

Abstract
Culture of endothelial cells (ECs) embedded in 3D scaffolds of denatured collagen has shown tremendous therapeutic potential in clinical trials of tissue repair. It is postulated that these matrix-embedded ECs (MEECs) attain a differential phenotype similar to early progenitor forms, which cannot be attained in 2D culture. MEECs are compared to 2D-ECs and endothelial progenitor cells (EPCs) by secretome, phenotype, and genetic fingerprint, and are found to be altered from 2D-ECs on all levels, adopting an EPC-like phenotype. This manifests in elevation of CD34 expression—a progenitor cell marker—and protein secretion and gene expression pro-files that are similar to EPCs. Even more striking is that EPCs in 2D lose their phenotype, evident by the loss of CD34 expression, but are able to regain expression over time when embedded in the same 3D matrices, suggesting that future in vitro EPC work should use ME-EPCs to recapitulate in vivo phenotype. These findings elucidate the relationship between EPCs and the substratum-dependent regulation imparted by ECs which is critical to understand in order to optimize MEEC therapy and propel it into the clinic.
Keywords: 3D, endothelial cells, endothelial progenitor cells, matrix embedding, scaffolds
1. Introduction
Cells reside in a complex 3D environment that drives cell differentiation and biology. As cell constructs are considered for diagnosis and therapy, increasing attention is being paid to creating substrata that mimic their native, in vivo environment.[1,2] Free, unattached cells are difficult to maintain in a stable phenotype or retain in forms that can provide consistent function. They often lose aspects of their function seen when in intact tissues and even become “foreign,” eliciting immune reactivity.[3–6] The heterogeneous effects of cells in different environments may not only allow for tuning and optimization for specific use but may also explain critical basic biological processes.[7,8] Substratum properties such as 3D structural variations and differences in stiffness are sensed by the cell, regulating transcriptional, translational, and protein secretome profiles.[3–5,9,10] Such changes are mediated by cytoskeletal alterations and various receptors, including integrins and, as such, dependence on substratum is not only comprehensible but also potentially modulatable.[3]
Endothelial cells (ECs) have been shown to be powerful regulators of tissue homeostasis and, moreover, matrix embedding has been shown to increase this regulation.[11,12] ECs embedded in denatured collagen matrices have been used as a therapeutic modality to control healing and vascular complications in dialysis patients undergoing insertion of arteriovenous vascular access grafting.[11] The collagen matrix readily enables cell adhesion, is relatively immunoquiescent, and can be digested with collagenase to model-controlled elimination or full ex vivo degradation to recover cells for examination. These constructs are powerful bioreactors that respond to local cues and, when in the perivascular space of vascular grafts, show potent repair-enhancing effects in animal and clinical trials.[11–14] The therapeutic capacity of these systems suggests that matrix-embedded ECs (MEECs) attain a phenotype not observed in 2D-EC, flat cell culture.
This study sought to explore the mechanism driving this substratum-dependent EC regulation. Two states exist in 2D-EC culture: confluent and subconfluent (the former reparative and the latter injurious). Two hypotheses emerged: the potency of MEECs derives from (1) attainment of confluence or (2) substratum-dependent transformation to a progenitor-like phenotype. Previous work analyzing the cytoskeletal state of MEECs suggested that confluence was not a dominating factor.[15] As such, this study compared MEECs and endothelial progenitor cells (EPCs) and found that MEECs, in fact, attain a progenitor-like phenotype, as defined by secretome, phenotype, and genetic fingerprint.[16,17] Furthermore, we found that EPCs cultured in 2D lose this phenotype, but regain it when matrix embedded. These findings show how dominant matrix embedding can be and how much these factors functionally transform cellular behavior and therapeutic potential.
2. Results
2.1. 3D Cell Growth, Morphology, and Cell–Cell Interaction Differ from 2D
ECs readily attach to and embed within porous scaffolds of denatured collagen within 1 h after seeding and proliferate until reaching a stable state 2–3 weeks later. Since 3D porous scaffolds have a large surface area, cell number increases 40-fold in 3 weeks, growing from 4 × 104 to 1.5 × 106 cells on a 1 × 0.5 × 0.3 cm scaffold. The equivalent surface area required to culture a similar number of ECs in a 2D flask would be ≈35 cm2, i.e., 70 times larger than the area of the 2D projection of the scaffold (0.5 cm2). While 2D-ECs on gelatin-coated tissue culture polystyrene assume a strained ovoid morphology (Figure 1A,B), MEECs line scaffold interstices, mold to the contours of scaffold struts, and project filopodia which make contact with adjacent cells (Figure 1C,D).
