

Abstract
Introduction
This multi-center phase II study assessed the combination of estramustine and weekly paclitaxel with metastatic castrate resistant prostate cancer (CRPC).
Methods
We enrolled 77 patients who had no prior chemotherapy for CRPC between 1998 and 2000. 74 were eligible. Each 8-week cycle included paclitaxel 90 mg/m2 IV weekly for 6 weeks, followed by 2 weeks off and oral estramustine 280 mg twice daily for 3 days beginning 24 hours prior to the first dose of paclitaxel. The primary endpoint was rate of objective or PSA response at 16 weeks. A 50% response rate was considered of further interest.
Results
Eligible patients received a median of three cycles (range of 1–10). The response rate among patients with measurable disease was 34% (95% CI 19% – 52%). The PSA response rate was 58% (95% CI 47% – 70%). Clinical benefit rate was 45% (95% CI 33% – 57%).The median progression-free survival was 5.9 (95% 4.4–6.7) months. The median overall survival was 17.6 (95% CI 14.6–20.8) months. The most common clinical grade 3–4 toxicities were fatigue (14%), and sensory neuropathy (7%). Grade 3–4 hematologic toxicities included lymphopenia (21%) and anemia (9%). There was one toxicity-related death. Quality of life scores improved by the 8th week but the change was not significant.
Conclusion
The combination has activity defined by PSA declines in CRPC but did not meet the protocol specified endpoint for efficacy as defined by objective response rate. Since this study was conducted, more effective, better tolerated regimens have been developed.
Introduction
Although prostate cancer is sensitive to the effects of medical or surgical castration, most patients will eventually become castration-resistant, requiring subsequent treatments. Treatments for metastatic castration-resistant prostate cancer (mCRPC) now include secondary hormonal therapy1–4, immunotherapy5, radiopharmaceuticals6 and chemotherapy. Two taxanes—docetaxel7,8 and cabazitaxel9 are now standard treatments. Estramustine is a nitrogen mustard derivate of estradiol 17 beta phosphate. The oral formulation of estramustine was U.S. FDA approved in 1981 for the palliative treatment of CRPC. Although the exact mechanism of action is unknown, it has been shown to have both hormonal and non-hormonal effects including inhibition of microtubule function10,11. Estramustine has been studied in combination with taxanes docetaxel and paclitaxel, as well as with vinblastine12.
We present the results of ECOG-ACRIN 1898, a multi-center phase II study of weekly paclitaxel and estramustine in mCRPC. This regimen was developed to improve on the efficacy and ease of administration of a previous paclitaxel and estramustine combination in which paclitaxel had been given as a 96 h IV infusion13. Phase 1 data of the combination showed found that daily estramustine had unacceptably toxicity14. Our primary objectives were to determine the activity of weekly paclitaxel plus estramustine on PSA response, to describe the toxicity of the combination, and to determine this regimen’s impact on pain, asthenia and quality of life.
Methods
Eligibility
Eligible patients were required to be at least 18 years of age and to have histologically proven adenocarcinoma of the prostate with evidence of progressive metastatic disease. Patients with an elevated serum prostate specific antigen (PSA) or serum acid phosphatase level as the only evidence of disease were ineligible. Patients with bone metastases only were required to have a PSA level of ≥ 20 ng/ml. Patients with soft tissue metastases (e.g., pelvic mass, lymph node, liver, or lung were required to have bidimensionally measurable disease or a PSA level ≥ 20 ng/ml.
Patients had to have prior treatment with bilateral orchiectomy or other primary hormonal therapy, with evidence of treatment failure. Patients treated with flutamide or nilutamide were required to discontinue these medications at least 4 weeks prior to registration, and discontinuation of bicalutamide was required at least 6 weeks prior to registration with continued evidence of progressive disease. Patients who had received prior treatment with chemotherapy or with radioisotope therapies were ineligible. Prior external beam radiotherapy was allowed if completed at least 4 weeks prior to study entry.
Other requirements included ECOG performance status of 0, 1, or 2, adequate bone marrow function, defined as WBC ≥ 4000/μL or granulocytes ≥ 2000/μL and platelet count ≥ 1000,000 μL, and adequate liver function (total bilirubin ≤ 1.5 mg/dl, transaminase levels (SGOT, SGPT) ≤ 2 times normal), and renal function (creatinine ≤ 2 mg/dl or calculated creatinine clearance ≥ 50 ml/min) within 4 weeks of study entry.
Patients with a history of venous thromboembolism, carcinomatous meningitis, or brain metastases were excluded. Patients with a history of prior malignancy were eligible provide they had been treated with curative intent and had been free of disease for the time period considered appropriate for cure of the specific cancer. All patients provided written, signed informed consent according to federal guidelines and those of their local Institutional Review Boards.
