phenotype (4.7% abnormality). This shows that this gene has a specific function in LR patterning (Fig. 3e).
Candidate genes exhibit global effects on early signaling pathways in LR development
An abnormal cardiac looping pattern can result from either disruption of early common laterality pathways or specific heart field effects. Based on the early expression patterns of the candidate genes and the patients’ clinical information of more than one organ malposition, we hypothesized that the abnormal cardiac looping phenotype results from disruption of early signaling pathways during LR development. To test this hypothesis, we examined the expression patterns in zebrafish morphants of pitx2 and lefty2, which are markers of the early common laterality pathway in the 18–22 somite stage. The pitx2 gene encodes a transcription factor that relays LR-patterning information necessary for proper organogenesis, whereas lefty2 encodes a protein of the TGF-β superfamily that inhibits nodal activation. The embryonic expression of pitx2 and lefty2 exhibited both normal (left side) and abnormal (right side, bilateral, absent) patterns (Fig. 4a, b). Negative control morphants exhibited approximately 9.1% abnormal pitx2 expression and 16.0% abnormal lefty2 expression. Morphants injected with galnt11 positive control exhibited 31.4% of pitx2 and 37.7% of lefty2 abnormality (P < 0.001). The numb, pacrg, tctn2, and dnah10 morphants exhibited significant abnormal pitx2 and lefty2 expression patterns (20.6–36.2 of pitx2 abnormality and 27.3–54.7% of lefty2 abnormality; P < 0.001). The rnf115 morphants showed 28.0% abnormality in pitx2 expression (P < 0.001) but did not exhibit significant abnormal lefty2 expression. Consistent with the results above, cfap46 morphants exhibited no significant abnormalities in either pitx2 or lefty2 expression (Fig. 4c, d).
Fig. 4.

Analysis of pitx2 and lefty2 expression patterns in the lateral plate mesoderm in 18–22 somites. a The expression of pitx2 exhibits four patterns, left, right, bilateral, or absent, in the posterior lateral plate mesoderm of zebrafish embryos. b Morphants show left, right, bilateral or absent lefty2 expression in the cardiac field. c, d Summary of abnormal pitx2 and lefty2 mRNA expression in zebrafish morphants. e, f Summary of abnormal pitx2 and lefty2 mRNA expression in zebrafish with mRNA over-expressed. Embryos are viewed dorsally with anterior to the top. Bars show the percent of abnormal pitx2 and lefty2 expression including three types: right, bilateral, absent expression. Standard control (StdCtrl or Std-RNA) is negative control. galnt11 is used as positive control. Error bars represent the SEM. *P < 0.05, **P < 0.01, ***P < 0.001, respectively, vs. standard control
Then, we analyzed the expression of pitx2 and lefty2 in embryos with over-expression of genes found in genic duplications (numb, pacrg, tctn2). The results showed that the embryonic expression of pitx2 and lefty2 also exhibited both normal (left side) and abnormal (right side, bilateral, absent) patterns. Negative control exhibited approximately 13.0% abnormal pitx2 expression and 17.7% abnormal lefty2 expression. Over-expression of galnt11 positive control exhibited 36.8% of pitx2 and 47.8% of lefty2 abnormality (P < 0.001). Over-expression of numb, pacrg, and tctn2 exhibited significant abnormal pitx2 and lefty2 expression patterns (24.4–40.1% of pitx2 abnormality and 36.9–46.5% of lefty2 abnormality; P < 0.01) (Fig. 4e, f).
Whole-exome sequencing and mutation screening
In order to further explore the relationship between the candidate genes and Htx, we screened the sequencing data of the five candidate genes for rare mutations in 65 Htx patients without mutations of other known laterality-related genes and rare CNVs. Filtering criteria were set as follows: (1) variants are located in exonic or splicing region, (2) exclude synonymous variants, (3) frequency is lower than 0.1% according to public variant databases 1000 Genomes and Exome Aggregation Consortium (ExAC), (4) exclude variants detected in our 100 normal Chinese individuals or 2000 non-heterotaxy patients, and (5) at least one scoring software analysis suggests that mutation is deleterious. Finally, six rare heterozygous mutations in DNAH10, RNF115, TCTN2, and NUMB were detected in six sporadic Htx patients (Additional file 2: Figure S5 and Additional file 1: Table S7).
