

Abstract
Mismatch repair (MMR) systems correct DNA mismatches that result from DNA polymerase misincorporation errors. Mismatches also appear in heteroduplex DNA intermediates formed during recombination between nearly identical sequences, and can be corrected by MMR or removed through an unwinding mechanism, known as anti-recombination or heteroduplex rejection. We review studies, primarily in baker's yeast, which support how specific factors can regulate the MMR/anti-recombination decision. Based on recent advances, we present models for how DNA structure, relative amounts of key repair proteins, the timely localization of repair proteins to DNA substrates and epigenetic marks can modulate this critical decision.
Keywords: heteroduplex rejection, DNA mismatch repair, Msh2, Msh6, Msh3, chromatin marks
The authors review studies, primarily in baker's yeast, which support how specific factors can regulate the MMR/anti-recombination decision.
INTRODUCTION
Mismatch repair (MMR) acts during DNA replication to remove polymerase misincorporation errors. A subset of proteins involved in this pathway is also involved in regulating homologous recombination (HR), one of several mechanisms that repairs double-strand breaks (DSBs) in DNA. Such breaks appear as the result of cellular exposure to exogenous or endogenous DNA-damaging agents. When encountered during DNA replication, these breaks can cause fork collapse, which if not repaired appropriately, lead to genome rearrangements. DNA mismatches generated during HR are corrected by the MMR pathway, or serve as substrates for anti-recombination activities (Kunkel and Erie 2015). In contrast to roles in promoting genome stability, a subset of MMR proteins promotes the expansion of trinucleotide repeat sequences. Expansions of these repeat sequences have been linked to neurological, neurodegenerative and neuromuscular diseases (reviewed in Slean et al. 2008; Schmidt and Pearson 2016). In this review, we will focus on how yeast MMR proteins interact with specific DNA substrates and other repair proteins to regulate the MMR/anti-recombination decision (Fig. 1). We also present hypotheses for how epigenetic mechanisms can influence this decision.
Figure 1.

Fate of DNA mismatches: factors that regulate the MMR/anti-recombination decision. DNA mismatches are created by DNA polymerase errors and during recombination between divergent DNA sequences. In baker's yeast, these mismatches are recognized by Msh2-Msh6 or Msh2-Msh3, depending on the type of mismatch generated, and can lead to either MMR or anti-recombination outcomes. The various factors that regulate this decision are discussed in the text and shown here.
A BRIEF OVERVIEW OF MMR DURING DNA REPLICATION
In the yeast Saccharomyces cerevisiae, DNA mismatches are recognized by the heterodimeric MutS homolog complexes Msh2-Msh6 (MutSα) and Msh2-Msh3 (MutSβ), which display partial redundancy. Msh2-Msh6 primarily recognizes base–base and small (1–2 nt) insertion deletion loop mismatches, and Msh2-Msh3 primarily recognizes small and large (up to ∼17 nt in size) insertion–deletion loops. Such loops can form as the result of slippage events occurring during the replication of repetitive DNA sequences such as microsatellites (Kolodner and Marsischky 1999; Jensen, Jauert and Kirkpatrick 2005; Modrich 2006; Kunkel and Erie 2015). In the sliding clamp model for MMR, once an MSH complex binds to a mismatch, it exchanges ADP for ATP, forming a clamp-like structure that dissociates from the mismatch and slides along DNA (Fig. 2; Gradia et al. 1999; Gradia, Acharya and Fishel 2000; Mazurek, Berardini and Fishel 2002; Acharya et al. 2003; Iyer et al. 2006). During this process, MSH proteins recruit heterodimeric MutL homologs, primarily Mlh1-Pms1 (MutLα) in yeast. The endonuclease activity of Mlh1-Pms1 is activated by the replication processivity clamp PCNA to nick the newly synthesized DNA strand in an ATP-dependent manner through a yet to be determined strand discrimination mechanism (Kunkel and Erie 2015). These nicks, 3′ or 5′ to the mismatch, serve as entry points for Exo1, which excises the newly synthesized strand containing the mismatch in a 5′ to 3′ direction. Alternatively, an Exo1 independent pathway, which is thought to involve strand-displacement synthesis by Polδ or Polɛ, can remove the nascent strand containing the mismatch (Kadyrov et al. 2009; Kunkel and Erie 2015). DNA polymerases subsequently resynthesize the excised DNA, culminating in a repair process that reduces the mutation rate on the order of 100–10000-fold (Sia et al. 1997; Tran et al. 1997; Earley and Crouse 1998; Drotschmann et al. 1999; Harfe and Jinks-Robertson 2000b).
Figure 2.
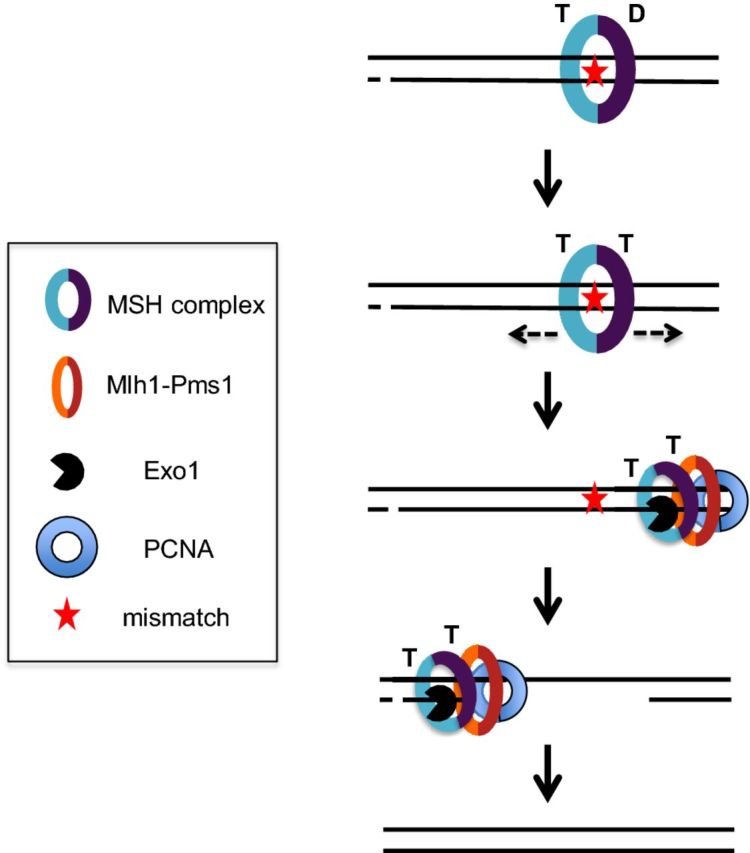
Post-replicative MMR. A mismatch in DNA formed due to DNA polymerase misincorporation during replication is shown. MSH complexes (Msh2-Msh6 or Msh2-Msh3) bind to the mismatch, resulting in ADP (D)→ATP (T) exchange, and both subunits of the MSH complex binding ATP. This causes a conformational change in the MSH complex to form a sliding clamp. The sliding clamp diffuses away from the mismatch to recruit, in a dynamic mechanism, downstream MMR factors such as Mlh1-Pms1, Exo1 and PCNA that excise the nascent strand containing the mismatch, using strand discrimination signals that are thought to involve pre-existing nicks. Final steps of MMR involve DNA synthesis and ligation. Adapted from Manhart and Alani (2016).
AN OVERVIEW OF MMR IN ANTI-RECOMBINATION
MMR proteins play vital roles in regulating the fidelity of HR during repair of DSBs. In this section, we provide a brief introduction to different types of HR mechanisms as well as roles for MMR proteins in HR and anti-recombination.
An overview of HR mechanisms
Cells often repair DSBs by HR, a process in which DSBs are repaired using a homologous DNA template (Fig. 3). In the initial steps of HR, DNA strands on each side of a DSB are resected in the 5′ to 3′ direction. In yeast, resection is initiated by Mre11-Rad50-Xrs2 (MRX) and Sae2, resulting in a limited resection that creates short 3′ overhangs (Cannavo and Cejka 2014; Symington 2014). A more extensive 5′ to 3′ resection is carried out by the Exo1 and Dna2 nucleases, with Dna2 activity being promoted by the Sgs1-Top3-Rmi1 (STR) helicase-topoisomerase complex. Replication protein A (RPA) binds to and protects the 3′ DNA tails, shielding them from degradation, while also stimulating resection of the other strand by Dna2 and STR (Cejka et al. 2010; Niu et al. 2010; Chen, Lisby and Symington 2013; Deng, Chen and Symington 2015). Rad51 then displaces RPA from the 3′ ends of ssDNA, a process mediated by Rad52, in order to facilitate a strand invasion process in which the Rad51 nucleoprotein filament invades homologous dsDNA and displaces one of the strands to pair with the complementary strand. The end result of this process is the formation of a displacement loop (D-loop). DNA synthesis then occurs at the 3′ end of the invading strand in the D-loop, using the homologous donor strand as a template, to create a single-end invasion intermediate.