Figure 1.
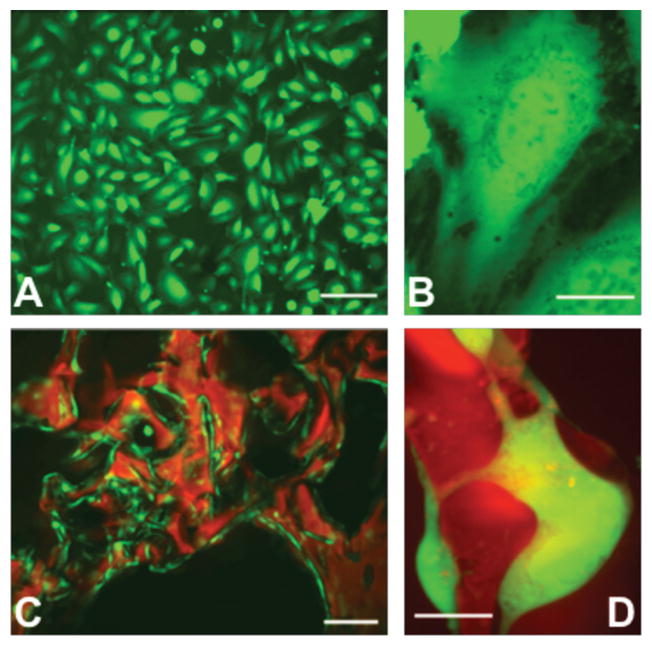
EC interaction with substratum affecting cellular morphology. A,B) In 2D culture, ECs (green) are flat and stretched; C,D) while on 3D denatured collagen matrices (red) the cell shape is complex and conforms to the material shape [scale bars: A) 50 μm; B, D) 25 μm; C) 100 μm].
2.2. EC Secretome is Altered by Matrix-Embedded Culture
We examined levels of secreted angiogenic factors and immune cytokines as a function of culture substratum given the divergent morphology of MEECs compared to 2D-ECs. The secretome is particularly important as MEECs are utilized as a cell therapy based on paracrine effects of cell-secreted factors. Seven proteins were significantly increased or decreased in conditioned media of MEECs compared to 2D-ECs, representing a wide array of functionalities that includes angiogenesis, immune modulation, chemotaxis, cell proliferation, and tissue remodeling (Figure 2). Five proteins were elevated in MEEC compared to 2D-EC conditioned media (Figure 2A): interleukin 8 (IL-8), Angiopoetin 2 (Ang2), platelet-derived growth factor BB (PDGF-BB), monocyte chemotactic protein 1 (MCP-1), and tissue inhibitor of metalloproteinases 1 (TIMP-1). These proteins represent a relatively wide array of biological functions (Figure 2B). IL-8 is a neutrophil chemotactic factor and a potent pro-angiogenic factor, as is Ang2. PDGF-BB is a mitogenic factor for cells of mesenchymal origin, while MCP-1 is a chemokine which attracts immune cells, and TIMP-1 is an inhibitor of matrix metalloproteinases but also promotes cell proliferation and has anti-apoptotic properties. The elevation in secretion of these proteins matches previous findings of the reparative capacity of MEECs.[12–14]
Figure 2.

Levels of cytokines in conditioned medium from 2D-ECs and MEECs at day 14 as analyzed by protein dot blot array. A) Five cytokines were upregulated and two were downregulated in MEEC compared to 2D-EC culture. B) These cytokines serve a variety of functions in EC behavior. Spot intensities were normalized, per manufacturer’s instructions, to positive control spots on each membrane and biological duplicates were used for each condition. n = 3; *p < 0.05.
Conversely, two proteins were reduced in MEEC conditioned media (Figure 2A): interleukin 6 (IL-6) and vascular endothelial growth factor (VEGF). IL-6 can play both the pro-inflammatory and anti-inflammatory roles, depending on the in vivo conditions; however, in blood vessels the role is usually considered pro-inflammatory. VEGF, on the other hand, is a pro-angiogenic factor, which can aid in repair but can also lead to unorganized vessel growth when disproportionately elevated. Reduction in both of these factors suggests that cells are in a less reactive state, which has been shown to be beneficial to MEEC-based tissue repair.