Discussion
Htx comprises a class of congenital disorders resulting from malformations in LR body patterning, but the underlying cause in the majority of patients remains unknown. In an effort to elucidate the molecular mechanism underlying the pathogenesis of Htx, we recruited 63 children with Htx but free of other syndromes. CNV analyses identified 19 rare CNVs. Further analyses revealed that six candidate genes associated with the 19 rare CNV segments were related with pathways reported to be involved in the regulation of LR development. Downregulation and over-expression of the candidate genes in zebrafish demonstrated that five genes (numb, pacrg, tcnt2, dnah10, and rnf115) strongly affect morphologic cardiac looping as well as the pattern of pitx2 and lefty2 expression. Moreover, we detected rare mutations in the coding sequence of candidate genes DNAH10, RNF115, TCTN2, and NUMB in Htx patients.
The study demonstrated that rare CNVs play an important role in the pathogenesis of Htx in patients. CytoScan HD microarray is a good method for copy-number analyses. However, balanced chromosomal rearrangements such as inversions or balanced translocations could be potentially missed on the CytoScan HD microarray. In our study, the percentage of subjects with rare CNVs (23.7%, 14 of 59 Htx subjects) was higher than that previously reported [15, 27]. The size of these rare CNVs varied from 57 to 1009 kb. One duplication segment at 10q26.3 (genomic coordinates 134,358,785–134,921,135, involving only five genes) identified in our study was included in previously published deletion segments associated with Htx: a terminal CNV beginning at 10q26.13 [15, 17]. The remaining 18 segments were never reported.
Among the five candidate genes we identified, the numb, pacrg, and tctn2 genes had been linked to LR development in animals but not in humans. The gene NUMB encodes an endocytic adaptor protein that plays a role in the determination of cell fate. Consistent with previous reports, zebrafish numb was expressed ubiquitously in the early developmental stage in the present study. Over-expression of numb has been shown to suppress Notch activity, thus causing bilateral distribution of lefty2 expression and disturbed heart tube looping [28]. This observation strongly supports our data showing that not only over-expression but also downregulation of numb in zebrafish leads to abnormal cardiac looping and a random distribution of lefty2 and pitx2 expression.
The genes pacrg and tctn2 are previously reported to regulate ciliary function [29–31]. Cilia play a pivotal role in earlier LR development. The gene pacrg is locally expressed in the KV in zebrafish, where LR asymmetry is initially established. Xenopus embryos injected with pacrg MO exhibit LR laterality defects as well as gastrulation and neural tube closure defects [29, 31]. TCTN2 encodes a type I membrane protein of the tectonic family. TCTN2 was linked to Joubert syndrome, a ciliopathy disease. Tctn2 knockout mice exhibit ventricular septal defects and a right-sided stomach, suggesting that Tctn2 plays a role in laterality defects [30]. Our data further show that over-expression of pacrg or tctn2 in zebrafish also leads to laterality defects. Moreover, this is the first report identifying CNVs of NUMB, PACRG, and TCTN2 in patients with Htx.
To date, no reports have linked DNAH10 and RNF115 with LR patterning in either humans or animals. The gene dnah10, expressed in cilia and flagella, is a component of the inner dynein arms, which are attached to the peripheral microtubule doublets; as a protein involved in ATP production, DNAH10 participates in protozoan flagellar motility [32, 33]. Recent studies have demonstrated that mutations in other cilia dynein heavy-chain genes (such as DNAH5, DNAH9, and DNAH11) can cause primary ciliary dyskinesia (PCD). PCD is a severe inherited disorder that results from defects in flagellar and ciliary axoneme substructures and is characterized by male infertility, respiratory diseases, and LR laterality in 50% of affected individuals [34–36]. We first identified the CNVs of DNAH10 in patients with Htx, and found that knockdown and mutation of dnah10 in zebrafish disturb the LR development and pitx2 and lefty2 expression patterns. Moreover, we found that dnah10 mRNA is highly expressed in the caudal notochord in the primordium 5 stage. The notochord breaks bilateral symmetry by altering cell shape and cilia distribution [37]. DNAH10 may regulate LR patterning by affecting the notochord, which could alter the function of cilia. In addition, both TCTN2 and DNAH10, located within the same CNV segment, can affect the development of LR patterning, and whether there is potential interaction between them needs further research.