Figure 3.

HR pathways and their regulation by MMR proteins. HR is initiated with the resection of a DSB (black DNA stretch) to create 3′ ssDNA ends, which then form nucleoprotein filaments by binding to Rad51. One of the 3′ ssDNA tails then invades homologous dsDNA (blue DNA stretch) and anneals to the complimentary strand, thereby displacing the other strand, which forms a D-loop (or displacement loop). (A) In the DSBR pathway, this is followed by branch migration (extension of the annealed regions), which facilitates capture of the second 3′ end of the broken chromosome to generate a dHJ intermediate, which can be resolved into CO or NCO products (arrows indicate the direction of resolution of the HJs in the examples shown). (B) In the SDSA pathway, second end capture does not occur and instead the Holliday junction is disassembled after DNA synthesis is initiated from the 3′ end. The two ends of the broken DNA molecule then anneal to each other and further DNA synthesis occurs to recover the missing information, followed by ligation, to repair the break. This pathway does not yield CO events. (C) When a DSB is formed between repetitive sequences, repair occurs via the SSA pathway. In SSA, resected DNA anneals at complementary sequences. Overhanging 3′ flaps are clipped, allowing DNA synthesis and ligation to repair the DSB by deleting sequences between the repeats. In all pathways, stages at which mismatches may arise as a result of divergent sequences attempting to recombine are highlighted in yellow. At each of these highlighted stages, MMR proteins will bind to the mismatches, providing the opportunity to promote anti-recombination or MMR.
Multiple subpathways act in double-strand break repair (DSBR). For detailed reviews, see Mehta and Haber (2014) and Symington, Rothstein and Lisby (2014). In the canonical DSBR pathway (Fig. 3A), initiation of strand synthesis at the invading strand end leads to branch migration, followed by second end capture, a process in which the displaced strand in the D-loop pairs with complementary sequences in the broken chromosome, and results in the formation of a double Holliday junction (dHJ). dHJ structures can be resolved by endonucleases into crossover (CO) or non-crossover (NCO) products. Resolution into COs is observed primarily in meiotic DSBR where a chromosome homolog serves as a repair template; in somatic DSBR, the sister chromatid primarily serves as a template, yielding NCO events.
A major alternative pathway, known as synthesis-dependent strand annealing (Fig. 3B), does not involve second end capture and promotes disassembly of the invasion intermediate after DNA extension off the first-strand invasion event. This extended strand then anneals back to the broken chromosome, after which DNA synthesis can occur to fill gaps prior to ligation. In the absence of the second end of the broken chromosome, a pathway known as break-induced replication can be utilized in which extensive DNA synthesis (conservative and through a migrating bubble) occurs following the initial strand invasion, often until the end of a chromosome.
A third HR pathway, known as single-strand annealing (SSA), can occur if a DSB forms between repetitive DNA sequences (Fig. 3C). In this case, once the repetitive sequences become single stranded following 5′ to 3′ strand resection, complementary sequences anneal to each other in a process facilitated by Rad52 and Rad59. This intrachromosomal repair mechanism does not require Rad51, and does not involve strand invasion. The annealing of the repetitive sequences generates 3′ non-homologous tails that arise from the resected DNA region between the repeats. The mismatch recognition heterodimer Msh2-Msh3 plays a critical role in SSA by stabilizing ssDNA-dsDNA junctions to permit Rad1-Rad10 mediated cleavage of the 3′ non-homologous tails. However, Msh2-Msh3′s role in tail removal is limited to SSA events in which the sequence repeats annealing to each other are relatively small (∼200 bp). SSA involving larger repeats (1 kbp) does not require Msh2-Msh3 for the 3′ tail clipping process, presumably because the longer homologous sequences that anneal to each other form more stable heteroduplex intermediates (Sugawara et al. 1997). Other types of recombination pathways may also require a 3′ tail removal step involving Msh2-Msh3 before DNA synthesis can be initiated. For example, the invading strand may have a non-homologous 3′ end that does not pair with a homologous DNA template, and this may need to be clipped before proceeding with subsequent DNA synthesis steps.
MMR proteins act in anti-recombination
During HR, it is critical that the broken chromosome recombines with a homologous template located in an allelic position in a donor chromosome. Recombination between divergent DNA sequences (also known as homeologous recombination) located in non-allelic positions can lead to gross chromosomal rearrangements that are often associated with diseases in humans such as cancer (for reviews, see George and Alani 2012; Liu et al. 2012; Zhang, Leibowitz and Pellman 2013). In this regard, the minimal efficient processing segment, defined as the smallest stretch of perfect homology needed for efficient recombination in vivo, becomes important in determining homology requirements to permit HR. This value is estimated to be ∼250 bp for yeast (Jinks-Robertson, Michelitch and Ramcharan 1993; Waldman 2008). MMR proteins play a crucial role in maintaining the fidelity of HR by preventing divergent sequences from recombining, a process known as anti-recombination, or heteroduplex rejection (Fig. 4). In this process, mismatches are created in heteroduplex DNA following DNA-strand exchange steps. These are then recognized by Msh2-Msh6 or Msh2-Msh3, depending on the type of mismatch formed, and the STR complex is then recruited to unwind the heteroduplex DNA joint molecule.
Figure 4.

Model for heteroduplex rejection during HR. Msh2-Msh6 or Msh2-Msh3 will locate a mismatch in a strand invasion intermediate involving divergent DNA sequences, and after ADP→ATP exchange, will enter a sliding clamp mode. This then results in the recruitment of Sgs1-Top3-Rmi1 at the ssDNA–dsDNA junction where it can load on to the DNA substrate and initiate unwinding of the heteroduplex DNA to enable a new homology search.
Several observations support the above model. (i) The absence of MSH proteins or Sgs1, a RecQ family helicase, causes a severe defect in heteroduplex rejection, as measured in SSA and recombination systems involving inverted repeats (Spell and Jinks-Robertson 2004; Sugawara et al. 2004; Goldfarb and Alani 2005). In such mutants, recombination between divergent sequences approaches the level seen between identical sequences. (ii) The helicase activity of Sgs1 is required for its role in heteroduplex rejection (Goldfarb and Alani 2005). (iii) Bailis, Arthur and Rothstein (1992) showed that top3 mutants exhibited hyperrecombination and genomic rearrangements, and Myung et al. (2001) showed that the absence of Top3 decreased the efficiency of heteroduplex rejection in a recombination system that involves inverted repeats. Lastly, temperature-sensitive mutations in Top3 and Rmi1, which together form a topoisomerase complex, confer defects in heteroduplex rejection during SSA, though the overall defects are weaker than conferred by sgs1 null mutations (Chakraborty et al. 2016). It is not clear if Top3 plays a catalytic role in unwinding the strands during heteroduplex rejection, or if it plays a structural role in stabilizing Sgs1.
The MMR components Mlh1-Pms1 and Exo1 play limited roles in promoting anti-recombination. A partial requirement for Mlh1-Pms1 was observed in promoting anti-recombination using an inverted repeat recombination reporter, suggesting a possible nucleolytic mechanism for suppressing homeologous recombination (Selva et al. 1995; Datta et al. 1996; Nicholson et al. 2000; Spell and Jinks-Robertson 2003, 2004). In SSA, Mlh1 was shown to play a minor role in preventing homeologous recombination, and Pms1 was shown to have no role (Sugawara et al. 2004). Exo1 was shown to play a minor role in heteroduplex rejection in the inverted repeat system, but no role in the SSA system (Nicholson et al. 2000; Sugawara et al. 2004). Thus, although MMR proteins play roles in heteroduplex rejection, how their activities are coordinated with other heteroduplex rejection proteins in different recombination systems is not well understood.
PATHWAY CHOICE FOR MMR PROTEINS: MMR VERSUS ANTI-RECOMBINATION
What factors influence the MMR pathway/anti-recombination decision, given that MSH complexes recognize mismatches in both pathways (Fig. 1)? This is a critical decision because implementing MMR instead of rejection during recombination would lead to gene conversion, loss of heterozygosity, and if involving non-allelic templates, genome rearrangements (Fig. 3). In contrast, the initiation of anti-recombination mechanisms at mismatches formed as the result of DNA replication errors would likely cause replication fork collapse and/or the formation of DNA lesions, resulting in chromosome loss or rearrangements. Thus, regulating this decision is critical to maintain genome stability. Factors that modulate pathway choice are shown in Fig. 1 and discussed below, and include (i) distinct protein components/interactions, (ii) structure of the DNA substrate, (iii) expression levels of MMR proteins, (iv) timely localization of proteins, (v) cell cycle phase, (vi) epigenetic modifications and chromatin conformation.
