

Supplemental Digital Content is available in the text.
Keywords: adaptive immunity, antigen presentation, brain injury, brain ischemia, T-cells
Abstract
Background and Purpose—
Autoimmune responses can occur when antigens from the central nervous system are presented to lymphocytes in the periphery or central nervous system in several neurological diseases. However, whether autoimmune responses emerge after brain ischemia and their impact on clinical outcomes remains controversial. We hypothesized that brain ischemia facilitates the genesis of autoimmunity and aggravates ischemic brain injury.
Methods—
Using a mouse strain that harbors a transgenic T-cell receptor to a central nervous system antigen, MOG35-55 (myelin oligodendrocyte glycoprotein) epitope (2D2), we determined the anatomic location and involvement of antigen-presenting cells in the development of T-cell reactivity after brain ischemia and how T-cell reactivity impacts stroke outcome. Transient middle cerebral artery occlusion and photothrombotic stroke models were used in this study. We also quantified the presence and status of T cells from brain slices of ischemic patients.
Results—
By coupling transfer of labeled MOG35-55-specific (2D2) T cells with tetramer tracking, we show an expansion in reactivity of 2D2 T cells to MOG91-108 and MOG103-125 in transient middle cerebral artery occlusion and photothrombotic stroke models. This reactivity and T-cell activation first occur locally in the brain after ischemia. Also, microglia act as antigen-presenting cells that effectively present MOG antigens, and depletion of microglia ablates expansion of 2D2 reactive T cells. Notably, the adoptive transfer of neuroantigen-experienced 2D2 T cells exacerbates Th1/Th17 responses and brain injury. Finally, T-cell activation and MOG-specific T cells are present in the brain of patients with ischemic stroke.
Conclusions—
Our findings suggest that brain ischemia activates and diversifies T-cell responses locally, which exacerbates ischemic brain injury.
Stroke is a devastating disorder that causes significant morbidity and mortality worldwide. Little progress has been made in finding new remedies for patients beyond the therapeutic window of tPA (tissue-type plasminogen activator).1,2 Autoimmune responses can occur when lymphocytes encounter brain antigens in the periphery or within the brain.3–5 Several studies during the past decade have attempted to determine how autoimmune responses to brain antigens can emerge in patients with stroke and in rodents after middle cerebral artery occlusion (MCAO).4–6 For example, both antibodies and circulating T cells can become sensitized to brain antigens, such as MBP (myelin basic protein) and related peptides,7–9 whereas other studies failed to demonstrate this.3,5 Therefore, development of autoimmunity to brain antigens in stroke remains debated.
Autoimmune responses are highly influenced by CD4+ T cells.10,11 In autoimmune diseases, the anatomic locations for activation of autoreactive T cells may include peripheral lymphoid organs, such as the spleen or cervical lymph nodes, as well as the central nervous system (CNS). Yet the timing, anatomic location, and antigen-presenting cells (APCs) possibly involved in the trigger of autoimmunity in stroke remain ill defined. Similarly, it is debated whether possible development of autoimmune responses can impact stroke outcomes. Although it has been reported that the adoptive transfer of lymphocytes against myelin antigen exacerbates stroke lesions12,13 and that proinflammatory lymphocytes are detrimental during early stage of ischemic brain injury,14 it is not known whether neuroantigen-specific T cells arising in vivo after stroke may play a detrimental or protective role on stroke outcomes. Here, we investigated the possibility that T-cell responses diversify after brain ischemia and that the expanded CNS antigen-specific T cells could promote brain injury.
Materials and Methods
This article adheres to the American Heart Association Journals implementation of the Transparency and Openness Promotion Guidelines. Details of materials and experimental procedures are available in the online-only Data Supplement. The data that support the findings of this study are available from the corresponding author on reasonable request.
Human Brain Sections
Human brain sections were obtained from the Department of Pathology of the Ohio State University (Columbus, OH) and Banner Boswell Medical Center (Sun City, AZ). The ethics consent was waived by the institutional review board because autopsy tissues were used. Details of human brain sections are given in Methods in the online-only Data Supplement.
Mice
We purchased male C57BL/6 (B6, H-2b) and Rag2–/– mice from Taconic (Santa Maria, CA). Ovalbumin (OVA) and 2D2 transgenic mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Details of mice used in this study are given in Methods in the online-only Data Supplement.
MCAO and Photothrombosis Procedure, Neurological Assessment, Neuroimaging, and Immunostaining
Adult male 10- to 12-week-old mice were subjected to 60 minutes of focal cerebral ischemia produced by transient intraluminal occlusion of the middle cerebral artery using a filament as described previously.15–17 Details of the MCAO and photothrombotic stroke procedures, neurological assessment, magnetic resonance imaging scan, and immunostaining are provided in Methods in the online-only Data Supplement.
Drug Administration
PLX3397 (Selleck Chemicals, Houston, Texas) was prepared and given as we described previously. Details are provided in Methods in the online-only Data Supplement.
Cell Isolation and Passive Transfer, CFSE Assay, Tetramer Staining, Flow Cytometry, and ELISA
Naive-like CD4+ T cells (CD4+CD44lowCD62Lhigh) were isolated from pooled splenocytes of WT (wild type), OVA, or 2D2 mice. Cell isolation, cell labeling, cell passive transfer, CFSE (carboxyfluorescein succinimidyl ester) assay, tetramer staining, flow cytometry, and ELISA were performed as described previously.15–17 Details are given in Methods in the online-only Data Supplement.