The only deleted gene identified in our study, RNF115, also known as breast cancer-associated gene 2 (BCA2), is a type of E3 ubiquitin ligase. E3 ubiquitin ligases play important roles in auto-ubiquitination activity, depending on their RING domain. RNF115 mRNA is expressed at moderate levels in the heart, skeletal muscle, and testis [38]. Previous findings indicated that ubiquitination by E3 ligases regulates a diverse array of cellular functions, such as cilia formation and assembly, and LR development-related signaling pathways (e.g., Nodal signaling) [22–24]. However, the relationship between RNF115 and LR asymmetry remains unknown. In our study, rnf115 morphant and mutant zebrafish exhibited disturbed cardiac looping, and rnf115 mRNA rescued the normal phenotype. Moreover, rnf115 morphants exhibited random distribution of pitx2 expression but minimal perturbations in the lefty2 expression pattern. According to the previously reported pathogenesis of Htx, asymmetric Nodal signaling activates pitx2 in the left lateral plate mesoderm via the Smad-FoxH1 pathway [4]. Our data suggest that rnf115 may function in the ubiquitination of Nodal-downstream genes (the genes in the Smad-FoxH1 pathway) or directly act on pitx2, to regulate LR patterning, which cannot alter the expression of lefty2. However, the specific mechanism through which DNAH10 and RNF115 direct LR axis development remains to be further investigated.
TTC40 encodes cilia and flagella associated protein 46, which is reported to play a role in the occurrence of nasopharyngeal carcinoma and acute myeloid leukemia [39, 40]. In our study, zebrafish with knockdown of this gene did not exhibit an abnormal phenotype in terms of LR patterning. Moreover, in the patient in our study with a CNV of CFAP46, Htx might have been caused by a nonsynonymous mutation (c. 841 A > G, p.Trp281Arg) in LEFTY1 instead.
Conclusions
Our results demonstrate that Htx in some sporadic patients may be attributed to rare CNVs. Moreover, we identified candidate genes in several novel segments of rare CNVs, some of which have never before been reported as related to LR patterning. Downregulation or over-expression of the candidate genes in zebrafish disturbed the development of LR asymmetry. We believe the results of our study advance the understanding of Htx and will aid in its diagnosis. However, according to previous literature and the results of our study [41, 42], we do not have sufficient evidence to show that the genes reported in this paper are causal for heterotaxy. They are only candidate genes for Htx. This is the limitation of our study. The five genes identified in our paper are reported for the first time in a heterotaxy population, and our study can be used as the first evidence for the future research of the pathogenic genes of Htx. This is hoped to be further confirmed in other populations. In addition, other rare CNVs that we did not study might also play significant roles in development of LR asymmetry. Two novel candidate genes identified in the present study, DANH10 and RNF115, should be examined in greater detail with respect to their role in the pathogenesis of defects in LR patterning.
Additional files
Table S1. MO sequences, injection doses, and total embryo numbers analyzed for heart looping and gene expression. Table S2. Specific primers and vector used to produce genes’ full-length mRNA. Table S3. Antisense RNA probes conducted for whole mount in situ hybridization. Table S4. The frequency of each candidate CNV in normal Chinese individuals and non-heterotaxy patients with developmental delay/intellectual disability. Table S5. The function of the genes associated with the 19 rare CNV segments. Table S6. The bioinformatics information on the variant of LEFTY1 in the patient with CNV of TTC40 (CFAP46). Table S7. The bioinformatics information on the variants of candidate genes. (PDF 369 kb)
Figure S1. Knockdown efficiency of splice blocking and translation blocking MOs in zebrafish embryos. Figure S2. The knockout efficiency of dnah10 and rnf115 by CRISPR/Cas9. Figure S3. Chromosomal view of rare CNVs in candidate Htx patients and the verified results of qPCR. Figure S4. Gene sequencing peak shows a nonsynonymous heterozygous mutation in LEFTY1 in the patient with CNV of TTC40 (CFAP46). Figure S5. Rare variations were detected in Htx patients. (PDF 792 kb)
Acknowledgements
We are grateful to Dr. J. L. Du for the zebrafish platform support and critical comments on the experiments. The authors would like to acknowledge Yifan Zhu for language editing.