MSH complexes interact with different sets of proteins in MMR and heteroduplex rejection
Although the MSH protein heterodimers are involved in mismatch recognition during both MMR and heteroduplex rejection, they interact with different proteins in the two pathways. For example, (i) Mlh1-Pms1 is critical in MMR as demonstrated by mlh1 or pms1 mutants displaying mutation rates similar to msh2; however, Mlh1-Pms1 appears less important for heteroduplex rejection, and separation of function alleles of PMS1 have been identified that reduce heteroduplex rejection efficiency but have little or no effect on the repair of replication errors (Strand et al. 1993; Greene and Jinks-Robertson 1997; Chen and Jinks-Robertson 1999; Nicholson et al. 2000; Harfe and Jinks-Robertson 2000a, b; Welz-Voegele et al. 2002; Sugawara et al. 2004). (ii) PCNA is known to play roles in both early and late stages of MMR, and its interactions with MSH complexes are important for this process (Umar et al. 1996; Clark et al. 2000; Flores-Rozas, Clark and Kolodner 2000; Lau, Flores-Rozas and Kolodner 2002; Hombauer et al. 2011a,b). However, PCNA, which interacts with both MSH and MLH proteins, plays little or no role in anti-recombination (Stone et al. 2008; Chakraborty et al. 2016). This finding is consistent with heteroduplex rejection not needing to be coupled to DNA replication (Hombauer et al. 2011b). (iii) The STR complex, components of which interact with MSH and MLH proteins, plays an important role in heteroduplex rejection, but not in MMR (Langland et al. 2001; Myung et al. 2001; Pedrazzi et al. 2001, 2003; Spell and Jinks-Robertson 2004; Sugawara et al. 2004; Goldfarb and Alani 2005; Chakraborty et al. 2016).
The structures of DNA substrates in MMR and heteroduplex rejection appear different and likely contribute to regulating pathway choice
Although interactions of MSH-mismatch complexes with other proteins appear important for pathway choice, it is not clear how the commitment to specific sets of interactions is made. A favored hypothesis is that the structure of the DNA substrate to which the MSH proteins are bound influences the anti-recombination/MMR decision. Consistent with this hypothesis, removal of 3′ non-homologous tails during SSA is thought to act as a temporal switch between heteroduplex rejection and MMR modes, where rejection is favored before 3′ tail removal and MMR is favored following removal (Chakraborty et al. 2016).
In current MMR models, the MSH complex recognizes mismatches and following ADP →ATP exchange, enters a sliding clamp diffusion mode (Fig. 2; Acharya et al. 2003; Kunkel and Erie 2005; Jiricny 2006). Recent studies suggest that the MSH sliding clamp can respond to different signals that yield MMR or anti-recombination outcomes. For example, studies have suggested that during SSA the presence of the 3′ tail creates an ssDNA-dsDNA splayed junction which is recognized by MSH proteins and permits recruitment of the STR complex (George and Alani 2012; Chakraborty et al. 2016). Additional biochemical data indicate that Y-shaped DNA structures act as substrates for binding to and unwinding by Sgs1 (Cejka and Kowalczykowski 2010). Also, Saydam et al. (2007) showed that hMSH2-hMSH6 enhances the unwinding of heteroduplex DNA by the WRN helicase (human homolog of Sgs1), but only on fork-shaped duplexes. In contrast, a continuous or nicked duplex DNA containing mismatches may be more suitable for recruiting downstream MMR proteins. These data suggest that 3′ non-homologous tails create loading sites for the STR complex, setting up heteroduplex rejection prior to tail removal.
In vitro systems have been developed with the goal of understanding how MSH complexes play different roles during MMR and heteroduplex rejection. In these systems, MSH complexes display distinct biochemical properties when interacting with different DNA substrates (Spies and Fishel 2015). Honda et al. (2014) reconstituted the initial steps of heteroduplex rejection using human proteins and compared the biochemical properties of hMSH2-hMSH6 bound to DNA substrates that mimic those formed in MMR to those formed in heteroduplex rejection. They observed that the hMSH2-hMSH6 complex binds to mismatch containing D-loops (heteroduplex rejection substrate) and mismatch containing linear dsDNA (MMR substrates) with similar affinities. hMSH2-hMSH6 undergoes ADP→ATP exchange on both DNA substrates and forms sliding clamps. Despite these similarities, they observed that the hMSH2-hMSH6 sliding clamp is less stable on a mismatch containing D-loop than on a mismatch containing linear duplex DNA substrate, indicating that mismatch processing is likely to be different during MMR and heteroduplex rejection. Their data suggest that hMSH2-hMSH6 likely undergoes a conformational change upon encountering ssDNA-dsDNA branch point junctions found in structures such as D-loops, and that this may direct the recruitment of downstream rejection factors such as the STR complex (Fig. 4). This idea is consistent with 3′ non-homologous tails forming such a junction during SSA, thus providing a platform for recruitment of the STR complex by Msh2-Msh6 to promote heteroduplex rejection (Chakraborty et al. 2016). Further biochemical analyses will be needed to understand how different downstream proteins of MMR and heteroduplex rejection are selected, and whether the structure of the DNA substrate drives this decision.
A recent in vitro study with bacterial MMR proteins showed that MutS binds to secondary structures in the displaced strand of the D-loop, in addition to mismatches in heteroduplex dsDNA in the D-loop, to suppress extensive heteroduplex DNA extension and branch migration (Tham et al. 2013; Tham, Kanaar and Lebbink 2016). The authors propose that MutS and MutL binding to the D-loop likely prevent free rotation of the recombining DNA strands with respect to each other. They also provide evidence that MutS tetramerization plays a role in heteroduplex rejection, suggesting that MutS proteins binding to multiple DNA strands or mismatch sites are important for heteroduplex rejection. Similar studies have yet to be performed with the eukaryotic MSH and MLH proteins; however, the detection of such activities would provide evidence for conserved modes of action of MMR proteins during heteroduplex rejection that are distinct from their roles in DNA MMR.
Levels of MSH proteins are tightly regulated by post-translational mechanisms
MMR protein concentrations appear to be carefully monitored in the cell, and altering their levels may affect critical genome stability pathways such as MMR and heteroduplex rejection. In yeast, the number of protein molecules per vegetative haploid cell is ∼1300 for Msh2, 1600–5000 for Msh6, ∼740 for Msh3, ∼320 for Mlh1 and ∼520 for Pms1 (Ghaemmaghami et al. 2003; Kumar et al. 2011; Kunkel and Erie 2015). A reduction in the amounts of MMR proteins is associated with elevated mutation rates and cancers in higher eukaryotes (Li 2008). In Saccharomyces cerevisiae, mutants lacking Msh2, Msh6, Msh3, Mlh1 or Pms1 display deficiencies in both MMR and anti-recombination pathways (Nicholson et al. 2000; Sugawara et al. 2004). In contrast, amplification of the MSH3 gene in methotrexate resistant leukemia cells causes reduced MMR of base–base mismatches due to sequestration of Msh2 by Msh3, leading to a reduction in Msh2-Msh6 levels (Drummond et al. 1997; Marra et al. 1998). In addition, Msh6 overexpression causes an increase in heteroduplex rejection efficiency at the expense of Msh2-Msh3 function during SSA. However, Msh6 overexpression disrupts heteroduplex rejection in a system where recombination is thought to occur during the replication phase of the cell cycle (Chakraborty et al. 2016).
Expression patterns of MMR proteins have been found to vary widely in a tissue-specific manner in the mouse, with higher levels of MMR proteins being present in proliferative tissues (Tomé et al. 2013). The above observations suggest that expression levels of MMR proteins and the relative concentrations of Msh3 and Msh6 are regulated in order to maintain various aspects of genome integrity.
In support of this idea, post-translational modifications have been shown to alter expression levels of MMR proteins. For example, a subset of microsatellite unstable (MIN+) sporadic colorectal tumors do not express hMLH1, but lack mutations in the hMLH1 coding region. In these cells, hypermethylation of the hMLH1 promoter causes epigenetic silencing and downregulation of hMLH1 (Kane et al. 1997; Grady and Markowitz 2002; Li 2008).