Statistical Analyses
Details of statistical analyses are given in Methods in the online-only Data Supplement. Significance was set at P<0.05. Data are shown as means±SEM.
Results
Expansion of T-Cell Reactivity to Neuroantigens Occurs in the Brain After Ischemia
Because of the heterogeneity/diversity in T-cell receptor (TCR) expression and the inherent technical limitations in measuring antigen-specific T-cell response in vivo, limited information is currently available on the dynamics of T-cell responses to neuroantigens after brain ischemia. Because MOG (myelin oligodendrocyte glycoprotein) is a neuroantigen that can be rapidly released into the periphery after brain ischemia,13,18 we used transgenic mice that harbor a CD4+ MOG35-55-specific T-cell clone (2D2). In these mice, the majority of CD4+ T cells express a Vα3.2 and Vβ11 TCR, and a large proportion of these mice spontaneously develop optic neuritis, suggesting preexisting CNS infiltration of autoreactive T cells and baseline CNS inflammation in these animals.
To measure the anatomic location and timing of T-cell changes after stroke, we adoptively transferred naive-like 2D2 CD4+ T cells (CD4+ CD44low CD62Lhigh Vα3.2+ Vβ11+) presorted from spleens of naive 2D2 mice that did not have any CNS inflammation (Figure 1A). The 2D2 CD4+ T cells were transferred into Rag2−/− recipients (lacking T and B cells) before MCAO or sham operations. After MCAO or sham procedures, 2D2 CD4+ T cells were isolated from spleen and brain of Rag2−/− recipient mice (Figure 1B). Thereafter, these cells were labeled with CFSE and cocultured with APCs in the presence of MOG peptides. Expansion of 2D2 CD4+ T cells was measured by dilution of CFSE fluorescence intensity by flow cytometry. Other tested brain antigens included NMDA (N-methyl-D-aspartate) receptor NR2A, S-100 calcium-binding protein β chain (S100β), MAP2 (microtubule-associated protein 2), MBP (myelin basic protein), and PLP (proteolipid protein). With the exception of MOG, none promoted proliferation of 2D2 CD4+ T cells (Figure I in the online-only Data Supplement). Of note, we observed a vigorous proliferation of 2D2 CD4+ T cells in response to MOG35-55 and other MOG epitopes (MOG91-108 and MOG103-125), especially in 2D2 CD4+ T cells isolated from brain tissues at day 4 after MCAO (Figure 1C; Figure I in the online-only Data Supplement). In contrast, proliferation of 2D2 CD4+ T cells in response to MOG35-55 and additional MOG epitopes was seen in 2D2 CD4+ T cells isolated from brain and spleen at day 7 after MCAO (Figure 1C).
Figure 1.
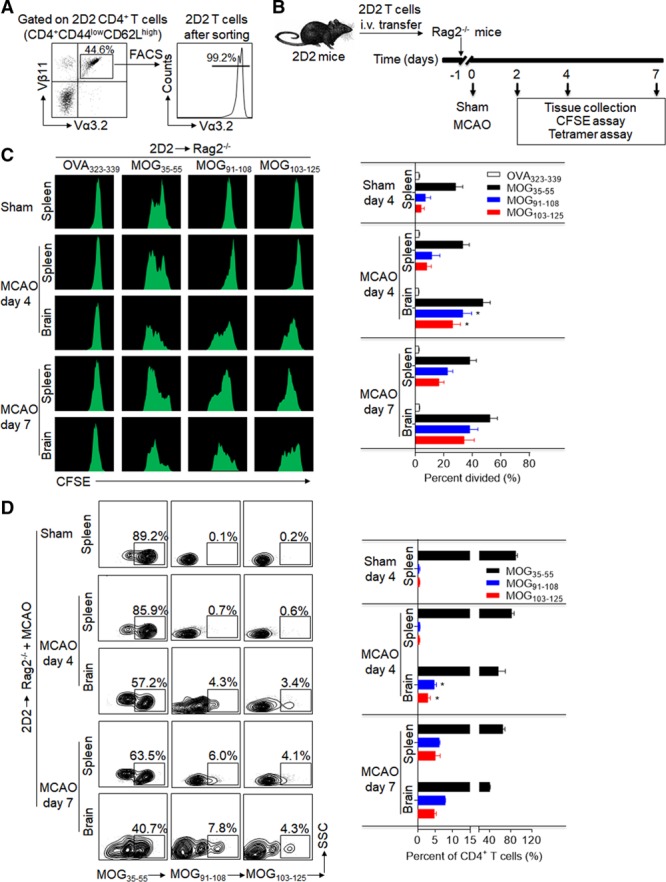
Development and expansion of 2D2 T-cell reactivity against neuroantigen epitopes within the ischemic brain. 2D2 CD4+ T cells (CD4+CD44lowCD62LhighVα3.2+Vβ11+) were sorted from naive 2D2 mice without any sign of central nervous system autoimmune disease and transferred intravenous (i.v.) into Rag2−/− mice after sham or 60-minute middle cerebral artery occlusion (MCAO) surgery. At day 4 or day 7 after surgery, 2D2 CD4+ T cells were reisolated from spleens and brains of Rag2−/− recipients. In the sham group, 2D2 CD4+ T cells were reisolated from spleens of Rag2−/− recipients at day 4 after surgery. A, Purity of sorted naive-like 2D2 T cells was validated by fluorescence-activated cell sorting (FACS). B, Schematic graph showing the experimental design. C, Flow cytometry plots and cumulative bar graph show CFSE (carboxyfluorescein succinimidyl ester) dilution (proliferation) of 2D2 CD4+ T cells in response to in vitro stimulation with OVA, MOG (myelin oligodendrocyte glycoprotein)35-55, MOG91-108, and MOG103-125 after coculture for 72 hours with antigen-presenting cells. D, After reisolation at day 4 or day 7 after surgery, 2D2 CD4+ T cells (Vα3.2+Vβ11+CD4+CD3+) were stained with PE-labeled MHC class II tetramers containing MOG35-55, MOG91-108, or MOG103-125 (to identify antigen-specific 2D2 T cells). Representative flow cytometry plots and bar graph show MOG epitope-specific T cells. All gates were set using fluorescence minus one controls. n=15 per group. Error bars represent SEM. *P<0.05, brain vs spleen.