Funding
The project was funded by grants from the National Natural Science Foundation of China (81270233), the Shanghai Municipal Commission of Health and Family Planning three-year action plan (GWIV-23), and the Science Committee of Shanghai (13JC1401705). The funding organizations had no role in study design, data collection and analysis, preparation of the manuscript, or in the decision to submit the article for publication.
Availability of data and materials
The CNV calls presented in Table 2 from Affymetrix CytoScan HD microarray can be accessed through the ArrayExpress repository (https://www.ebi.ac.uk/arrayexpress/) under accession number E-MTAB-6820.
The raw data of next generation sequencing of related genes in heterotaxy patients generated during the current study can be accessed through the Sequence Read Archive (SRA) database [https://www.ncbi.nlm.nih.gov/Traces/study/] under accession number SRP145525. Other data generated or analyzed during this study are included in the main paper or its additional files.
Abbreviations
- CNV
Copy number variant
- d-loop
Dextral loop
- gRNA
Guide RNA
- hpf
Hours post fertilization
- Htx
Heterotaxy
- kb
Kilobase
- KV
Kupffer’s vesicle
- LR
Left-right
- Mb
Megabase
- MO
Morpholino oligo
- PCD
Primary ciliary dyskinesia
- qPCR
Quantitative real-time polymerase chain reaction
- s-loop
Sinistral loop
- WMISH
Whole mount in situ hybridization
Authors’ contributions
CL, RC, RX, and KS were involved in conceptualization; CL and RC performed the experiments; FL, SC, and KS provided the clinical information of the patients; YX and TL analyzed the sequencing data; CL wrote the main manuscript text; KS and RX were involved in supervision, project administration, and funding acquisition. All authors reviewed the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The research has been performed in accordance with the principles of the Declaration of Helsinki. The study was also approved by the ethics committees of Xinhua Hospital (XHEC-C-2012-018, Shanghai, China) and Shanghai Children’s Medical Center (SCMC-201004). Parents of the children gave consent for their participation. All zebrafish experiments were conducted at the Institute of Neuroscience, Chinese Academy of Sciences. In the design and process of the zebrafish experiments involved in the study, we have followed relevant guidelines.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Chunjie Liu and Ruixue Cao contributed equally to this work.
Electronic supplementary material
The online version of this article (10.1186/s13073-018-0549-y) contains supplementary material, which is available to authorized users.
Contributor Information
Rang Xu, Email: rangxu@shsmu.edu.cn.
Kun Sun, Email: sunkun@xinhuamed.com.cn.
References
- 1.Jacobs JP, Anderson RH, Weinberg PM, Walters HL, 3rd, Tchervenkov CI, Del Duca D, et al. The nomenclature, definition and classification of cardiac structures in the setting of heterotaxy. Cardiol Young. 2007;17 Suppl 2:1–28. doi: 10.1017/S1047951107001138. [DOI] [PubMed] [Google Scholar]
- 2.Taketazu M, Lougheed J, Yoo SJ, Lim JS, Hornberger LK. Spectrum of cardiovascular disease, accuracy of diagnosis, and outcome in fetal heterotaxy syndrome. Am J Cardiol. 2006;97(5):720–724. doi: 10.1016/j.amjcard.2005.09.119. [DOI] [PubMed] [Google Scholar]
- 3.Shiraishi I, Ichikawa H. Human heterotaxy syndrome — from molecular genetics to clinical features, management, and prognosis. Circ J. 2012;76(9):2066–2075. doi: 10.1253/circj.CJ-12-0957. [DOI] [PubMed] [Google Scholar]
- 4.Zhu L, Belmont JW, Ware SM. Genetics of human heterotaxias. Eur J Hum Genet. 2006;14(1):17–25. doi: 10.1038/sj.ejhg.5201506. [DOI] [PubMed] [Google Scholar]
- 5.Lin X, Xu X. Distinct functions of Wnt/beta-catenin signaling in KV development and cardiac asymmetry. Development. 2009;136(2):207–217. doi: 10.1242/dev.029561. [DOI] [PubMed] [Google Scholar]
