

Abstract
Background
Multilevel data integration is becoming a major area of research in systems biology. Within this area, multi-‘omics datasets on complex diseases are becoming more readily available and there is a need to set standards and good practices for integrated analysis of biological, clinical and environmental data. We present a framework to plan and generate single and multi-‘omics signatures of disease states.
Methods
The framework is divided into four major steps: dataset subsetting, feature filtering, ‘omics-based clustering and biomarker identification.
Results
We illustrate the usefulness of this framework by identifying potential patient clusters based on integrated multi-‘omics signatures in a publicly available ovarian cystadenocarcinoma dataset. The analysis generated a higher number of stable and clinically relevant clusters than previously reported, and enabled the generation of predictive models of patient outcomes.
Conclusions
This framework will help health researchers plan and perform multi-‘omics big data analyses to generate hypotheses and make sense of their rich, diverse and ever growing datasets, to enable implementation of translational P4 medicine.
Electronic supplementary material
The online version of this article (10.1186/s12918-018-0556-z) contains supplementary material, which is available to authorized users.
Keywords: Molecular signatures, ‘Omics data, Stratification, Systems medicine
Background
Since the early days of medicine, practitioners have always combined their observations from patient examinations with their medical knowledge and experience to diagnose medical conditions and find treatments tailored to the patient [1]. Nowadays, this rationale includes the integration of molecular, clinical, imaging information and other data sources to inform diagnosis and prognosis [2] or in other words, personalised medicine.
Various data integration methods developed through systems biology and computer science are now available to researchers. These methods aim at bridging the gap between the vast amounts of data generated in an ever-cheaper way [3] and our understanding of biology reflecting the complexity of biological systems [4]. Promises of data integration are the reduced cost of clinical trials, better statistical power, more accurate hypothesis generation and ultimately, individualised and cheaper healthcare [2].
However, a lack of communication exists between the fields of clinical medicine and systems biology, bioinformatics and biostatistics, as suggested by the reluctance or distrust to recent developments of personalised medicine by the medical community [1, 5, 6]. To address this issue, we developed a computational/analysis framework that aims at facilitating communication between healthcare professionals, computational biologists and bioinformaticians.
Among several ways of integrating data across biological levels, one of the components is multi-omics data integration. The identification of molecular signatures has been a focus of the biology and bioinformatics communities for over three decades. Early studies focused on a small number of molecules, paving the way for larger studies, eventually supporting the emergence of the ‘omics’ concept in the late 1990’s, starting with ‘genomics’ [7, 8]. Owing to both technical and biological advances, many classes of molecules have been studied by ‘omics technologies such as transcriptomics [9–11], proteomics [12, 13], lipidomics [14, 15], metabolomics (first mentioned in [16, 17]), the composition of the exhaled breath by breathomics (first mentioned in [18]) [19], and interactomics [20, 21], among others.
Consequently, bioinformatics tools have been developed to analyse this new wealth of biological data, as reviewed in [22]. The concept of systems biology was developed first in the 1960’s [23, 24] to study biological organisms as complete and complex systems, integrating various sources of information (phenotypic data, molecular data, etc.) in combination with pathway/network analysis and mathematical modelling [25–33]. These systems approaches are highly suitable for the discovery of disease phenotypes (based on empirical recognition of observed characteristics) and so-called endotypes (capturing complex causative mechanisms in disease) [34]. The logical next step was to apply systems biology tools to improve clinical diagnosis, refine the endotypes leading to diseases, develop a comprehensive approach to the human body and assess an individual’s health in light of its ‘omics status. In this way the ‘systems medicine’ concept was born [35–41]. The systems medicine rationale is outlined in Fig. 1.
Fig. 1.

Outline of the Systems Medicine rationale. Represented in orange are the steps linked to quality data production, followed by curation in grey, identification of interesting features through statistical analysis in blue and hypothesis generation and their validation in green. Modelling and knowledge representation methods can inform the hypotheses generated through statistical analysis of generated hypotheses on their own (in purple). Outputs of this exercise are represented in red: drug repurposing, new drugs and improved diagnostics, with the help of clinical trials
Any meaningful experiment relies on a robust, bias-controlled study design [42] using appropriate technologies, leading to the production of trustworthy quality-checked data. Data curation then aims at organising, annotating, integrating and preserving data from various sources for reuse and further integration. The next step is to identify relevant molecular features using statistical evidence. A tremendous and constantly growing number of methods is available for this purpose, making the process of method selection a crucial and challenging task. We provide some guidelines here but recommend that the reader turns to specialised reviews (such as [43]) for more insights on the relevance and appropriateness of individual methods. Once features are statistically selected, their annotation is required to interpret results and produce a single ‘omic signature. Annotation is a complex task that links identifiers from the technological platforms to existing entities (i.e. genes, peptides, metabolites, lipids, etc.) [44, 45]. If the data permit, information from several ‘omics platforms is integrated into multi-‘omics signatures. Single and multi ‘omics signatures ultimately serve to identify molecular mechanisms driving pathobiology.
Contextualisation of signatures with existing knowledge is now standard practice (e.g. ontology, enrichment and pathway analysis [46]), or performed with more advanced tools for data integration and visualisation such as a disease map [47]. Exploratory analysis using network-based information is valuable, with tools such as the STRING database [48], among many others. Hypotheses can then be formulated and tested in two ways, with external datasets and/or new experiments; or by modelling and knowledge representation (see review in [49] and disease maps examples in [47, 50–52]). With the help of systems pharmacology (see [53]), outcomes of this whole exercise are enabling: (i) identification of new potential drug targets associated with newly identified patient clusters, (ii) elucidation of potential biomarkers for diagnosis, (iii) repurposing of existing drugs and, ultimately, (iv) changes in diagnostic processes and development of new drugs and treatments for disease management. The key step in the systems medicine process is pattern recognition, for which a robust and step-wise framework is required.
Definitions
Our article focuses on the identification of disease mechanisms through statistical analysis of raw data, annotation with up-to-date ontologies to generate fingerprints (biomarker signatures derived from data collected from a single technical platform), handprints (biomarker signatures derived from data collected within multiple technical platforms, either by fusion of multiple fingerprints or by direct integration of several data types) and interpretation on a pathway level to identify disease-driving mechanisms.
One way to better define the different endotypes is to generate molecular fingerprints (e.g. blood cell transcriptomics analysis yields genes differentially expressed between clinical populations [54]) and handprints (e.g. mRNA expression, DNA methylation and miRNA expression data fused to generate clusters of cancer patients [55]). The latter can be combined to study patients e.g. at the ‘blood biological compartment’ level, and linked with specific disease markers to better define the underlying biology, hence providing new avenues for therapy.
Despite the wealth of ‘omics analyses, little consensus exist on which statistical or bioinformatics methods to apply on each type of data set, nor on the ‘best’ integrative methods for their combined analysis (although standards exist for some data types, see [22]). Here, we present a generic framework to perform statistical and bioinformatics analyses of ‘omics measurements, starting from raw data management to multi-platform data integration, pathway and network modelling that has been adopted by the Innovative Medicines Initiative (IMI) U-BIOPRED Consortium (Unbiased BIOmarkers for the PREDiction of respiratory disease outcomes, http://www.ubiopred.eu) and extended in the eTRIKS Consortium (https://www.etriks.org/) to support a large number of national and European translational medicine projects. This article is not a review of the very large body of literature on relevant bioinformatics methods. Instead it describes generic steps in ‘omics data analysis to which many methods can be mapped to help multidisciplinary teams comprising clinical experts, wet-lab researchers, bioinformaticians, biostatisticians and computational systems biologists share a common understanding and communicate effectively throughout the systems medicine process [56].
We illustrate our pragmatic approach to the design and implementation of the analysis pipeline through a handprint analysis using the TCGA Research Network (The Cancer Genome Atlas – http://cancergenome.nih.gov/) Ovarian serous cystadenocarcinoma (OV) dataset.
Data preparation: Quality control, correction for possible batch effects, missing data handling, and outlier detection
Quality Control (QC) comprises several important steps in data preparation. First, the platform-specific technical QC and normalisation are performed according to the standards of the respective fields of each particular technological platform.
Batch effects are a technical bias arising during study design and data production, due to variability in production platforms, staff, batches, reagent lots, etc. Their impact can be assessed using descriptive methods such as Principal Component Analysis (PCA) and graphical displays. Tools such as ComBat [57] and methodologies developed by van der Kloet [58] can be used to adjust for batch effects when necessary.
Missing data are features of all biological studies and arise for a variety of reasons. If the source of the missingness is unrelated to phenotype or biology, the missing data points can be classified as Missing Completely At Random (MCAR). Such missing values may be handled through imputation (to the mean, mode, mean of nearest neighbours, or by multiple imputation etc.) or by simple deletion [59].
Additional non-random missing data may arise due to assay- or platform-specific performances. For example, the measurement of abundances can fall below the lower limit of detection or quantitation (LLQ) of the instrument. In such instances, imputation is generally applied. Common methods include imputation to zero, LLQ, LLQ/2, or LLQ/√2; extrapolation and maximum likelihood estimation (MLE) can also be used [59].
Particular difficulty occurs in the analysis of mass spectrometry data, when it is impossible to distinguish MCAR data points from those below the LLQ of the technique. The combined levels of missing data often far exceed 10%. For these, the process depicted in the Fig. 2 is proposed.
Fig. 2.
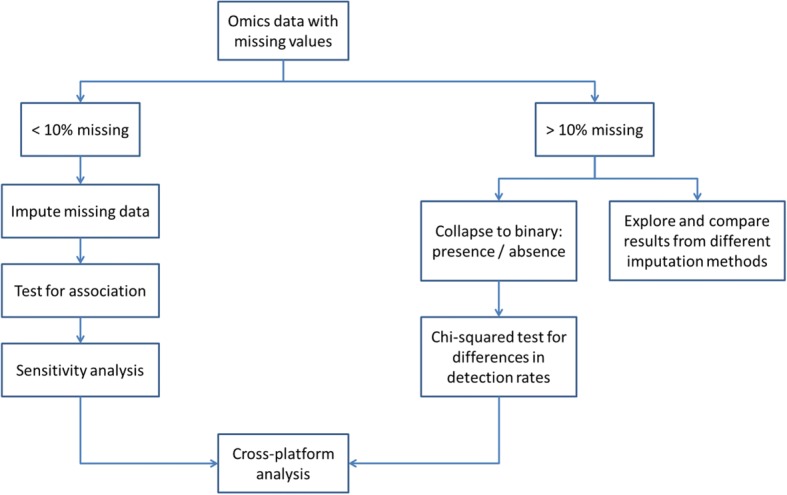
Process proposed for handling high levels of non-random missing data. If there are less than 10% missing values, data imputation is used, then tested for association (artificial associations might arise from the imputation process, which would then skew the analysis downstream) and submitted to a sensitivity analysis. If there are more than 10% missing values, we either collapse the feature/patient to a binary (presence/absence) scheme and run a χ2 test for difference in detection rates, or explore several imputation methods with highly cautious interpretation
Critical appraisal of the pattern of missingness is crucial. Where extensive imputation is applied, the robustness of imputation needs to be assessed by re-analysis, using a second imputation method, or by discarding the imputed values.
Outliers are expected in any biological/platform data. When these are clearly seen to arise due to technical artefacts (differences by many orders of magnitude, etc.), they should be discarded. Otherwise and in general, outlying values in biological data should be retained, flagged and subjected to statistical analysis.
When there is no community-wide consensus on a specific quality threshold for a particular biological data type, the research group generating the data applies quality filters on the basis of their knowledge and experience. Precise description of each data processing step should accompany each dataset to inform colleagues performing downstream analysis.
Methods
The framework concept
Several key generic steps in data analysis were identified and are highlighted in Fig. 3 below.
Fig. 3.

Overview of the framework. Starting from quality-checked and pre-processed ‘omics data, four key generic steps are highlighted: (a) dataset subsetting, including formulation of the biological question to be answered and data preparation, (b) feature filtering (optional step) where features that are uninformative in relation to the question can be removed, (c) ‘omics-based unsupervised clustering (optional step) aiming at finding groups of participants arising from the data structure using the (optionally filtered) features, and finally d) biomarker identification, including feature selection by bioinformatics means and machine learning algorithms for prediction
Step 1: Dataset subsetting
This first box of Fig. 3 3 comprises two major steps: 1) formulating the biological question to be addressed and 2) preparing the data.
Formulating the biological question
Several types of biological questions can be tackled, leading to different partitions of the dataset(s) to study. A partitioning scheme may rely on cohort definitions based on current state of the art, a specific biological question (e.g. comparing highly atopic to non-atopic severe asthmatics), or clustering results, obtained with clinical variables alone, distinct specific ‘omic or multi-‘omics clustering, etc.
Data preparation
Depending on the question formulated at the previous step, data are then subsetted when appropriate. Then, an additional outlier detection check, data transformation and normalisation step can be performed, with methods described above. In this step, the statistical power that the analyst can expect (or the effect size that can be expected to be discovered) can be investigated (for more details on the computation of statistical power in ‘omics data analysis, see [60]). A decision on whether to split the datasets into training and validation sets is also made at this point (see section 4, replication of findings).
Step 2: Feature filtering
Given the complexity and large amount of clinical and ‘omics data in a complex dataset, the number of features measured is vastly superior to the number of replicates creating various statistical challenges, i.e.. the ‘curse of dimensionality’ [61, 62]. Feature filtering (Fig. 3b) is therefore often used to select a subset of features relevant to the biological question studied, remove noise from the dataset and reduce the computing power and time needed [63–65].
Features can be filtered according to specific criteria, based for example on nominal p-values arising from comparison between groups. Indeed, several methods exist to perform feature filtering, based on mean expression values, p-values, fold changes, correlation values [66, 67], information content measures [68, 69], network-based metrics (connectivity, centrality [70, 71]) or using a non-linear machine learning algorithm [72]. We redirect the reader to the following reviews for more details [33, 73–75]. As this step might introduce bias into the downstream analyses, it is not always applied.
Step 3: ‘Omics-based clustering
Clustering analysis groups elements so that objects in the same group are more similar to each other than to those in other groups (Fig. 3c). All methods available rely on similarity or distance measures and a clustering algorithm [76–78]. The most classical clustering methods may be categorized as ‘partitioning’ (constructing k clusters) or ‘hierarchical’ (seeking to build a hierarchy of clusters), and either agglomerative (each observation starts in its own cluster, and pairs of clusters are merged as one moves up the hierarchy, ending in a single cluster) or divisive (all observations start in the same cluster and splits are performed recursively as one moves down the hierarchy, ending with clusters containing one single observation).
It is important to note that clustering techniques are descriptive in nature and will yield clusters, whether they represent reality or not [76]. One way of finding out whether clusters represent reality is to assess their stability, with the consensus clustering approach [79] for example. Using different stable clustering algorithms on the same dataset and comparing them with the meta-clustering rationale [80] is a further step to assess if clusters represent accurately and reproducibly the biological situation in the data.
When several ‘omics datasets on the same patients are available, a handprint analysis can be performed with the Similarity Network Fusion (SNF) method to derive a patient-wise multi-‘omics similarity matrix [55]. Other methods for data integration in the context of subtype discovery are available such as iCluster [81], Multiple Dataset Integration [82], or Patient-Specific Data Fusion [83], further discussed in [84] or under development, for example by the European Stategra FP7 project (http://www.stategra.eu).
Step 4: Biomarker identification
Steps 1 to 3 aim at finding groups of patients to best describe the biological condition(s), with respect to the questions addressed. Step 4 aims at 1) finding the smallest set of molecular features whose difference in abundance between these patient groups (Fig. 3d) enable their distinction (biomarkers) and 2) building classification models through machine-learning techniques, some of which use both feature reduction and classification model building together. The outcome is a fingerprint or handprint, depending on the number of different ‘omics datasets included in the analysis.
Over-fitting and false-discovery rate control
As already mentioned, ‘omics technologies suffer from what is known as the ‘curse of dimensionality’, typically due to the large number of features (p) and low number of samples (n). As statistical methods were historically developed for a situation where the dimensions were n >> > p instead of the p >> > n situation, methods adjustments had to be made. The main issue in statistical analysis is the high type I error rate (false positives) in null hypothesis testing. Several ways of correcting for this have been developed, the most well-known and used being the Bonferroni correction and the Benjamini-Hochberg False Discovery Rate (FDR) controlling procedure [85]. Discussions are still ongoing in the statistics community as to which method is best to control the false positive rates in the context of ‘omics data analysis [46, 86, 87]. We therefore advise to split the data in testing and validation groups. Tests made within each group are corrected for FDR with the Benjamini-Hochberg’s procedure whenever possible or advised by domain experts, and only features detected in both groups should be considered for further analysis and interpretation.