Treatment
Each 8-week treatment cycle consisted of paclitaxel administered by 1-hour IV infusion at a dose of 90 mg/m2 each week for 6 weeks, followed by 2 weeks off. As prophylaxis for hypersensitivity reactions, a premedication regimen consisting of dexamethasone, H2 blockers, and diphenhydramine was administered prior to each dose of paclitaxel.
Estramustine was administered by mouth at a fixed dose of 280 mg twice a day for 3 days beginning 24 hours prior to the first dose of paclitaxel. This 3-day sequence was repeated each week for 6 weeks, and was followed by 2 weeks of rest. Doses of estramustine that were omitted were not to be added on to later doses.
In response to hematologic toxicity, the doses of paclitaxel and estramustine were based on the CBC obtained on the day of treatment, with 25% dose reduction of paclitaxel dose if granulocyte count was 1200–1999/μl, or platelets were 75,000–99,000/μl. For granulocytes < 1200/μl or platelets <75,000, both paclitaxel and estramustine were held until recovery to minimal levels required for re-treatment as noted above. If blood counts were not recovered by day 15 from the last dose, the patient was withdrawn from the study. In response to non-hematologic toxicity, both agents were reduced by 25% for grade 2 toxicity, and for grade 3–4 toxicity, they were held until toxicity resolved to grade 0–1, and then reduced to 75% of the doses for both. Exceptions for nausea and edema were specified in the protocol.
Patients who had not undergone orchiectomy were required to continue LHRH therapy for the duration of protocol therapy. Prophylactic use of G-CSF, or use of G-CSF for uncomplicated febrile neutropenia was not permitted. Patients were not given warfarin or other anticoagulants for prophylaxis of venous thromboembolism.
Quality of Life
Prior to treatment and at week 4, week 8, week 20 and week 24, patients completed the Brief Pain Inventory Short Form, the Functional Assessment of Cancer Therapy for Prostate Cancer (FACT-P) quality of life survey, and the Schwartz Cancer Fatigue Scale. Patients were required to complete a pain medication diary during the 48-hour period prior to each of these assessment times. The primary comparison of interest for the patient-reported outcomes was a comparison of baseline and week 8 assessments.
Toxicity
Toxicities were assessed at every infusion and graded according to the NCI Common Toxicity Criteria, version 2.
Response
The primary endpoint was the proportion of patients with objective response or PSA response by 16 weeks. Response was assessed separately for patients with and without measurable disease. For patients without measurable disease, response was defined as a PSA decrease by 50% or greater from baseline measured at two successive visits 4 weeks apart. Progressive disease in these patients was defined as an increase in serum PSA above the nadir value achieved by at least 50% for three successive measurements at least 4 weeks apart. Patients who did not meet criteria for response or progression for at least ninety days were considered to have stable disease.
For patients with measurable disease, a complete response was the complete disappearance of all clinically detectable malignant disease for at least 4 weeks. Partial response was a decrease greater than or equal to 50% tumor area (defined as multiplication of the longest diameter by the greatest perpendicular diameter) or 50% decrease in the sum of the products of the perpendicular diameters of multiple lesions in the same organ site for at least 4 weeks duration, with no increase in size of any malignant disease or appearance of new lesions. Disappearance of bone lesions were noted, but were not considered as response.
Progressive disease was defined as a 25% increase in the area of any malignant lesions greater than 2 cm2 or 50% increase in the size of smaller lesions, or the appearance of new lesions. Patients who developed new symptomatic bone lesions were considered to have progressed. In addition, deterioration in ECOG PS greater than or 1 level, related to malignancy was also considered progression.
Stable disease was defined as no CR, PR, or progressive disease as defined above for at least 12 weeks.
Statistical Considerations and Study Design
The primary response endpoint was the proportion of patients who by week 16 achieved reduction in serum PSA of 50%, confirmed by a second measurement 4 weeks later, or who had a PR or CR in bidimensionally measurable disease. A two-stage accrual design was adopted. In the first stage, 15 eligible patients were to be enrolled. If 5 or more responses were observed, the design called for an additional 37 eligible patients to be enrolled. The study with this design had 8% probability of rejecting the treatment if it was effective (50% or better response rate) and over 99% probability of rejecting the treatment if it was ineffective (20% or worse response rate). If criteria for efficacy were met (20 or more responses among 52 patients), additional patients with measurable disease were to be enrolled to better define the response rate such that the total with measurable disease was 38 patients. No formal protocol specific criteria were established for bone disease.
Progression-free survival was defined as time from study registration until first progression of any applicable type (measurable disease, PSA, or bone) or death, whichever came first. Patients alive without progression were censored at the last date known to be progression-free. For patients who only progressed in PSA, time to progression was defined as time from registration until the first measurement showing at least 50% increase from the nadir value. Overall survival was defined as time from study registration until death from any cause.
The primary quality of life endpoint was change in FACT-P score at week 8. It was assumed that 70%, or approximately 36 patients, would provide both baseline and 8-week measurements. Assuming that the baseline FACT-P score would be 109 with a standard deviation of 18.6 points, the study had 80% power to detect a ½ standard deviation, or 9-point change, from baseline to week 8.