- 6.Bamford RN, Roessler E, Burdine RD, Saplakoglu U, dela Cruz J, Splitt M, et al. Loss-of-function mutations in the EGF-CFC gene CFC1 are associated with human left-right laterality defects. Nat Genet. 2000;26(3):365–369. doi: 10.1038/81695. [DOI] [PubMed] [Google Scholar]
- 7.Mohapatra B, Casey B, Li H, Ho-Dawson T, Smith L, Fernbach SD, et al. Identification and functional characterization of NODAL rare variants in heterotaxy and isolated cardiovascular malformations. Hum Mol Genet. 2009;18(5):861–871. doi: 10.1093/hmg/ddn411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kosaki R, Gebbia M, Kosaki K, Lewin M, Bowers P, Towbin JA, et al. Left-right axis malformations associated with mutations in ACVR2B, the gene for human activin receptor type IIB. Am J Med Genet. 1999;82(1):70–76. doi: 10.1002/(SICI)1096-8628(19990101)82:1<70::AID-AJMG14>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 9.Kosaki K, Bassi MT, Kosaki R, Lewin M, Belmont J, Schauer G, et al. Characterization and mutation analysis of human LEFTY A and LEFTY B, homologues of murine genes implicated in left-right axis development. Am J Hum Genet. 1999;64(3):712–721. doi: 10.1086/302289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaasinen E, Aittomaki K, Eronen M, Vahteristo P, Karhu A, Mecklin JP, et al. Recessively inherited right atrial isomerism caused by mutations in growth/differentiation factor 1 (GDF1) Hum Mol Genet. 2010;19(14):2747–2753. doi: 10.1093/hmg/ddq164. [DOI] [PubMed] [Google Scholar]
- 11.Ware SM, Peng J, Zhu L, Fernbach S, Colicos S, Casey B, et al. Identification and functional analysis of ZIC3 mutations in heterotaxy and related congenital heart defects. Am J Hum Genet. 2004;74(1):93–105. doi: 10.1086/380998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhian S, Belmont J, Maslen CL. Specific association of missense mutations in CRELD1 with cardiac atrioventricular septal defects in heterotaxy syndrome. Am J Med Genet A. 2012;158A(8):2047–2049. doi: 10.1002/ajmg.a.35457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Izumi K, Noon S, Wilkens A, Krantz ID. NKX2.5 mutation identification on exome sequencing in a patient with heterotaxy. Eur J Med Genet. 2014;57(10):558–561. doi: 10.1016/j.ejmg.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 14.Sutherland MJ, Ware SM. Disorders of left-right asymmetry: heterotaxy and situs inversus. Am J Med Genet C Semin Med Genet. 2009;151C(4):307–317. doi: 10.1002/ajmg.c.30228. [DOI] [PubMed] [Google Scholar]
- 15.Fakhro KA, Choi M, Ware SM, Belmont JW, Towbin JA, Lifton RP, Brueckner M, et al. Rare copy number variations in congenital heart disease patients identify unique genes in left-right patterning. Proc Natl Acad Sci U S A. 2011;108(7):2915–2920. doi: 10.1073/pnas.1019645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hagen EM, Sicko RJ, Kay DM, Rigler SL, Dimopoulos A, Ahmad S, Mills JL, et al. Copy-number variant analysis of classic heterotaxy highlights the importance of body patterning pathways. Hum Genet. 2016;135(12):1355–1364. doi: 10.1007/s00439-016-1727-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rigler SL, Kay DM, Sicko RJ, Fan R, Liu A, Caggana M, Mills JL, et al. Novel copy-number variants in a population-based investigation of classic heterotaxy. Genet Med. 2015;17(5):348–357. doi: 10.1038/gim.2014.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu PC, Gu SY, Bu JW, Du JL. TRPC1 is essential for in vivo angiogenesis in zebrafish. Circ Res. 2010;106(7):1221–1232. doi: 10.1161/CIRCRESAHA.109.207670. [DOI] [PubMed] [Google Scholar]
- 19.Xu B, Zhang Y, Du XF, Li J, Zi HX, Bu JW, et al. Neurons secrete miR-132-containing exosomes to regulate brain vascular integrity. Cell Res. 2017;27(7):882–897. doi: 10.1038/cr.2017.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thisse C, Thisse B, Schilling TF, Postlethwait JH. Structure of the zebrafish snail1 gene and its expression in wild-type, spadetail and no tail mutant embryos. Development. 1993;119(4):1203–1215. doi: 10.1242/dev.119.4.1203. [DOI] [PubMed] [Google Scholar]