To confirm the above findings, we measured MOG35-55-, MOG91-108-, and MOG103-125-specific T cells via MHC (major histocompatibility complex) class II tetramers. At day 4 after MCAO, higher frequencies of MOG91-108- and MOG103-125-specific T cells were seen in the brains but fewer in the spleens of Rag2−/− mice receiving 2D2 CD4+ T cells (Figure 1D) or earlier in the brain and spleen, that is, at day 2 after MCAO (Figures II and III in the online-only Data Supplement). A similar finding was seen in WT mice receiving adoptive transfer of naive-like 2D2 CD4+ T cells (Figure IV in the online-only Data Supplement). However, MOG91-108- and MOG103-125-specific T cells were observed in higher numbers both in the spleen and brain at day 7 after MCAO (Figure 1D). Immunostaining of brain sections from Rag2−/− mice receiving 2D2 CD4+ T cells also showed the presence of MOG35-55-, MOG91-108-, and MOG103-125-specific T cells at day 4 after MCAO and reperfusion (Figure V in the online-only Data Supplement). Together, these results suggest that the expanded reactivity of 2D2 T cells to brain antigens emerges primarily in the brain after cerebral ischemia.
Antigen-Specific T Cells Are Activated Locally in the Brain After Ischemia
Little is known about the anatomic location where T cells are activated in response to brain antigens. To define the location of T-cell activation and the timing of the autoreactive T-cell response after cerebral ischemia, we transferred CFSE-labeled MOG35-55-specific CD4+ transgenic (2D2) T cells into Rag2−/− recipient mice, followed by 60 minutes of MCAO (Figure 1B). 2D2 CD4+ T cells proliferated robustly in the ischemic brains of Rag2−/− recipient mice (Figure 2A and 2B). In contrast, proliferation of transferred 2D2 CD4+ T cells was weak in spleen, lymph nodes, and circulation (Figure 2A and 2B). There was an increase of CD25+CD4+ and CD69+CD4+ 2D2 T cells in the brain but not in the spleen, lymph nodes, and circulation at day 4 after reperfusion (Figure 2C and 2D). These data suggest that 2D2 CD4+ T cells are activated primarily in the brain at day 4 after reperfusion.
Figure 2.
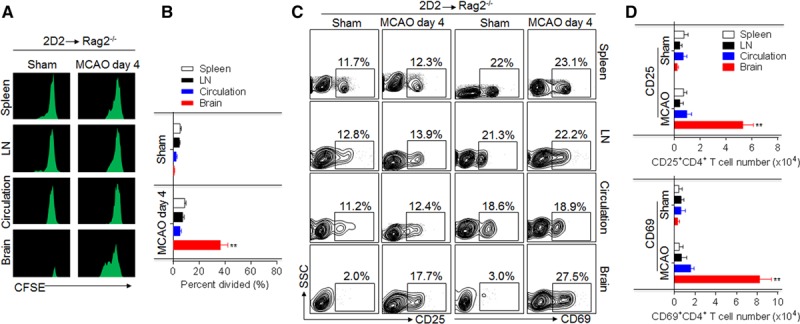
Activation and proliferation of 2D2 T cells occur primarily in the brain after brain ischemia. A and B, 2D2 CD4+ T cells were isolated from splenocytes of 2D2 mice without any sign of central nervous system autoimmune disease and adoptively transferred into Rag2−/− mice followed by sham or 60-minute middle cerebral artery occlusion (MCAO) procedures. At day 4 after sham or MCAO, 2D2 CD4+ T cells were reisolated from spleens, lymph nodes (LNs), peripheral blood, and brains of Rag2−/− recipients. Reactivity of 2D2 CD4+ T cells was assessed by CFSE assay. Bar graph shows quantification of 2D2 CD4+ T-cell reactivity. C and D, Flow cytometry plots and quantification of CD4+ cell activation (CD25+CD4+, CD69+CD4+) in single-cell suspensions reisolated from spleens, LNs, circulating blood, and brains of passively transferred mice subjected to MCAO. All gates were set using fluorescence minus one controls. n=10 per group. Error bars represent SEM. **P<0.01, brain vs spleen, LN, or circulation.
Lastly, we found a dramatic increase of TCRvβ usage in brain-infiltrating 2D2 CD4+ T cells (Figure VI in the online-only Data Supplement). In particular, the usage of TCRvβ 5.1, 5.2, 13, and 3 was significantly higher (Figure VI in the online-only Data Supplement), suggesting the presence of clonal expansions in brain-infiltrating 2D2 CD4+ T cells after ischemia. Together, these results suggest that the entry of 2D2 CD4+ T cells into the ischemic brain is not stochastic but antigen driven.