Descriptive statistics were used to characterize patients at study entry. The method of Kaplan and Meier was used to describe progression-free survival and overall survival. The paired t-test was used to test for differences in quality of life between baseline and week 8. This study was approved by the Institutional Review Boards at all sites.
Results
Patient and recruitment timeline
After reaching the study met its first stage accrual goal, it was suspended for response assessment on April 29, 1999, and reactivated on June 24, 1999 after the necessary responses were observed. It was suspended again on October 20, 1999 after meeting its accrual goal of 67 patients. After determining that 20 patients had experienced a response, it was reopened to patients with measureable disease on June 14, 2000. After accruing 11 additional patients with measurable disease, the study was terminated on December 15, 2000.
One patient was registered twice, resulting in 77 unique patients. Of these, three patients were ineligible, primarily because of baseline laboratory values that did not meet inclusion criteria. Patients were accrued from 21 ECOG (now ECOG-ACRIN) institutions and affiliates. Pre-treatment characteristics of the 74 eligible patients are summarized in Table 1.
Table 1.
Patient Characteristics
| N | Percent | ||
|---|---|---|---|
| Total Eligible | 74 | ||
| Age | Median (Range) | 71 (48–90) | |
| Race | Caucasian | 66 | 89.2 |
| African American | 7 | 9.5 | |
| Asian | 1 | 1.4 | |
| ECOG Performance | 0 | 31 | 41.9 |
| 1 | 39 | 52.7 | |
| 2 | 4 | 5.4 | |
| Metastatic Disease | Bidimensional, Measurable | 35 | 47.3 |
| Osseous, Evaluable | 63 | 85.1 | |
| Disease Sites | Bone | 64 of 74 | 86.5 |
| Lymph Nodes | 26 of 68 | 38.2 | |
| Lung | 9 of 69 | 13.0 | |
| Liver | 8 of 72 | 11.1 | |
| Elevated PSA (ng/ml) | 73 | 98.7 | |
| Median | 151.1 | ||
| Interquartile Range | 63.7 – 536.0 | ||
| Hemoglobin (U/L) | Median | 12.8 | |
| Interquartile Range | 11.6–13.8 | ||
| Radiotherapy | To Metastatic Site | 17 | 23.0% |
Treatment
A total of 270 cycles of treatment were administered to the 74 eligible patients. Patients received a median of three 8-week cycles (range of 1–10 cycles). Sixty one percent of cycles were administered without dose reduction.
Patients were removed from treatment due to progressive disease (44.3%), patient request (20.3%), excessive toxicity (14.9%), other complications (2.7%), death without progression (2.7%), and for other reasons (12.2%).
Toxicity
Table 2 shows toxicities of each type that were considered to be possibly, probably, or definitely related to treatment and were observed in more than 1 patient at grade 3 or higher, or in at least 1 patient at grade 4 or higher. Worst degree toxicities for each patient are tabulated. This table includes all 77 treated patients, regardless of eligibility status. The most common clinical toxicities were fatigue, affecting 14% of patients at grade 3 or 4, and sensory neuropathy, affecting 7% at grades 3–4. Hematologic toxicities included lymphopenia (21% grade 3–4) and anemia (9%). One patient died following one cycle of treatment after experiencing renal failure and pulmonary emboli.
Table 2. Worst Grade Toxicities – All Treated Patients.
Includes all events considered possibly, probably or definitely related to treatment and occurring in at least 2 patients at grade 3 or in at least 1 patient at grade 4 or higher, assessed via CTC Version 2.0.
| Patients (n=77) | |||||
|---|---|---|---|---|---|
| Grade | |||||
| 1,2 | 3 | 4 | 5 | ||
| (n) | (n) | (n) | (n) | % with grade 3–5 | |
| Hemoglobin | 64 | 7 | – | – | 9.1% |
| Lymphopenia | 39 | 16 | – | – | 20.8% |
| Neutrophils | 11 | 2 | 1 | – | 3.9% |
| Cardiac–ischemia | – | – | 2 | – | 2.6% |
| Edema | 22 | 3 | – | – | 3.9% |
| Hypotension | 5 | – | 1 | – | 1.3% |
| Thrombosis/embolism | – | 5 | 1 | – | 7.8% |
| Fatigue | 48 | 10 | 1 | – | 14.3% |
| Weight loss | 13 | 2 | – | – | 2.6% |
| Diarrhea | 18 | 4 | – | – | 5.2% |
| Nausea | 38 | 3 | – | – | 3.9% |
| Alkaline phosphatase | 23 | 4 | – | – | 5.2% |
| Bilirubin | 6 | 1 | 1 | – | 2.6% |
| SGOT | 10 | 2 | – | – | 2.6% |
| SGPT | 5 | 2 | – | – | 2.6% |
| Infection w/o neutropenia | 7 | 3 | – | – | 3.9% |
| Hyperglycemia | 4 | – | 1 | – | 1.3% |
| Cerebrovascular ischemia | – | – | 1 | – | 1.3% |
| Dizziness/lightheadedness | 6 | 1 | 1 | – | 2.6% |
| Neuropathy–sensory | 28 | 4 | 1 | – | 6.5% |
| Syncope | 1 | 2 | – | – | 2.6% |
| Arthralgia | 9 | 2 | – | – | 2.6% |
| Dyspnea | 7 | 1 | 1 | – | 2.6% |
| Pneumonitis/pulmonary infiltrates | – | 2 | – | – | 2.6% |
| Creatinine | 8 | 2 | – | – | 2.6% |
| Renal failure | – | – | – | 1 | 1.3% |
| WORST DEGREE | 29 | 38 | 9 | 1 | 62.3% |
Enrollment
After reaching the first stage accrual goal, it was suspended for response assessment on April 29, 1999, and reactivated on June 24, 1999 after the necessary responses were observed. It was suspended again on October 20, 1999 after meeting its accrual goal of 67 patients. After determining that 20 patients had experienced a response, the stud was reopened to patients with measureable disease on June 14, 2000. After accruing 11 additional patients with measurable disease, the study was terminated on December 15, 2000.