- 21.Hugh D, Allen DJD, Shaddy RE, Feltes TF. Moss & Adams’ heart disease in infants, children, and adolescents, including the fetus and young adult. Baltimore: Lippincott Williams and Wilkins; 2012. [Google Scholar]
- 22.Villumsen BH, Danielsen JR, Povlsen L, Sylvestersen KB, Merdes A, Beli P, et al. A new cellular stress response that triggers centriolar satellite reorganization and ciliogenesis. EMBO J. 2013;32(23):3029–3040. doi: 10.1038/emboj.2013.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cota CD, Bagher P, Pelc P, Smith CO, Bodner CR, Gunn TM. Mice with mutations in Mahogunin ring finger-1 (Mgrn1) exhibit abnormal patterning of the left-right axis. Dev Dyn. 2006;235(12):3438–3447. doi: 10.1002/dvdy.20992. [DOI] [PubMed] [Google Scholar]
- 24.Upadhyay A, Amanullah A, Chhangani D, Mishra R, Prasad A, Mishra A. Mahogunin Ring Finger-1 (MGRN1), a multifaceted ubiquitin ligase: recent unraveling of neurobiological mechanisms. Mol Neurobiol. 2016;53(7):4484–4496. doi: 10.1007/s12035-015-9379-8. [DOI] [PubMed] [Google Scholar]
- 25.Noel ES, Verhoeven M, Lagendijk AK, Tessadori F, Smith K, Choorapoikayil S, et al. A nodal-independent and tissue-intrinsic mechanism controls heart-looping chirality. Nat Commun. 2013;4:2754. doi: 10.1038/ncomms3754. [DOI] [PubMed] [Google Scholar]
- 26.Boskovski MT, Yuan S, Pedersen NB, Goth CK, Makova S, Clausen H, et al. The heterotaxy gene GALNT11 glycosylates Notch to orchestrate cilia type and laterality. Nature. 2013;504(7480):456–459. doi: 10.1038/nature12723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cowan JR, Tariq M, Shaw C, Rao M, Belmont JW, Lalani SR, et al. Copy number variation as a genetic basis for heterotaxy and heterotaxy-spectrum congenital heart defects. Philos Trans R Soc Lond Ser B Biol Sci. 2016;371(1710) [DOI] [PMC free article] [PubMed]
- 28.Niikura Y, Tabata Y, Tajima A, Inoue I, Arai K, Watanabe S. Zebrafish Numb homologue: phylogenetic evolution and involvement in regulation of left-right asymmetry. Mech Dev. 2006;123(5):407–414. doi: 10.1016/j.mod.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 29.Thumberger T, Hagenlocher C, Tisler M, Beyer T, Tietze N, Schweickert A, et al. Ciliary and non-ciliary expression and function of PACRG during vertebrate development. Cilia. 2012;1(1):13. doi: 10.1186/2046-2530-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sang L, Miller JJ, Corbit KC, Giles RH, Brauer MJ, Otto EA, et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell. 2011;145(4):513–528. doi: 10.1016/j.cell.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loucks CM, Bialas NJ, Dekkers MP, Walker DS, Grundy LJ, Li C, et al. PACRG, a protein linked to ciliary motility, mediates cellular signaling. Mol Biol Cell. 2016;27(13):2133–2144. doi: 10.1091/mbc.e15-07-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maiti AK, Mattei MG, Jorissen M, Volz A, Zeigler A, Bouvagnet P. Identification, tissue specific expression, and chromosomal localisation of several human dynein heavy chain genes. Eur J Hum Genet. 2000;8(12):923–932. doi: 10.1038/sj.ejhg.5200555. [DOI] [PubMed] [Google Scholar]
- 33.Zukas R, Chang AJ, Rice M, Springer AL. Structural analysis of flagellar axonemes from inner arm dynein knockdown strains of Trypanosoma brucei. Biocell. 2012;36(3):133–141. [PubMed] [Google Scholar]
- 34.Pazour GJ, Agrin N, Walker BL, Witman GB. Identification of predicted human outer dynein arm genes: candidates for primary ciliary dyskinesia genes. J Med Genet. 2006;43(1):62–73. doi: 10.1136/jmg.2005.033001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwabe GC, Hoffmann K, Loges NT, Birker D, Rossier C, de Santi MM, et al. Primary ciliary dyskinesia associated with normal axoneme ultrastructure is caused by DNAH11 mutations. Hum Mutat. 2008;29(2):289–298. doi: 10.1002/humu.20656. [DOI] [PubMed] [Google Scholar]