2D2 T Cells Are Activated by Local APCs in the Brain After Cerebral Ischemia
Although CD4+ T cells are activated by MHC II presentation of epitopes by APCs, recent data also suggest alternative pathways that may not depend on MHC II.14,19 To determine whether MHC II/TCR interaction is required for brain ischemia-induced expansion of 2D2 CD4+ T cells to MOG epitopes, we used MHC II knockout mice (H2-Ab1–/–H2-Aa–/–H2-Eb1–/–H2-Eb2–/–H2-Ea–/– mice). We transferred 2D2 CD4+ T cells into Rag2−/−MHC II–/– mice (Figure 3A). Although the transferred 2D2 CD4+ T cells were unable to be maintained in the periphery because of lack of MHC II, as reported,19,20 these cells survived for at least a week in Rag2−/−MHC II–/– recipients (Figure 3B and 3C). The occurrence of the new reactivity against additional MOG epitopes (MOG91-108 and MOG103-125) was reduced in 2D2 T cells after transfer into Rag2−/−MHC II–/– mice. A similar finding was seen in MHC II–/– mice receiving adoptive transfer of 2D2 CD4+ T cells (Figure VII in the online-only Data Supplement). Together, these results suggest the requirement of MHC class II/TCR interaction for the expansion of T cells to neuroantigens (Figure 3B and 3C).
Figure 3.
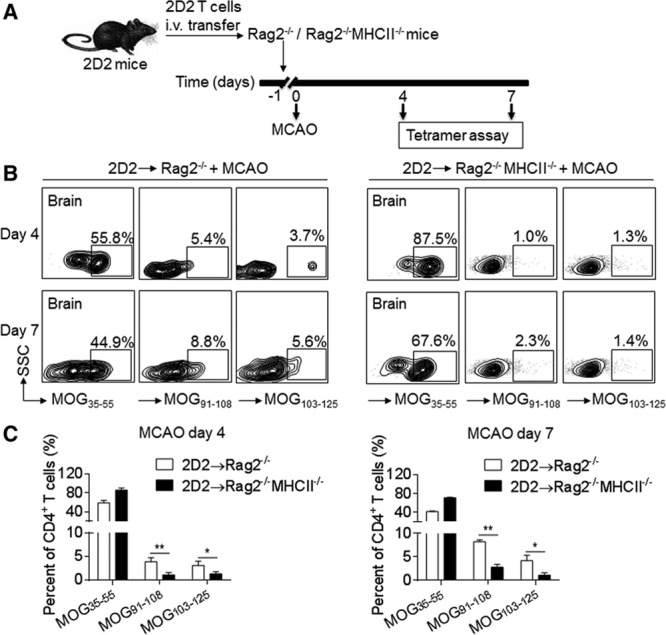
The occurrence of new reactivity of 2D2 T cells depends on MHC II. 2D2 CD4+ T cells were sorted from spleen of 2D2 mice without central nervous system autoimmune disease, and transferred intravenous (i.v.) into Rag2−/− or Rag2−/−MHC II−/− recipient mice before 60 min of middle cerebral artery occlusion (MCAO) surgery. A, Schematic graph showing the experimental design. B, At day 4 or day 7 after reperfusion, 2D2 CD4+ T cells were reisolated from brains of Rag2−/− or Rag2−/−MHC II−/− mice and then stained with PE-labeled tetramers containing MOG (myelin oligodendrocyte glycoprotein)35-55, MOG91-108, or MOG103-125 epitopes. Flow cytometry plots show MOG epitope-specific 2D2 CD4+ T cells. All gates were set using fluorescence minus one controls. C, Bar graph shows quantification of surface expression of antigen-specific T-cell receptors at day 4 and day 7 after reperfusion. n=15 per group. Error bars represent SEM. *P<0.05, **P<0.01.
Because activation and proliferation of 2D2 CD4+ T cells is predominantly initiated in the brain but not peripheral sites after ischemia, we determined which APCs presented brain antigens to 2D2 CD4+ T cells. Microglia-like cells had the highest expression of MHC II in the brain of Rag2−/− mice at day 4 after MCAO (Figure VIII in the online-only Data Supplement). Also, among the cells positive for MHC II and MOG35-55, microglia-like cells had the highest numbers (Figure VIII in the online-only Data Supplement). Finally, some 2D2 CD4+ T cells were in close proximity to Iba1+ cells in brain sections from Rag2−/− mice at day 4 after MCAO (Figure IX in the online-only Data Supplement), and MOG35-55 colocalized with Iba1+ cells (Figure X in the online-only Data Supplement), suggesting that MOG may be processed by these cells and presented to the infiltrating 2D2 CD4+ T cells.
Next, CFSE-labeled 2D2 CD4+ T cells were cocultured with purified microglia-like cells (CD11b+CD45int), macrophages (CD11b+CD45hi F4/80+), or dendritic cells (CD11c+CD45hiF4/80−) in either presence or absence (to assess endogenous presentation) of MOG35-55 peptide (Figure VIII in the online-only Data Supplement). In the presence of exogenous peptide, all 3 populations could induce proliferation of 2D2 CD4+ T cells. Without added peptide, all 3 APC populations also induced proliferation of 2D2 CD4+ T cells. However, only microglia-like cells induced robust proliferation of 2D2 CD4+ T cells. In contrast, the proliferation of 2D2 CD4+ T cells induced by either macrophage or dendritic cells was to a lesser extent (Figure VIII in the online-only Data Supplement). These data demonstrate a strong capacity of microglia to present brain antigen peptides to T cells.