2.3. The Progenitor Marker CD34 is Elevated in MEECs
The proven therapeutic effects of MEEC regulation and the understanding that EC morphology and secretome are significantly modified when matrix embedded led us to hypothesize that these cells may have a progenitor-like cellular phenotype. This prompted examination of ECs and EPCs (both in 2D and matrix-embedded) for associated cellular markers. As expected, none of the cells expressed immune (CD14), hematopoietic (CD45), or stem cell markers (CD133) (not shown). Furthermore, all the cells expressed the endothelial marker, CD31. This further exemplifies the plasticity of EC in response to their spatiotemporal and physiochemical environment.
Most interestingly, CD34, a transmembrane protein usually found on progenitor cells including EPCs, but not on mature macrovascular ECs,[16–18] was detected in MEECs (Figure 3). 2D-ECs expressed very low levels of CD34 (1.5% CD34+ cells at day 14) similar to mature macrovascular ECs in vivo,[18] but expression increased in MEECs as a function of culture duration from 1.6% at day 3 to 21.2% at day 14. The high expression of CD34 in MEECs at day 14 is similar to that of EPCs cultured in the same scaffold (ME-EPC, 14.4%) and that of cord blood EPCs in vivo.[17] EPCs are isolated precisely by selection for CD34, but lost this expression when cultured in 2D (2.5% at day 14); however, embedding these cells in 3D matrices allowed them to regain CD34 expression over time (Figure 3A,B). Therefore, matrix embedding allows ECs to attain a progenitor-like phenotype and prevents EPCs from losing their phenotype.
Figure 3.


Expression of CD34 (an EPC marker) in ECs and EPCs as a function of time and substratum by FACS. A) Expression in MEECs and ME-EPCs at 3 d is as low as that of 2D-ECs and 2D-EPCs; however, expression increases in both cell types in 3D as a function of time. B) While mature ECs in the vessel wall have only ≈1% CD34+ cells,[18] MEECs 14 d in culture have a CD34 expression that matches and even surpasses that of ME-EPCs. Conversely, EPCs are highly CD34-expressing in vivo,[17] but lose this marker when cultured in 2D, resulting in expression levels that are comparable to 2D-ECs. n = 3; *p < 0.05.
2.4. Gene Expression of MEECs is Similar to that of EPCs
The marked similarity in CD34 expression profiles between MEECs and EPCs motivated an analysis of the expression of 84 EC biology genes in MEECs, ME-EPCs, 2D-ECs, and 2D-EPCs to better understand the effects of substratum on EC phenotype (Figure 4) and the genetic similarities between MEECs and EPCs (Figure 5).
Figure 4.

Differential expression of endothelial biology genes as a function of substratum. A) Six genes are significantly upregulated in both ECs and EPCs in matrix-embedded culture compared to 2D. C) Fourteen further genes are upregulated specifically in MEECs compared to 2D-ECs. B,D) For each set of genes, the interaction maps show connections between these identified genes (circles) and neighboring downstream/upstream genes (diamonds). E) The 20 genes encompass a variety of endothelial functions. All genes shown were significantly upregulated (p < 0.05) by more than twofold after comparison of three samples per cell–substrate condition.
Figure 5.

Expression profile of endothelial biology genes in ME-EPCs compared to MEECs and 2D-ECs. A) While 14 genes had significantly increased expression in ME-EPCs compared to 2D-ECs, B) only 3 genes were upregulated in ME-EPCs compared to MEECs, C) with 2 of those common to both comparisons. D) Of the 14 genes differentially expressed between EPCs and 2D-ECs, 8 of these are indeed also seen when comparing MEECs and 2D-ECs. Unlike 2D-ECs, then MEECs attain a genetic fingerprint similar to ME-EPCs. E) The pathways of MAPK1, TGFB1, and HIF1A, which are central to this expression profile (Circles: genes probed; Diamonds: neighboring upstream/downstream genes). F) The genes covers a variety of functions. All genes shown were significantly upregulated (p < 0.05) by more than twofold after comparison of three samples per cell–substrate condition.