Studies have shed light on how levels of some MMR proteins are regulated in the cell. MSH2 concentrations in humans are regulated by ubiquitination and deubiquitination, which have opposite effects on MSH2 stability. HDAC6 is a histone deacetylase protein that encodes two deacetylase domains and a zinc finger containing domain that binds to monoubiquitin or polyubiquitin, as well as ubiquitinated proteins (Seigneurin-Berny et al. 2001; Hook et al. 2002; Boyault et al. 2006). Zhang et al. (2014) showed that HDAC6 interacts with MSH2 and regulates its turnover via sequential deacetylation and ubiquitination, ultimately leading to its degradation. HDAC6 confers cellular resistance to DNA-damaging agents such as 6-thioguanine (6-TG) and N-methyl-N'-nitro-N-nitrosoguanidine (MNNG), and reduces cellular DNA MMR activities by downregulating MSH2. On the other hand, ubiquitin-specific peptidase 10 (USP10) interacts with and stabilizes MSH2 by deubiquitinating it, thus essentially counteracting the effect of HDAC6 on MSH2 levels (Zhang et al. 2016). Depletion of USP10 decreases cellular sensitivity to MNNG and 6-TG and decreases DNA MMR activities.
In addition, protein kinase C (PKC) regulates expression levels of human MMR proteins such as MSH2, MSH6 and PMS2. Higher levels of PKC activation resulted in higher expression of MMR proteins (Humbert et al. 2002). Similarly, inhibition of PKC is associated with lower levels of MMR protein expression and leads to a decrease in MMR function. Furthermore, levels of PKCzeta, an atypical isoform of PKC, positively correlate with hMSH2-hMSH6 levels and MMR activity. PKCzeta stabilizes hMSH2-hMSH6 by suppressing its degradation by the ubiquitin-proteosome pathway (Hernandez-Pigeon et al. 2005).
Timely localization of MMR or rejection factors is likely to regulate pathway choice
Both MMR and anti-recombination pathways are initiated by the recognition of mismatches by MSH complexes. It is possible that other proteins involved in the two pathways are present on or near the substrate prior to the mismatch recognition step and play roles in biasing pathway choice. For example, the STR complex acts in 5′ to 3′ strand resection after the formation of a DSB, and Sgs1 interacts with Msh2-Msh6 (Pedrazzi et al. 2003; Chakraborty et al. 2016). Thus, it is possible that Sgs1 plays a role in recruiting Msh2-Msh6 to the site of HR. If recombination is occurring between divergent sequences, this could bias the pathway choice towards rejection rather than MMR. For example, rapid recruitment of the MSH complex to the site of recombination between divergent sequences could ensure that the structure of the DNA substrate remains suitable for heteroduplex rejection and has not yet been extensively processed by downstream recombination steps. In the SSA pathway, rapid recruitment of the MSH complexes by STR would ensure MSH presence prior to the clipping of the 3′ tails to recruit downstream rejection factors. Furthermore, the STR complex, being physically present at the recombination site prior to the formation of mismatches by strand invasion, may have a temporal advantage over downstream MMR proteins such as Mlh1-Pms1 in interacting with the MSH complex.
Msh2-Msh6 is coupled to the moving replication fork via its interaction with PCNA (Kleczkowska et al. 2001; Hombauer et al. 2011a; Haye and Gammie 2015). Through this interaction PCNA may play a role in biasing mismatch recognition to the MMR pathway. For example, PCNA activates the endonuclease function of Mlh1-Pms1 during MMR (Kadyrov et al. 2006, 2007; Pluciennik et al. 2010; Kunkel and Erie 2015). In addition, trimethylation of lysine 36 in histone 3 of the histone octamer (H3K36me3) recruits human hMSH2-hMSH6 to chromatin prior to or during the early S-phase of the cell cycle, before replication initiates, to bring MMR factors in close proximity to newly replicated DNA (Li et al. 2013). Such early recruiters of the MSH proteins may also play roles in determining pathway choice post-mismatch recognition. For example, such early recruitment of MSH proteins may not exist or may be carried out by different proteins/histone marks during HR. Although there is no direct evidence, we speculate that such early recruiters of MSH complexes may regulate the recruitment of downstream interactors of MSH proteins by (i) physically mediating an interaction with downstream factors; (ii) causing distinct conformational/post-translational modifications in the MSH complex that determine downstream interactions; and (iii) making sure that the timing of MSH complex recruitment is coordinated with the appearance of specific DNA substrate structures, which in turn dictates which downstream factors are recruited.
Cell cycle phase as a possible regulator of pathway choice
Hombauer et al. (2011b) showed that MMR, but not heteroduplex rejection, is temporally coupled to DNA replication. This observation suggests that cell cycle specific features (e.g. specific protein expression patterns and localization, as well as histone marks) play a role in the decision to reject or repair a recombination substrate. In support of this idea, moderate overexpression of Msh6 had very different effects on heteroduplex rejection in two recombination systems thought to occur in different phases of the cell cycle (Chen and Jinks-Robertson 1998; Mehta and Haber 2014; Chakraborty et al. 2016). Msh6 overexpression significantly improved heteroduplex rejection in an SSA repair event that can occur at any point in the cell cycle. Msh6 overexpression, however, severely impaired heteroduplex rejection in recombination events involving inverted repeats. These recombination events are thought to occur via gene conversion during the replication phase of the cell cycle (Chen and Jinks-Robertson 1998; Chakraborty et al. 2016). In the latter scenario, the impairment of rejection was dependent upon Msh6 interacting with PCNA, which had no effect on anti-recombination in the SSA assay. The above data indicate that heteroduplex rejection may be regulated differently, depending on when the initiating event occurs in the cell cycle.
To explain the impairment of heteroduplex rejection as a result of Msh6 overexpression in the inverted repeats recombination system, Chakraborty et al. (2016) suggested that interactions between PCNA and excess Msh6 at replication centers sequestered anti-recombination factors, preventing them from acting in heteroduplex rejection. This idea was tested by overexpressing a mutant msh6 protein displaying a PCNA interaction defect. Consistent with the above hypothesis, overexpression of the mutant protein restored and in fact improved heteroduplex rejection. We hypothesize that MSH proteins are present in two pools: one coupled to replication sites (see Hombauer et al. 2011a; Li et al. 2013; Haye and Gammie 2015) and a second soluble pool available for recombination and heteroduplex rejection. In this framework, levels of MSH proteins are likely to affect their availability and localization as well as their distribution between the two pools (Fig. 5A–C). It would be interesting to explore if these pools differ in ways other than localization, such as post-translational modifications or binding to distinct factors.
Figure 5.

Models for regulating the heteroduplex rejection/MMR decision through epigenetic mechanisms and chromatin conformation during homeologous recombination. In this cartoon, histone marks act to recruit MSH complexes to chromatin, prior to DSB formation (A), which in conjunction with post-translational modifications facilitate localization of the MSH complexes to DSB sites immediately after they are formed (B). This rapid localization of MSH proteins to the DSB site may be essential to recruit other factors and promote anti-recombination, since subsequent DNA structures that form may not be favorable for anti-recombination. In this model, the cell is primed to deal with homeologous recombination events. MSH protein levels are tightly regulated in cells by various epigenetic mechanisms, affecting their availability and localization in (A), (B) and (C). This can regulate the efficiency of anti-recombination and modulate the anti-recombination/MMR decision. Factors promoting nucleosome assembly such as CAF1 interact with PCNA and deposit nucleosomes in a DNA synthesis coupled manner (D). Deposition of nucleosomes inhibits Msh2-Msh6 and stabilizes the D-loop, thus preventing anti-recombination. Thus, nucleosome deposition may limit the time frame in which anti-recombination can be performed, playing a role in the anti-recombination/MMR decision (D). If anti-recombination fails to occur, these substrates are subject to MMR.
Several studies in yeast and mammalian cells have reported that the levels of some MMR proteins and Sgs1 change during the cell cycle (Marra et al. 1996; Cho et al. 1998; Frei and Gasser 2000; Klingler et al. 2002; Iwanaga, Komori and Ohtani 2004; Schroering et al. 2007; Tennen et al. 2013), further supporting the idea that the cell cycle may play a role in indirectly regulating heteroduplex rejection and pathway choice by affecting levels or localization of MMR and rejection proteins.
Impact of epigenetic modifications and chromatin conformation on the function of MMR proteins and pathway choice
The role of histone marks and post-translational modifications in DNA repair processes and genome stability is receiving greater attention. Chromatin structure influences MMR and mutation rates (Hawk et al. 2005; Schuster-Bockler and Lehner 2012; Li et al. 2016). Advances have been made in understanding the role of epigenetic factors such as chromatin structure and post-translational modifications in the regulation of specific MMR proteins and the pathways they are involved in. However, roles for chromatin marks and epigenetic factors in regulating the decision to repair versus reject mismatches during homeologous recombination remain unknown. Recent findings regarding epigenetic regulation of MMR proteins are discussed in this section, along with speculations on their possible roles in influencing the MMR/anti-recombination decision (Fig. 5).