Over-fitting may occur when a statistical model includes too many parameters relative to the number of observations. The over-fitted model describes random error instead of the underlying relationship of interest and performs poorly with independent data. In deriving prediction models therefore, a guiding principle is that there should be at least ten observations (or events) per predictor element [88] while simple models with few parameters should be favoured whenever possible.
All in all, the combination of internal replication, FDR correction and conservative over-fitting considerations allows the detection of interesting ‘omics features with a reference statistical foundation.
Replication of findings
When a large number of statistical tests have been planned, a comprehensive adjustment for multiple testing can be detrimental to statistical power. Validation and replication of findings is therefore essential in order to avoid the widespread unvalidated biomarker syndrome that has plagued the vast majority of claimed biomarkers. Indeed, fewer than 1/1000 have proved clinically useful and approved by regulatory authorities [89–94]. For each combination of platform and sample type, an assessment can be made as to whether the data should be split into training and validation sets, or instead analysed as a single pool.
The predictive value of a biomarker identified after proper internal replication applies to the dataset in which it was discovered. Replication of findings in additional sample sets is a crucial step in producing clinically usable biomarkers and predictive models [95, 96] and should thus always be sought.
Once the feature filtering step is performed, the next step is to make sense of the results, either in a biological or mathematical manner. Biological annotation can be performed using pathways (see review in [97]) or functional categories (reviewed in [98]); however, this kind of analysis is hampered by factors such as statistical considerations (which method to use, independence between genes and between pathways, how to take into account the magnitude of the changes) and pathway architecture considerations (pathways can cross and overlap, meaning that if one pathway is truly affected, one may observe other pathways being significantly affected due to the set of overlapping genes and proteins involved) [99]. One way of overcoming those limitations is to use the complete genome-scale network of protein-protein interactions to define affected sub-regions of the network, with available academic [100, 101] and commercial solutions (e.g. MetaCore™ Thomson Reuters, IPA Ingenuity Pathway Analysis). A recent proposed solution is the disease map concept, following the examples of the Parkinson’s disease map [47], the Atlas of Cancer Signalling Networks [50] and the AlzPathway [51, 52] where an exhaustive set of relevant interactions to a particular disease are represented in details as a single network, which can then be analysed biologically and mathematically, with the supervision of domain experts for coverage and specificity [102].
Results
Application to a public domain dataset: TCGA OV dataset for handprint analysis
The Cancer Genome Atlas (TCGA, http://cancergenome.nih.gov/) is a joint effort of the National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI) in the USA. It aims to accelerate our understanding of the molecular basis of cancer through application of genome analysis technologies. Among other functionalities, TCGA offers a freely available database of multi-‘omics datasets (including clinical data, imaging, DNA, mRNA and miRNA sequencing, protein, gene exon and miRNA expression, DNA methylation and copy number variation (CNV)) for several cancer types, with patient numbers ranging from a few dozens to above a thousand.
As a use case, the ovarian cancer OV dataset was chosen, as it comprises several ‘omics measurements for a large group of patients; this dataset has already been well characterized in several publications but without a data fusion analysis, in contrast to the glioblastoma TCGA dataset, for example [55]. It comprises data from a total of 586 patients, along with several ‘omics datasets (such as SNP, Exome, methylation…), as shown in the Table 1. below. All data matrices were downloaded using the Broad Institute FireBrowse TCGA interface (http://firebrowse.org/?cohort=OV&download_dialog=true#); the results shown here are based upon data generated by the TCGA Research Network.
Table 1.
This table shows the number of cases in each ‘omics platform available for the TCGA Ovarian Serous Cystadenocarcinoma dataset (source: https://gdc.cancer.gov/)
| Ovarian serous cystadenocarcinoma | Total | Exome | SNP | Methylation | mRNA | miRNA | Clinical |
|---|---|---|---|---|---|---|---|
| Cases | 586 | 536 | 579 | 584 | 574 | 582 | 584 |
Data preparation
We used the clinical, methylation, mRNA and miRNA data matrices from the 453 patients (out of a total of 586 patients) for which all four data types were available. The overview of the analysis is summarized in the Fig. 4.
Fig. 4.

Framework outline for the TCGA handprint analysis with additional feature filtering. Each dataset was separately filtered based on nominal p-values < 0.05 when comparing alive versus deceased patients at the end of the study taking into account the total amount of days alive. A total of 6753 features were selected: 899 differentially methylated genes, 37 miRNAs and 5817 differentially expressed probesets. Consensus clustering on the fused similarity matrices determined the number of stable clusters that were viewed in a Kaplan-Meyer plot and tested for differential survival. Machine learning was then performed to identify candidate features predicting the identified groups: Recursive Feature Elimination (RFE) on a linear Support-Vector-Machine (SVM) model to identify informative features, followed by a Random Forest (RF) model building in parallel with DIABLO sPLS-DA on those features
Feature selection
Preliminary analysis without feature selection was performed (data not shown). Briefly, this analysis led to the identification of four stable clusters, mainly differentiated by lymphatic and venous invasion status and clinical stage. Biologically speaking, the comparison of clusters led to the highlighting of well-known ovarian cancer biomarkers and pathways.
In order to produce a handprint more focused on the survival status of patients in the dataset, each ‘omics dataset was treated separately to identify features associated with survival status at the end of the study and overall survival time. The latter was obtained by summing the age (in days) of the participants at enrolment in the study and the post-study survival time, both values available in the clinical variables from the TCGA website. After data preparation including imputation of missing data in methylation and normalisation, linear models testing for survival status with survival time as a cofactor were fitted feature-wise and p-values for differential expression/abundance were derived. All features with a nominal p-value < 0.05 were selected. This yielded a total of 899 features in the methylation dataset, 37 miRNAs and 5817 probesets in transcriptomics.
‘Omics-based clustering
Similarity matrices were derived from each filtered ‘omics dataset, which were fused with SNF, and spectral clustering with a consensus clustering step was applied to detect stable clusters, as shown in Fig. 5 below. The choice of the optimal number of stable clusters is based on two mathematical parameters: the deviation from ideal stability (DIS, a measure of the deviation from horizontality of the CDF curves in the left panel of the Fig. 5, the formulation of which can be found in the supplementary material of [103]), and the number of patients assigned in each cluster (clusters with fewer than 10 patients should be avoided [58]). The DIS across the number of clusters can be found in the Additional file 1. The DIS shows a minimal value for k = 3 clusters, but very similar values can be seen for k = 6, 7, 9, 10, 11 and 12. As it is clinically interesting to distinguish a higher number of clusters and to define clusters with different survival status, we chose the number of clusters associated with low DIS, no clusters with fewer than 10 patients, and statistically significant differences in survival status and survival time of patients, k = 9.
Fig. 5.

Consensus clustering results for the handprint analysis with feature filtering. A number of stable clustering schemes are available (k = 3, 6, 7, 8, 9). Nine clusters were chosen as the most informative, while keeping a low value of the deviation from ideal stability index and with clinical characteristics of the clusters statistically different in both survival time and survival status between clusters
The clinical characteristics of the nine clusters are shown in Table 2. Survival curves are also shown in the Kaplan-Meyer plot (Fig. 6). Survival status and survival time differ between the nine clusters, showing for example that patients in cluster 1 have a higher mortality rate.
Table 2.
Clinical characteristics of the nine clusters found in the focused handprint analysis
| Variables/clusters | C1 (n = 49) | C2 (n = 30) | C3 (n = 75) | C4 (n = 41) | C5 (n = 47) | C6 (n = 52) | C7 (n = 46) | C8 (n = 56) | C9 (n = 57) | P-value |
|---|---|---|---|---|---|---|---|---|---|---|
| Age at initial pathologic diagnosis (Yr) | 57.6 ± 13.2 | 53.5 ± 8.16 | 59.8 ± 10.7 | 61.1 ± 12 | 60.2 ± 9.67 | 63.4 ± 11.8 | 59.8 ± 12.5 | 59.4 ± 11.6 | 60 ± 11.4 | 3.40E-02 2 |
| Days from birth (Days) | −21,200 ± 4830 | −19,700 ± 3030 | −21,900 ± 3870 | −22,700 ± 4260 | −22,200 ± 2580 | − 23,300 ± 4290 | −22,000 ± 4560 | −21,900 ± 4240 | −22,200 ± 4140 | 3.15E-02 2 |
| Days to death (Days (IQR)) | 1220 (725–1490) | 1480 (1210–2360) | 997 (404–1230) | 949 (563–1360) | 787 (512–1340) | 1090 (680–1580) | 978 (536–1450) | 1070 (340–1440) | 1290 (731–1700) | 2.11E-02 1 |
| Days to last followup (Days (IQR)) | 1090 (689–1460) | 1200 (688–1550) | 664 (238–1120) | 763 (272–1820) | 676 (185–1560) | 804 (339–1560) | 651 (347–1370) | 816 (223–1370) | 1280 (605–1690) | 3.74E-02 1 |
| Initial pathologic diagnosis method | Cytology: 9; Excisional biopsy: 2; Fine needle aspiration biopsy: 2; Incisional biopsy: 4; Tumor resection: 32 | Cytology: 3; Excisional biopsy: 0; Fine needle aspiration biopsy: 0; Incisional biopsy: 0; Tumor resection: 27 | Cytology: 12; Excisional biopsy: 0; Fine needle aspiration biopsy: 3; Incisional biopsy: 0; Tumor resection: 59; NA: 1 | Cytology: 2; Excisional biopsy: 0; Fine needle aspiration biopsy: 2; Incisional biopsy: 1; Tumor resection: 36 | Cytology: 9; Excisional biopsy: 2; Fine needle aspiration biopsy: 0; Incisional biopsy: 2; Tumor resection: 33; NA: 1 | Cytology: 6; Excisional biopsy: 0; Fine needle aspiration biopsy: 1; Incisional biopsy: 0; Tumor resection: 44; NA: 1 | Cytology: 2; Excisional biopsy: 0; Fine needle aspiration biopsy: 0; Incisional biopsy: 0; Tumor resection: 44 | Cytology: 9; Excisional biopsy: 1; Fine needle aspiration biopsy: 0; Incisional biopsy: 3; Tumor resection: 43 | Cytology: 5; Excisional biopsy: 0; Fine needle aspiration biopsy: 1; Incisional biopsy: 0; Tumor resection: 51 | 3.28E-03 3 |
| Lymphatic invasion | No: 4; Yes: 9; NA: 36 | No: 6; Yes: 10; NA: 14 | No: 7; Yes: 19; NA: 49 | No: 13; Yes: 5; NA: 23 | No: 1; Yes: 17; NA: 29 | No: 13; Yes: 6; NA: 33 | No: 8; Yes: 21; NA: 17 | No: 4; Yes: 8; NA: 44 | No: 5; Yes: 14; NA: 38 | 2.43E-02 3 |
| Neoplasm histologic grade | G1: 1; G2: 13; G3: 33; G4: 0; Gb: 1; Gx: 1 | G1: 0; G2: 5; G3: 24; G4: 0; Gb: 0; Gx: 0; NA: 1 | G1: 0; G2: 5; G3: 70; G4: 0; Gb: 0; Gx: 0 | G1: 0; G2: 5; G3: 36; G4: 0; Gb: 0; Gx: 0 | G1: 0; G2: 6; G3: 39; G4: 0; Gb: 0; Gx: 2 | G1: 0; G2: 6; G3: 44; G4: 1; Gb: 0; Gx: 1 | G1: 0; G2: 8; G3: 38; G4: 0; Gb: 0; Gx: 0 | G1: 0; G2: 1; G3: 53; G4: 0; Gb: 0; Gx: 2 | G1: 0; G2: 6; G3: 49; G4: 0; Gb: 0; Gx: 1; NA: 1 | 1.89E-02 1 |
| Ethnicity | American Indian or Alaska native: 1; Asian: 1; Black or African American: 3; White: 43; NA: 1 | American Indian or Alaska native: 0; Asian: 1; Black or African American: 2; White: 27; NA: 0 | American Indian or Alaska native: 0; Asian: 3; Black or African American: 2; White: 68; NA: 2 | American Indian or Alaska native: 0; Asian: 1; Black or African American: 3; White: 37; NA: 0 | American Indian or Alaska native: 1; Asian: 1; Black or African American: 0; White: 41; NA: 4 | American Indian or Alaska native: 0; Asian: 2; Black or African American: 4; White: 44; NA: 2 | American Indian or Alaska native: 0; Asian: 3; Black or African American: 1; White: 41; NA: 1 | American Indian or Alaska native: 0; Asian: 3; Black or African American: 2; White: 49; NA: 2 | American Indian or Alaska native: 0; Asian: 0; Black or African American: 4; White: 51; NA: 2 | 6.72E-01 3 |
| Clinical stage | iia: 0; iib: 0; iic: 0; iiia: 1; iiib: 0; iiic: 38; iv: 10; NA: 0 | iia: 0; iib: 0; iic: 1; iiia: 0; iiib: 1; iiic: 24; iv: 3; NA: 1 | iia: 0; iib: 0; iic: 3; iiia: 1; iiib: 3; iiic: 51; iv: 16; NA: 1 | iia: 0; iib: 0; iic: 3; iiia: 4; iiib: 4; iiic: 22; iv: 7; NA: 1 | iia: 0; iib: 1; iic: 1; iiia: 0; iiib: 2; iiic: 33; iv: 9; NA: 1 | iia: 0; iib: 0; iic: 2; iiia: 1; iiib: 5; iiic: 38; iv: 6; NA: 0 | iia: 1; iib: 1; iic: 2; iiia: 0; iiib: 4; iiic: 34; iv: 4; NA: 0 | iia: 0; iib: 0; iic: 1; iiia: 0; iiib: 1; iiic: 42; iv: 12; NA: 0 | iia: 2; iib: 2; iic: 4; iiia: 0; iiib: 1; iiic: 41; iv: 7; NA: 0 | 2.65E-02 1 |
| Tumor residual disease | > 20 mm: 10; 1–10 mm: 26; 11–20 mm: 6; no macroscopic disease: 4; NA: 3 | > 20 mm: 5; 1–10 mm: 17; 11–20 mm: 5; no macroscopic disease: 12; NA: 4 | > 20 mm: 17; 1–10 mm: 29; 11–20 mm: 5; no macroscopic disease: 12; NA: 12 | > 20 mm: 6; 1–10 mm: 18; 11–20 mm: 1; no macroscopic disease: 12; NA: 4 | > 20 mm: 11; 1–10 mm: 21; 11–20 mm: 4; no macroscopic disease: 3; NA: 8 | > 20 mm: 4; 1–10 mm: 24; 11–20 mm: 5; no macroscopic disease: 12; NA: 7 | > 20 mm: 8; 1–10 mm: 15; 11–20 mm: 5; no macroscopic disease: 13; NA: 5 | > 20 mm: 6; 1–10 mm: 29; 11–20 mm: 2; no macroscopic disease: 14; NA: 5 | > 20 mm: 11; 1–10 mm: 25; 11–20 mm: 2; no macroscopic disease: 14; NA: 5 | 6.13E-02 1 |
| Tumor tissue site | Omentum: 0; Ovary: 48; Peritoneum ovary: 1 | Omentum: 0; Ovary: 30; Peritoneum ovary: 0 | Omentum: 1; Ovary: 74; Peritoneum ovary: 0 | Omentum: 0; Ovary: 41; Peritoneum ovary: 0 | Omentum: 1; Ovary: 46; Peritoneum ovary: 0 | Omentum: 0; Ovary: 52; Peritoneum ovary: 0 | Omentum: 0; Ovary: 46; Peritoneum ovary: 0 | Omentum: 0; Ovary: 56; Peritoneum ovary: 0 | Omentum: 0; Ovary: 57; Peritoneum ovary: 0 | 5.01E-01 3 |
| Venous invasion | No: 3; Yes: 3; NA: 43 | No: 3; Yes: 10; NA: 17 | No: 8; Yes: 7; NA: 60 | No: 12; Yes: 3; NA: 26 | No: 1; Yes: 10; NA: 36 | No: 10; Yes: 5; NA: 37 | No: 7; Yes: 20; NA: 19 | No: 3; Yes: 1; NA: 52 | No: 3; Yes: 10; NA: 44 | 7.24E-02 3 |
| Vital status | Alive: 9; Dead: 40, NA: 0 | Alive: 14; Dead: 16; NA: 0 | Alive: 33; Dead: 42; NA:0 | Alive: 18; Dead: 23; NA: 0 | Alive: 20; Dead: 27; NA: | Alive: 20; Dead: 31; NA: 1 | Alive: 28; Dead: 18; NA: 0 | Alive: 31; Dead: 25; NA: 0 | Alive: 27; Dead: 30; NA: 0 | 1.90E-03 3 |
| Primary therapy outcome success | Complete remission/response: 24; Partial remission/response: 12; Progressive disease: 3; Stable disease: 1; NA: 9 | Complete remission/response: 17; Partial remission/response: 3; Progressive disease: 4; Stable disease: 2; NA: 4 | Complete remission/response: 41; Partial remission/response: 7; Progressive disease: 2; Stable disease: 4; NA: 21 | Complete remission/response: 24; Partial remission/response: 4; Progressive disease: 2; Stable disease: 0; NA: 11 | Complete remission/response: 24; Partial remission/response: 8; Progressive disease: 4; Stable disease: 3; NA: 8 | Complete remission/response: 29; Partial remission/response: 6; Progressive disease: 1; Stable disease: 5; NA: 11 | Complete remission/response: 27; Partial remission/response: 5; Progressive disease: 4; Stable disease: 6; NA: 4 | Complete remission/response: 36; Partial remission/response: 4; Progressive disease: 7; Stable disease: 2; NA: 7 | Complete remission/response: 35; Partial remission/response: 5; Progressive disease: 5; Stable disease: 1; NA: 11 | 5.08E-01 1 |
| Days lived known | 22,300 ± 4750 | 21,100 ± 3150 | 22,800 ± 3930 | 23,800 ± 4050 | 23,300 ± 3840 | 24,500 ± 4140 | 23,000 ± 4490 | 22,800 ± 4430 | 23,400 ± 4240 | 3.85E-02 2 |
Nominally statistically significant differences (p < 0.05) are shown in italic. Interestingly, significant differences are detected in lymphatic invasion, clinical stage at diagnosis, vital status and the overall number of days alive
Fig. 6.