Comparing effects associated directly with changes in substratum, six genes were found upregulated in matrix embedding regardless of cell type (MEEC vs 2D-EC and ME-EPC vs 2D-EPC) (Figure 4A). These genes are involved in angiogenesis (platelet and endothelial cell adhesion molecule 1, PECAM1; and fms related tyrosine kinase 1, FLT1); inflammation (arachidonate 5-lipoxygenase, ALOX5; and phospholipase A2 group IVC, PLA2G4C); vasodilation (prostaglandin I2 synthase, PTGIS); and extracellular matrix construction (collagen type XVIII alpha 1 chain, COL18A1). In terms of interactions, the central genes are VEGF type A (VEGFA), kinase insert domain receptor (KDR), a VEGF receptor; and protein tyrosine phosphatase non-receptor type 11 (PTPN11), involved in cell growth, differentiation, and mitotic cycle (Figure 4B,E). The proteins encoded by these genes are in interaction with VEGF and Src which, among other functions, protect ECs from apoptosis.[19]
Focusing specifically on EC response to substratum, 20 genes were found significantly upregulated in MEECs compared to 2D-ECs (Figure 4C), including most essentially PLA2G4C; C-X3-C motif chemokine ligand 1 (CXC3L1); and natriuretic peptide receptor 1 (NPR1). These genes are tied to signaling, inflammation, adhesion, and vascular tone (Figure 4E), and are themselves upregulated by transforming growth factor beta (TGF-β), nuclear factor kappa B (NF-κ B), tumor necrosis factor (TNF), and mitogen-activated protein kinase 1 (MAPK1), which are regulatory proteins central to hypoxia, proliferation, differentiation, and many more (Figure 4D). Interestingly, MEECs showed upregulation of the Kit proto-oncogene receptor tyrosine kinase (KIT), which encodes for the cytokine receptor CD117, a marker of hematopoietic stem cells involved in cell survival, proliferation, and differentiation.
Having explored the substratum-related differences, we compared EC and EPC genetic profiles: while 14 genes across were significantly different in ME-EPCs compared to 2D-ECs (Figure 5A), only 3 genes separated ME-EPCs from MEECs (Figure 5B). PTGIS and endothelin receptor typa A (EDNRA)—genes central to vascular tone—were the only two genes common to both comparisons, i.e., higher in ME-EPCs compared to ECs regardless of substratum; however, differential expression between EPCs and MEECs (2.6-fold, PTGIS; 6.8-fold, EDNRA) was less than between EPCs and 2D-ECs (17.1-fold, PTGIS; 8.4-fold, EDNRA) (Figure 5C). The sole gene expressed differentially between ME-EPCs and MEECs alone was matrix metallopeptidase 1 (MMP1)––a gene associated with tissue modification. Furthermore, of the 14 genes differentially expressed between EPCs and 2D-ECs (Figure 5A), 8 of these are also seen when comparing MEECs and 2D-ECs (Figure 5D).
In short, MEECs more closely resemble EPCs than 2D-ECs with increases in progenitor-like gene expression relevant to vascular repair including angiogenesis, vasodilation, coagulation, and tissue modulation, many of these with highly interconnected pathway interactions (Figure 5E,F). The most interconnected genes include FLT1 and KDR (both VEGF receptors), VEGFA, as well as two extracellular matrix modulators matrix metallopeptidase 2 (MMP2) and fibronectin 1 (FN1); and nitric oxide synthase (NOS2) which catalyzes nitric oxide, a potent vasodilator (Figure 5E). Upstream genes that are regulators of these genes include MAPK1, of the MAP kinase family; TGF-β, a potent growth factor; and hypoxia inducible factor 1 alpha subunit (HIF-1α), a master cell regulator in response to hypoxia. Two of these upstream regulatory genes (MAPK1 and TGF-β) are tied to gene regulation via the cytoskeleton as a result of substratum properties.[20,21]
3. Discussion
Cell–substratum interactions have long been an important area of research, especially for cells such as ECs, which have a definitive up or apical aspect that is exposed to flow, and a down or basal aspect that is anchored to underlying basement membrane. The a priori assumption has been that cell–substratum interactions affect cell biology. Hence, the plethora of studies creating specific 3D structures supports ECs. The current study is different––we examined the effects of embedding ECs in matrices following a posteriori empirical in vivo evidence that MEECs possess unique reparative properties. Indeed, MEECs control proliferation, hemostasis, and immunoreactivity, and elicit no immune reaction, unlike injection of isolated mature allogeneic ECs.[12,22]
These matrices offer tremendous advantages for cell therapy: they serve as a vehicle for handling of cells at precise and reproducible numbers over and again, preservation over months, and positioning in tissue spaces without dispersion. Yet, the question we asked is not whether these 3D structures are supportive or even if they are “better” than 2D culture but rather how and why MEECs attain their reparative phenotype, in this case by obtaining an EPC-like phenotype. Cells in 2D, on poly-styrene alone or coated with matrix materials, can become regulatory––their conditioned media can limit injury and induce repair when ECs are confluent. Along with several others, we have shown that this is because the ECs join in a physically contiguous monolayer that has contact inhibition and powerful countercurrent cell–cell interchange that governs cell state.[23,24] However, confluence is a delicate, metastable state that often degenerates into post-confluent overgrowth or inevitable cell loss and subconfluent regression. When not confluent, the same cells in the same environment will have the reverse effects, promoting injury and inhibiting repair.