- 36.Bartoloni L, Blouin JL, Pan Y, Gehrig C, Maiti AK, Scamuffa N, et al. Mutations in the DNAH11 (axonemal heavy chain dynein type 11) gene cause one form of situs inversus totalis and most likely primary ciliary dyskinesia. Proc Natl Acad Sci U S A. 2002;99(16):10282–10286. doi: 10.1073/pnas.152337699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Compagnon J, Barone V, Rajshekar S, Kottmeier R, Pranjic-Ferscha K, Behrndt M, et al. The notochord breaks bilateral symmetry by controlling cell shapes in the zebrafish laterality organ. Dev Cell. 2014;31(6):774–783. doi: 10.1016/j.devcel.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 38.Burger AM, Gao Y, Amemiya Y, Kahn HJ, Kitching R, Yang Y, et al. A novel RING-type ubiquitin ligase breast cancer-associated gene 2 correlates with outcome in invasive breast cancer. Cancer Res. 2005;65(22):10401–10412. doi: 10.1158/0008-5472.CAN-05-2103. [DOI] [PubMed] [Google Scholar]
- 39.Ayadi W, Allaya N, Frikha H, Trigui E, Khabir A, Ghorbel A, et al. Identification of a novel methylated gene in nasopharyngeal carcinoma: TTC40. Biomed Res Int. 2014;2014:691742. doi: 10.1155/2014/691742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saleki R, Christensen T, Liu W, Wang X, Chen QC, Aakre M, et al. A novel TTC40-MSI2 fusion in de novo acute myeloid leukemia with an unbalanced 10;17 translocation. Leuk Lymphoma. 2015;56(4):1137–1139. doi: 10.3109/10428194.2014.947611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.MacArthur DG, Manolio TA, Dimmock DP, Rehm HL, Shendure J, Abecasis GR, et al. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014;508(7497):469–476. doi: 10.1038/nature13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Strande NT, Riggs ER, Buchanan AH, Ceyhan-Birsoy O, DiStefano M, Dwight SS, et al. Evaluating the clinical validity of gene-disease associations: an evidence-based framework developed by the Clinical Genome Resource. Am J Hum Genet. 2017;100(6):895–906. doi: 10.1016/j.ajhg.2017.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. MO sequences, injection doses, and total embryo numbers analyzed for heart looping and gene expression. Table S2. Specific primers and vector used to produce genes’ full-length mRNA. Table S3. Antisense RNA probes conducted for whole mount in situ hybridization. Table S4. The frequency of each candidate CNV in normal Chinese individuals and non-heterotaxy patients with developmental delay/intellectual disability. Table S5. The function of the genes associated with the 19 rare CNV segments. Table S6. The bioinformatics information on the variant of LEFTY1 in the patient with CNV of TTC40 (CFAP46). Table S7. The bioinformatics information on the variants of candidate genes. (PDF 369 kb)
Figure S1. Knockdown efficiency of splice blocking and translation blocking MOs in zebrafish embryos. Figure S2. The knockout efficiency of dnah10 and rnf115 by CRISPR/Cas9. Figure S3. Chromosomal view of rare CNVs in candidate Htx patients and the verified results of qPCR. Figure S4. Gene sequencing peak shows a nonsynonymous heterozygous mutation in LEFTY1 in the patient with CNV of TTC40 (CFAP46). Figure S5. Rare variations were detected in Htx patients. (PDF 792 kb)
Data Availability Statement
The CNV calls presented in Table 2 from Affymetrix CytoScan HD microarray can be accessed through the ArrayExpress repository (https://www.ebi.ac.uk/arrayexpress/) under accession number E-MTAB-6820.
The raw data of next generation sequencing of related genes in heterotaxy patients generated during the current study can be accessed through the Sequence Read Archive (SRA) database [https://www.ncbi.nlm.nih.gov/Traces/study/] under accession number SRP145525. Other data generated or analyzed during this study are included in the main paper or its additional files.