Response
The primary response endpoint was the proportion of patients who by week 16 achieved reduction in serum PSA of 50%, confirmed by a second measurement 4 weeks later, or who had a PR or CR in bidimensionally measurable disease. These two endpoints were assessed separately. Response based on assessment (measurable disease, PSA or bone disease) are shown in Table 3.
Table 3.
Response Based on Disease Category
| Response | N | % |
|---|---|---|
| Measurable disease (n=35) | ||
| Complete response | 2 | 5.7 |
| Partial response | 9 | 25.7 |
| Stable disease | 10 | 28.6 |
| Progression | 9 | 25.7 |
| Unevaluable | 5 | 14.3 |
| PSA (n=73) | ||
| Response | 43 | 58.9 |
| Stable disease | 15 | 20.5 |
| Progression | 5 | 6.8 |
| Unevaluable | 10 | 13.7 |
| Bone disease (n=63) | ||
| Response | 2 | 3.2 |
| Stable disease | 38 | 60.3 |
| Progression | 10 | 15.9 |
| Unevaluable | 13 | 20.6 |
Measureable disease
Among the 35 patients who had measurable disease, 12 (34%, 95% exact binomial confidence interval 19% – 52%) exhibited response to treatment.
PSA response
Among 73 patients with elevated PSA, 43 (58%, 95% exact binomial confidence interval 47% –70%) met the criteria for PSA response. As noted, no formal response criteria was noted for bone lesions. However, four of these patients exhibited progression in bone.
Clinical Benefit
To construct a consolidated measure of clinical benefit, we considered patients with measurable disease who exhibited an objective response, regardless of PSA status, to be responders, provided there was no evidence of worsening bone disease. Patients without measurable disease who had the required improvement in PSA were considered responders, again provided there was no evidence of worsening bone disease. Using this consolidated measure, 33 of the 74 eligible, treated patients exhibited response (45%, 95% exact binomial confidence interval 33% – 57%). Median duration of response among these 33 patients was 6.8 months (95% confidence interval, 4.8 to 8.8 months). Response duration among the 12 patients whose response was based on measurable disease was median 6.5 months (95% confidence interval, 3.8 to 8.8 months). Among the 21 patients whose response was based on PSA improvement, median duration of response was 6.9 months (95% confidence interval, 4.8 to 8.9 months).
Survival and Progression-free Survival
At the time follow-up ceased (March 2, 2004), 74 patients had died. All patients had experienced disease progression. Median progression-free survival was 5.9 months (95% confidence interval, 4.4 months to 6.7 months)(Figure 1).
Figure 1.