ECs in 2D then can find themselves in one of three states as a function of confluence and external stimuli: (1) quiescence––the natural state of equilibrium in the absence of external force or stimulus; (2) activating––wherein cells respond to heal injury and respond to stimuli through a growth and acute response phase; and (3) deactivating––where the cells sensing that growth and reactivity are no longer necessary dampen these processes.[23,25–27] Vessels with an intact monolayer of quiescent ECs are relatively resistant to injury and powerful stimuli; however, there is a rampant response when the endothelial integrity is violated and/or denuded. The question then is whether the same is true when matrix embedded––will MEECs attain a regulatory phenotype when in contact with other or is there another mechanism at play?
Our data suggest otherwise. Yes, there are areas where MEECs appear confluent but they do not make an intact sheet, nor do MEECs align to create a monolithic plane. Rather, MEECs conform to the architecture of the substratum, assuming a shape that minimizes strain and cytoskeletal contortion.[15] Moreover, we now also show that these MEECs attain a state that is not the inhibitory, confluent, contact-inhibited phenotype but rather EPC-like. This is the difference—MEECs are far more like EPCs than 2D-ECs, even when confluent.
We must be careful here. MEECs are not likely transformed to EPCs nor can one be definitive about what an EPC is when ex vivo and in particular once cultured. EPC-derived ECs are also less immunogenic than aortic ECs.[28] EPCs and often ECs must be grown in culture from whatever numbers can be isolated directly from cord blood. The 2D culture in turn causes EPCs, as for most if not all cell types, to lose their native biological properties, most evident in the loss of CD34 expression. The exact biological role of CD34 remains unclear. Some suggest that this protein directly modulates proliferation of hematopoietic progenitor cells, promotion of lymphocyte adhesion, and immune regulation.[29,30] However, since CD34 is a membrane protein and given that MEECs are clinically active even as matrix embedded and perivascular, it would appear that the CD34 does not play a direct functional role in this context. Rather, we posit that CD34 is a marker that when expressed by a larger percentage of cells suggest a degree of cell population dedifferentiation with a modified secretome.
What we can say then is that matrix embedding enables EPCs to retain their physiologic expression, e.g., CD34 and immunomodulatory (e.g., leptin, IL-6, IL-8, and MCP-1) profile, and MEECs appear much like EPCs. For this reason, we consider the ME-EPC as being biologically similar to native EPC in terms of their biological properties and function. This may very well be a universal 3D effect that is amplified for “sided” cells. An important distinction can now be made. We can still strive to preserve endothelial monolayer integrity ex vivo to limit injury from mechanical, noxious, and pharmacology stress but when we envision how ECs in complex environments behave or how tissue-engineered constructs might be optimized we focus on attaining an EPC-like state rather than a confluent structure. Denuding procedures like angioplasty, endovascular stenting, vascular bypass, or injury from circulating toxins abrade and denude the arterial endothelial lining, inducing intimal hyperplasia following phases of inflammation, thrombosis and proliferation, and migration of vascular smooth muscle cells until the monolayer is restored. But when extravascular MEEC constructs control these same processes even in the presence of persistent denudation, they do so because the ECs are in a phenotype that resembles EPCs. This is a metric we can use to define native EC biology and optimize such tissue-engineered constructs.
4. Conclusions
ECs show a significantly different and intriguing phenotype when cultured in 3D matrices as compared to 2D surfaces. These biological changes include differences on the gene and secretome levels and marker expression and are primarily the result of the difference in substratum properties. Matrix embedding transforms ECs to an EPC-like biological profile which suggests that they may de-differentiate to an extent when cultured in the denatured collagen matrix environment. These findings are of importance with regard to understanding the mechanism of action and optimization of matrix-embedded cell therapy. In a wider context, these findings highlight the significance of cell–substratum interactions on cell biology, not only in terms of differentiation pathways, but also on all biological levels from gene to secreted protein milieu. Furthermore, these findings suggest that one can utilize materials science as a tool to program cell phenotype and behavior and to maintain a stable phenotype post-implantation.