Kaplan-Meyer plot of survival for patients from the nine clusters revealed with the consensus clustering analysis. The x axis bears the total amount of days that patients have lived, i.e. the sum of their age at enrolment in the study plus the recorded amount of days they survived during the study, censored to the right by the end of measurements in the study (enrolment plus 4624 days)
Biomarker identification
Enrichment analysis
In order to detect differentially expressed features that are specific to one group, each of the nine clusters was compared to the rest of the dataset. Table 3 shows the summary of statistically different features (p-value < 0.05, 5% FDR correction) identified in each comparison.
Table 3.
Number of statistically significant different features obtained when comparing each cluster against all other patients in the dataset, for each platform. P-values were computed by a linear model in each ‘omics platform independently, and Benjamini-Hochberg FDR corrected
| 1 vs Rest (49 vs 404) | 2 vs Rest (30 vs 423) | 3 vs Rest (75 vs 378) | 4 vs Rest (41 vs 412 | 5 vs Rest (47 vs 406 | 6 vs Rest (52 vs 401 | 7 vs Rest (46 vs 407 | 8 vs Rest (56 vs 397 | 9 vs Rest (57 vs 396) | |
|---|---|---|---|---|---|---|---|---|---|
| mRNA | 1861 | 245 | 4101 | 1073 | 2480 | 3617 | 2557 | 4620 | 1843 |
| Methylation | 335 | 550 | 4 | 388 | 498 | 233 | 387 | 528 | 75 |
| miRNA | 18 | 0 | 1 | 9 | 24 | 1 | 8 | 14 | 11 |
Enrichment analysis of features differentially expressed/abundant between the clusters was then performed. Complete results are presented in the Additional file 2; an overview of results for which there is already evidence in the literature is presented below in Table 4.
Table 4.
Enrichment analysis for each comparison across all ‘omics types, with q-values, and the literature references mentioning involvement of the terms in ovarian cancer development. Q-values are the minimal false discovery rate at which the test may be called significant, or in other words, the p-value threshold to satisfy the FDR criteria set by the Benjamini-Hochberg procedure
| Term | Term type | ‘Omic type | Contrast | q-value | Reference of implication in ovarian cancer |
|---|---|---|---|---|---|
| E2F | Transcription factor | Transcriptomics | 1 vs Rest | 8.17E-48 | [123, 124] |
| Sp1 | Transcription factor | Transcriptomics | 1 vs Rest | 1.95E-35 | [125] |
| Mitochondrial translation | Reactome | Transcriptomics | 1 vs Rest | 9.02E-21 | [126] |
| hsa-miR-193a-5p | miRNA | Transcriptomics | 1 vs Rest | 4.33E-09 | [127] |
| CREM | Transcription factor | Methylation | 1 vs Rest | 2.45E-03 | [128] |
| hsa-miR-940 | miRNA | Transcriptomics | 1 vs Rest | 6.80E-03 | [129] |
| hsa-miR-601 | miRNA | Transcriptomics | 1 vs Rest | 6.81E-03 | [129] |
| hsa-miR-503 | miRNA | Transcriptomics | 1 vs Rest | 1.41E-02 | [129] |
| AP-1 | Transcription factor | Methylation | 1 vs Rest | 1.52E-02 | [130] |
| TCF-4 | Transcription factor | Methylation | 1 vs Rest | 2.04E-02 | [131] |
| hsa-miR-361-3p | miRNA | Transcriptomics | 1 vs Rest | 2.53E-02 | [129] |
| C/EBP | Transcription factor | Methylation | 2 vs Rest | 1.13E-05 | [132] |
| LMXB1 | Transcription factor | Methylation | 2 vs Rest | 9.32E-05 | [133] |
| hsa-miR-330-5p | miRNA | Transcriptomics | 2 vs Rest | 7.57E-03 | [134] |
| Chemical carcinogenesis | KEGG pathways | Transcriptomics | 2 vs Rest | 1.77E-02 | [135–137] |
| hsa-miR-335 | miRNA | Transcriptomics | 2 vs Rest | 3.95E-02 | [138] |
| MZF-1 | Transcription factor | Transcriptomics | 3 vs Rest | 4.06E-39 | [139] |
| SREBP-1 | Transcription factor | Transcriptomics | 3 vs Rest | 5.29E-38 | [140] |
| AP-2gamma | Transcription factor | Transcriptomics | 3 vs Rest | 1.79E-36 | [141] |
| GPCR ligand binding | Reactome | Transcriptomics | 3 vs Rest | 8.14E-10 | [142] |
| hsa-miR-328 | miRNA | Transcriptomics | 3 vs Rest | 9.92E-10 | [129] |
| hsa-miR-370 | miRNA | Transcriptomics | 3 vs Rest | 1.09E-08 | [129] |
| hsa-miR-601 | miRNA | Transcriptomics | 3 vs Rest | 1.07E-07 | [129] |
| hsa-miR-423-5p | miRNA | Transcriptomics | 3 vs Rest | 1.36E-06 | [129] |
| hsa-miR-139-3p | miRNA | Transcriptomics | 3 vs Rest | 2.28E-05 | [129] |
| hsa-miR-769-5p | miRNA | Transcriptomics | 3 vs Rest | 9.05E-05 | [129] |
| hsa-miR-339-3p | miRNA | Transcriptomics | 3 vs Rest | 2.16E-04 | [129] |
| hsa-miR-940 | miRNA | Transcriptomics | 3 vs Rest | 2.94E-04 | [129] |
| hsa-miR-542-5p | miRNA | Transcriptomics | 3 vs Rest | 8.13E-04 | [129] |
| hsa-miR-483-5p | miRNA | Transcriptomics | 3 vs Rest | 1.50E-03 | [129] |
| hsa-miR-361-3p | miRNA | Transcriptomics | 3 vs Rest | 7.88E-03 | [129] |
| hsa-miR-449a | miRNA | Transcriptomics | 3 vs Rest | 4.87E-02 | [129] |
| T cell aggregation | GO Biological Process | Transcriptomics | 4 vs Rest | 1.94E-38 | [143] |
| T cell activation | GO Biological Process | Transcriptomics | 4 vs Rest | 1.94E-38 | [144] |
| Natural killer cell mediated cytotoxicity | KEGG pathways | Transcriptomics | 4 vs Rest | 8.60E-14 | [145] |
| Cell adhesion molecules (CAMs) | KEGG pathways | Transcriptomics | 4 vs Rest | 2.37E-11 | [146] |
| Hedgehog ‘on’ state | Reactome | Transcriptomics | 4 vs Rest | 7.21E-05 | [147] |
| HIC1 | Transcription factor | Methylation | 4 vs Rest | 2.46E-04 | [148] |
| hsa-miR-328 | miRNA | Transcriptomics | 4 vs Rest | 1.49E-02 | [129] |
| AP-2gamma | Transcription factor | Transcriptomics | 4 vs Rest | 3.00E-02 | [141] |
| T cell activation | GO Biological Process | Transcriptomics | 5 vs Rest | 1.94E-38 | [144] |
| T cell aggregation | GO Biological Process | Transcriptomics | 5 vs Rest | 2.25E-22 | [143] |
| Natural killer cell mediated cytotoxicity | KEGG pathways | Transcriptomics | 5 vs Rest | 8.60E-14 | [145] |
| Antigen processing and presentation | KEGG pathways | Transcriptomics | 5 vs Rest | 4.33E-11 | [149] |
| Interferon alpha/beta signalling | Reactome | Transcriptomics | 5 vs Rest | 6.11E-08 | [150] |
| hsa-miR-423-5p | miRNA | Transcriptomics | 5 vs Rest | 3.09E-05 | [129] |
| hsa-miR-328 | miRNA | Transcriptomics | 5 vs Rest | 5.23E-04 | [129] |
| VEGFA-VEGFR2 Pathway | Reactome | Transcriptomics | 5 vs Rest | 2.57E-03 | [151, 152] |
| Hedgehog ‘off’ state | Reactome | Transcriptomics | 5 vs Rest | 1.21E-02 | [153] |
| hsa-miR-139-3p | miRNA | Transcriptomics | 5 vs Rest | 1.35E-02 | [129] |
| NF- κB signalling pathway | KEGG pathways | Transcriptomics | 5 vs Rest | 1.53E-02 | [154] |
| hsa-miR-601 | miRNA | Transcriptomics | 5 vs Rest | 2.71E-02 | [129] |
| Jak-STAT signalling pathway | KEGG pathways | Transcriptomics | 5 vs Rest | 3.54E-02 | [155] |
| hsa-miR-375 | miRNA | Transcriptomics | 5 vs Rest | 3.74E-02 | [129] |
| Signalling by GPCR | Reactome | Transcriptomics | 6 vs Rest | 1.24E-14 | [156] |
| hsa-miR-328 | miRNA | Transcriptomics | 6 vs Rest | 1.47E-08 | [129] |
| hsa-miR-601 | miRNA | Transcriptomics | 6 vs Rest | 6.94E-07 | [129] |
| hsa-miR-370 | miRNA | Transcriptomics | 6 vs Rest | 2.46E-06 | [129] |
| hsa-miR-423-5p | miRNA | Transcriptomics | 6 vs Rest | 4.81E-06 | [129] |
| hsa-miR-423-3p | miRNA | Transcriptomics | 6 vs Rest | 1.77E-05 | [129] |
| cAMP metabolic process | GO Biological Process | Transcriptomics | 6 vs Rest | 9.22E-05 | [157] |
| hsa-miR-769-5p | miRNA | Transcriptomics | 6 vs Rest | 5.13E-04 | [129] |
| hsa-miR-139-3p | miRNA | Transcriptomics | 6 vs Rest | 2.70E-03 | [129] |
| hsa-miR-483-5p | miRNA | Transcriptomics | 6 vs Rest | 4.90E-03 | [129] |
| hsa-miR-940 | miRNA | Transcriptomics | 6 vs Rest | 5.05E-03 | [129] |
| T cell selection | GO Biological Process | Transcriptomics | 6 vs Rest | 1.41E-02 | [158] |
| Arachidonic acid metabolism | KEGG pathways | Transcriptomics | 6 vs Rest | 1.42E-02 | [135] |
| hsa-miR-542-5p | miRNA | Transcriptomics | 6 vs Rest | 1.73E-02 | [129] |
| Oxidative phosphorylation | KEGG pathways | Transcriptomics | 7 vs Rest | 9.49E-13 | [159] |
| Stabilization of p53 | Reactome | Transcriptomics | 7 vs Rest | 1.06E-07 | [160] |
| Spliceosome | KEGG pathways | Transcriptomics | 7 vs Rest | 1.59E-07 | [161] |
| NF-kB signalling pathway | Reactome | Transcriptomics | 7 vs Rest | 3.97E-05 | [154] |
| hsa-miR-542-5p | miRNA | Transcriptomics | 7 vs Rest | 2.53E-03 | [129] |
| hsa-miR-601 | miRNA | Transcriptomics | 7 vs Rest | 2.62E-03 | [129] |
| hsa-miR-423-5p | miRNA | Transcriptomics | 7 vs Rest | 5.88E-03 | [129] |
| hsa-let-7c | miRNA | Transcriptomics | 7 vs Rest | 2.67E-02 | [129] |
| Regulation of HIF by oxygen | Reactome | Transcriptomics | 7 vs Rest | 3.32E-02 | [162] |
| hsa-miR-361-3p | miRNA | Transcriptomics | 7 vs Rest | 4.16E-02 | [129] |
| hsa-miR-328 | miRNA | Transcriptomics | 8 vs Rest | 9.25E-15 | [129] |
| hsa-miR-370 | miRNA | Transcriptomics | 8 vs Rest | 3.60E-11 | [129] |
| hsa-miR-940 | miRNA | Transcriptomics | 8 vs Rest | 1.37E-10 | [129] |
| hsa-miR-423-5p | miRNA | Transcriptomics | 8 vs Rest | 4.29E-10 | [129] |
| hsa-miR-423-3p | miRNA | Transcriptomics | 8 vs Rest | 7.47E-09 | [129] |
| hsa-miR-139-3p | miRNA | Transcriptomics | 8 vs Rest | 5.08E-07 | [129] |
| hsa-miR-601 | miRNA | Transcriptomics | 8 vs Rest | 9.47E-07 | [129] |
| hsa-miR-542-5p | miRNA | Transcriptomics | 8 vs Rest | 4.72E-04 | [129] |
| hsa-miR-361-3p | miRNA | Transcriptomics | 8 vs Rest | 1.07E-03 | [129] |
| hsa-miR-483-5p | miRNA | Transcriptomics | 8 vs Rest | 1.32E-03 | [129] |
| hsa-miR-769-5p | miRNA | Transcriptomics | 8 vs Rest | 1.68E-03 | [129] |
| Potassium signalling pathway | Reactome | Transcriptomics | 8 vs Rest | 1.15E-02 | [163] |
| hsa-miR-99b | miRNA | Transcriptomics | 8 vs Rest | 1.93E-02 | [129] |
| hsa-miR-339-3p | miRNA | Transcriptomics | 8 vs Rest | 2.28E-02 | [129] |
| T cell lineage commitment | GO Biological Process | Transcriptomics | 8 vs Rest | 3.80E-02 | [164] |
| hsa-miR-139-3p | miRNA | Transcriptomics | 9 vs Rest | 3.58E-09 | [129] |
| hsa-miR-423-5p | miRNA | Transcriptomics | 9 vs Rest | 5.89E-09 | [129] |
| hsa-miR-328 | miRNA | Transcriptomics | 9 vs Rest | 2.32E-08 | [129] |
| hsa-miR-370 | miRNA | Transcriptomics | 9 vs Rest | 4.83E-08 | [129] |
| hsa-miR-423-3p | miRNA | Transcriptomics | 9 vs Rest | 3.89E-06 | [129] |
| hsa-miR-940 | miRNA | Transcriptomics | 9 vs Rest | 5.37E-06 | [129] |
| hsa-miR-769-5p | miRNA | Transcriptomics | 9 vs Rest | 1.07E-04 | [129] |
| hsa-miR-339-3p | miRNA | Transcriptomics | 9 vs Rest | 0.000173 | [129] |
| hsa-miR-601 | miRNA | Transcriptomics | 9 vs Rest | 2.05E-04 | [129] |
| hsa-miR-483-5p | miRNA | Transcriptomics | 9 vs Rest | 7.33E-03 | [129] |
| Calcium signalling pathway | KEGG pathways | Transcriptomics | 9 vs Rest | 1.55E-02 | [165] |
| hsa-miR-542-5p | miRNA | Transcriptomics | 9 vs Rest | 1.69E-02 | [129] |
| cAMP signalling pathway | KEGG pathways | Transcriptomics | 9 vs Rest | 2.33E-02 | [166] |
| Ion transfer | GO Biological Process | Transcriptomics | 9 vs Rest | 3.43E-02 | [167] |
In short, the biological functions enriched in each cluster are as follows: cluster 1 is mostly enriched in mitochondrial translation and energy metabolism, cell cycle regulation, negative regulation of apoptosis and DNA damage response. In addition, several miRNAs and transcription factors are enriched; the details can be found in the Additional file 2.
Cluster 2 is associated with chemical carcinogenesis, miR-330-5p, miR-693-5p and the Pax-2 transcription factor. Other transcription factors are also highlighted through the methylation measurements.