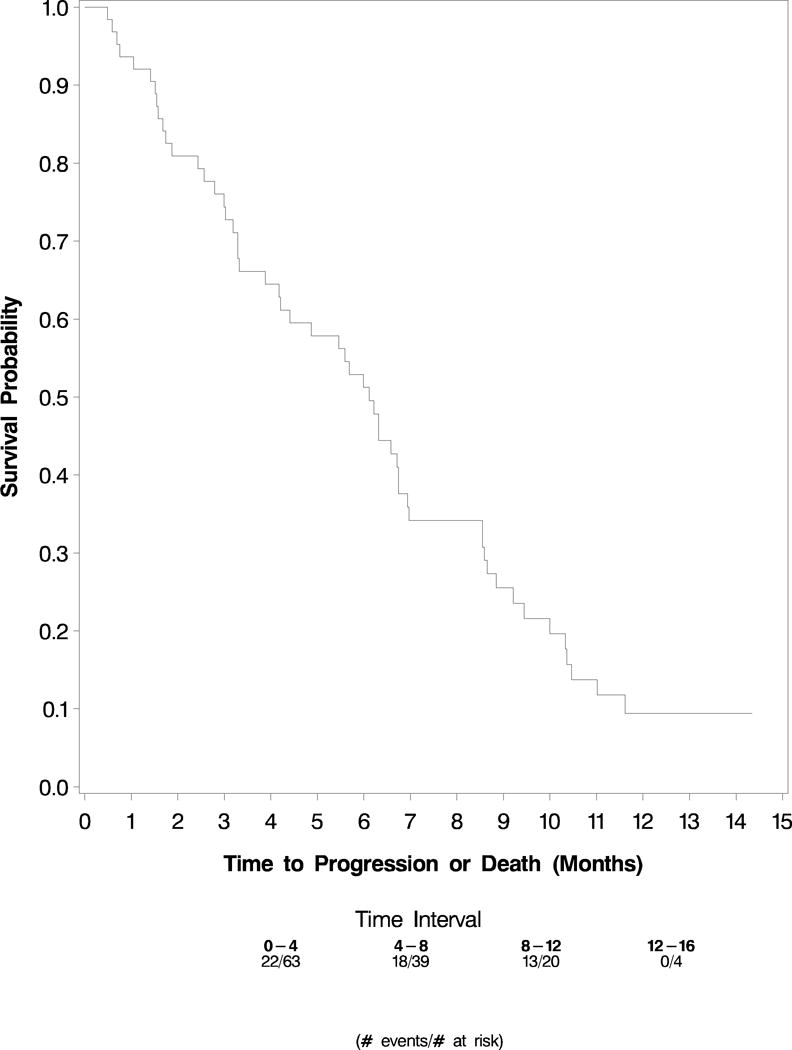
PFS
Overall survival was defined as time from study registration until death from any cause. Median overall survival is 17.6 months (95% confidence interval, 14.6 months to 20.8 months)(Figure 2).
Figure 2.
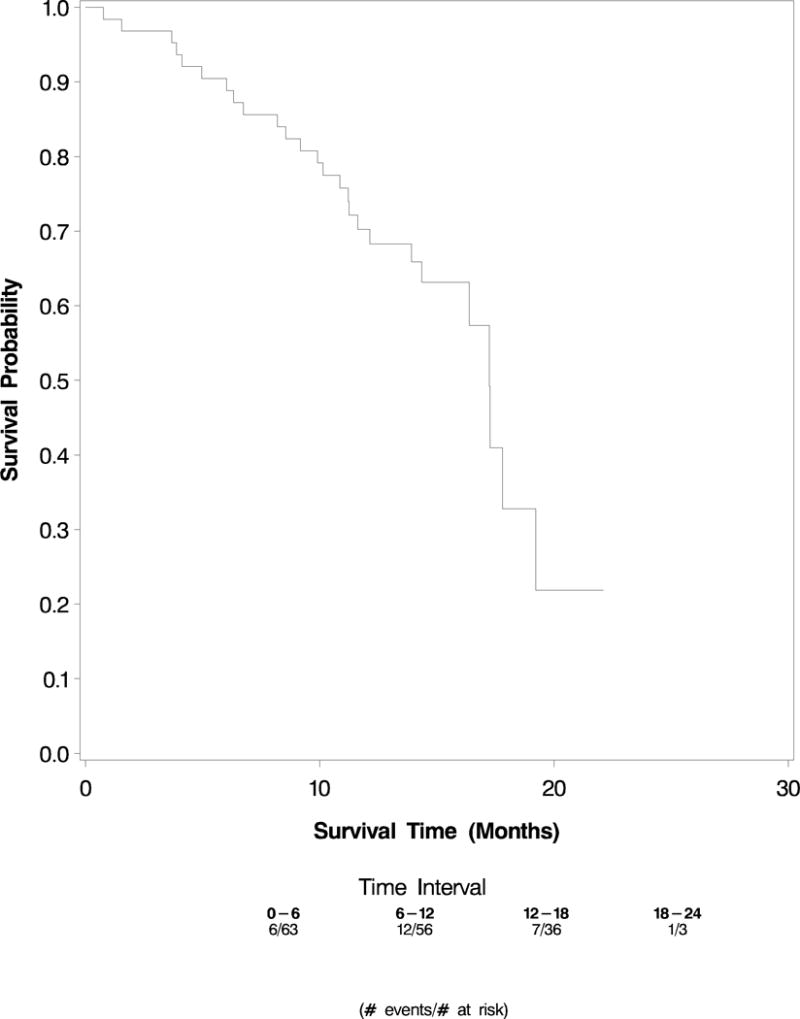
OS
Quality of Life, Pain and Fatigue
Results are shown in Table 4. Sixty-four patients completed the baseline and 8 week FACT-P surveys. The mean baseline score was 100.4, with standard deviation of 19.6 points. There was a 4-point improvement in FACT-P score from baseline to 4 weeks, a significant improvement (p=0.02). This difference had diminished somewhat by week 8, to a 3-point improvement (p=0.18) (Figure 3).