Early association of MMR proteins with chromatin prior to mismatch formation
Li et al. (2013) showed that the histone mark H3K36me3 is required in mammalian cells to recruit hMSH2-hMSH6 by directly interacting with a PWWP motif in hMSH6. This recruitment of hMSH2-hMSH6 to chromatin occurs right before or during early S-phase, ensuring that mismatch recognition proteins are localized to chromatin before mismatches arise during DNA replication. In the absence of the H3K36 trimethyltransferase SETD2, hMSH2-hMSH6 is compromised for recruitment to chromatin, resulting in microsatellite instability and elevated spontaneous mutation frequency phenotypes, also seen in MMR-deficient cells. Although yeast Msh6 and human MSH3 proteins do not have PWWP motifs, it is possible, as the authors suggest, that these proteins are recruited by other histone marks to chromatin. It is not known whether similar mechanisms exist for recruiting MMR proteins to act in anti-recombination. It is tantalizing to imagine the existence of such a mechanism because it would ensure the early presence of MMR proteins at the site of homeologous recombination to maintain recombination fidelity. Such a possibility may involve recruitment of MMR proteins to chromatin prior to DSB formation (Fig. 5A), which may then facilitate immediate localization of the MSH proteins to a DSB site once a break arises (Fig. 5B). This could be achieved by specific histone marks recruiting MMR proteins to chromatin prior to the replication phase in which DSBs form as the result of damage to DNA occurring during DNA replication. This would be analogous to what Li et al. (2013) found, where hMSH2-hMSH6 is recruited to chromatin right before or during early S-phase, in order to be prepared to initiate MMR even before mismatches are formed due to replication errors. Alternately, in order to be prepared for DSBs that arise from exogenous sources at any phase in the cell cycle, a fraction of MMR proteins may be associated with chromatin at all times.
MSH proteins have been shown to rapidly localize to DSBs in yeast and mammals, and play a role in the DNA damage response (Evans et al. 2000; Hong et al. 2008; Burdova et al. 2015). MSH proteins recruited in this fashion may undergo post-translational modifications, such as phosphorylation, which could mark them for anti-recombination functions, possibly by dictating interactions with specific partner proteins. In support of such a possibility, hMSH6 has been shown to be phosphorylated following gamma irradiation (Matsuoka et al. 2007). In this manner, association of MSH proteins to chromatin and their early recruitment to damage sites may influence the anti-recombination/MMR choice by regulating the timely localization of these proteins to the break sites, as well as potentially ‘marking’ them for one pathway versus the other. Further studies are needed to explore these possibilities.
Acetylation/deacetylation of H3K56 is important for genomic stability
In S. cerevisiae, deacetylation and acetylation on histone H3K56 regulates mutation avoidance mechanisms that cooperate with MMR and proofreading activities of replicative DNA polymerases (Kadyrova et al. 2013). Simultaneous loss of the H3K56 deacetylases Hst3 and Hst4 causes spontaneous gross chromosomal rearrangements, base substitutions, 1-bp insertions/deletions and complex mutations. In addition, Kadyrova et al. (2013) suggested that the formation of the majority of gross chromosomal rearrangements in hst3Δhst4Δ strains involves both Msh2-Msh6 and Msh2-Msh3, suggesting that Hst3 and Hst4 are required to suppress inappropriate MMR functions. As the authors discuss, MMR has been previously implicated in promoting deleterious and pathogenic outcomes such as destabilizing DNA triplet repeats. It is possible that a compromised chromatin environment due to the absence of the H3K56 deacetylases causes MMR proteins to act in a deleterious way, and/or that these chromatin modifications impact the role of MMR proteins in anti-recombination.
Kinetics of nucleosome reassembly regulate MSH protein functions
A number of studies have shown that MMR and nucleosome assembly on newly replicated DNA during replication are mutually inhibitory processes (Li et al. 2009; Kadyrova, Blanko and Kadyrov 2011; Schöpf et al. 2012; Blanko, Kadyrova and Kadyrov 2016). Nucleosome assembly on newly replicated DNA involves the loading of the (H3-H4)2 tetramer onto DNA, followed by the addition of two H2A–H2B dimers to the (H3-H4)2 tetramer (Loyola and Almouzni 2004). Histone H3-H4 chaperones such as chromatin assembly factor 1 (CAF-1) and anti-silencing function 1(Asf1) act in a concerted manner to facilitate nucleosome assembly onto newly replicated DNA. CAF-1 is known to interact with the replication fork via PCNA (Shibahara and Stillman 1999) and promotes replication coupled nucleosome deposition. In addition, the acetylation of H3K56 by Rtt109 is important for effective association of the (H3-H4)2 tetramer with CAF-1, and CAF-1 with PCNA (Li et al. 2008). MMR during replication must occur in the short time frame between the formation of the mismatch and the assembly of nucleosomes in the newly replicated DNA (Li et al. 2009; Blanko, Kadyrova and Kadyrov 2016). Recent in vitro studies with human proteins show that hMSH2-hMSH6 interacts with CAF-1 (Schöpf et al. 2012). In addition, hMSH2-hMSH6 inhibits CAF-1 and ASF1A-dependent packaging of a DNA mismatch into a nucleosome, and deposition of the (H3-H4)2 tetramers on DNA protects the discontinuous daughter strand from unnecessary degradation during MMR (Blanko, Kadyrova and Kadyrov 2016).
Chromatin assembly has been shown to be coupled with DNA repair and is known to stabilize D-loops during HR. CAF-1 mediated nucleosome assembly during DNA repair, like DNA replication, is coupled with DNA synthesis and requires an interaction with PCNA (Gaillard et al. 1996; Tyler et al. 1999; Linger and Tyler 2005; Polo, Roche and Almouzni 2006; Pietrobon et al. 2014). An intriguing possibility is that the dynamics of nucleosome assembly play a role in regulating MMR protein functions during heteroduplex rejection, in a manner analogous to their crosstalk during MMR. For example MSH complexes may have to initiate heteroduplex rejection during HR prior to CAF-1 mediated nucleosome assembly, the latter being coupled to the DNA synthesis step. Thus, MMR proteins and nucleosome assembly factors may inhibit and compete with each other in a regulated manner to maintain a proper balance between heteroduplex rejection and repair of a DSB (Fig. 5D). Nucleosome assembly may act as a temporal commitment step between rejection and repair requiring MMR components to reach the recombination substrate rapidly in order to perform anti-recombination. This would in turn affect the anti-recombination/MMR balance; by regulating the time available for heteroduplex rejection, nucleosome assembly may partially dictate the fraction of homeologous recombination events that will be successfully rejected, with the remaining events subject to MMR.
CONCLUSIONS
MMR proteins play critical roles in MMR and in regulating recombination fidelity in prokaryotes and eukaryotes. Mechanisms ensuring the appropriate choice between these distinct functions are only beginning to be understood. Deregulating these pathways will likely have deleterious effects on genome stability; thus, it is critical to understand what factors influence the decision to implement anti-recombination versus MMR. In addition, epigenetic control mechanisms of MMR proteins are starting to be understood. Further studies are needed to decode the crosstalk between genetic and epigenetic mechanisms to fully understand how misregulation of MMR proteins can lead to genome instability and cancer.
Acknowledgments
We are grateful to the Alani lab for fruitful discussions.
FUNDING
UC and EA were supported by NIH GM53085. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
Conflict of interest. None declared.