We also depleted microglia using PLX3397—an inhibitor of colony stimulating factor 1 receptor, which specifically depletes microglia.21 We found that reduction of microglia abolished the increase of MOG91-108- or MOG103-125-specific 2D2 T cells (Figure VIII in the online-only Data Supplement). These data imply that brain-resident microglia are key APCs for the expansion of 2D2 T-cell reactivity to brain antigens in the ischemic brain.
Diversified T-Cell Responses Exacerbate Ischemic Brain Injury
To evaluate the impact of brain ischemia-induced diversification of T-cell response on stroke outcomes, we used 2D2 CD4+ T cells from spleens of sham 2D2 mice and antigen-experienced 2D2 CD4+ T cells from spleens of 2D2 mice at day 7 after MCAO or photothrombosis. We transferred WT, OVA-specific, 2D2, or antigen-experienced 2D2 CD4+ T cells into Rag2−/− mice before MCAO or photothrombosis (Figure 4A). More severe neurodeficits and significantly larger infarct volumes were seen in Rag2−/− mice receiving antigen-experienced 2D2 CD4+ T cells as compared with WT mice, OVA-specific, or 2D2 CD4+ T cells from naive 2D2 mice, starting at day 4 after MCAO (Figure 4B) or at day 7 after photothrombotic stroke (Figure 4C). Increased neurodeficits and brain infarction volumes persisted at least until day 7 after MCAO (Figure 4B) or photothrombotic stroke (Figure 4C). Thus, brain ischemia-induced diversification of T-cell responses has a detrimental impact on ischemic brain injury.
Figure 4.
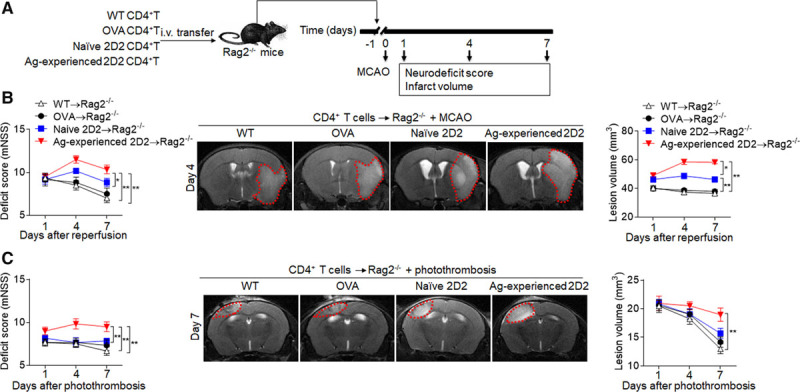
Diversified T-cell responses exacerbate neurodeficits and brain infarction. CD4+ T cells (CD4+CD44lowCD62Lhigh) were obtained from spleens of wild-type mice (WT→Rag2−/−) and OVA mice (OVA→Rag2−/−) at day 7 after sham surgery, naive 2D2 mice (naive 2D2→Rag2−/−) at day 7 after sham surgery, or 2D2 mice at day 7 after middle cerebral artery occlusion (MCAO) or photothrombosis (antigen [Ag]-experienced 2D2→Rag2−/−). These T cells were injected intravenous (i.v.) into Rag2−/− recipients and followed by 60 min of MCAO or photothrombotic stroke procedures at 24 hours after injection. A, Schematic graph showing the experimental design. B, The neurological deficit scores (mNSS) were assessed in different groups at the indicated days after MCAO (B) or photothrombosis (C). 7T magnetic resonance imaging shows brain lesions in different groups at day 4 after MCAO (B) or photothrombosis (C). Cumulative results show quantifications of lesion volume. n=12 mice per group. *P<0.05, **P<0.01.
To visualize brain-infiltrating antigen-experienced 2D2 T cells, we noninvasively tracked 2D2 CD4+ T cells by adoptive transfer of Molday ION Rhodamine B-labeled cells into Rag2−/− mice before MCAO. After reperfusion, we found increased hypointensive spots in Rag2−/− mice receiving antigen-experienced 2D2 CD4+ T cells (Figure 5A). In addition, significantly increased levels of IFN-γ (interferon gamma), interleukin (IL)-17, TNF-α (tumor necrosis factor-α), IL-1β, and IL-6 were seen in brain homogenates of Rag2−/− mice receiving antigen-experienced 2D2 CD4+ T cells (Figure 5B). The analysis of Th1 and Th17 responses in Rag2−/− mice receiving antigen-experienced 2D2 CD4+ T cells showed significantly higher numbers of brain-infiltrating CD4+, IFN-γ+ CD4+, and IL-17+CD4+ T cells in Rag2−/− mice receiving antigen-experienced 2D2 CD4+ T cells versus WT, OVA-specific, or naive 2D2 CD4+ T cells (Figure 5C), suggesting that antigen-experienced T cells exacerbate brain inflammation after cerebral ischemia.
Figure 5.