Table 4.
Quality of Life, Fatigue and Pain scores
| Baslined | Week 4 | Week 8 | Week 20 | Week 24 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | Mean | Std Dev | N | Mean | Std Dev | N | Mean | Std Dev | N | Mean | Std Dev | N | Mean | Std Dev | |
| FACT-P | 74 | 100.4 | 19.6 | 64 | 104.7 | 20.3 | 58 | 103.8 | 18.3 | 37 | 103.1 | 20.3 | 32 | 104.4 | 20.7 |
| BPI – Worst Pain | 71 | 3.8 | 2.9 | 59 | 2.8 | 2.6 | 52 | 3.2 | 3.2 | 32 | 3.1 | 2.5 | 29 | 3.4 | 2.9 |
| BPI – Average Pain | 69 | 2.7 | 2.0 | 59 | 2.0 | 2.0 | 52 | 2.3 | 2.4 | 31 | 2.3 | 2.0 | 29 | 2.2 | 2.0 |
| BPI – Relief from Pain | 59 | 57.2 | 33.2 | 51 | 60.4 | 33.7 | 36 | 60.6 | 36.0 | 23 | 59.6 | 34.4 | 18 | 60.0 | 36.6 |
| SCFS-6 Fatigue | 72 | 2.0 | 0.9 | 61 | 2.2 | 0.8 | 58 | 2.0 | 0.8 | 37 | 2.0 | 0.7 | 30 | 2.0 | 0.7 |
Figure 3.
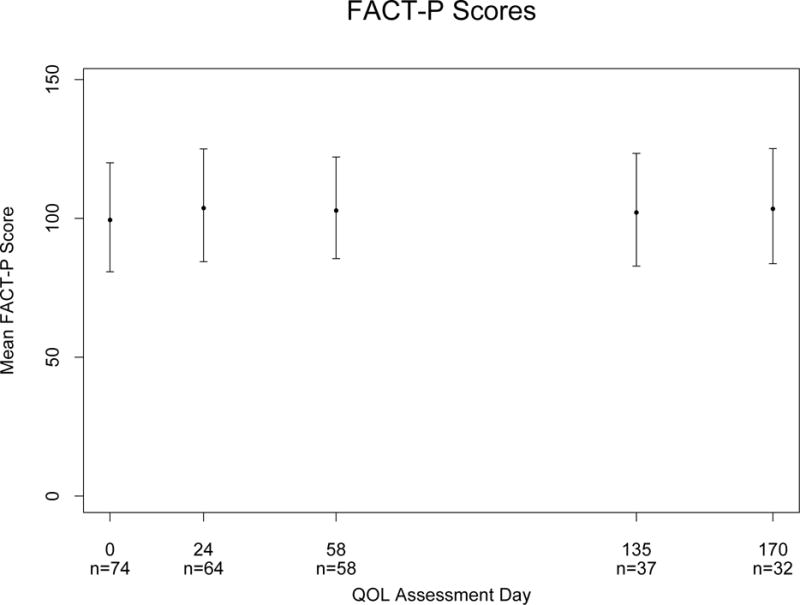
FACT P
Seventy one patients completed the BPI at baseline, and 52 of these patients reported worst pain of 2/10 or higher. 43 of these 52 patients completed the brief pain inventory at 4 weeks. Nineteen (44%) had at least a 2-point decrease on the 10 point pain scale. Five additional patients decreased narcotic dose by at least 50% without increase in pain score. Thus, 59% of patients had either decreased pain or narcotic dose.
Seventy-two and fifty-eight patients completed the Schwarz Cancer Fatigue Scale at baseline and eight weeks respectively. The mean score (2) was consistent across the two time points, and remained stable through the study.
Discussion
This multi-institutional phase II trial characterizes the activity of paclitaxel given together with oral estramustine in patients with hormone refractory prostate cancer. The median progression free survival and overall survival for this regimen were 5.9 months and 17.9 months, respectively.
This weekly paclitaxel-estramustine regimen was associated with a low incidence of severe toxicity. As anticipated, fatigue was the most common toxicity, but was of grade 3 or 4 severity in only 10 and 1 patients, respectively. The 8% incidence of venous thrombosis was similar to the 7% reported in a meta-analysis of 23 estramustine containing regimens studied in 896 patients15. Patient reported outcomes of fatigue using validated instruments confirmed that fatigue was prevalent, but not intrusive for the majority, even after multiple cycles of treatment. Similarly, patient reported measures did not demonstrate a decrease of QOL over time. In addition, treatment with weekly paclitaxel and oral estramustine led to significant pain relief or reduction of narcotic dosage for 59% of patients with narcotic-requiring pain at baseline.