Cluster 3 is associated with immune system regulation (T cell-related processes, and more precisely CD4 and CD8-T cells lineages-related processes…), cell-cell signalling, cAMP signalling, cytokine-cytokine interaction, G-Protein coupled receptor (GPCR) ligand binding and neuronal and muscle-related pathways (potassium and calcium channels, other ion channels and synapses). Again, several miRNAs and transcription factors are highlighted.
Cluster 4 is also associated with the immune response, and key functions such as lymphocyte activation, T cell aggregation, differentiation, proliferation and activation, adaptive immune system, regulation of lymphocyte cell-cell activation, immune response-regulating signalling pathway, cytokine-cytokine receptor interaction, antigen processing and presentation, hematopoietic cell lineage and hematopoiesis and B cell activation. Primary immunodeficiency pathway and cell adhesion molecules, along with miR-938 and several transcription factors are also enriched.
Cluster 5 is related to immune response, enriched in lymphocyte activation, T cell aggregation, differentiation, activation and proliferation, leukocyte differentiation, aggregation and activation, positive regulation of cell-cell adhesion, antigen processing and presentation, cytokine production, inflammatory response, NK cell-mediated cytotoxicity and cytokine-cytokine receptor interaction. Other processes involved are NF-κB signalling, Jak-STAT signalling, Interferon α/β signalling, TCR signalling, VEGF signalling, VEGFR2-mediated cell proliferation, Hedgehog ‘off’ state, along with several miRNAs and transcription factors.
Cluster 6 is enriched in several signalling pathways, such as cAMP, GPCR signalling, arachidonic acid metabolism and fatty acids metabolism, as well as positive T cell selection, several miRNAs and transcription factors.
Cluster 7 is linked with respiratory metabolism, p53 and cell cycle regulation, splicing regulation as well as signalling by NF-κB and miRNAs and transcription factors.
Cluster 8 is enriched with T cell lineage commitment, potassium channels, miRNAs and transcription factors.
Cluster 9 is associated with ion transport (including synaptic, calcium and potassium channels), cAMP signalling, nicotine addiction, as well as miRNAs and transcription factors.
Each cluster is linked with one or several of the well-known hallmarks of cancer such as regulation of the cell cycle (clusters 1 and 7), energy metabolism (cluster 1 and 7), immune system (clusters 3, 4, 5 and 8), epithelial-to-mesenchymal transition (cluster 4) or angiogenesis (cluster 5) [104–106]. Interestingly, our analysis based on ‘omics profiles is able to identify clusters that seem to separate some of those hallmarks out, while an analysis taking into account only the clinical data cannot. As seen above, cluster 6 is associated with a higher rate of survival. It would therefore be interesting to further explore the signalling networks enriched in the comparison between cluster 6 and the other clusters to identify the molecular mechanisms responsible for the extended survival.
Machine-learning predictive modelling
The next step in the analysis is to establish a model that can predict which cluster a patient belongs to, based on the ‘omics measurements alone. Machine-learning techniques (reviewed in [107, 108]), available in the caret R package [109] and in the MixOmics R packages [110, 111] were used.
Two models were built in parallel, on the same dataset.
A Recursive Feature Elimination (RFE) procedure was performed to identify the smallest number of features from the three ‘omics platforms that allow satisfactory separation of the clusters. This procedure was controlled by Leave-Group-Out Cross Validation (LGOCV) with 100 iterations (this number was chosen to ensure convergence of the validation procedure) and using between 1 and 50 predictors, with the addition of the whole set of 6753 features. A Random Forest (RF) model was built with the features identified in the previous step. To avoid overfitting, the RF model was built using LGOCV with 100 iterations and in three quarters of the samples available (N = 300) and then tested in the remaining quarter of samples (N = 153). More details can be found in the Additional file 3.
Concatenation-based integration of data combines multiple datasets into a single large dataset, with the aim to predict an outcome. However, this approach does not account for or model relationships between datasets and thus limits our understanding of molecular interactions at multiple functional levels. This is the rationale behind the development of novel integrative modelling methods, such as the DIABLO sPLSDA method [112]. A DIABLO model was built using the same dataset as the SNF analysis described above. A DIABLO model is a type of partial least square (sparse PLS Discriminant Analysis) regression model, which uses multiple ‘omics platform measurements on the same samples to predict an outcome, with a biomarkers selection step (sparse) to select necessary and sufficient features to predict the groups (discriminant analysis) within the outcome. Details of this analysis can be found in the Additional file 4. In short, this analysis was run as follows: the datasets were split in 2/3 training and 1/3 testing sets. The DIABLO model was then trained with boundaries set on the number of features allowed per component (gene expression and methylation between 50 and 110 features, and between 5 and 35 miRNA features). The performances were then estimated within the training model by 10 repeats of 10-fold validation and the prediction power estimated in the testing set.
Topological data analysis
In order to visualize the patients’ relationships as measured by their ‘omics profiles, we used Topology Data Analysis (TDA), a general framework to analyse high-dimensional, incomplete and noisy data in a manner that is less sensitive to the particular metric that is chosen, and provides dimensionality reduction and robustness to noise. TDA is embedded in the software produced by the Ayasdi company to which the data were uploaded [113]. As shown in Fig. 7, the network of patients’ similarities obtained through TDA analysis and then colored by the vital status of the patients at the end of the study shows a higher level of complexity than is identified by the clustering analysis, suggesting that statistical and/or technical limitations of the clustering methods prevent us to accurately represent reality.
Fig. 7.
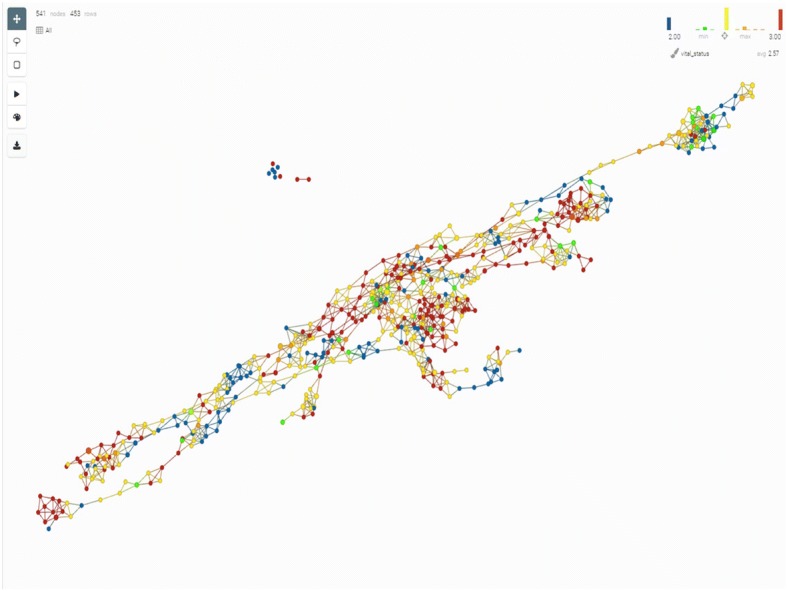
Network of patients shown in the TDA platform. The network is constructed as ‘bins’ grouping patients who are similar based on their ‘omics profiles. Each dot in the network represents a bin. The bins are overlapping by an adaptable percentage, and if at least one patient is present in the overlap of two bins, the two bins will be linked in the network. The survival status of the patients is then translated as a color scheme (blue representing deceased patients and red alive patients). Using this technique, it is easy to identify ‘islands’ of good and poor survival among the patients, and equally easy to acknowledge that there are more such islands than is identified through the clustering technique. Thorough analysis of such networks can lead to insights into biology, as detailed in [168]
Discussion
Multi-omics data integration is, among other components of biological data integration, a very promising and emerging field. We show a structured and effective way to combine ‘omics data from multiple sources to search for molecular profiles of patients. This process allowed for the classification of a well-studied dataset of OV. Other studies have been performed, either on this same dataset [114–118], or on the same disease [119].
Tothill et al. in 2015 identified six clusters of patients, based on mRNA, immunohistochemistry and clinical data from a cohort of 285 Australian and Dutch participants, with a consensus clustering analysis of mRNA data alone. The TCGA consortium produced their own dataset in 2011, identifying four clusters based on combined mRNA, miRNA and DNA methylation data (data combined by summarising to the gene-level all datasets through a factor analysis) and using a non-negative matrix factorisation to identify clusters [120]. Further analysis of the same dataset was then performed by Zhang et al. [118], Jin et al. [115] and Kim et al. [116] (with some variations), but these authors did not look for new phenotypes in their analysis, rather comparing data based on clinical endpoints (survival time, histological grades and stage of disease). Gevaert et al. [114] used an original algorithm to combine DNA methylation, Copy Number Variation (CNV) and gene expression data, using the clusters defined in the TCGA original paper. Those studies showed different ways of analysing the data, leading to the identification of clinically relevant clusters in the case of Tothill and TCGA original paper [117, 119]. It is however the first time in this paper that TCGA mRNA, miRNA and methylation data were fused with an advanced data integration method to identify robust subtypes of disease.
The number of clusters found in the same dataset differs between the TCGA analysis and our analysis. We believe that the higher number of clusters we found is the result of more up-to-date and powerful methods for subtype discovery, as shown in the SNF original paper [55]. Moreover, the subtypes identified in this analysis do allow for a more in-depth classification of patients linked with specific molecular subtypes than was previously reported. Building predictive models based on multiple ‘omics profiles also contributes to the novelty of this approach as other reported studies did not produce such a model, with the exception of the Tothill et al. study [119] in which the authors developed a class prediction model based on transcriptomics data only.
Clinically speaking, classifications are most useful when they allow the identification of a subset of patients with a clinically relevant outcome, such as low or high survival rate, thus indicating where efforts may be focused to develop new drugs, therapies and procedures. In our analysis, the groups identified after feature reduction are statistically different in terms of survival rate and time. For example, cluster 6 shows the highest rate of survival among the 9 clusters identified and is associated with the GPCR signalling pathway, cAMP, ion channels, arachidonic acid metabolism and a number of miRNAs (see Table 4 or the Additional file 2 for more details).
Interestingly, while the two sets of groups defined with or without feature reduction show differences in invasion and clinical stage, statistically significant differences in vital status are only detected amongst groups defined with feature reduction. The reduced data also allows for the definition of a higher number of stable groups (9 instead of 4), thereby pointing to the usefulness of performing feature reduction prior to clustering analysis.
The biological functions highlighted by enrichment analysis between the clusters indicate that these are associated with different biological mechanisms leading to the development of cancer in patients, ranging from immune system disorders, cell cycle dysregulation, impaired response to DNA damage, modified energy metabolism, etc.
The predictive models that were trained and tested with two different methods gave mixed power results. In the Random Forest case, the model could predict quite well when patients did not belong to the clusters, but not so well when patients did belong to them; in other words, the model is specific but not sensitive. In the case of the DIABLO PLS, the model is able to predict fairly accurately the clusters 4 and 8 and less accurately cluster 5. Moreover, in the case of the DIABLO analysis, the model showed that the clusters have different ‘omics patterns, with clusters 2 and 8 showing distinct methylation profiles, and cluster 4 showing different methylation and transcriptomics profiles.
The results presented in this manuscript are not perfectly predictive, however. It seems that the cluster definitions are not as stable as they could be; the predictive models are not accurate in all clusters and the survival status of the clusters are not clear cut. This reflects the fact shown in Fig. 7, that there seems to be much more complexity within the dataset than what the clustering analysis is able to detect.
This is due to multiple factors: the recurring issue of low number of patients, which in turn influences the number of clusters we can find with statistical confidence – a point which is not taken into account in the TDA analysis discussed here – and highlighting the need for better stratification methods in the context of personalized medicine where, ideally, each patient is his/her own cluster (n = 1); sub-optimal clustering methods and algorithms also play a part in this result and it is our hope that continuous methods development will allow for better classification. Clustering analysis is descriptive in nature: applying a clustering algorithm to a dataset will always yield clusters, whether real clusters exist or not. Analytical methods exist to ascertain cluster ‘reality’, among which stability in patients through bootstrapping, stability in time through cluster identification from time-series experiments [121], meta clustering across several studies, yet only replication studies may confirm the existence of these clusters. Such replication effort however lies outside the scope of this manuscript.
Despite the use of most recent databases and tools, the biological interpretation of the differences between the clusters remains challenging. The main issues stem from the overlapping nature of pathways described in literature and the non-unicity of relationships between biological entities, leading to a high false positive rate in the results of pathway analysis [97]. Efforts are made in the systems biology community to correct these shortcomings, among which the disease maps mentioned above.
This underlines the variability in biological events potentially leading to the development of cancer and metastasis and the need for a more personalised care for patients suffering from complex diseases, such as cancer. It is our hope that this methodology will be repeated on other datasets, diseases and clinical situations as it is one more step towards establishing a true personalised data analysis pipeline.
The clusters that were found in this analysis are interesting hypotheses. They would however require further validation to become clinically useful, as detailed in the replication of findings section above. We encourage other researchers to use our findings in their research towards a cross-validated and clinically useful stratification of ovarian cancer, towards a better and more personalized care.
Conclusion
This article presents an overview of the integrative systems biology analyses developed, performed and validated in the IMI U-BIOPRED and eTRIKS projects, proposing a template for other researchers wishing to perform similar analyses for other diseases. We demonstrate the usefulness of generating hypotheses through a fingerprint/handprint analysis by applying to a well-studied dataset of ovarian carcinoma, identifying a higher number of robust groups than previously reported, potentially improving our understanding of this disease. Better characterisation of the clusters found in the handprint analyses and validation of the predictive model obtained by machine learning are both ongoing. We believe that handprint analyses, performed on large scale ‘omics datasets will allow researchers to identify subtypes of disease (phenotypes and endotypes) [34] with greater confidence, providing better diagnosis tools for the clinicians, new avenues for drug development for the pharmaceutical industry and deeper insights into disease mechanisms. To be effective, handprint analyses need to be performed on the same subjects with multiple ‘omics platforms. They suffer from some limitations, such as the decreasing but nevertheless still elevated cost of ‘omics data production and the protocol standardisation requirements to avoid time-consuming data pre-processing, the rather large technical, human resources and expertise requirements to perform the analyses (particularly the machine-learning analysis) or the lack of accurate and independent benchmarking tools to identify the most powerful and/or best-suited method to analyse a particular dataset.
Additional work is therefore needed to make the framework and the analyses proposed here more accessible to a broad audience of health researchers. Efforts of the bioinformatics community are shifting in this direction; for instance, the eTRIKS European project (http://www.etriks.org) or the Galaxy project hosted in the USA (https://galaxyproject.org) mandate the delivery of user-friendly interfaces to advanced bioinformatics resources. Implementation of P4 medicine across the entire health spectrum [122] will be leveraged through promotion of advanced analytical tools available to the larger multidisciplinary community. The methods and results demonstrated in this paper should contribute to pave this promising road.