5. Experimental Section
Cell Culture
Primary human aortic ECs (HAECs, Lonza) and cord blood EPCs (Lonza) were isolated[17] and cultured in Endothelial Growth Medium-2 (EGM-2, Lonza) supplemented with an additional 8% (10% total) fetal bovine serum (FBS), penicillin (100 U mL−1) and streptomycin (100 μg mL−1), on gelatin-coated tissue culture polystyrene (gTCPS) plates (0.1% gelatin type A, Sigma, St. Louis, MO) and used between passages 2 and 6. All EGM-2 growth media referenced below was supplemented in the same fashion. Cells were passaged by detachment with trypsin (0.05% trypsin/0.04% ethylenediaminetetraacetic acid) and split 1:4. EC-conditioned media were generated from confluent HAEC and EPC monolayers by 48 h of culture in EGM-2. Cells and debris were removed by centrifugation (5 min, 500 × g), and media were aliquoted and stored at −80 °C. Cells were cultured under standard conditions (37 °C, 5% CO2).
MEECs were generated by culturing ECs within sterile-compressed denatured collagen matrices (Pfizer, New York, NY). Matrices were cut into 1 × 0.5 × 0.3 cm blocks and hydrated in EGM-2 at 37 °C for 4–24 h. 2 × 104 ECs were suspended in 50 μL of medium, seeded onto hydrated denatured collagen matrices, and allowed to attach for 1.5 h. Then the contralateral side was seeded with an equal number of cells (4 × 104 cells total). After additional 1.5 h for cell attachment, matrices with cells were placed in 50 mL polypropylene tubes containing 6 ml of EGM-2 (two blocks per tube). MEECs were cultured for up to 2 weeks, with media changed every 48–72 h, under standard culture conditions. Samples from each lot were digested with collagenase type I (2 mg mL−1, Worthington Biochemicals), and cell number was determined with a Z1 Coulter particle counter (Beckman Coulter). Cell viability was assessed by trypan blue exclusion.
To facilitate scaffold imaging, prior to cell seeding collagen matrices were incubated in a corresponding volume of 0.2 M sodium bicarbonate buffer and 1.7% (v/v) Texas Red (T6134, Invitrogen) dissolved in dimethyl sulfoxide (DMSO). The matrices were incubated for 2 h at room temperature and then washed twice for 1 h in PBS and then once overnight in PBS to remove all traces of unconjugated Texas Red.
Generation and Identification of Cells Labeled with enhanced Green Fluorescent Protein (GFP)
HAECs (Invitrogen) were transduced with retroviral particles expressing enhanced GFP generated in HEK293T cells. To generate viral particles, subconfluent HEK293T cells were co-transfected with packaging and capsid plasmids along with pGEP retroviral plasmid using Lipofectamine 2000 per manufacturer’s instructions. The medium harvested after 48 h was cleared of the cell debris and ultracentrifuged for 90 min at 60,000 × g at 4 °C. The viral pellet flesh frozen was used to transduce target cells using polybrene (Sigma). To generate stable GFP expression, the highest 5% of GFP-expressing cells were selected by fluorescence-activated cell sorting (FACSAria, BD). These cells were expanded and seeded on gTCPS or matrices as described. Microscopy was performed using a Zeiss LSM 510 meta laser scanning confocal microscope.
RNA Expression Measurement and Analysis
RNA extraction for reverse-transcription polymerase chain reaction (RT-PCR) was done using the RNeasy kit (Qiagen). ECs and EPCs were isolated by flash freezing collagen matrices with cells, adding 500 μL RLT buffer (Qiagen) and physically disrupting the gelatin by pipetting with a 1-ml syringe. Prior to adding RNA to the RNeasy column, the resulting mix was strained through a 40 μm cell strainer cap (BD). RNA from cells grown on gTCPS was processed as specified in the Qiagen RNeasy protocol. RNA purity was verified using a Bioanalyzer (Agilent). RT-PCR was done using the human endothelial cell biology PCR Array (SAbiosciences) according to the kit instructions and accompanying materials for first strand RNA production and SYBR green qPCR master mix. Results were obtained from a LightCycler 480 (Roche). For each cell–substrate condition, three samples were tested three separate times—only genes that were more than twofold upregulated and had a p-value of less than 0.05 were considered as significantly up- or downregulated. Gene and pathway interactions were analyzed using GNCPro (SAbiosciences).