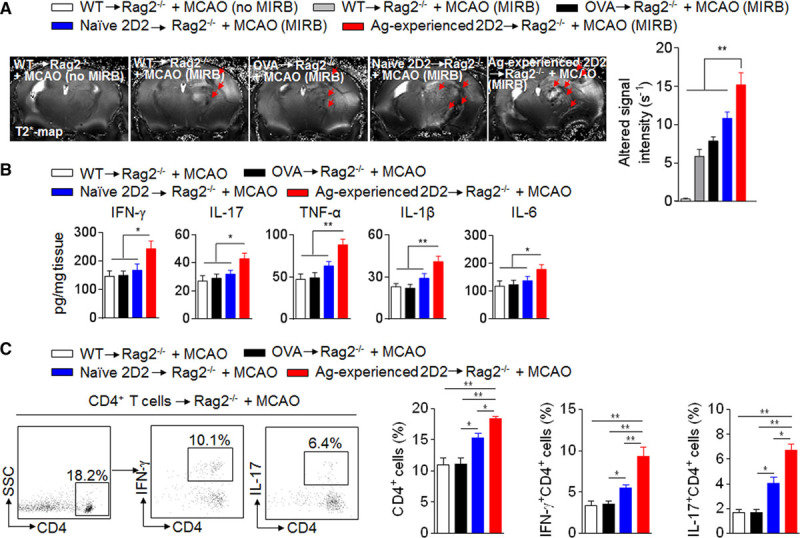
Diversified T-cell responses exacerbate brain inflammation after ischemia. A, CD4+ T cells were obtained from spleen tissues of wild-type mice, OVA mice, naive 2D2 mice, or 2D2 mice at day 7 after middle cerebral artery occlusion (MCAO). After in vitro incubation with Molday ION Rhodamine B (MIRB) for 24 hours, MIRB-labeled CD4+ T cells were transferred intravenous into Rag2−/− recipient mice followed by 60 min of MCAO. The CD4+ T cells from wild-type mice without MIRB labeling were intravenous transferred into Rag2−/− recipient mice as an unlabeled control. At day 4 after reperfusion, MIRB-labeled CD4+ T cells were visualized using a 7T magnetic resonance imaging with a Multislice Gradient Echo sequence and acquired T2* map. MIRB signal intensity was measured by quantifying R2* values. Bar graph shows quantification of MIRB signal intensity at day 4 after MCAO. B and C. CD4+ T cells were obtained from spleens of wild-type mice, OVA mice, naive 2D2 mice, or 2D2 mice at day 4 after MCAO. These T cells were injected intravenous into groups of Rag2−/− recipient mice and followed by 60 min of MCAO. At day 4 after MCAO and reperfusion, brain homogenates were prepared to measure cytokine expression. Bar graphs show cytokine levels measured with a Multi-Analyte ELISArray kit (B). Representative flow cytometry dot plots and summarized bar graph show brain-infiltrating IFN-γ+CD4+ or IL-17+CD4+ T cells at day 4 after MCAO and reperfusion (C). n=8 per group. Error bars represent SEM. * P<0.05, **P<0.01.
These results imply that CD4+ T cells enter the brain and become activated after ischemia, where multiple brain antigens presented by local APCs diversify the T-cell responses that exacerbate brain infarction.
Presence of Neuroantigen-Specific T Cells in the Brain of Patients With Stroke
Brain slices from patients with acute middle cerebral artery ischemic stroke showed that CD4+ T cells infiltrated the infarct and peri-infarct areas. Some of these cells resided in close vicinity to ischemic neurons (Figure XI in the online-only Data Supplement). Of interest, MOG35-55-, MOG91-108-, and MOG103-125-specific CD4+ T cells were seen in the peri-infarct areas (Figure XI in the online-only Data Supplement), indicating the presence of neuroantigen-specific CD4+ T cells in human stroke lesions.
Discussion
In this study, we precisely tracked the myelin-reactive T cells with tetramers and revealed that the antigen spectrum was expanded with the progression of ischemia-induced brain injury. To our knowledge, this study provides the first definitive evidence on the activation of CD4+ T cells, which subsequently acquire new reactivity within the brain.
The finding of the 2D2 T-cell specificities to MOG91-108 and MOG103-125 suggests the expansion of T-cell reactivity against new brain antigen epitopes after stroke. This finding is supported by a previous study showing that T cells bearing the same Vα and Vβ genes can have different antigen specificity.22 However, one might argue that new specificities could derive from carryover clones from the 2D2 donors or derive from MOG35-55-specific clones with cross-reactivity to the other 2 MOG peptides. Yet, MOG35-55-specific T cells isolated from naive 2D2 mice only had minimal reaction to MOG91-108 and MOG103-125, suggesting that MOG35-55-specific 2D2 T cells were specific, as also supported by the diversity of Vβ expression that supports the expansion of T-cell response in the poststroke environment. One recent study showed that stroke induced a secondary, dynamic autoimmune response to neuronal antigens, maintained through 10 days in both lymph nodes and spleen.9 In line with those findings, our results suggest that the expansion of T-cell response could occur both in the periphery and CNS after ischemia. However, the activation and proliferation of naive-like 2D2 T cells were more prominent in the brain than in peripheral lymphoid organs, suggesting that the expansion of T cells could be initiated in the brain, possibly because of the stroke-induced environment. In support of this, another report suggested that the expansion of T-cell clones may occur first in the ischemic brain and subsequently in secondary lymphoid organs.23 Thus, activation of T cells would occur predominantly in the CNS after stroke.