Additional files
AUC of consensus clustering. (XLSX 13 kb)
Complete results of the enrichment analysis between clusters. (XLSX 4293 kb)
Table S7. Estimated accuracy and standard deviation of the RFE procedure. Table S8. Accuracy and Kappa values of the Random Forest models in the training set. Table S9. Performances values for the Random Forest model in the testing set. Figure S11. Relative importance of the top 20 predictors building the final model of the RF. The importance axis is scaled, with the mRNA expression of CD3D scaled to 100% and the methylation state of POLA2 to 0% (not shown). (DOCX 18 kb)
DIABLO sPLSDA model results. (DOCX 18966 kb)
Acknowledgements
The U-BIOPRED study group consists of Ian M. Adcock (Imperial College, London, UK), Nora Adriaens (University of Amsterdam, The Netherlands), Hassan Ahmed (EISBM, Lyon, France), Antonios Aliprantis (Merck Research, Boston, USA), Kjell Alving (Uppsala University, Sweden), Charles Auffray (EISBM, Lyon, France), Philipp Badorrek (Fraunhofer ITEM, Hannover, Germany), Cornelia Faulenbach (Fraunhofer ITEM, Hannover, Germany), Per Bakke (University of Bergen, Norway), David Balgoma (Karolinska Institutet, Stockholm, Sweden), Aruna T. Bansal (Acclarogen Ltd. Cambridge, UK), Clair Barber (University of Southampton, UK), Frédéric Baribaud (Janssen R & D, Springhouse, USA), An Bautmans (MSD Brussels, Belgium), Annelie F. Behndig (Umeå University, Sweden), Elisabeth Bel (University of Amsterdam, The Netherlands), Jorge Beleta (Almirall S.A., Barcelona, Spain), Ann Berglind (Karolinska Institutet, Stockholm, Sweden), Alix Berton (AstraZeneca, Mölndal, Sweden), Jeanette Bigler (Amgen Inc., Seattle, USA), Hans Bisgaard, University of Copenhagen, Denmark), Grazyna Bochenek, Jagiellonian University, Krakow, Poland), Michael J. Boedigheimer (Amgen Inc., Seattle, USA), Klaus Bønnelykke (University of Copenhagen, Denmark), Joost Brandsma, (University of Southampton, UK), Armin Braun (Fraunhofer ITEM, Hannover, Germany), Paul Brinkman (University of Amsterdam, The Netherlands), Dominic Burg (University of Southampton, UK), Davide Campagna (University of Catania, Italy), Leon Carayannopoulos, (MSD, USA), Massimo Caruso (University of Catania, Italy), Pedro Carvalho da Purificacão Rocha João Pedro (Royal Brompton and Harefield NHS Fondation Trust, UK), Amphun Chaiboonchoe (EISBM, Lyon, France), Romanas Chaleckis (Karolinska Institutet, Stockholm, Sweden), Pascal Chanez (University of Aix Marseille, France), Kiang Fan Chung, Imperial College London, UK), Courtney Coleman (Asthma UK, London, UK), Chris Compton (GSK, UK), Julie Corfield (Arateva R & D, Nottingham, UK), Arnaldo D’Amico (University of Rome ‘Tor Vergata’, Rome, Italy), Barbro Dahlén (Karolinska Institutet, Stockholm, Sweden), Sven-Erik Dahlén (Karolinska Institutet, Stockhlom, Sweden), Jorge De Alba (Almirall S.A., Barcelona, Spain), Pim de Boer (Londfonds, Amersfoort, The Netherlands), Inge De Lepeleire (MSD, Brussels, Belgium), Bertrand De Meulder (EISBM, Lyon, France), Tamara Dekker (University of Amsterdam, The Netherlands), Ingrid Delin (Karolinska Institutet, Stockholm, Sweden), Patrick Dennison (University of Southampton, UK), Annemiek Dijkhuis (University of Amsterdam, The Netherlands), Ratko Djukanovic (University of Southampton, UK), Aleksandra Draper (BioSci Consulting, Maasmechelen, Belgium), Jessica Edwards (Asthma UK, London, UK), Rosalia Emma (University of Catania, Italy), Magnus Ericsson (Karolinska University Hospital, Stockholm, Sweden), Veit Erpenbeck (Novartis Institutes for Biomedical Research, Basel, Switzerland), Damijan Erzen (Boehringer Ingelheim Pharma GmbH & Co. KKKG; Biberach, Germany), Klaus Fichtner (Boehringer Ingelheim Pharma GmbH & Co. KKKG; Biberach, Germany), Neil Fitch (BioSci Consulting, Maasmechelen, Belgium), Louise J. Fleming (Imperial College London, UK), Breda Flood (Asthma UK, London, UK), Stephen J. Fowler (Manchester Academic Health Sciences Center, Manchester, UK), Urs Frey (University Children’s Hospital, Basel, Switzerland), Martina Gahlemann (Boehringer Ingelheim GmbH, Switzerland), Gabriella Galffy (Semmelweis University, Budapest, Hungary), Hactor Gallart (Karolinska Institutet, Stockholm, Sweden), Trevor Garret (BioSci Consulting, Maasmechelen, Belgium), Thomas Geiser (University Hospital Bern, Switzerland), Julaiha Gent (Royal Brompton and Harefield NHS Fondation Trust, London, UK), Maria Gerhardsson de Verdier (AstraZeneca Molndal, Sweden), David Gibeon (Imperial College, London, UK), Cristina Gomez (Karolinska Institutet, Stockholm, Sweden), Kerry Gove (NIHR Southampton Respiratory Biomedical Research Unit and Clinical and Experimental Sciences, Southampton, UK), Neil Gozzard (UCB, UK), Yi-ke Guo (Imperial College, London, UK), Simone Hashimoto (University of Amsterdam, The Netherlands), John Haughney (International Primary Care Respiratory Group, Aberdeen, Scotland), Gunilla Hedlin (Karolinska Institutet, Stockholm, Sweden), Pieter-Paul Hekking (University of Amsterdam, The Netherlands), Elisabeth Henriksson (Karolinska Institutet, Stockholm, Sweden), Lorraine Hewitt (NIHR Southampton Respiratory Biomedical Research Unit, Southampton, UK), Tim Higgenbottam (Allergy Therapeutics, West Sussex, UK), Uruj Hoda (Imperial College, London, UK), Jans Hohlfeld (Fraunhofer ITEM, Hannover, Germany), Cecile Holweg (Genentech, San Francisco, USA), Ildiko Horvath (Semmelweis University, Budapest, Hungary), Peter Howarth (NIHR Southampton Respiratory Biomedical Research Unit, Southampton, UK), Richard Hu (Amgen Inc., Seattle, USA), Sile Hu (Imperial College London, UK), Xugang Hu (Amgen Inc., Seattle, USA), Val Hudson (Asthma UK, London, UK), Anna J. James (Karolinska Institutet, Stockholm, Sweden), Juliette Kamphuis (Longfonds, Amersfoort, The Netherlands), Erika J. Kennington (Asthma UK, London, UK), Dyson Kerry (CromSource, Stirling, UK), Matthias Klüglich (Boehringer Ingelheim Pharma GmbH & Co. KG, Biberach, Germany), Hugo Knobel (Philips Research Laboratories, Eindhoven, The Netherlands), Richard Knowles (Arachos Pharma, UK), Alan Know (University of Nottingham, UK), Johan Kolmert (Karolinska Institutet, Stockholm, Sweden), Jon Konradsen (Karolinska Institutet, Stockholm, Sweden), Maxim Kots (Chiesi Pharmaceutical, Parma, Italy), Linn Krueger (University Children’s Hospital, Bern, Switzerland), Norbert Krug (Fraunhofer ITEM, Hannover, Germany), Scott Kuo (Imperial College, London, UK), Maciej Kupczyk (Karolinska Institutet, Stockholm, Sweden), Bart Lambrecht (University of Gent, Belgium), Ann-Sofie Lantz (Karolinska Institutet, Stockholm, Sweden), Lars Lazarinis (Karolinska Institutet, Stockholm, Sweden), Diane Lefaudeux (EISBM, Lyon, France), Saeeda Lone-Latif (University of Amsterdam, The Netherlands), Matthew J. Loza (Janssen R & D, Springhouse, USA), Rene Lutter (University of Amsterdam, The Netherlands), Lisa Marouzet (NIHR Southampton Respiratory Biomedical Research Unit, Southampton, UK), Jane Martin (NIHR Southampton Respiratory Biomedical Research Unit, Southampton, UK), Sarah Masefield (European Lung Fondation, Shefield, UK), Caroline Mathon (Karolinska Institutet, Stockholm, Sweden), John G. Matthews (Genentech, San Francisco, USA), Alexander Mazein (EISBM, Lyon, France), Sally Meah (Imperial College, London, UK), Andrea Meiser (Imperial College, London, UK), Andrew Manzies-Gow (Royal Brompton and Harefield NHS Fondation Trust, London, UK), Leanne Metcalf (Asthma UK, London, UK), Roelinde Middelveld (Karolinska Institutet, Stockholm, Sweden), Maria Mikus (Science for Life Laboratory, Stockholm, Sweden), Montse Miralpeix (Almirall, Barcelona, Spain), Philip Monk (Synairgen Research Ltd., Southampton, UK), Paolo Montuschi (Università Cattolica del Sacro Cuore, Rome, Italy), Nadia Mores (Università Cattolica del Sacro Cuore, Rome, Italy), Clare S. Murray (University of Manchester, UK), Jacek Musial (Jagiellonian University Medical College, Krakow, Poland), David Myles (GSK, UK), Shama Naz (Karolinska Institutet, Stockholm, Sweden), Katja Nething (Boehringer Ingelheim Pharma GmbH & Co. KG, Biberach, Germany), Ben Nicholas (University of Southampton, UK), Ulf Nihlen (AstraZeneca, Molndal, Sweden), Peter Nilsson (Science for Life Laboratory, Stockholm, Sweden), Björn Nordlund (Karolinska Institutet, Stockholm, Sweden), Jörgen Östling (AstraZeneca, Molndal, Sweden), Antonio Pacino (Lega Italiano Anti Fumo, Catania, Italy), Laurie Pahus (Aix-Marseille University, Marseille, France), Susanna Palkonen (European Federation of Allergy and Airways Diseases Patient’s Associations, Brussels, Belgium), Ioannis Pandis (Imperial College London, UK), Stelios Pavlidis (Imperial College London, UK), Giorgio Pennazza (University of Rome ‘Tor Vergata’, Rome, Italy), Anne Petrén (Karolinska Institutet, Stockholm, Sweden), Sandy Pink (NIHR Southampton Respiratory Biomedical Research Unit, Southampton, UK), Anthony Postle (University of Southampton, UK), Pippa Powel (European Lung Fondation, Sheffield, UK), Malayka Rahman-Amin (Asthma UK, London, UK), Navin Rao (Janssen R & D, La Jolla, USA), Lara Ravanetti (University of Amsterdam, The Netherlands), Emma Ray (NIHR Southampton Respiratory Biomedical Research Unit, Southampton, UK), Stacey Reinke (Karolinska Institutet, Stockholm, Sweden), Leanne Reynolds (Asthma UK, London, UK), Kathrin Riemann (Boehringer Ingelheim Pharma GmbH & Co. KG, Biberach, Germany), John Riley (GSK, UK), Martine Robberechts (MSD, Brussels, Belgium), Amanda Roberts (Asthma UK, London, UK), Graham Roberts (NIHR Southampton Respiratory Biomedical Research Unit, Southampton, UK), Christos Rossios (Imperial College London, UK), Anthony Rowe (Janssen R & D, UK), Kirsty Russel (Imperial College London, UK), Michael Rutgers (Longfonds, Amersfoort, The Netherlands), Thomas Sandström (Umeå University, Sweden), Giuseppe Santini (Università Cattolica del Sacro Cuore, Italy), Marco Santoninco (University of Rome ‘Tor Vergata’, Rome, Italy), Corinna Schoelch (Boehringer Ingelheim Pharma GmbH & Co. KG, Biberach, Germany), James P.R. Schofield (University of Southampton, UK), Wolfgang Seibold (Boehringer Ingelheim Pharma GmbH & Co. KG, Biberach, Germany), Dominick E. Shaw (University of Nottingham, UK), Ralf Sigmund (Boehringer Ingelheim Pharma GmbH & Co. KG, Biberach, Germany), Florian Singer (University Children’s Hospital, Zurich, Switzerland), Marcus Sjödin (Karolinska Institutet, Stockholm, Sweden), Paul J. Skipp (University of Southampton, UK), Barbara Smids (University of Amsterdam, The Netherlands), Caroline Smith (NIHR Southampton Respiratory Biomedical Research Unit, Southampton, UK), Jessica Smith (Asthma UK, London, UK), Katherine M. Smith (University of Nottingham, UK), Päivi Söderman, Karolinska Institutet, Stockholm, Sweden), Adesimbo Sogbesan (Royal Brompton and Harefield NHS Fondation Trust, London, UK), Ana R. Sousa (GSK, UK), Doroteya Staykova (University of Southampton, UK), Peter J. Sterk (University of Amsterdam, The Netherlands), Karin Strandberg (Karolinska Institutet, Stockholm, Sweden), Kai Sun (Imperial College, London, UK), David Supple (Asthma UK, London, UK), Marton Szentkereszty (Semmelweis University, Budapest, Hungary), Lilla Tamasi (Semmelweis University, Budapest, Hungary), Kamran Tariq (University of Southampton, UK), John-Olof Thörngren (Karolinska University Hospital, Stockholm, Sweden), Bob Thornton (MSD, USA), Jonathan Thorsen (University of Copenhagen, Denmark), Salvatore Valente (Università Cattolica del Sacro Cuore, Rome, Italy), Wim van Aalderen (University of Amsterdam, The Netherlands), Marianne van de Pol (University of Amsterdam, The Netherlands), Kees van Drunen (University of Amsterdam, The Netherlands), Marleen van Drunen (University of Amsterdam, The Netherlands), Jenny Versnel (Asthma UK, London, UK), Jorgen Vestbo (Manchester Academic Health Sciences Centre, Manchester, UK), Anton Vink (Philips Research Laboratories, Eindhoven, The Netherlands), Nadja Vising (University of Copenhagen, Denmark), Christophe von Garnier (University Hospital, Bern, Switzerland), Ariane Wagener (University of Amsterdam, The Netherlands), Scott Wagers (BioSci Consulting, Maasmechelen, Belgium), Frans Wald (Boehringer Ingelheim Pharma GmbH & Co. KG, Biberach, Germany), Samantha Walker (Asthma UK, London, UK), Jonathan Ward (University of Southampton, UK), Zsoka Weiszhart (Semmelweis University, Budapest Hungary), Kristiane Wetzel (Boehringer Ingelheim Pharma GmbH & Co. KG, Biberach, Germany), Craig E. Wheelock (Karolinska Institutet, Stockholm, Sweden), Coen Wiegman (Imperial College London, UK), Siân Williams (International Primary Care Respiratory Group, Aberdeen, Scotland), Susan J. Wilson (University of Southampton, UK), Ashley Woodcock (Manchester Academic Health Science Centre, Manchester, UK), Xian Yang (Imperial College London, UK), Elizabeth Yeyasingham (GSK, UK), Wen Yu (Amgen Inc., Seattle, USA), Wilhelm Zetterquist (Karolinska Institutet, Stockholm, Sweden), Koos Zwinderman (University of Amsterdam, The Netherlands). The eTRIKS consortium members are: Alireza Tamaddoni Nezhad (Imperial College London, UK), Adriano Barbosa da Silva (University of Luxemburg, Luxemburg), Alexander Mazein (EISBM, Lyon, France), Andreas Tielmann (Merck), Angela Gaudette (Pfizer), Anna Silberberg (Pfizer), Antigoni (Anna) Elefsinioti (Bayer), Axel Oehmichen (Imperial College London, UK), Maria Biryukov (University of Luxemburg, Luxemburg), Bertrand De Meulder (EISBM, Lyon, France), Jen Birgitte (Lundbeck), Bron Kisler (CDISC), Anna Maria Carusi, Charles Auffray (EISBM, Lyon, France), Diana O’Malley (Imperial College London, UK), David Henderson (Bayer), Dorina Bratfalean (CDISC), Diane Lefaudeux (EISBM, Lyon, France), Denny Verbeeck (Janssen), Ejner Knud Moltzen (Lundbeck), Eva Lindgren (Astra Zeneca), Florian Guitton (Imperial College London, UK), Fabien Richard (EISBM, Lyon, France), Francisco Bonachela Capdevila (Janssen), Ghita Rahal (CNRS, Lyon, France), Heike Dagmar Schuermann (Sanofi), Ibrahim Emam (Imperial College London, UK), Irina Balaur (EISBM, Lyon, France), Ingrid Sofie Harbo (Lundbeck), Jay Bergeron (Pfizer), Kai Sun (Imperial College London, UK), Laurence Mazuranok (Sanofi), Laurence Painell’s (IDBS), Manfred Hendlich (Sanofi), Gino Marchetti (CNRS, Lyon, France), Derek Marren (Lilly), Jaroslav Martasek (Lilly), Martin Romacker (Roche), Michael Braxenthaler (Roche), Maria Manuela Nogueira (EISBM, Lyon, France), Mansoor Saqi (EISBM, Lyon, France), Neil Fitch (BioSci Consulting), Nesrine Taibi (EISBM, Lyon, France), Odile Brasier (EISBM, Lyon, France), Paul Agapow (Imperial College London, UK), Peter Rice (Imperial College London, UK), Paul Houston (CDISC), Philippe Rocca-Serra (University of Oxford, UK), Reinhard Schneider (University of Luxemburg, Luxemburg), James Rimell (Lilly), Stelios Pavlidis (Imperial College London, UK), Susanna-Assunta Sansone (University of Oxford, UK), Sally Miles (Imperial College London, UK), Samiul Hasan (GSK), Sascha Herzinger (University of Luxemburg, Luxemburg), Scott Wagers (BioSci Consulting), Sikander Hayat (Bayer), Tomas Dalentoft (Astra Zeneca), Vahid Elyasigomari (Imperial College London, UK), Venkata Satagopam (University of Luxemburg, Luxemburg), Wei Gu (University of Luxemburg, Luxemburg), Xian Yang (Imperial College London, UK), Yi-Ke Guo (Imperial College London, UK).
Funding
This work was supported through the Innovative Medicines Initiative U-BIOPRED and eTRIKS projects (IMI n°115010 and IMI n°115446 respectively).
Availability of data and materials
The datasets analysed in this study are available in the NIH National Cancer Institute repository (https://portal.gdc.cancer.gov/) [117].