REFERENCES
- Acharya S, Foster PL, Brooks P, et al. The coordinated functions of the E. coli MutS and MutL proteins in mismatch repair. Mol Cell. 2003;12:233–46. doi: 10.1016/s1097-2765(03)00219-3. [DOI] [PubMed] [Google Scholar]
- Bailis AM, Arthur L, Rothstein R. Genome rearrangement in top3 mutants of Saccharomyces cerevisiae requires a functional RAD1 excision repair gene. Mol Cell Biol. 1992;12:4988–93. doi: 10.1128/mcb.12.11.4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanko ER, Kadyrova LY, Kadyrov FA. DNA mismatch repair interacts with CAF-1- and ASF1A-H3-H4-dependent histone (H3-H4)2 tetramer deposition. J Biol Chem. 2016;291:9203–17. doi: 10.1074/jbc.M115.713271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyault C, Gilquin B, Zhang Y, et al. HDAC6-p97/VCP controlled polyubiquitin chain turnover. EMBO J. 2006;25:3357–66. doi: 10.1038/sj.emboj.7601210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdova K, Mihaljevic B, Sturzenegger A, et al. The Mismatch-binding factor MutSbeta can mediate ATR Activation in response to DNA double-strand breaks. Mol Cell. 2015;59:603–14. doi: 10.1016/j.molcel.2015.06.026. [DOI] [PubMed] [Google Scholar]
- Cannavo E, Cejka P. Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature. 2014;514:122–5. doi: 10.1038/nature13771. [DOI] [PubMed] [Google Scholar]
- Cejka P, Cannavo E, Polaczek P, et al. DNA end resection by Dna2–Sgs1–RPA and its stimulation by Top3–Rmi1 and Mre11–Rad50–Xrs2. Nature. 2010;467:112–6. doi: 10.1038/nature09355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejka P, Kowalczykowski SC. The full-length Saccharomyces cerevisiae Sgs1 protein is a vigorous DNA helicase that preferentially unwinds holliday junctions. J Biol Chem. 2010;285:8290–301. doi: 10.1074/jbc.M109.083196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty U, George CM, Lyndaker AM, et al. A delicate balance between repair and replication factors regulates recombination between divergent DNA sequences in Saccharomyces cerevisiae. Genetics. 2016;202:525–40. doi: 10.1534/genetics.115.184093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Lisby M, Symington LS. RPA coordinates DNA end resection and prevents formation of DNA hairpins. Mol Cell. 2013;50:589–600. doi: 10.1016/j.molcel.2013.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Jinks-Robertson S. Mismatch repair proteins regulate heteroduplex formation during mitotic recombination in yeast. Mol Cell Biol. 1998;18:6525–37. doi: 10.1128/mcb.18.11.6525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Jinks-Robertson S. The role of the mismatch repair machinery in regulating mitotic and meiotic recombination between diverged sequences in yeast. Genetics. 1999;151:1299–313. doi: 10.1093/genetics/151.4.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho RJ, Campbell MJ, Winzeler EA, et al. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol Cell. 1998;2:65–73. doi: 10.1016/s1097-2765(00)80114-8. [DOI] [PubMed] [Google Scholar]
- Clark AB, Valle F, Drotschmann K, et al. Functional interaction of proliferating cell nuclear antigen with MSH2-MSH6 and MSH2-MSH3 complexes. J Biol Chem. 2000;275:36498–501. doi: 10.1074/jbc.C000513200. [DOI] [PubMed] [Google Scholar]
- Datta A, Adjiri A, New L, et al. Mitotic crossovers between diverged sequences are regulated by mismatch repair proteins in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:1085–93. doi: 10.1128/mcb.16.3.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng SK, Chen H, Symington LS. Replication protein A prevents promiscuous annealing between short sequence homologies: implications for genome integrity. Bioessays. 2015;37:305–13. doi: 10.1002/bies.201400161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drotschmann K, Clark AB, Tran HT, et al. Mutator phenotypes of yeast strains heterozygous for mutations in the MSH2 gene. P Natl Acad Sci USA. 1999;96:2970–5. doi: 10.1073/pnas.96.6.2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond JT, Genschel J, Wolf E, et al. DHFR/MSH3 amplification in methotrexate-resistant cells alters the hMutSalpha/hMutSbeta ratio and reduces the efficiency of base-base mismatch repair. P Natl Acad Sci USA. 1997;94:10144–9. doi: 10.1073/pnas.94.19.10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley MC, Crouse GF. The role of mismatch repair in the prevention of base pair mutations in Saccharomyces cerevisiae. P Natl Acad Sci USA. 1998;95:15487–91. doi: 10.1073/pnas.95.26.15487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans E, Sugawara N, Haber JE, et al. The Saccharomyces cerevisiae Msh2 mismatch repair protein localizes to recombination intermediates in vivo. Mol Cell. 2000;5:789–99. doi: 10.1016/s1097-2765(00)80319-6. [DOI] [PubMed] [Google Scholar]
- Flores-Rozas H, Clark D, Kolodner RD. Proliferating cell nuclear antigen and Msh2p-Msh6p interact to form an active mispair recognition complex. Nat Genet. 2000;26:375–8. doi: 10.1038/81708. [DOI] [PubMed] [Google Scholar]
- Frei C, Gasser SM. The yeast Sgs1p helicase acts upstream of Rad53p in the DNA replication checkpoint and colocalizes with Rad53p in S-phase-specific foci. Genes Dev. 2000;14:81–96. [PMC free article] [PubMed] [Google Scholar]
- Gaillard PH, Martini EM, Kaufman PD, et al. Chromatin assembly coupled to DNA repair: a new role for chromatin assembly factor I. Cell. 1996;86:887–96. doi: 10.1016/s0092-8674(00)80164-6. [DOI] [PubMed] [Google Scholar]
- George CM, Alani E. Multiple cellular mechanisms prevent chromosomal rearrangements involving repetitive DNA. Crit Rev Biochem Mol. 2012;47:297–313. doi: 10.3109/10409238.2012.675644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh W-K, Bower K, et al. Global analysis of protein expression in yeast. Nature. 2003;425:737–41. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- Goldfarb T, Alani E. Distinct roles for the Saccharomyces cerevisiae mismatch repair proteins in heteroduplex rejection, mismatch repair and nonhomologous tail removal. Genetics. 2005;169:563–74. doi: 10.1534/genetics.104.035204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradia S, Acharya S, Fishel R. The role of mismatched nucleotides in activating the hMSH2-hMSH6 molecular switch. J Biol Chem. 2000;275:3922–30. doi: 10.1074/jbc.275.6.3922. [DOI] [PubMed] [Google Scholar]
- Gradia S, Subramanian D, Wilson T, et al. hMSH2-hMSH6 forms a hydrolysis-independent sliding clamp on mismatched DNA. Mol Cell. 1999;3:255–61. doi: 10.1016/s1097-2765(00)80316-0. [DOI] [PubMed] [Google Scholar]
- Grady WM, Markowitz SD. Genetic and epigenetic alterations in colon cancer. Annu Rev Genom Hum G. 2002;3:101–28. doi: 10.1146/annurev.genom.3.022502.103043. [DOI] [PubMed] [Google Scholar]
- Greene CN, Jinks-Robertson S. Frameshift intermediates in homopolymer runs are removed efficiently by yeast mismatch repair proteins. Mol Cell Biol. 1997;17:2844–50. doi: 10.1128/mcb.17.5.2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harfe BD, Jinks-Robertson S. DNA mismatch repair and genetic instability. Annu Rev Genet. 2000a;34:359–99. doi: 10.1146/annurev.genet.34.1.359. [DOI] [PubMed] [Google Scholar]
- Harfe BD, Jinks-Robertson S. Sequence composition and context effects on the generation and repair of frameshift intermediates in mononucleotide runs in Saccharomyces cerevisiae. Genetics. 2000b;156:571–8. doi: 10.1093/genetics/156.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawk JD, Stefanovic L, Boyer JC, et al. Variation in efficiency of DNA mismatch repair at different sites in the yeast genome. P Natl Acad Sci USA. 2005;102:8639–43. doi: 10.1073/pnas.0503415102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haye JE, Gammie AE. The eukaryotic mismatch recognition complexes track with the replisome during DNA synthesis. PLoS Genet. 2015;11:e1005719. doi: 10.1371/journal.pgen.1005719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Pigeon H, Quillet-Mary A, Louat T, et al. hMutS alpha is protected from ubiquitin-proteasome-dependent degradation by atypical protein kinase C zeta phosphorylation. J Mol Biol. 2005;348:63–74. doi: 10.1016/j.jmb.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Hombauer H, Campbell CS, Smith CE, et al. Visualization of eukaryotic DNA mismatch repair reveals distinct recognition and repair intermediates. Cell. 2011a;147:1040–53. doi: 10.1016/j.cell.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hombauer H, Srivatsan A, Putnam CD, et al. Mismatch repair, but not heteroduplex rejection, is temporally coupled to DNA replication. Science. 2011b;334:1713–6. doi: 10.1126/science.1210770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda M, Okuno Y, Hengel SR, et al. Mismatch repair protein hMSH2-hMSH6 recognizes mismatches and forms sliding clamps within a D-loop recombination intermediate. P Natl Acad Sci USA. 2014;111:E316–25. doi: 10.1073/pnas.1312988111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Z, Jiang J, Hashiguchi K, et al. Recruitment of mismatch repair proteins to the site of DNA damage in human cells. J Cell Sci. 2008;121:3146–54. doi: 10.1242/jcs.026393. [DOI] [PubMed] [Google Scholar]