The findings of 2D2 T cells reactive to other MOG epitopes in the ischemic brain and its dependence on MHC II implicate antigen presentation. In the context of stroke, the CNS harbors both peripheral APCs homing into the ischemic brain and CNS-resident APCs. Our findings of higher numbers of MHC II-expressing microglia-like cells colocalizing with MOG peptide, together with their stronger capacity to present endogenous MOG, suggest a major role of microglia in the expansion of 2D2 T cells. Indeed, depletion of microglia ablated occurrence of new reactivity to additional MOG epitopes after stroke. Past studies reported the expression of MHC II on microglia on their activation and myelin debris phagocytosed by microglia in neuroinflammatory conditions.24–26 More recent findings suggest that resident microglia are the first responders to brain ischemia and that phagocytosis of neural debris by microglia takes place in the early stages of stroke, before the arrival of infiltrating peripheral macrophages.26,27 Of note, microglia possess multiple functions, including antigen presentation and production of inflammatory factors, which enable microglia to interact with several cell types other than T cells. These diverse features suggest a profound role of microglia in brain injury depending on the settings of animal models and disease conditions.
Some have suggested that myelin-reactive lymphocytes are detrimental in rodent stroke models,13 and clinical studies have linked autoreactive T-cell responses to worsened outcomes in patients with stroke.28 Our study suggests a series of events in which antigen presentation promotes T-cell expansion that fuels focal inflammation during the evolution of brain infarction and neurodeficit. This raises the possibility that preexisting or newly emerged brain antigen-specific T cells could favor brain injury in patients with first-ever or recurrent stroke attacks, implying possible therapeutic blockade of the recruitment and activation of T cells to attenuate ischemic brain injury.
Acknowledgments
Drs Liu and Shi formulated the study concept and designed the studies; K. Wood and Drs Jin and Liu performed the studies; Y. Feng and Drs La Cava, Chai, and Gonzales advised on the design, execution of experiments, and interpretation of results; and Drs Jin, Liu, Shi, La Cava, Gonzales, and Dong wrote and edited the manuscript.
Sources of Funding
This study was supported, in part, by the American Heart Association grant 16SDG27250236, a Valley Research Partnership grant, and an Arizona Alzheimer’s Research Consortium grant.
Disclosures
None.
Supplementary Material
Footnotes
The online-only Data Supplement is available with this article at http://stroke.ahajournals.org/lookup/suppl/doi:10.1161/STROKEAHA.118.020203/-/DC1.
References
- 1.Albers GW, Bates VE, Clark WM, Bell R, Verro P, Hamilton SA. Intravenous tissue-type plasminogen activator for treatment of acute stroke: the Standard Treatment with Alteplase to Reverse Stroke (STARS) study. JAMA. 2000;283:1145–1150. doi: 10.1001/jama.283.9.1145. [DOI] [PubMed] [Google Scholar]
- 2.Lees KR, Bluhmki E, von Kummer R, Brott TG, Toni D, Grotta JC, et al. ECASS, ATLANTIS, NINDS and EPITHET rt-PA Study Group. Time to treatment with intravenous alteplase and outcome in stroke: an updated pooled analysis of ECASS, ATLANTIS, NINDS, and EPITHET trials. Lancet. 2010;375:1695–1703. doi: 10.1016/S0140-6736(10)60491-6. doi: 10.1016/S0140-6736(10)60491-6. [DOI] [PubMed] [Google Scholar]
- 3.Becker KJ. Sensitization and tolerization to brain antigens in stroke. Neuroscience. 2009;158:1090–1097. doi: 10.1016/j.neuroscience.2008.07.027. doi: 10.1016/j.neuroscience.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808. doi: 10.1038/nm.2399. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gill D, Veltkamp R. Dynamics of T cell responses after stroke. Curr Opin Pharmacol. 2016;26:26–32. doi: 10.1016/j.coph.2015.09.009. doi: 10.1016/j.coph.2015.09.009. [DOI] [PubMed] [Google Scholar]
- 6.Becker KJ. Activation of immune responses to brain antigens after stroke. J Neurochem. 2012;123(suppl 2):148–155. doi: 10.1111/j.1471-4159.2012.07953.x. doi: 10.1111/j.1471-4159.2012.07953.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bornstein NM, Aronovich B, Korczyn AD, Shavit S, Michaelson DM, Chapman J. Antibodies to brain antigens following stroke. Neurology. 2001;56:529–530. doi: 10.1212/wnl.56.4.529. [DOI] [PubMed] [Google Scholar]
- 8.Becker KJ, Kindrick DL, Lester MP, Shea C, Ye ZC. Sensitization to brain antigens after stroke is augmented by lipopolysaccharide. J Cereb Blood Flow Metab. 2005;25:1634–1644. doi: 10.1038/sj.jcbfm.9600160. doi: 10.1038/sj.jcbfm.9600160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ortega SB, Noorbhai I, Poinsatte K, Kong X, Anderson A, Monson NL, et al. Stroke induces a rapid adaptive autoimmune response to novel neuronal antigens. Discov Med. 2015;19:381–392. [PMC free article] [PubMed] [Google Scholar]