Authors’ contributions
All authors read and approved the final version of the manuscript. BDM wrote the main body of the manuscript, performed the analyses presented within and contributed to the development of the data analysis plan, as a member of U-BIOPRED and eTRIKS projects. DL contributed to the writing of the manuscript, to the planning and the performing of the analyses within and contributed to the development of the data analysis plan, as a member of U-BIOPRED and eTRIKS projects. ATB contributed to the design of the analyses presented within along with all statistical concerns during the development of the data analysis plan, as a member of the U-BIOPRED project. AMaz contributed to the enrichment analysis parts of the manuscript, as a member of U-BIOPRED and eTRIKS projects. AC contributed to the design of the data analysis plans and to the clustering parts of the manuscript as a member of the U-BIOPRED project. HA contributed to the design of the data analysis plans and to the clustering parts of the manuscript as a member of the U-BIOPRED project. IB contributed to the enrichment analysis and machine-learning parts of the manuscript as a member of the eTRIKS project. MS contributed to the enrichment analysis and machine-learning parts of the manuscript as a member of the eTRIKS project. JP contributed to the data preparation parts and to the visualisations of the manuscript. SB contributed to the design of the data analysis plan and to the clustering, data integration and enrichment analysis parts of the manuscript. NL contributed to the data preparation parts of the manuscript. KS contributed to the data managements aspects of the manuscript as a member of the eTRIKS project. IP contributed to the data managements aspects of the manuscript as a member of the eTRIKS project. XY contributed to the data managements aspects of the manuscript as a member of the eTRIKS project. MB contributed to the data managements and clustering aspects of the manuscript as a member of the U-BIOPRED project. KK contributed to the development of the data analysis plan and related parts in the manuscript as a member of the U-BIOPRED project. JvE contributed to the development of the data analysis plan and related parts in the manuscript as a member of the U-BIOPRED project. AB contributed to the development of the data analysis plan and related parts in the manuscript as a member of the U-BIOPRED project. TD contributed to the development of the data analysis plan and related parts in the manuscript as a member of the U-BIOPRED project. PD contributed to the development of the data analysis plan and related parts in the manuscript as a member of the U-BIOPRED project. CL contributed to the development of the data analysis plan and related parts in the manuscript as a member of the U-BIOPRED project. AP contributed to the development of the data analysis plan and related parts in the manuscript as a member of the U-BIOPRED project. JC contributed to the development of the data analysis plan and related parts in the manuscript as a member of the U-BIOPRED project. RD contributed to the development of the data analysis plan and related parts in the manuscript as a member of the U-BIOPRED project. KFC contributed to the overall design of the study as a member of the U-BIOPRED project. IMA contributed to the overall design of the study as a member of the U-BIOPRED project. YG contributed to the data management aspects of the manuscript as a member of the eTRIKS project. PJS contributed to the overall design of the study as a member of the U-BIOPRED project. AMan contributed to the development of the data analysis plan and co-led the systems biology work package of the U-BIOPRED project. AR contributed to the development of the data analysis plan and co-led the systems biology work package of the U-BIOPRED and eTRIKS projects. FB contributed to the development of the data analysis plan and co-led the systems biology work package of the U-BIOPRED project. CA contributed to the overall design and supervision of the study, to the development of the data analysis plan and co-led the systems biology work package of the U-BIOPRED project and its extension in the eTRIKS project.
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
ATB received fees from Acclarogen Ltd. KK received fees from UCB Celltech Ltd. JvE received fees from UCB Pharma S.A. AB received fees from Roche Products Ltd. TD received fees from Janssen R & D High Wycombe Ltd. PD received fees from AstraZeneca Ltd. CL received fees from GSK Ltd. JC received fees from Areteva R & D Ltd. AMan received fees from Roche Diagnostics GmbH, AR received fees from Janssen R & D High Wycombe Ltd. FB received fees from Janssen R & D Springhouse LLC.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12918-018-0556-z) contains supplementary material, which is available to authorized users.
Bertrand De Meulder and Diane Lefaudeux contributed equally to this work.
Contributor Information
Bertrand De Meulder, Email: bdemeulder@eisbm.org.
Diane Lefaudeux, Email: dlefaudeux@eisbm.org.
Aruna T. Bansal, Email: aruna.t.bansal@acclarogen.com
Alexander Mazein, Email: amazein@eisbm.org.
Amphun Chaiboonchoe, Email: ac4793@nyu.edu.
Hassan Ahmed, Email: hrafat@gmail.com.
Irina Balaur, Email: ibalaur@eisbm.org.
Mansoor Saqi, Email: msaqi@eisbm.org.
Johann Pellet, Email: jpellet@eisbm.org.
Stéphane Ballereau, Email: stephane.ballereau@cruk.cam.ac.uk.
Nathanaël Lemonnier, Email: nathanael.lemonnier@univ-grenoble-alpes.fr.
Kai Sun, Email: kai.sun08@imperial.ac.uk.
Ioannis Pandis, Email: pandis83@gmail.com.
Xian Yang, Email: xian.yang08@imperial.ac.uk.
Manohara Batuwitage, Email: m.kannangara-batuwitage@imperial.ac.uk.
Kosmas Kretsos, Email: kkretsos@hotmail.com.
Jonathan van Eyll, Email: jonathan.vaneyll@ucb.com.
Alun Bedding, Email: alun.bedding@roche.com.
Timothy Davison, Email: timothy.davison@almacgroup.com.
Paul Dodson, Email: paul.dodson@astrazeneca.com.
Christopher Larminie, Email: christopher.g.larminie@gsk.com.
Anthony Postle, Email: a.d.postle@soton.ac.uk.
Julie Corfield, Email: juliecorfield@areteva.com.
Ratko Djukanovic, Email: r.djukanovic@soton.ac.uk.
Kian Fan Chung, Email: f.chung@imperial.ac.uk.
Ian M. Adcock, Email: ian.adcock@imperial.ac.uk
Yi-Ke Guo, Email: y.guo@imperial.ac.uk.
Peter J. Sterk, Email: p.j.sterk@amc.nl
Alexander Manta, Email: alexander.manta@gmail.com.
Anthony Rowe, Email: arowe4@its.jnj.com.
Frédéric Baribaud, Email: fbaribau@its.jnj.com.
Charles Auffray, Email: cauffray@eisbm.org.
References
- 1.Jameson JL, Longo DL. Precision medicine--personalized, problematic, and promising. N Engl J Med. 2015;372(23):2229–2234. doi: 10.1056/NEJMsb1503104. [DOI] [PubMed] [Google Scholar]
- 2.Chen R, Snyder M. Promise of personalized omics to precision medicine. Wiley Interdiscip Rev Syst Biol Med. 2013;5(1):73–82. doi: 10.1002/wsbm.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Viceconti M, Hunter P, Hose R. Big data, big knowledge: big data for personalized healthcare. IEEE J Biomed Health Inform. 2015;19(4):1209–1215. doi: 10.1109/JBHI.2015.2406883. [DOI] [PubMed] [Google Scholar]
- 4.Ritchie MD, Holzinger ER, Li R, Pendergrass SA, Kim D. Methods of integrating data to uncover genotype-phenotype interactions. Nat Rev Genet. 2015;16(2):85–97. doi: 10.1038/nrg3868. [DOI] [PubMed] [Google Scholar]
- 5.Berger B, Gaasterland T, Lengauer T, Orengo C, Gaeta B, Markel S, Valencia A. ISCB's initial reaction to the New England journal of medicine editorial on data sharing. PLoS Comput Biol. 2016;12(3):e1004816. doi: 10.1371/journal.pcbi.1004816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Longo DL, Drazen JM. Data Sharing. N Engl J Med. 2016;374(3):276–277. doi: 10.1056/NEJMe1516564. [DOI] [PubMed] [Google Scholar]
- 7.Hawkins TL, McKernan KJ, Jacotot LB, MacKenzie JB, Richardson PM, Lander ES. A magnetic attraction to high-throughput genomics. Science. 1997;276(5320):1887–1889. doi: 10.1126/science.276.5320.1887. [DOI] [PubMed] [Google Scholar]
- 8.MacKenzie S. High-throughput interpretation of pathways and biology. Drug News Perspect. 2001;14(1):54–57. [PubMed] [Google Scholar]
- 9.Pietu G, Mariage-Samson R, Fayein NA, Matingou C, Eveno E, Houlgatte R, Decraene C, Vandenbrouck Y, Tahi F, Devignes MD, et al. The Genexpress IMAGE knowledge base of the human brain transcriptome: a prototype integrated resource for functional and computational genomics. Genome Res. 1999;9(2):195–209. [PMC free article] [PubMed] [Google Scholar]
- 10.Velculescu VE, Zhang L, Zhou W, Vogelstein J, Basrai MA, Bassett DE, Jr, Hieter P, Vogelstein B, Kinzler KW. Characterization of the yeast transcriptome. Cell. 1997;88(2):243–251. doi: 10.1016/S0092-8674(00)81845-0. [DOI] [PubMed] [Google Scholar]
- 11.DeRisi JL, Iyer VR, Brown PO. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 1997;278(5338):680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- 12.Wilkins MR, Pasquali C, Appel RD, Ou K, Golaz O, Sanchez JC, Yan JX, Gooley AA, Hughes G, Humphery-Smith I, et al. From proteins to proteomes: large scale protein identification by two-dimensional electrophoresis and amino acid analysis. Biotechnology (N Y) 1996;14(1):61–65. doi: 10.1038/nbt0196-61. [DOI] [PubMed] [Google Scholar]
- 13.James P. Protein identification in the post-genome era: the rapid rise of proteomics. Q Rev Biophys. 1997;30(4):279–331. doi: 10.1017/S0033583597003399. [DOI] [PubMed] [Google Scholar]
- 14.Kishimoto K, Urade R, Ogawa T, Moriyama T. Nondestructive quantification of neutral lipids by thin-layer chromatography and laser-fluorescent scanning: suitable methods for “lipidome” analysis. Biochem Biophys Res Commun. 2001;281(3):657–662. doi: 10.1006/bbrc.2001.4404. [DOI] [PubMed] [Google Scholar]
- 15.Han X, Gross RW. Global analyses of cellular lipidomes directly from crude extracts of biological samples by ESI mass spectrometry: a bridge to lipidomics. J Lipid Res. 2003;44(6):1071–1079. doi: 10.1194/jlr.R300004-JLR200. [DOI] [PubMed] [Google Scholar]
- 16.Oliver SG, Winson MK, Kell DB, Baganz F. Systematic functional analysis of the yeast genome. Trends Biotechnol. 1998;16(9):373–378. doi: 10.1016/S0167-7799(98)01214-1. [DOI] [PubMed] [Google Scholar]
- 17.Tweeddale H, Notley-McRobb L, Ferenci T. Effect of slow growth on metabolism of Escherichia coli, as revealed by global metabolite pool (“metabolome”) analysis. J Bacteriol. 1998;180(19):5109–5116. doi: 10.1128/jb.180.19.5109-5116.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sterk PJ. Towards the Physionomics of asthma and COPD. Copenhagen: European Respiratory Society Annual Congress; 2005. pp. 17–21. [Google Scholar]
- 19.Machado RF, Laskowski D, Deffenderfer O, Burch T, Zheng S, Mazzone PJ, Mekhail T, Jennings C, Stoller JK, Pyle J, et al. Detection of lung cancer by sensor array analyses of exhaled breath. Am J Respir Crit Care Med. 2005;171(11):1286–1291. doi: 10.1164/rccm.200409-1184OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sanchez C, Lachaize C, Janody F, Bellon B, Roder L, Euzenat J, Rechenmann F, Jacq B. Grasping at molecular interactions and genetic networks in Drosophila melanogaster using FlyNets, an internet database. Nucleic Acids Res. 1999;27(1):89–94. doi: 10.1093/nar/27.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cesareni G, Ceol A, Gavrila C, Palazzi LM, Persico M, Schneider MV. Comparative interactomics. FEBS Lett. 2005;579(8):1828–1833. doi: 10.1016/j.febslet.2005.01.064. [DOI] [PubMed] [Google Scholar]
- 22.Mayer B. Bioinformatics for omics data : methods and protocols. New York: Humana Press; 2011. [Google Scholar]
- 23.Mesarovic MD. Case institute of technology. Systems research center.: systems theory and biology. Proceedings of the 3rd systems symposium at case institute of technology. Berlin: Springer; 1968. [Google Scholar]
- 24.Noble D. Cardiac action and pacemaker potentials based on the Hodgkin-Huxley equations. Nature. 1960;188:495–497. doi: 10.1038/188495b0. [DOI] [PubMed] [Google Scholar]
- 25.Auffray C, Imbeaud S, Roux-Rouquie M, Hood L. From functional genomics to systems biology: concepts and practices. C R Biol. 2003;326(10–11):879–892. doi: 10.1016/j.crvi.2003.09.033. [DOI] [PubMed] [Google Scholar]
- 26.Auffray C, Noble D. Origins of systems biology in William Harvey's masterpiece on the movement of the heart and the blood in animals. Int J Mol Sci. 2009;10(4):1658–1669. doi: 10.3390/ijms10041658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Auffray C, Nottale L. Scale relativity theory and integrative systems biology: 1. Founding principles and scale laws. Prog Biophys Mol Biol. 2008;97(1):79–114. doi: 10.1016/j.pbiomolbio.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 28.Davidson EH, Rast JP, Oliveri P, Ransick A, Calestani C, Yuh CH, Minokawa T, Amore G, Hinman V, Arenas-Mena C, et al. A genomic regulatory network for development. Science. 2002;295(5560):1669–1678. doi: 10.1126/science.1069883. [DOI] [PubMed] [Google Scholar]
- 29.Ideker T, Galitski T, Hood L. A new approach to decoding life: systems biology. Annu Rev Genomics Hum Genet. 2001;2:343–372. doi: 10.1146/annurev.genom.2.1.343. [DOI] [PubMed] [Google Scholar]
- 30.Kitano H. Looking beyond the details: a rise in system-oriented approaches in genetics and molecular biology. Curr Genet. 2002;41(1):1–10. doi: 10.1007/s00294-002-0285-z. [DOI] [PubMed] [Google Scholar]
- 31.Noble D. Modeling the heart--from genes to cells to the whole organ. Science. 2002;295(5560):1678–1682. doi: 10.1126/science.1069881. [DOI] [PubMed] [Google Scholar]
- 32.Nottale L, Auffray C. Scale relativity theory and integrative systems biology: 2. Macroscopic quantum-type mechanics. Prog Biophys Mol Biol. 2008;97(1):115–157. doi: 10.1016/j.pbiomolbio.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 33.Prokop A, Csukas B. Systems biology - integrative biology and simulation tools. Dordrecht: Springer; 2013.