- Hook SS, Orian A, Cowley SM, et al. Histone deacetylase 6 binds polyubiquitin through its zinc finger (PAZ domain) and copurifies with deubiquitinating enzymes. P Natl Acad Sci USA. 2002;99:13425–30. doi: 10.1073/pnas.172511699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert O, Hermine T, Hernandez H, et al. Implication of protein kinase C in the regulation of DNA mismatch repair protein expression and function. J Biol Chem. 2002;277:18061–8. doi: 10.1074/jbc.M103451200. [DOI] [PubMed] [Google Scholar]
- Iwanaga R, Komori H, Ohtani K. Differential regulation of expression of the mammalian DNA repair genes by growth stimulation. Oncogene. 2004;23:8581–90. doi: 10.1038/sj.onc.1207976. [DOI] [PubMed] [Google Scholar]
- Iyer RR, Pluciennik A, Burdett V, et al. DNA mismatch repair: functions and mechanisms. Chem Rev. 2006;106:302–23. doi: 10.1021/cr0404794. [DOI] [PubMed] [Google Scholar]
- Jensen LE, Jauert PA, Kirkpatrick DT. The large loop repair and mismatch repair pathways of Saccharomyces cerevisiae act on distinct substrates during meiosis. Genetics. 2005;170:1033–43. doi: 10.1534/genetics.104.033670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinks-Robertson S, Michelitch M, Ramcharan S. Substrate length requirements for efficient mitotic recombination in Saccharomyces cerevisiae. Mol Cell Biol. 1993;13:3937–50. doi: 10.1128/mcb.13.7.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol. 2006;7:335–46. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- Kadyrov FA, Dzantiev L, Constantin N, et al. Endonucleolytic function of MutLalpha in human mismatch repair. Cell. 2006;126:297–308. doi: 10.1016/j.cell.2006.05.039. [DOI] [PubMed] [Google Scholar]
- Kadyrov FA, Genschel J, Fang Y, et al. A possible mechanism for Exonuclease 1-independent eukaryotic mismatch repair. P Natl Acad Sci USA. 2009;106:8495–500. doi: 10.1073/pnas.0903654106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadyrov FA, Holmes SF, Arana ME, et al. Saccharomyces cerevisiae MutLα is mismatch repair endonuclease. J Biol Chem. 2007;282:37181–90. doi: 10.1074/jbc.M707617200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadyrova LY, Blanko ER, Kadyrov FA. CAF-I-dependent control of degradation of the discontinuous strands during mismatch repair. P Natl Acad Sci USA. 2011;108:2753–8. doi: 10.1073/pnas.1015914108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadyrova LY, Mertz TM, Zhang Y, et al. A reversible histone H3 acetylation cooperates with mismatch repair and replicative polymerases in maintaining genome stability. PLoS Genet. 2013;9:e1003899. doi: 10.1371/journal.pgen.1003899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane MF, Loda M, Gaida GM, et al. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57:808–11. [PubMed] [Google Scholar]
- Kleczkowska HE, Marra G, Lettieri T, et al. hMSH3 and hMSH6 interact with PCNA and colocalize with it to replication foci. Genes Dev. 2001;15:724–36. doi: 10.1101/gad.191201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingler H, Hemmerle C, Bannwart F, et al. Expression of the hMSH6 mismatch-repair protein in colon cancer and Hela cells. Swiss Med Wkly. 2002;132:57–63. doi: 10.4414/smw.2002.09855. [DOI] [PubMed] [Google Scholar]
- Kolodner RD, Marsischky GT. Eukaryotic DNA mismatch repair. Curr Opin Genet Dev. 1999;9:89–96. doi: 10.1016/s0959-437x(99)80013-6. [DOI] [PubMed] [Google Scholar]
- Kumar C, Piacente SC, Sibert J, et al. Multiple factors insulate Msh2– Msh6 mismatch repair activity from defects in Msh2 domain I. J Mol Biol. 2011;411:765–80. doi: 10.1016/j.jmb.2011.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- Kunkel TA, Erie DA. Eukaryotic mismatch repair in relation to DNA replication. Ann Rev Genet. 2015;49:291–313. doi: 10.1146/annurev-genet-112414-054722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langland G, Kordich J, Creaney J, et al. The BLM helicase interacts with hMLH1 but is not required for DNA mismatch repair. J Biol Chem. 2001;276:30031–5. doi: 10.1074/jbc.M009664200. [DOI] [PubMed] [Google Scholar]
- Lau PJ, Flores-Rozas H, Kolodner RD. Isolation and characterization of new proliferating cell nuclear antigen (POL30) mutator mutants that are defective in DNA mismatch repair. Mol Cell Biol. 2002;22:6669–80. doi: 10.1128/MCB.22.19.6669-6680.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Mao G, Tong D, et al. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSα. Cell. 2013;153:590–600. doi: 10.1016/j.cell.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Ortega J, Gu L, et al. Regulation of mismatch repair by histone code and posttranslational modifications in eukaryotic cells. DNA Repair. 2016;38:68–74. doi: 10.1016/j.dnarep.2015.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Tian L, Gu L, et al. Evidence that nucleosomes inhibit mismatch repair in eukaryotic cells. J Biol Chem. 2009;284:33056–61. doi: 10.1074/jbc.M109.049874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18:85–98. doi: 10.1038/cr.2007.115. [DOI] [PubMed] [Google Scholar]
- Li Q, Zhou H, Wurtele H, et al. Acetylation of histone H3 lysine 56 regulates replication-coupled nucleosome assembly. Cell. 2008;134:244–55. doi: 10.1016/j.cell.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linger J, Tyler JK. The yeast histone chaperone chromatin assembly factor 1 protects against double-strand DNA-damaging agents. Genetics. 2005;171:1513–22. doi: 10.1534/genetics.105.043000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Carvalho CM, Hastings PJ, et al. Mechanisms for recurrent and complex human genomic rearrangements. Curr Opin Genet Dev. 2012;22:211–20. doi: 10.1016/j.gde.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loyola A, Almouzni G. Histone chaperones, a supporting role in the limelight. Biochim Biophys Acta. 2004;1677:3–11. doi: 10.1016/j.bbaexp.2003.09.012. [DOI] [PubMed] [Google Scholar]
- Manhart CM, Alani E. Roles for mismatch repair family proteins in promoting meiotic crossing over. DNA Repair. 2016;38:84–93. doi: 10.1016/j.dnarep.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra G, Chang CL, Laghi LA, et al. Expression of human MutS homolog 2 (hMSH2) protein in resting and proliferating cells. Oncogene. 1996;13:2189–96. [PubMed] [Google Scholar]
- Marra G, Iaccarino I, Lettieri T, et al. Mismatch repair deficiency associated with overexpression of the MSH3 gene. P Natl Acad Sci USA. 1998;95:8568–73. doi: 10.1073/pnas.95.15.8568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S, Ballif BA, Smogorzewska A, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–4. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- Mazurek A, Berardini M, Fishel R. Activation of human MutS homologs by 8-oxo-guanine DNA damage. J Biol Chem. 2002;277:8260–6. doi: 10.1074/jbc.M111269200. [DOI] [PubMed] [Google Scholar]
- Mehta A, Haber JE. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb Perspect Biol. 2014;6:a016428. doi: 10.1101/cshperspect.a016428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrich P. Mechanisms in eukaryotic mismatch repair. J Biol Chem. 2006;281:30305–9. doi: 10.1074/jbc.R600022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung K, Datta A, Chen C, et al. SGS1, the Saccharomyces cerevisiae homologue of BLM and WRN, suppresses genome instability and homeologous recombination. Nat Genet. 2001;27:113–6. doi: 10.1038/83673. [DOI] [PubMed] [Google Scholar]
- Nicholson A, Hendrix M, Jinks-Robertson S, et al. Regulation of mitotic homeologous recombination in yeast: functions of mismatch repair and nucleotide excision repair genes. Genetics. 2000;154:133–46. doi: 10.1093/genetics/154.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu H, Chung WH, Zhu Z, et al. Mechanism of the ATP-dependent DNA end-resection machinery from Saccharomyces cerevisiae. Nature. 2010;467:108–11. doi: 10.1038/nature09318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrazzi G, Bachrati CZ, Selak N, et al. The Bloom's syndrome helicase interacts directly with the human DNA mismatch repair protein hMSH6. Biol Chem. 2003;384:1155–64. doi: 10.1515/BC.2003.128. [DOI] [PubMed] [Google Scholar]