- 10.Dittel BN. CD4 T cells: balancing the coming and going of autoimmune-mediated inflammation in the CNS. Brain Behav Immun. 2008;22:421–430. doi: 10.1016/j.bbi.2007.11.010. doi: 10.1016/j.bbi.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lancaster E, Dalmau J. Neuronal autoantigens–pathogenesis, associated disorders and antibody testing. Nat Rev Neurol. 2012;8:380–390. doi: 10.1038/nrneurol.2012.99. doi: 10.1038/nrneurol.2012.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zierath D, Schulze J, Kunze A, Drogomiretskiy O, Nhan D, Jaspers B, et al. The immunologic profile of adoptively transferred lymphocytes influences stroke outcome of recipients. J Neuroimmunol. 2013;263:28–34. doi: 10.1016/j.jneuroim.2013.07.014. doi: 10.1016/j.jneuroim.2013.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ren X, Akiyoshi K, Grafe MR, Vandenbark AA, Hurn PD, Herson PS, et al. Myelin specific cells infiltrate MCAO lesions and exacerbate stroke severity. Metab Brain Dis. 2012;27:7–15. doi: 10.1007/s11011-011-9267-5. doi: 10.1007/s11011-011-9267-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kleinschnitz C, Schwab N, Kraft P, Hagedorn I, Dreykluft A, Schwarz T, et al. Early detrimental T-cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood. 2010;115:3835–3842. doi: 10.1182/blood-2009-10-249078. doi: 10.1182/blood-2009-10-249078. [DOI] [PubMed] [Google Scholar]
- 15.Gan Y, Liu Q, Wu W, Yin JX, Bai XF, Shen R, et al. Ischemic neurons recruit natural killer cells that accelerate brain infarction. Proc Natl Acad Sci USA. 2014;111:2704–2709. doi: 10.1073/pnas.1315943111. doi: 10.1073/pnas.1315943111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin WN, Shi SX, Li Z, Li M, Wood K, Gonzales RJ, et al. Depletion of microglia exacerbates postischemic inflammation and brain injury. J Cereb Blood Flow Metab. 2017;37:2224–2236. doi: 10.1177/0271678X17694185. doi: 10.1177/0271678X17694185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li M, Li Z, Yao Y, Jin WN, Wood K, Liu Q, et al. Astrocyte-derived interleukin-15 exacerbates ischemic brain injury via propagation of cellular immunity. Proc Natl Acad Sci USA. 2017;114:E396–E405. doi: 10.1073/pnas.1612930114. doi: 10.1073/pnas.1612930114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dirnagl U, Klehmet J, Braun JS, Harms H, Meisel C, Ziemssen T, et al. Stroke-induced immunodepression: experimental evidence and clinical relevance. Stroke. 2007;38(suppl 2):770–773. doi: 10.1161/01.STR.0000251441.89665.bc. doi: 10.1161/01.STR.0000251441.89665.bc. [DOI] [PubMed] [Google Scholar]
- 19.Walsh JT, Hendrix S, Boato F, Smirnov I, Zheng J, Lukens JR, et al. MHCII-independent CD4+ T cells protect injured CNS neurons via IL-4. J Clin Invest. 2015;125:2547. doi: 10.1172/JCI82458. doi: 10.1172/JCI82458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takeda S, Rodewald HR, Arakawa H, Bluethmann H, Shimizu T. MHC class II molecules are not required for survival of newly generated CD4+ T cells, but affect their long-term life span. Immunity. 1996;5:217–228. doi: 10.1016/s1074-7613(00)80317-9. [DOI] [PubMed] [Google Scholar]
- 21.Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82:380–397. doi: 10.1016/j.neuron.2014.02.040. doi: 10.1016/j.neuron.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buenafe AC, Tsu RC, Bebo B, Jr, Vandenbark AA, Offner H. Myelin basic protein-specific and TCR V beta 8.2-specific T-cell lines from TCR V beta 8.2 transgenic mice utilize the same V alpha and V beta genes: specificity associated with the V alpha CDR3-J alpha region. J Neurosci Res. 1997;47:489–499. [PubMed] [Google Scholar]
- 23.Liesz A, Karcher S, Veltkamp R. Spectratype analysis of clonal T cell expansion in murine experimental stroke. J Neuroimmunol. 2013;257:46–52. doi: 10.1016/j.jneuroim.2013.01.013. doi: 10.1016/j.jneuroim.2013.01.013. [DOI] [PubMed] [Google Scholar]
- 24.Aloisi F. Immune function of microglia. Glia. 2001;36:165–179. doi: 10.1002/glia.1106. [DOI] [PubMed] [Google Scholar]
- 25.Mack CL, Vanderlugt-Castaneda CL, Neville KL, Miller SD. Microglia are activated to become competent antigen presenting and effector cells in the inflammatory environment of the Theiler’s virus model of multiple sclerosis. J Neuroimmunol. 2003;144:68–79. doi: 10.1016/j.jneuroim.2003.08.032. [DOI] [PubMed] [Google Scholar]
- 26.Neumann H, Kotter MR, Franklin RJ. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain. 2009;132(pt 2):288–295. doi: 10.1093/brain/awn109. doi: 10.1093/brain/awn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fu R, Shen Q, Xu P, Luo JJ, Tang Y. Phagocytosis of microglia in the central nervous system diseases. Mol Neurobiol. 2014;49:1422–1434. doi: 10.1007/s12035-013-8620-6. doi: 10.1007/s12035-013-8620-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Becker KJ. Modulation of the postischemic immune response to improve stroke outcome. Stroke. 2010;41(suppl 10):S75–S78. doi: 10.1161/STROKEAHA.110.592881. doi: 10.1161/STROKEAHA.110.592881. [DOI] [PMC free article] [PubMed] [Google Scholar]