- 34.Anderson GP. Endotyping asthma: new insights into key pathogenic mechanisms in a complex, heterogeneous disease. Lancet. 2008;372(9643):1107–1119. doi: 10.1016/S0140-6736(08)61452-X. [DOI] [PubMed] [Google Scholar]
- 35.Auffray C, Chen Z, Hood L. Systems medicine: the future of medical genomics and healthcare. Gen Med. 2009;1(1):2. doi: 10.1186/gm2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Auffray C, Charron D, Hood L. Predictive, preventive, personalized and participatory medicine: back to the future. Gen Med. 2010;2(8):57. doi: 10.1186/gm178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Auffray C, Hood L. Editorial: systems biology and personalized medicine - the future is now. Biotechnol J. 2012;7(8):938–939. doi: 10.1002/biot.201200242. [DOI] [PubMed] [Google Scholar]
- 38.Hood L, Auffray C. Participatory medicine: a driving force for revolutionizing healthcare. Gen Med. 2013;5(12):110. doi: 10.1186/gm514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hood L, Balling R, Auffray C. Revolutionizing medicine in the 21st century through systems approaches. Biotechnol J. 2012;7(8):992–1001. doi: 10.1002/biot.201100306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sobradillo P, Pozo F, Agusti A. P4 medicine: the future around the corner. Arch Bronconeumol. 2011;47(1):35–40. doi: 10.1016/j.arbres.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 41.Wolkenhauer O, Auffray C, Jaster R, Steinhoff G, Dammann O. The road from systems biology to systems medicine. Pediatr Res. 2013;73(4 Pt 2):502–507. doi: 10.1038/pr.2013.4. [DOI] [PubMed] [Google Scholar]
- 42.Leek JT, Scharpf RB, Bravo HC, Simcha D, Langmead B, Johnson WE, Geman D, Baggerly K, Irizarry RA. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat Rev Genet. 2010;11(10):733–739. doi: 10.1038/nrg2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McDonald JH. Handbook of biological statistics. 3. Baltimore: Sparky House Publishing; 2014. [Google Scholar]
- 44.Lapatas V, Stefanidakis M, Jimenez RC, Via A, Schneider MV. Data integration in biological research: an overview. J Biol Res Thessalon. 2015;22:1–16. doi: 10.1186/s40709-015-0032-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rhee SY, Wood V, Dolinski K, Draghici S. Use and misuse of the gene ontology annotations. Nat Rev Genet. 2008;9(7):509–515. doi: 10.1038/nrg2363. [DOI] [PubMed] [Google Scholar]
- 46.Reimand J, Arak T, Vilo J. G:profiler--a web server for functional interpretation of gene lists (2011 update) Nucleic Acids Res. 2011;39(Web Server issue):W307–W315. doi: 10.1093/nar/gkr378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fujita KA, Ostaszewski M, Matsuoka Y, Ghosh S, Glaab E, Trefois C, Crespo I, Perumal TM, Jurkowski W, Antony PM, et al. Integrating pathways of Parkinson's disease in a molecular interaction map. Mol Neurobiol. 2014;49(1):88–102. doi: 10.1007/s12035-013-8489-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(Database issue):D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vallabhajosyula RR, Raval A. Computational modeling in systems biology. Methods Mol Biol. 2010;662:97–120. doi: 10.1007/978-1-60761-800-3_5. [DOI] [PubMed] [Google Scholar]
- 50.Kuperstein I, Bonnet E, Nguyen HA, Cohen D, Viara E, Grieco L, Fourquet S, Calzone L, Russo C, Kondratova M, et al. Atlas of cancer Signalling network: a systems biology resource for integrative analysis of cancer data with Google maps. Oncogene. 2015;4:e160. doi: 10.1038/oncsis.2015.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mizuno S, Iijima R, Ogishima S, Kikuchi M, Matsuoka Y, Ghosh S, Miyamoto T, Miyashita A, Kuwano R, Tanaka H. AlzPathway: a comprehensive map of signaling pathways of Alzheimer's disease. BMC Syst Biol. 2012;6:52. doi: 10.1186/1752-0509-6-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ogishima S, Mizuno S, Kikuchi M, Miyashita A, Kuwano R, Tanaka H, Nakaya J. AlzPathway, an updated map of curated signaling pathways: towards deciphering Alzheimer's disease pathogenesis. Methods Mol Biol. 2016;1303:423–432. doi: 10.1007/978-1-4939-2627-5_25. [DOI] [PubMed] [Google Scholar]
- 53.Zhao S, Iyengar R. Systems pharmacology: network analysis to identify multiscale mechanisms of drug action. Annu Rev Pharmacol Toxicol. 2012;52:505–521. doi: 10.1146/annurev-pharmtox-010611-134520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bigler J, Hu X, Boedigheimer M, Rowe A, Chung F, Djukanovic R, Sousa A, Corfield J, Adcock I, Sterk P, et al. Whole transcriptome analysis in peripheral blood from asthmatic and healthy subjects in the U-BIOPRED study. Eur Respir J. 2014;44(Suppl 58):2027.
- 55.Wang B, Mezlini AM, Demir F, Fiume M, Tu Z, Brudno M, Haibe-Kains B, Goldenberg A. Similarity network fusion for aggregating data types on a genomic scale. Nat Methods. 2014;11(3):333–337. doi: 10.1038/nmeth.2810. [DOI] [PubMed] [Google Scholar]
- 56.Auffray C, Balling R, Barroso I, Bencze L, Benson M, Bergeron J, Bernal-Delgado E, Blomberg N, Bock C, Conesa A, et al. Making sense of big data in health research: towards an EU action plan. Gen Med. 2016;8(1):71. doi: 10.1186/s13073-016-0323-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8(1):118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 58.van der Kloet FM, Bobeldijk I, Verheij ER, Jellema RH. Analytical error reduction using single point calibration for accurate and precise metabolomic phenotyping. J Proteome Res. 2009;8(11):5132–5141. doi: 10.1021/pr900499r. [DOI] [PubMed] [Google Scholar]
- 59.Gelman A, Hill J. Data analysis using regression and multilevel/hierarchical models. Cambridge: Cambridge University Press; 2007. [Google Scholar]
- 60.Guo Y, Graber A, McBurney RN, Balasubramanian R. Sample size and statistical power considerations in high-dimensionality data settings: a comparative study of classification algorithms. BMC Bioinformatics. 2010;11:447. doi: 10.1186/1471-2105-11-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Michiels S, Kramar A, Koscielny S. Multidimensionality of microarrays: statistical challenges and (im) possible solutions. Mol Oncol. 2011;5(2):190–196. doi: 10.1016/j.molonc.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee JA, Verleysen M. Nonlinear dimensionality reduction. New York: Springer; 2007. [Google Scholar]
- 63.Calza S, Raffelsberger W, Ploner A, Sahel J, Leveillard T, Pawitan Y. Filtering genes to improve sensitivity in oligonucleotide microarray data analysis. Nucleic Acids Res. 2007;35(16):e102. doi: 10.1093/nar/gkm537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stanberry L, Mias GI, Haynes W, Higdon R, Snyder M, Kolker E. Integrative analysis of longitudinal metabolomics data from a personal multi-omics profile. Meta. 2013;3(3):741–760. doi: 10.3390/metabo3030741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ideker T, Dutkowski J, Hood L. Boosting signal-to-noise in complex biology: prior knowledge is power. Cell. 2011;144(6):860–863. doi: 10.1016/j.cell.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Varshavsky R, Gottlieb A, Linial M, Horn D. Novel unsupervised feature filtering of biological data. Bioinformatics. 2006;22(14):e507–e513. doi: 10.1093/bioinformatics/btl214. [DOI] [PubMed] [Google Scholar]
- 68.Bonev B, Escolano F, Cazorla MA. A novel information theory method for filter feature selection. Lect Notes Artif Int. 2007;4827:431–440. [Google Scholar]
- 69.Meyer PE. The rank Minrelation coefficient. Qual Technol Quant M. 2014;11(1):61–70. doi: 10.1080/16843703.2014.11673325. [DOI] [Google Scholar]
- 70.Scardoni G, Petterlini M, Laudanna C. Analyzing biological network parameters with CentiScaPe. Bioinformatics. 2009;25(21):2857–2859. doi: 10.1093/bioinformatics/btp517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27(3):431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cannistraci CV, Ravasi T, Montevecchi FM, Ideker T, Alessio M. Nonlinear dimension reduction and clustering by minimum Curvilinearity unfold neuropathic pain and tissue embryological classes. Bioinformatics. 2010;26(18):i531–i539. doi: 10.1093/bioinformatics/btq376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Estevez PA, Tesmer M, Perez CA, Zurada JM. Normalized mutual information feature selection. IEEE Trans Neural Netw. 2009;20(2):189–201. doi: 10.1109/TNN.2008.2005601. [DOI] [PubMed] [Google Scholar]
- 74.Guyon I, Elisseeff A. An introduction to variable and feature selection. J Mach Learn Res. 2003;3:1157–1182. [Google Scholar]
- 75.Saeys Y, Inza I, Larranaga P. A review of feature selection techniques in bioinformatics. Bioinformatics. 2007;23(19):2507–2517. doi: 10.1093/bioinformatics/btm344. [DOI] [PubMed] [Google Scholar]
- 76.Jain AK, Murty MN, Flynn PJ. Data clustering: a review. ACM Comput Surv. 1999;31(3):264–323. doi: 10.1145/331499.331504. [DOI] [Google Scholar]
- 77.Ronan T, Qi Z, Naegle KM. Avoiding common pitfalls when clustering biological data. Sci Signal. 2016;9(432):re6. doi: 10.1126/scisignal.aad1932. [DOI] [PubMed] [Google Scholar]
- 78.Shirkhorshidi AS, Aghabozorgi S, Teh YW, Herawan T. Big Data Clustering: A Review. Computational Science and Its Applications. 2014;8583:707–720. [Google Scholar]
- 79.Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26(12):1572–1573. doi: 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Caruana R, Elhawary M, Nguyen N, Smith C. Meta clustering. Ieee Data Mining. 2006:107–18.
- 81.Shen R, Mo Q, Schultz N, Seshan VE, Olshen AB, Huse J, Ladanyi M, Sander C. Integrative subtype discovery in glioblastoma using iCluster. PLoS One. 2012;7(4):e35236. doi: 10.1371/journal.pone.0035236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kirk P, Griffin JE, Savage RS, Ghahramani Z, Wild DL. Bayesian correlated clustering to integrate multiple datasets. Bioinformatics. 2012;28(24):3290–3297. doi: 10.1093/bioinformatics/bts595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yuan Y, Savage RS, Markowetz F. Patient-specific data fusion defines prognostic cancer subtypes. PLoS Comput Biol. 2011;7(10):e1002227. doi: 10.1371/journal.pcbi.1002227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bersanelli M, Mosca E, Remondini D, Giampieri E, Sala C, Castellani G, Milanesi L. Methods for the integration of multi-omics data: mathematical aspects. BMC Bioinformatics. 2016;17(Suppl 2):15. doi: 10.1186/s12859-015-0857-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J Roy Stat Soc B Met. 1995;57(1):289–300. [Google Scholar]
- 86.Noble WS. How does multiple testing correction work? Nat Biotechnol. 2009;27(12):1135–1137. doi: 10.1038/nbt1209-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xie J, Cai TT, Maris J, Li H. Optimal false discovery rate control for dependent data. Stat Interface. 2011;4(4):417–430. doi: 10.4310/SII.2011.v4.n4.a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Peduzzi P, Concato J, Feinstein AR, Holford TR. Importance of events per independent variable in proportional hazards regression analysis. II. Accuracy and precision of regression estimates. J Clin Epidemiol. 1995;48(12):1503–1510. doi: 10.1016/0895-4356(95)00048-8. [DOI] [PubMed] [Google Scholar]
- 89.Auffray C. Sharing knowledge: a new frontier for public-private partnerships in medicine. Genome Med. 2009;1(3):29. doi: 10.1186/gm29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lindpaintner K. Biomarkers: call on industry to share. Nature. 2011;470(7333):175. doi: 10.1038/470175d. [DOI] [PubMed] [Google Scholar]
- 91.McShane LM, Cavenagh MM, Lively TG, Eberhard DA, Bigbee WL, Williams PM, Mesirov JP, Polley MY, Kim KY, Tricoli JV, et al. Criteria for the use of omics-based predictors in clinical trials: explanation and elaboration. BMC Med. 2013;11:220. doi: 10.1186/1741-7015-11-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.McShane LM, Cavenagh MM, Lively TG, Eberhard DA, Bigbee WL, Williams PM, Mesirov JP, Polley MY, Kim KY, Tricoli JV, et al. Criteria for the use of omics-based predictors in clinical trials. Nature. 2013;502(7471):317–320. doi: 10.1038/nature12564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Poste G. Bring on the biomarkers. Nature. 2011;469(7329):156–157. doi: 10.1038/469156a. [DOI] [PubMed] [Google Scholar]
- 94.Sung J, Wang Y, Chandrasekaran S, Witten DM, Price ND. Molecular signatures from omics data: from chaos to consensus. Biotechnol J. 2012;7(8):946–957. doi: 10.1002/biot.201100305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Altman DG, Vergouwe Y, Royston P, Moons KG. Prognosis and prognostic research: validating a prognostic model. BMJ. 2009;338:b605. doi: 10.1136/bmj.b605. [DOI] [PubMed] [Google Scholar]
- 96.Hemingway H, Riley RD, Altman DG. Ten steps towards improving prognosis research. BMJ. 2009;339:b4184. doi: 10.1136/bmj.b4184. [DOI] [PubMed] [Google Scholar]
- 97.Jin L, Zuo XY, Su WY, Zhao XL, Yuan MQ, Han LZ, Zhao X, Chen YD, Rao SQ. Pathway-based analysis tools for complex diseases: a review. Genomics Proteomics Bioinformatics. 2014;12(5):210–220. doi: 10.1016/j.gpb.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Khatri P, Draghici S. Ontological analysis of gene expression data: current tools, limitations, and open problems. Bioinformatics. 2005;21(18):3587–3595. doi: 10.1093/bioinformatics/bti565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Khatri P, Sirota M, Butte AJ. Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput Biol. 2012;8(2):e1002375. doi: 10.1371/journal.pcbi.1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Croft D, Mundo AF, Haw R, Milacic M, Weiser J, Wu G, Caudy M, Garapati P, Gillespie M, Kamdar MR, et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2014;42(Database issue):D472–D477. doi: 10.1093/nar/gkt1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Milacic M, Haw R, Rothfels K, Wu G, Croft D, Hermjakob H, D'Eustachio P, Stein L. Annotating cancer variants and anti-cancer therapeutics in reactome. Cancers. 2012;4(4):1180–1211. doi: 10.3390/cancers4041180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mizuno S, Ogishima S, Kitatani K, Kikuchi M, Tanaka H, Yaegashi N, Nakaya J. Network analysis of a comprehensive knowledge repository reveals a dual role for ceramide in alzheimer's disease. PlosOne 2016;11(2):e0148431. [DOI] [PMC free article] [PubMed]
- 103.Lefaudeux D, De Meulder B, Loza MJ, Peffer N, Rowe A, Baribaud F, Bansal AT, Lutter R, Sousa AR, Corfield J, et al. U-BIOPRED clinical adult asthma clusters linked to a subset of sputum -omics. J Allergy Clin Immunol. 2016; In press [DOI] [PubMed]
- 104.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 105.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 106.Bast RC, Jr, Hennessy B, Mills GB. The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer. 2009;9(6):415–428. doi: 10.1038/nrc2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Angermueller C, Parnamaa T, Parts L, Stegle O. Deep learning for computational biology. Mol Syst Biol. 2016;12(7):878. doi: 10.15252/msb.20156651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sommer C, Gerlich DW. Machine learning in cell biology - teaching computers to recognize phenotypes. J Cell Sci. 2013;126(Pt 24):5529–5539. doi: 10.1242/jcs.123604. [DOI] [PubMed] [Google Scholar]
- 109.Kuhn M, Wing J, Weston S, Williams A, Keefer C, Engelhardt A. Caret: classification and regression training. 2012. pp. 15–044. [Google Scholar]
- 110.Le Cao KA, Gonzalez I, Dejean S. Integromics: an R package to unravel relationships between two omics datasets. Bioinformatics. 2009;25(21):2855–2856. doi: 10.1093/bioinformatics/btp515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Le Cao KA, Rohart F4, Gonzalez I, Dejean S, Gautier B, Bartolo F, Monget P, Coquery J, Yao FBL. mixOmics: omics data integration project: R package version; 2016. p. 6.1.1.
- 112.Singh ABG, Shannon C, Vacher M, Rohart F, Tebutt S, Le Cao KA. DIABLO - an integrative, multi-omics, multivariate method for multi-group classification: bioRxiv; 2016.