Although these data were collected in a different era of prostate cancer treatment, this is important to summarize and put into context. Fortunately, the clinical landscape for treatment of metastatic castrate resistant prostate cancer has changed significantly. Docetaxel has been used as first line chemotherapy since 2004 when two studies reported a survival benefit of docetaxel based chemotherapy compared to mitoxantrone and prednisone. In TAX 327 the experimental arm received either docetaxel given on a weekly or every three week basis in addition to prednisone8. In SWOG 9916 the experimental arm received docetaxel 60 mg/m2 day 1 every three weeks in combination with estramustine (280 mg three times per day on days 1–5)7.
While associated with increased efficacy compared to the mitoxantrone arm, the estramustine-containing arm in SWOG 9916 was also associated with increased toxicity including thromboembolic events and gastrointestinal symptoms. Since the every three week docetaxel and prednisone arm of TAX 327 had similar efficacy but less toxicity, docetaxel-prednisone has been adopted as a standard of care for men with castrate resistant prostate cancer. The American Society of Clinical Oncology practice guidelines now advises against the use of estramustine due to increased toxicity without improvement in survival or symptom palliation16. Along with reports of poor GI tolerance, the risk of thromboembolism has been cited as high as 12 percent15. Investigators have explored its use through both prospective clinical trials and retrospective studies across a variety of disease states, and doses both with and without thromboprophylaxis17–21. These studies have yielded mixed results regarding efficacy and toxicity profile. As a result, there is no clinical setting or dose of estramustine where the clinical benefits have been found to outweigh the risks.
This study also highlights the progress made in the design and interpretation of studies of castrate resistant prostate cancer since this study was developed. As described, PSA and disease measurements were used to characterize response to treatment in E1898. Although the protocol did not include formal criteria for describing bone response, it did note that the development of new symptomatic lesions represented progressive disease. Since then, the Prostate Cancer Working Group (PCWG), has developed formal response criteria for bone metastases using the “2+2 rule” to distinguish tumor progression from tumor flare after starting treatment. In addition, RECIST definitions of response and progression in patients with measurable disease have been developed since this study was developed. The PCWG 3 guidelines also include criteria to define progression in small lymph nodes, which is important given the high incidence of lymph node metastases in men with prostate cancer.22
It is unlikely that our results would have differed significantly if the study were evaluated under newer response criteria; in particular, the bone criteria noted progression if new painful metastases were noted in spite of a declining PSA. Our results were similar to contemporary studies of estramustine-taxane based therapy21.
In summary, we found that paclitaxel and estramustine treatment in patients with castrate resistant prostate cancer is active but does not provide sufficient clinical benefit he results in this uncontrolled clinical trial were not better than those noted in later randomized phase III studies of docetaxel-based therapies. As newer therapies with more favorable toxicity profiles have since been approved for CRPC, we cannot recommend the further development of this regimen.
Clinical Practice Points.
Estramustine has been shown to have both hormonal and non-hormonal effects, including inhibition of microtubule function. It has been studied in combination with both paclitaxel and docetaxel for castrate resistant prostate cancer.
In this study, the combination of estramustine and paclitaxel did not meet protocol specified endpoints for response.
Practice guidelines from the American Society of Clinical Oncology practice guidelines advise against the use of estramustine due to increased toxicity without improvement in survival or symptom palliation.
Since the study was conducted, more effective and better tolerated regimens for CRPC have been developed.
Acknowledgments
This study was coordinated by the ECOG-ACRIN Cancer Research Group (Peter F. O’Dwyer MD and Mitchell D. Schnall, MD, PhD, Group Co-Chairs) and supported by the National Cancer Institute of the National Institutes of Health under the following award numbers: CA180820, CA180794, CA189828, CA180795, CA180799, CA180802, CA180847, CA180858, CA189956, CA180857. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Parker C, Sartor O. Abiraterone and increased survival in metastatic prostate cancer. The New England journal of medicine. 2011;365(8):767. doi: 10.1056/NEJMc1107198. [DOI] [PubMed] [Google Scholar]
- 2.Scher HI, Heller G, Molina A, et al. Evaluation of circulating tumor cell (CTC) enumeration as an efficacy response biomarker of overall survival (OS) in metastatic castration-resistant prostate cancer (mCRPC): Planned final analysis (FA) of COU-AA-301, a randomized, double-blind, placebo-controlled, phase III study of abiraterone acetate (AA) plus low-dose prednisone (P) post docetaxel. J Clin Oncol. 2011;29(18_suppl):LBA4517. [Google Scholar]