Cell Markers and Flow Cytometry (FC)
EC and EPCs were harvested from 2D gTCPS using trypsin and from 3D scaffolds using collagenase (as described). Cells were then pelleted, washed, and resuspended in FC buffer (Phosphate buffered saline with 1% FBS, 0.1% sodium azide [Sigma]). After washing three times with FC buffer, cells were incubated with the appropriate primary and secondary antibodies. Following 30-min incubation at 4 °C, samples were again washed twice with cold FC buffer, and 5 × 105 cells from three replicate samples of each cell–substrate condition were analyzed by flow cytometry (LSR II Cytometer, BD Biosciences). The antibodies used were conjugated to either phycoerythrin (PE) or fluorescein isothiocyanate (FITC), and were as follows: anti-human CD31 PE, anti-human CD144 FITC, anti-mouse IgG1k PE, anti-human CD34 FITC, anti-human CD309 PE, anti-mouse IgG1k FITC (BD Biosciences); anti-mouse IgG2a FITC, anti-mouse IgG2a PE, and anti-human CD45 PE (Miltenyi); and anti-human CD14 FITC, anti-human CD133 PE, and anti-mouse IgG1 PE (Biolegend).
Protein Array
Conditioned media were analyzed for the expression of 43 protein analytes with a membrane dot blot (Human Angiogenesis Array C1000, RayBiotech, Norcross, GA) per the manufacturer’s instructions, using three samples per cell–substrate condition and unconditioned medium as a cell-free control. Spot intensity for each analyte was normalized to positive control spots on each membrane and biological duplicates were used for each condition.
Statistics
One-way analysis of variance for independent samples followed by Tukey’s Honestly Significant Difference (HSD) Test was used for analysis of cell markers. Analysis of RT2 gene array was conducted using the online RT2 profiler PCR array data analysis version 3.5. For all statistical analysis, the value of p < 0.05 was taken as statistically significant.
Acknowledgments
E.A., J.F., V.C., N.A., and E.R.E designed and performed research. E.A., O.G., J.F., N.A., and E.R.E. analyzed data. E.A., O.G., N.A., and E.R.E. wrote the paper. E.R.E. was supported in part by a grant from the NIH (R01 GM49039) and E.A. was supported by the NIH Ruth L. Kirschstein postdoctoral training fellowship, Harvard Medical School Training Program in Molecular Hematology.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Dr. Eytan Abraham, Institute for Medical Engineering and Science, Massachusetts Institute of Technology, E25-438, Cambridge, MA 02139, USA. Department of Medicine, Brigham and Women’s Hospital, Cardiovascular Division, Harvard Medical School, Boston, MA 02115, USA
Or Gadish, Institute for Medical Engineering and Science, Massachusetts Institute of Technology, E25-438, Cambridge, MA 02139, USA.
Dr. Joseph W. Franses, Institute for Medical Engineering and Science, Massachusetts Institute of Technology, E25-438, Cambridge, MA 02139, USA
Prof. Vipul C. Chitalia, Renal Section, Department of Medicine, Boston University School of Medicine, Boston, MA 02118, USA
Prof. Natalie Artzi, Institute for Medical Engineering and Science, Massachusetts Institute of Technology, E25-438, Cambridge, MA 02139, USA. Department of Medicine, Division of Engineering in Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA
Prof. Elazer R. Edelman, Institute for Medical Engineering and Science, Massachusetts Institute of Technology, E25-438, Cambridge, MA 02139, USA. Department of Medicine, Brigham and Women’s Hospital, Cardiovascular Division, Harvard Medical School, Boston, MA 02115, USA
References
- 1.Justice BA, Badr NA, Felder RA. Drug Discovery Today. 2009;14:102. doi: 10.1016/j.drudis.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 2.Elliott NT, Yuan F. J Pharm Sci. 2011;100:59. doi: 10.1002/jps.22257. [DOI] [PubMed] [Google Scholar]
- 3.Engler AJ, Sen S, Sweeney HL, Discher DE. Cell. 2006;126:677. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 4.Wells RG. Hepatology. 2008;47:1394. doi: 10.1002/hep.22193. [DOI] [PubMed] [Google Scholar]