- Pedrazzi G, Perrera C, Blaser H, et al. Direct association of Bloom's syndrome gene product with the human mismatch repair protein MLH1. Nucleic Acids Res. 2001;29:4378–86. doi: 10.1093/nar/29.21.4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrobon V, Fréon K, Hardy J, et al. The chromatin assembly factor 1 promotes Rad51-dependent template switches at replication forks by counteracting D-loop disassembly by the RecQ-type Helicase Rqh1. PLoS Biol. 2014;12:e1001968. doi: 10.1371/journal.pbio.1001968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluciennik A, Dzantiev L, Iyer RR, et al. PCNA function in the activation and strand direction of MutLα endonuclease in mismatch repair. P Natl Acad Sci USA. 2010;107:16066–71. doi: 10.1073/pnas.1010662107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo SE, Roche D, Almouzni G. New histone incorporation marks sites of UV repair in human cells. Cell. 2006;127:481–93. doi: 10.1016/j.cell.2006.08.049. [DOI] [PubMed] [Google Scholar]
- Saydam N, Kanagaraj R, Dietschy T, et al. Physical and functional interactions between Werner syndrome helicase and mismatch-repair initiation factors. Nucleic Acids Res. 2007;35:5706–16. doi: 10.1093/nar/gkm500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt MH, Pearson CE. Disease-associated repeat instability and mismatch repair. DNA Repair. 2016;38:117–26. doi: 10.1016/j.dnarep.2015.11.008. [DOI] [PubMed] [Google Scholar]
- Schöpf B, Bregenhorn S, Quivy J-P, et al. Interplay between mismatch repair and chromatin assembly. P Natl Acad Sci USA. 2012;109:1895–900. doi: 10.1073/pnas.1106696109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroering AG, Edelbrock MA, Richards TJ, et al. The cell cycle and DNA mismatch repair. Exp Cell Res. 2007;313:292–304. doi: 10.1016/j.yexcr.2006.10.018. [DOI] [PubMed] [Google Scholar]
- Schuster-Bockler B, Lehner B. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature. 2012;488:504–7. doi: 10.1038/nature11273. [DOI] [PubMed] [Google Scholar]
- Seigneurin-Berny D, Verdel A, Curtet S, et al. Identification of components of the murine histone deacetylase 6 complex: link between acetylation and ubiquitination signaling pathways. Mol Cell Biol. 2001;21:8035–44. doi: 10.1128/MCB.21.23.8035-8044.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selva EM, New L, Crouse GF, et al. Mismatch correction acts as a barrier to homeologous recombination in Saccharomyces cerevisiae. Genetics. 1995;139:1175–88. doi: 10.1093/genetics/139.3.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibahara K, Stillman B. Replication-dependent marking of DNA by PCNA facilitates CAF-1-coupled inheritance of chromatin. Cell. 1999;96:575–85. doi: 10.1016/s0092-8674(00)80661-3. [DOI] [PubMed] [Google Scholar]
- Sia EA, Kokoska RJ, Dominska M, et al. Microsatellite instability in yeast. Dependence on repeat unit size and DNA mismatch repair genes. Mol Cell Biol. 1997;17:2851–8. doi: 10.1128/mcb.17.5.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slean MM, Panigrahi GB, Ranum LP, et al. Mutagenic roles of DNA ‘repair’ proteins in antibody diversity and disease-associated trinucleotide repeat instability. DNA Repair. 2008;7:1135–54. doi: 10.1016/j.dnarep.2008.03.014. [DOI] [PubMed] [Google Scholar]
- Spell RM, Jinks-Robertson S. Role of mismatch repair in the fidelity of Rad51- and Rad59-dependent recombination in Saccharomyces cerevisiae. Genetics. 2003;165:1733–44. doi: 10.1093/genetics/165.4.1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spell RM, Jinks-Robertson S. Examination of the roles of Sgs1 and Srs2 helicases in the enforcement of recombination fidelity in Saccharomyces cerevisiae. Genetics. 2004;168:1855–65. doi: 10.1534/genetics.104.032771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spies M, Fishel R. Mismatch repair during homologous and homeologous recombination. Cold Spring Harb Perspect Biol. 2015;7:a022657. doi: 10.1101/cshperspect.a022657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JE, Ozbirn RG, Petes TD, et al. Role of proliferating cell nuclear antigen interactions in the mismatch repair-dependent processing of mitotic and meiotic recombination intermediates in yeast. Genetics. 2008;178:1221–36. doi: 10.1534/genetics.107.085415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strand M, Prolla TA, Liskay RM, et al. Destabilization of tracts of simple repetitive DNA in yeast by mutations affecting DNA mismatch repair. Nature. 1993;365:274–6. doi: 10.1038/365274a0. [DOI] [PubMed] [Google Scholar]
- Sugawara N, Goldfarb T, Studamire B, et al. Heteroduplex rejection during single-strand annealing requires Sgs1 helicase and mismatch repair proteins Msh2 and Msh6 but not Pms1. P Natl Acad Sci USA. 2004;101:9315–20. doi: 10.1073/pnas.0305749101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara N, Pâques F, Colaiácovo M, et al. Role of Saccharomyces cerevisiae Msh2 and Msh3 repair proteins in double-strand break-induced recombination. P Natl Acad Sci USA. 1997;94:9214–9. doi: 10.1073/pnas.94.17.9214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS. DNA repair: making the cut. Nature. 2014;514:39–40. doi: 10.1038/nature13751. [DOI] [PubMed] [Google Scholar]
- Symington LS, Rothstein R, Lisby M. Mechanisms and regulation of mitotic recombination in Saccharomyces cerevisiae. Genetics. 2014;198:795–835. doi: 10.1534/genetics.114.166140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennen RI, Haye JE, Wijayatilake HD, et al. Cell-cycle and DNA damage regulation of the DNA mismatch repair protein Msh2 occurs at the transcriptional and post-transcriptional level. DNA Repair. 2013;12:97–109. doi: 10.1016/j.dnarep.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tham KC, Hermans N, Winterwerp HH, et al. Mismatch repair inhibits homeologous recombination via coordinated directional unwinding of trapped DNA structures. Mol Cell. 2013;51:326–37. doi: 10.1016/j.molcel.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tham KC, Kanaar R, Lebbink JHG. Mismatch repair and homeologous recombination. DNA Repair. 2016;38:75–83. doi: 10.1016/j.dnarep.2015.11.010. [DOI] [PubMed] [Google Scholar]
- Tomé S, Simard JP, Slean MM, et al. Tissue-specific mismatch repair protein expression: MSH3 is higher than MSH6 in multiple mouse tissues. DNA Repair. 2013;12:46–52. doi: 10.1016/j.dnarep.2012.10.006. [DOI] [PubMed] [Google Scholar]
- Tran HT, Keen JD, Kricker M, et al. Hypermutability of homonucleotide runs in mismatch repair and DNA polymerase proofreading yeast mutants. Mol Cell Biol. 1997;17:2859–65. doi: 10.1128/mcb.17.5.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler JK, Adams CR, Chen SR, et al. The RCAF complex mediates chromatin assembly during DNA replication and repair. Nature. 1999;402:555–60. doi: 10.1038/990147. [DOI] [PubMed] [Google Scholar]
- Umar A, Buermeyer AD, Simon JA, et al. Requirement for PCNA in DNA mismatch repair at a step preceding DNA resynthesis. Cell. 1996;87:65–73. doi: 10.1016/s0092-8674(00)81323-9. [DOI] [PubMed] [Google Scholar]
- Waldman AS. Ensuring the fidelity of recombination in mammalian chromosomes. Bioessays. 2008;30:1163–71. doi: 10.1002/bies.20845. [DOI] [PubMed] [Google Scholar]
- Welz-Voegele C, Stone JE, Tran PT, et al. Alleles of the yeast Pms1 mismatch-repair gene that differentially affect recombination- and replication-related processes. Genetics. 2002;162:1131–45. doi: 10.1093/genetics/162.3.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CZ, Leibowitz ML, Pellman D. Chromothripsis and beyond: rapid genome evolution from complex chromosomal rearrangements. Genes Dev. 2013;27:2513–30. doi: 10.1101/gad.229559.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Hu C, Tong D, et al. Ubiquitin-specific Peptidase 10 (USP10) deubiquitinates and stabilizes MutS homolog 2 (MSH2) to regulate cellular sensitivity to DNA damage. J Biol Chem. 2016;291:10783–91. doi: 10.1074/jbc.M115.700047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Xiang S, Joo HY, et al. HDAC6 Deacetylates and Ubiquitinates MSH2 to maintain proper levels of MutSα. Mol Cell. 2014;55:31–46. doi: 10.1016/j.molcel.2014.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]