- 3.Beer TM, Tombal B. Enzalutamide in metastatic prostate cancer before chemotherapy. The New England journal of medicine. 2014;371(18):1755–1756. doi: 10.1056/NEJMc1410239. [DOI] [PubMed] [Google Scholar]
- 4.Ryan CJ, Smith MR, de Bono JS, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. The New England journal of medicine. 2013;368(2):138–148. doi: 10.1056/NEJMoa1209096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. The New England journal of medicine. 2010;363(5):411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 6.Parker C, Nilsson S, Heinrich D, et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. The New England journal of medicine. 2013;369(3):213–223. doi: 10.1056/NEJMoa1213755. [DOI] [PubMed] [Google Scholar]
- 7.Petrylak DP, Tangen CM, Hussain MH, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. The New England journal of medicine. 2004;351(15):1513–1520. doi: 10.1056/NEJMoa041318. [DOI] [PubMed] [Google Scholar]
- 8.Tannock IF, de Wit R, Berry WR, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. The New England journal of medicine. 2004;351(15):1502–1512. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 9.de Bono JS, Oudard S, Ozguroglu M, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376(9747):1147–1154. doi: 10.1016/S0140-6736(10)61389-X. [DOI] [PubMed] [Google Scholar]
- 10.Benson R, Hartley-Asp B. Mechanisms of action and clinical uses of estramustine. Cancer Invest. 1990;8(3-4):375–380. doi: 10.3109/07357909009012056. [DOI] [PubMed] [Google Scholar]
- 11.Perry CM, McTavish D. Estramustine phosphate sodium. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy in prostate cancer. Drugs Aging. 1995;7(1):49–74. doi: 10.2165/00002512-199507010-00006. [DOI] [PubMed] [Google Scholar]
- 12.Hudes G, Einhorn L, Ross E, et al. Vinblastine versus vinblastine plus oral estramustine phosphate for patients with hormone-refractory prostate cancer: A Hoosier Oncology Group and Fox Chase Network phase III trial. J Clin Oncol. 1999;17(10):3160–3166. doi: 10.1200/JCO.1999.17.10.3160. [DOI] [PubMed] [Google Scholar]
- 13.Hudes GR, Nathan F, Khater C, et al. Phase II trial of 96-hour paclitaxel plus oral estramustine phosphate in metastatic hormone-refractory prostate cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1997;15(9):3156–3163. doi: 10.1200/JCO.1997.15.9.3156. [DOI] [PubMed] [Google Scholar]
- 14.Haas N, Roth B, Garay C, et al. Phase I trial of weekly paclitaxel plus oral estramustine phosphate in patients with hormone-refractory prostate cancer. Urology. 2001;58(1):59–64. doi: 10.1016/s0090-4295(01)01011-1. [DOI] [PubMed] [Google Scholar]
- 15.Lubiniecki GM, Berlin JA, Weinstein RB, Vaughn DJ. Thromboembolic events with estramustine phosphate-based chemotherapy in patients with hormone-refractory prostate carcinoma: results of a meta-analysis. Cancer. 2004;101(12):2755–2759. doi: 10.1002/cncr.20673. [DOI] [PubMed] [Google Scholar]
- 16.Basch E, Loblaw DA, Oliver TK, et al. Systemic therapy in men with metastatic castration-resistant prostate cancer:American Society of Clinical Oncology and Cancer Care Ontario clinical practice guideline. J Clin Oncol. 2014;32(30):3436–3448. doi: 10.1200/JCO.2013.54.8404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fizazi K, Le Maitre A, Hudes G, et al. Addition of estramustine to chemotherapy and survival of patients with castration-refractory prostate cancer: a meta-analysis of individual patient data. Lancet Oncol. 2007;8(11):994–1000. doi: 10.1016/S1470-2045(07)70284-X. [DOI] [PubMed] [Google Scholar]
- 18.Fizzazi K, Lesaunier F, Delva R, et al. A phase III trial of docetaxel-estramustine in high-risk localised prostate cancer: a planned analysis of response, toxicity and quality of life in the GETUG 12 trial. Eur J Cancer. 2012;48(2):209–217. doi: 10.1016/j.ejca.2011.10.015. [DOI] [PubMed] [Google Scholar]
- 19.Inoue T, Ogura K, Kawakita M, et al. Effective and Safe Administration of Low-Dose Estramustine Phosphate for Castration-Resistant Prostate Cancer. Clin Genitourin Cancer. 2016;14(1):e9–e17. doi: 10.1016/j.clgc.2015.08.008. [DOI] [PubMed] [Google Scholar]
- 20.Petrioli R, Roviello G, Fiaschi AI, et al. Low-Dose Estramustine Phosphate and Concomitant Low-Dose Acetylsalicylic Acid in Heavily Pretreated Patients With Advanced Castration-Resistant Prostate Cancer. Clin Genitourin Cancer. 2015;13(5):441–446. doi: 10.1016/j.clgc.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 21.Machiels JP, Mazzeo F, Clausse M, et al. Prospective randomized study comparing docetaxel, estramustine, and prednisone with docetaxel and prednisone in metastatic hormone-refractory prostate cancer. J Clin Oncol. 2008;26(32):5261–5268. doi: 10.1200/JCO.2008.16.9524. [DOI] [PubMed] [Google Scholar]
- 22.Scher HI, Morris MJ, Stadler WM, et al. Trial Design and Objectives for Castration-Resistant Prostate Cancer: Updated Recommendations From the Prostate Cancer Clinical Trials Working Group 3. J Clin Oncol. 2016;34(12):1402–1418. doi: 10.1200/JCO.2015.64.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]