- 5.Winer JP, Janmey PA, McCormick ME, Funaki M. Tissue Eng Part A. 2009;15:147. doi: 10.1089/ten.tea.2007.0388. [DOI] [PubMed] [Google Scholar]
- 6.Frith JE, Thomson B, Genever PG. Tissue Eng, Part C. 2010;16:735. doi: 10.1089/ten.TEC.2009.0432. [DOI] [PubMed] [Google Scholar]
- 7.Shin L, Peterson DA. Stem Cells Transl Med. 2013;2:33. doi: 10.5966/sctm.2012-0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seib FP, Prewitz M, Werner C, Bornhauser M. Biochem Biophys Res Commun. 2009;389:663. doi: 10.1016/j.bbrc.2009.09.051. [DOI] [PubMed] [Google Scholar]
- 9.Hong H, Stegemann JP. J Biomater Sci, Polym Ed. 2008;19:1279. doi: 10.1163/156856208786052380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mishra DK, Sakamoto JH, Thrall MJ, Baird BN, Blackmon SH, Ferrari M, Kurie JM, Kim MP. PLoS One. 2012;7:e45308. doi: 10.1371/journal.pone.0045308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nugent HM, Ng YS, White D, Groothius A, Kanner G, Edelman ER. J Vasc Interv Radiol. 2009;20:1617. doi: 10.1016/j.jvir.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gadish O, Edelman ER. In: Translational Regenerative Medicine. Atala A, Allickson JG, editors. Ch. 14 Elsevier Academic Press; Amsterdam, The Netherlands: 2015. [Google Scholar]
- 13.Nathan A, Nugent MA, Edelman ER. Proc Natl Acad Sci USA. 1995;92:8130. doi: 10.1073/pnas.92.18.8130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Methe H, Nugent HM, Groothuis A, Seifert P, Sayegh MH, Edelman ER. Circulation. 2005;112:I89. doi: 10.1161/01.CIRCULATIONAHA.105.524991. [DOI] [PubMed] [Google Scholar]
- 15.Indolfi L, Baker AB, Edelman ER. Biomaterials. 2012;33:7019. doi: 10.1016/j.biomaterials.2012.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, Witzenbichler B, Schatteman G, Isner JM. Science. 1997;275:964. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 17.Hirschi KK, Ingram DA, Yoder MC. Arterioscler, Thromb, Vasc Biol. 2008;28:1584. doi: 10.1161/ATVBAHA.107.155960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fina L, Molgaard HV, Robertson D, Bradley NJ, Monaghan P, Delia D, Sutherland DR, Baker MA, Greaves MF. Blood. 1990;75:2417. [PubMed] [Google Scholar]
- 19.Eliceiri BP, Paul R, Schwartzberg PL, Hood JD, Leng J, Cheresh DA. Mol Cell. 1999;4:915. doi: 10.1016/s1097-2765(00)80221-x. [DOI] [PubMed] [Google Scholar]
- 20.Pullikuth AK, Catling AD. Cell Signaling. 2007;19:1621. doi: 10.1016/j.cellsig.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wells RG, Discher DE. Sci Signaling. 2008;1:pe13. doi: 10.1126/stke.110pe13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Methe H, Hess S, Edelman ER. Thromb Haemostasis. 2007;98:278. [PubMed] [Google Scholar]
- 23.Mi LY, Ettenson DS, Edelman ER. Cell Proliferation. 2008;41:671. doi: 10.1111/j.1365-2184.2008.00544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ettenson DS, Koo EW, Januzzi JL, Edelman ER. J Cell Physiol. 2000;184:93. doi: 10.1002/(SICI)1097-4652(200007)184:1<93::AID-JCP10>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 25.Nugent MA, Nugent HM, Iozzo RV, Sanchack K, Edelman ER. Proc Natl Acad Sci USA. 2000;97:6722. doi: 10.1073/pnas.97.12.6722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liebner S, Cavallaro U, Dejana E. Arterioscler, Thromb, Vasc Biol. 2006;26:1431. doi: 10.1161/01.ATV.0000218510.04541.5e. [DOI] [PubMed] [Google Scholar]
- 27.Dejana E. Nat Rev Mol Cell Biol. 2004;5:261. doi: 10.1038/nrm1357. [DOI] [PubMed] [Google Scholar]
- 28.Ladhoff J, Fleischer B, Hara Y, Volk HD, Seifert M. Cardiovasc Res. 2010;88:121. doi: 10.1093/cvr/cvq109. [DOI] [PubMed] [Google Scholar]
- 29.Nielsen JS, McNagny KM. J Cell Sci. 2008;121:3683. doi: 10.1242/jcs.037507. [DOI] [PubMed] [Google Scholar]
- 30.Drew E, Merzaban JS, Seo W, Ziltener HJ, McNagny KM. Immunity. 2005;22:43. doi: 10.1016/j.immuni.2004.11.014. [DOI] [PubMed] [Google Scholar]