- 113.Lum PY, Singh G, Lehman A, Ishkanov T, Vejdemo-Johansson M, Alagappan M, Carlsson J, Carlsson G. Extracting insights from the shape of complex data using topology. Sci Rep. 2013;3:1236. doi: 10.1038/srep01236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gevaert O, Villalobos V, Sikic BI, Plevritis SK. Identification of ovarian cancer driver genes by using module network integration of multi-omics data. Interface Focus. 2013;3(4):20130013. doi: 10.1098/rsfs.2013.0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jin N, Wu H, Miao Z, Huang Y, Hu Y, Bi X, Wu D, Qian K, Wang L, Wang C, et al. Network-based survival-associated module biomarker and its crosstalk with cell death genes in ovarian cancer. Sci Rep. 2015;5:11566. doi: 10.1038/srep11566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kim D, Joung JG, Sohn KA, Shin H, Park YR, Ritchie MD, Kim JH. Knowledge boosting: a graph-based integration approach with multi-omics data and genomic knowledge for cancer clinical outcome prediction. J Am Med Inform Assoc. 2015;22(1):109–120. doi: 10.1136/amiajnl-2013-002481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Network TCGAR. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang Q, Burdette JE, Wang JP. Integrative network analysis of TCGA data for ovarian cancer. BMC Syst Biol. 2014;8:1338. doi: 10.1186/s12918-014-0136-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tothill RW, Tinker AV, George J, Brown R, Fox SB, Lade S, Johnson DS, Trivett MK, Etemadmoghadam D, Locandro B, et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin Cancer Res. 2008;14(16):5198–5208. doi: 10.1158/1078-0432.CCR-08-0196. [DOI] [PubMed] [Google Scholar]
- 120.Brunet JP, Tamayo P, Golub TR, Mesirov JP. Metagenes and molecular pattern discovery using matrix factorization. P Natl Acad Sci USA. 2004;101(12):4164–4169. doi: 10.1073/pnas.0308531101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Paparrizos J, Gravano L. K-shape: efficient and accurate clustering of time series in: SIGMOD international conference on Management of Data: June 4, 2015. Melbourne: Australia: Edited by ACM; 2015. pp. 1855–1870. [Google Scholar]
- 122.Sagner M, McNeil A, Puska P, Auffray C, Price ND, Hood L, Lavie CJ, Han ZG, Chen Z, Brahmachari SK, et al. The P4 health Spectrum - a predictive, preventive, personalized and participatory continuum for promoting Healthspan. Prog Cardiovasc Dis. 2017;59(5):506–521. doi: 10.1016/j.pcad.2016.08.002. [DOI] [PubMed] [Google Scholar]
- 123.Reimer D, Sadr S, Wiedemair A, Goebel G, Concin N, Hofstetter G, Marth C, Zeimet AG. Expression of the E2F family of transcription factors and its clinical relevance in ovarian cancer. Ann N Y Acad Sci. 2006;1091:270–281. doi: 10.1196/annals.1378.073. [DOI] [PubMed] [Google Scholar]
- 124.Xanthoulis A, Tiniakos DG. E2F transcription factors and digestive system malignancies: how much do we know? World J Gastroenterol. 2013;19(21):3189–3198. doi: 10.3748/wjg.v19.i21.3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Miyata K, Yotsumoto F, Nam SO, Odawara T, Manabe S, Ishikawa T, Itamochi H, Kigawa J, Takada S, Asahara H, et al. Contribution of transcription factor, SP1, to the promotion of HB-EGF expression in defense mechanism against the treatment of irinotecan in ovarian clear cell carcinoma. Cancer Med. 2014;3(5):1159–1169. doi: 10.1002/cam4.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Permuth-Wey J, Chen YA, Tsai YY, Chen Z, Qu X, Lancaster JM, Stockwell H, Dagne G, Iversen E, Risch H, et al. Inherited variants in mitochondrial biogenesis genes may influence epithelial ovarian cancer risk. Cancer Epidemiol Biomark Prev. 2011;20(6):1131–1145. doi: 10.1158/1055-9965.EPI-10-1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nakano H, Yamada Y, Miyazawa T, Yoshida T. Gain-of-function microRNA screens identify miR-193a regulating proliferation and apoptosis in epithelial ovarian cancer cells. Int J Oncol. 2013;42(6):1875–1882. doi: 10.3892/ijo.2013.1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Archer MC. Role of sp transcription factors in the regulation of cancer cell metabolism. Genes Cancer. 2011;2(7):712–719. doi: 10.1177/1947601911423029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Li Y, Yao L, Liu F, Hong J, Chen L, Zhang B, Zhang W. Characterization of microRNA expression in serous ovarian carcinoma. Int J Mol Med. 2014;34(2):491–498. doi: 10.3892/ijmm.2014.1813. [DOI] [PubMed] [Google Scholar]
- 130.Hein S, Mahner S, Kanowski C, Loning T, Janicke F, Milde-Langosch K. Expression of Jun and Fos proteins in ovarian tumors of different malignant potential and in ovarian cancer cell lines. Oncol Rep. 2009;22(1):177–183. doi: 10.3892/or_00000422. [DOI] [PubMed] [Google Scholar]
- 131.Wang JX, Zeng Q, Chen L, Du JC, Yan XL, Yuan HF, Zhai C, Zhou JN, Jia YL, Yue W, et al. SPINDLIN1 promotes cancer cell proliferation through activation of WNT/TCF-4 signaling. Mol Cancer Res. 2012;10(3):326–335. doi: 10.1158/1541-7786.MCR-11-0440. [DOI] [PubMed] [Google Scholar]
- 132.Sundfeldt K, Ivarsson K, Carlsson M, Enerback S, Janson PO, Brannstrom M, Hedin L. The expression of CCAAT/enhancer binding protein (C/EBP) in the human ovary in vivo: specific increase in C/EBPbeta during epithelial tumour progression. Br J Cancer. 1999;79(7–8):1240–1248. doi: 10.1038/sj.bjc.6690199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.He L, Guo L, Vathipadiekal V, Sergent PA, Growdon WB, Engler DA, Rueda BR, Birrer MJ, Orsulic S, Mohapatra G. Identification of LMX1B as a novel oncogene in human ovarian cancer. Oncogene. 2014;33(33):4226–4235. doi: 10.1038/onc.2013.375. [DOI] [PubMed] [Google Scholar]
- 134.White NM, Chow TF, Mejia-Guerrero S, Diamandis M, Rofael Y, Faragalla H, Mankaruous M, Gabril M, Girgis A, Yousef GM. Three dysregulated miRNAs control kallikrein 10 expression and cell proliferation in ovarian cancer. Br J Cancer. 2010;102(8):1244–1253. doi: 10.1038/sj.bjc.6605634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Downie D, McFadyen MC, Rooney PH, Cruickshank ME, Parkin DE, Miller ID, Telfer C, Melvin WT, Murray GI. Profiling cytochrome P450 expression in ovarian cancer: identification of prognostic markers. Clin Cancer Res. 2005;11(20):7369–7375. doi: 10.1158/1078-0432.CCR-05-0466. [DOI] [PubMed] [Google Scholar]
- 136.Gambineri A, Tomassoni F, Munarini A, Stimson RH, Mioni R, Pagotto U, Chapman KE, Andrew R, Mantovani V, Pasquali R, et al. A combination of polymorphisms in HSD11B1 associates with in vivo 11{beta}-HSD1 activity and metabolic syndrome in women with and without polycystic ovary syndrome. Eur J Endocrinol. 2011;165(2):283–292. doi: 10.1530/EJE-11-0091. [DOI] [PubMed] [Google Scholar]
- 137.Howells REJ, Dhar KK, Hoban PR, Jones PW, Fryer AA, Redman CWE, Strange RC. Association between glutathione-S-transferase GSTP1 genotypes, GSTP1 over-expression, and outcome in epithelial ovarian cancer. Int J Gynecol Cancer. 2004;14(2):242–250. doi: 10.1111/j.1048-891X.2004.014207.x. [DOI] [PubMed] [Google Scholar]
- 138.Cao J, Cai J, Huang D, Han Q, Yang Q, Li T, Ding H, Wang Z. miR-335 represents an invasion suppressor gene in ovarian cancer by targeting Bcl-w. Oncol Rep. 2013;30(2):701–706. doi: 10.3892/or.2013.2482. [DOI] [PubMed] [Google Scholar]
- 139.Tsai SJ, Hwang JM, Hsieh SC, Ying TH, Hsieh YH. Overexpression of myeloid zinc finger 1 suppresses matrix metalloproteinase-2 expression and reduces invasiveness of SiHa human cervical cancer cells. Biochem Bioph Res Co. 2012;425(2):462–467. doi: 10.1016/j.bbrc.2012.07.125. [DOI] [PubMed] [Google Scholar]
- 140.Nie LY, Lu QT, Li WH, Yang N, Dongol S, Zhang X, Jiang J. Sterol regulatory element-binding protein 1 is required for ovarian tumor growth. Oncol Rep. 2013;30(3):1346–1354. doi: 10.3892/or.2013.2575. [DOI] [PubMed] [Google Scholar]
- 141.Odegaard E, Staff AC, Kaern J, Florenes VA, Kopolovic J, Trope CG, Abeler VM, Reich R, Davidson B. The AP-2gamma transcription factor is upregulated in advanced-stage ovarian carcinoma. Gynecol Oncol. 2006;100(3):462–468. doi: 10.1016/j.ygyno.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 142.Hudson LG, Zeineldin R, Silberberg M, Stack MS. Activated epidermal growth factor receptor in ovarian cancer. Cancer Treat Res. 2009;149:203–226. doi: 10.1007/978-0-387-98094-2_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Landskron J, Helland O, Torgersen KM, Aandahl EM, Gjertsen BT, Bjorge L, Tasken K. Activated regulatory and memory T-cells accumulate in malignant ascites from ovarian carcinoma patients. Cancer Immunol Immunother. 2015;64(3):337–347. doi: 10.1007/s00262-014-1636-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gavalas NG, Karadimou A, Dimopoulos MA, Bamias A. Immune response in ovarian cancer: how is the immune system involved in prognosis and therapy: potential for treatment utilization. Clin Dev Immunol. 2010;2010:791603. doi: 10.1155/2010/791603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Carlsten M, Norell H, Bryceson YT, Poschke I, Schedvins K, Ljunggren HG, Kiessling R, Malmberg KJ. Primary human tumor cells expressing CD155 impair tumor targeting by down-regulating DNAM-1 on NK cells. J Immunol. 2009;183(8):4921–4930. doi: 10.4049/jimmunol.0901226. [DOI] [PubMed] [Google Scholar]
- 146.Bellone S, Siegel ER, Cocco E, Cargnelutti M, Silasi DA, Azodi M, Schwartz PE, Rutherford TJ, Pecorelli S, Santin AD. Overexpression of epithelial cell adhesion molecule in primary, metastatic, and recurrent/chemotherapy-resistant epithelial ovarian cancer: implications for epithelial cell adhesion molecule-specific immunotherapy. Int J Gynecol Cancer. 2009;19(5):860–866. doi: 10.1111/IGC.0b013e3181a8331f. [DOI] [PubMed] [Google Scholar]
- 147.Szkandera J, Kiesslich T, Haybaeck J, Gerger A, Pichler M. Hedgehog signaling pathway in ovarian cancer. Int J Mol Sci. 2013;14(1):1179–1196. doi: 10.3390/ijms14011179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Feng Q, Deftereos G, Hawes SE, Stern JE, Willner JB, Swisher EM, Xi L, Drescher C, Urban N, Kiviat N. DNA hypermethylation, Her-2/neu overexpression and p53 mutations in ovarian carcinoma. Gynecol Oncol. 2008;111(2):320–329. doi: 10.1016/j.ygyno.2008.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Clarke B, Tinker AV, Lee CH, Subramanian S, van de Rijn M, Turbin D, Kalloger S, Han G, Ceballos K, Cadungog MG, et al. Intraepithelial T cells and prognosis in ovarian carcinoma: novel associations with stage, tumor type, and BRCA1 loss. Mod Pathol. 2009;22(3):393–402. doi: 10.1038/modpathol.2008.191. [DOI] [PubMed] [Google Scholar]
- 150.Powell CB, Manning K, Collins JL. Interferon-alpha (IFN alpha) induces a cytolytic mechanism in ovarian carcinoma cells through a protein kinase C-dependent pathway. Gynecol Oncol. 1993;50(2):208–214. doi: 10.1006/gyno.1993.1194. [DOI] [PubMed] [Google Scholar]
- 151.Adham SA, Sher I, Coomber BL. Molecular blockade of VEGFR2 in human epithelial ovarian carcinoma cells. Lab Investig. 2010;90(5):709–723. doi: 10.1038/labinvest.2010.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Chen H, Ye D, Xie X, Chen B, Lu W. VEGF, VEGFRs expressions and activated STATs in ovarian epithelial carcinoma. Gynecol Oncol. 2004;94(3):630–635. doi: 10.1016/j.ygyno.2004.05.056. [DOI] [PubMed] [Google Scholar]
- 153.Chen Q, Gao G, Luo S. Hedgehog signaling pathway and ovarian cancer. Chin J Cancer Res. 2013;25(3):346–353. doi: 10.3978/j.issn.1000-9604.2013.06.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Darb-Esfahani S, Sinn BV, Weichert W, Budczies J, Lehmann A, Noske A, Buckendahl AC, Muller BM, Sehouli J, Koensgen D, et al. Expression of classical NF-kappaB pathway effectors in human ovarian carcinoma. Histopathology. 2010;56(6):727–739. doi: 10.1111/j.1365-2559.2010.03544.x. [DOI] [PubMed] [Google Scholar]
- 155.Wang H, Xie X, Lu WG, Ye DF, Chen HZ, Li X, Cheng Q. Ovarian carcinoma cells inhibit T cell proliferation: suppression of IL-2 receptor beta and gamma expression and their JAK-STAT signaling pathway. Life Sci. 2004;74(14):1739–1749. doi: 10.1016/j.lfs.2003.07.051. [DOI] [PubMed] [Google Scholar]
- 156.Hurst JH, Hooks SB. Regulator of G-protein signaling (RGS) proteins in cancer biology. Biochem Pharmacol. 2009;78(10):1289–1297. doi: 10.1016/j.bcp.2009.06.028. [DOI] [PubMed] [Google Scholar]
- 157.Leung PC, Choi JH. Endocrine signaling in ovarian surface epithelium and cancer. Hum Reprod Update. 2007;13(2):143–62. [DOI] [PubMed]
- 158.Townsend KN, Spowart JE, Huwait H, Eshragh S, West NR, Elrick MA, Kalloger SE, Anglesio M, Watson PH, Huntsman DG, et al. Markers of T cell infiltration and function associate with favorable outcome in vascularized high-grade serous ovarian carcinoma. PLoS One. 2013;8(12):e82406. doi: 10.1371/journal.pone.0082406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Matassa DS, Amoroso MR, Lu H, Avolio R, Arzeni D, Procaccini C, Faicchia D, Maddalena F, Simeon V, Agliarulo I, et al. Oxidative metabolism drives inflammation-induced platinum resistance in human ovarian cancer. Cell Death Differ. 2016; [DOI] [PMC free article] [PubMed]
- 160.Corney DC, Flesken-Nikitin A, Choi J, Nikitin AY. Role of p53 and Rb in ovarian cancer. Adv Exp Med Biol. 2008;622:99–117. doi: 10.1007/978-0-387-68969-2_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sampath J, Long PR, Shepard RL, Xia X, Devanarayan V, Sandusky GE, Perry WL, 3rd, Dantzig AH, Williamson M, Rolfe M, et al. Human SPF45, a splicing factor, has limited expression in normal tissues, is overexpressed in many tumors, and can confer a multidrug-resistant phenotype to cells. Am J Pathol. 2003;163(5):1781–1790. doi: 10.1016/S0002-9440(10)63538-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Daponte A, Ioannou M, Mylonis I, Simos G, Minas M, Messinis IE, Koukoulis G. Prognostic significance of hypoxia-inducible factor 1 alpha (HIF-1 alpha) expression in serous ovarian cancer: an immunohistochemical study. BMC Cancer. 2008;8:335. doi: 10.1186/1471-2407-8-335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kim JH, Karnovsky A, Mahavisno V, Weymouth T, Pande M, Dolinoy DC, Rozek LS, Sartor MA. LRpath analysis reveals common pathways dysregulated via DNA methylation across cancer types. BMC Genomics. 2012;13:526. doi: 10.1186/1471-2164-13-526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ye J, Livergood RS, Peng G. The role and regulation of human Th17 cells in tumor immunity. Am J Pathol. 2013;182(1):10–20. doi: 10.1016/j.ajpath.2012.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Leung CS, Yeung TL, Yip KP, Pradeep S, Balasubramanian L, Liu J, Wong KK, Mangala LS, Armaiz-Pena GN, Lopez-Berestein G, et al. Calcium-dependent FAK/CREB/TNNC1 signalling mediates the effect of stromal MFAP5 on ovarian cancer metastatic potential. Nat Commun. 2014;5:5092. doi: 10.1038/ncomms6092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lengyel E. Ovarian cancer development and metastasis. Am J Pathol. 2010;177(3):1053–1064. doi: 10.2353/ajpath.2010.100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Frede J, Fraser SP, Oskay-Ozcelik G, Hong Y, Ioana Braicu E, Sehouli J, Gabra H, Djamgoz MB. Ovarian cancer: ion channel and aquaporin expression as novel targets of clinical potential. Eur J Cancer. 2013;49(10):2331–2344. doi: 10.1016/j.ejca.2013.03.016. [DOI] [PubMed] [Google Scholar]
- 168.Bigler J, Boedigheimer M, Schofield JPR, Skipp PJ, Corfield J, Rowe A, Sousa AR, Timour M, Twehues L, Hu X, et al. A severe asthma disease signature from gene expression profiling of peripheral blood from U-BIOPRED cohorts. Am J Respir Crit Care Med. 2017;195(10):1311–1320. doi: 10.1164/rccm.201604-0866OC. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
AUC of consensus clustering. (XLSX 13 kb)
Complete results of the enrichment analysis between clusters. (XLSX 4293 kb)
Table S7. Estimated accuracy and standard deviation of the RFE procedure. Table S8. Accuracy and Kappa values of the Random Forest models in the training set. Table S9. Performances values for the Random Forest model in the testing set. Figure S11. Relative importance of the top 20 predictors building the final model of the RF. The importance axis is scaled, with the mRNA expression of CD3D scaled to 100% and the methylation state of POLA2 to 0% (not shown). (DOCX 18 kb)
DIABLO sPLSDA model results. (DOCX 18966 kb)
Data Availability Statement
The datasets analysed in this study are available in the NIH National Cancer Institute repository (https://portal.gdc.cancer.gov/) [117].