

Abstract
Stem cell transplantation, as used clinically, suffers from low retention and engraftment of the transplanted cells. Inspired by the ability of platelets to recruit stem cells to sites of injury on blood vessels, we hypothesized that platelets might enhance the vascular delivery of cardiac stem cells (CSCs) to sites of myocardial infarction injury. Here, we show that CSCs with platelet nanovesicles fused onto their surface membranes express platelet surface markers that are associated with platelet adhesion to injury sites. We also find that the modified CSCs selectively bind collagen-coated surfaces and endothelium-denuded rat aortas, and that in rat and porcine models of acute myocardial infarction the modified CSCs increase retention in the heart and reduce infarct size. Platelet-nanovesicle-fused CSCs thus possess the natural targeting and repairing ability of their parental cell types. This stem cell manipulation approach is fast, straightforward and safe, does not require genetic alteration of the cells, and should be generalizable to multiple cell types.
The mortality of cardiovascular disease poses an immense burden on society1. New therapeutic strategies including stem cell therapies and tissue engineering products hold the potential to alter the trajectory of disease progression after an initial insult such as acute myocardial infarction (MI)2,3. One of the big challenges is targeting the injected stem cells to the injury site. Therapeutic benefits are hampered by the low cell retention in the target tissue4. For example, it has been reported that more than 90% of transplanted cells are ‘washed out’ hours after transplantation regardless of cell type and delivery route5,6. Vascular routes (such as intravenous or intracoronary) are relatively safe but have even poorer cell retention rates as compared to direct muscle injection. This partially explains the inconsistent and marginal therapeutic benefits seen in meta-analysis of stem cell therapy outcomes for heart diseases7. Novel approaches are urgently needed to better target infused stem cells to the MI injury site6.
The vascular endothelium provides a barrier between the subendothelial matrix and circulating cells such as haematocytes and platelets. It has been established that ischaemic heart injuries such as acute MI can induce vascular damage and expose components of the subendothelial matrix including collagen, fibronectin and von Willebrand factor (vWF) to recruit platelets. Platelets can also accumulate and bind directly to injured endothelial cells. Various platelet surface molecules such as glycoprotein (GP)VI, GPIV, GPIb, GPIX, GPV and GPIIb/IIIa are involved in platelet recruitment8. It has previously been reported that platelets could form co-aggregates with circulating CD34+ progenitors in patients with acute coronary syndromes, and these co-aggregates improve prognosis by promoting peripheral recruitment of CD34+ cells in the ischaemic microcirculatory area and boosting their adhesion to the vascular lesion9.
Over the past seven years the regenerative potential of cardiosphere-derived cardiac stem cells (CSCs) as a treatment for MI has been investigated in laboratory animal model studies10–14 and a recently completed phase I clinical trial15,16. However, similarly to other cell types, CSCs suffer from low cell retention in the heart after delivery5.
In this study, we sought to harness the natural MI-homing ability of platelets to enhance the vascular delivery of CSCs to the site of MI injury. We came up with a design of decorating platelet nanovesicles (PNVs) onto the surface of CSCs. Such decoration was nontoxic as it did not alter the viability and functions of CSCs, but augmented the targeting of the engineered PNV-fused CSCs to the MI for enhanced therapeutic outcomes.
Results
Intravenously injected platelets target myocardial infarction
To evaluate the natural MI-homing ability of platelets, we intravenously injected DiI-labelled platelets through the tail vein in animals with recent ischaemia/reperfusion-induced MI (Fig. 1a). Ex vivo fluorescent imaging at 1 hr post injection revealed that a larger number of injected platelets were retained in the MI heart as compared to the Sham heart (no MI) (Fig. 1b). Histology further confirmed platelets concentrated at the region of injured myocardium (Fig. 1c). These results confirmed the MI-homing ability of platelets and suggested the potential of targeting PNV-engineered stem cells to the MI region.
Fig. 1. Platelet binding to myocardial infarction sites and the derivation of platelet nanovesicles.
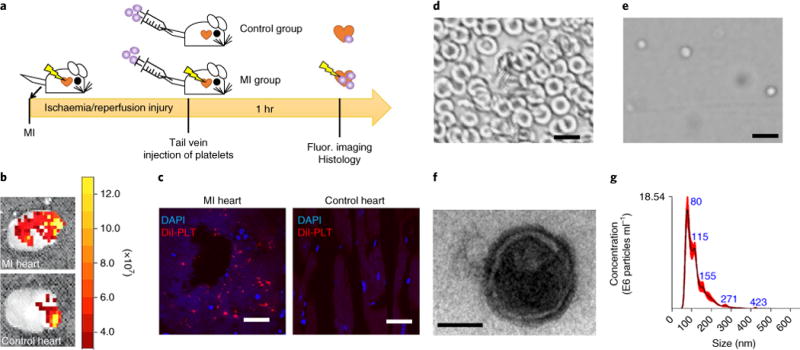
a, A schematic showing the animal study design to test the innate binding ability of platelets to sites of myocardial infarction (MI). b, Representative ex vivo fluorescent imaging showing binding of intravenously injected DiI-labelled platelets in hearts with or without ischaemia/reperfusion (I/R) injury. c, Representative fluorescent microscopic images showing the targeting of Dil-labelled platelets (red) to the MI area (DAPI, nuclei). Scale bars, 100 μm. d,e, Collected rat red blood cells (d) as seen under a light microscope, demonstrating a distinctive morphology compared to platelets (e). Scale bars, 10 μm. f, A transmission electron micrograph of a platelet nanovesicle. Scale bar, 100 nm. g, Size examination of platelet membrane nanovesicles by NanoSight.
Characterization of PNVs and CSCs
To keep the platelet adhesion molecules but avoid the use of live platelets (which are pro-thrombotic), we derived PNVs from intact platelets. Bright-field imaging showed distinctive morphologies of red blood cells (Fig. 1d) and platelets (Fig. 1e). Transmission electron microscopy showed the morphology of PNVs (Fig. 1f). NanoSight analysis revealed the size distribution of PNVs (Fig. 1g) with an approximate average size of 100 nm. Rat CSCs were derived from rat myocardium using the cardiosphere method (Supplementary Fig. 1).
Characterization of PNV-CSCs
PNVs were derived from platelets and decorated onto the surface of CSCs to form PNV-decorated CSCs (PNV-CSCs) through membrane fusion (Fig. 2a). Fluorescent microscopy showed red fluorescent DiI-labelled CSC (red, Fig. 2b) were decorated with green fluorescent DiO-labelled PNVs (yellow, Fig. 2c). To demonstrate that the fluorescence overlapping was not simply from dye transfer, we co-cultured PNV-CSCs (yellow) with control CSCs (red) for 24 hrs. There was no evidence of dye transfer from PNV-CSCs to CSCs (Fig. 2d). Western blot analysis further revealed distinct platelet surface markers including CD42b (GPIbα), GPVI and CD36 (GPIV) expression in platelets, PNVs and PNV-CSCs, but not in control CSCs (Fig. 2e and Supplementary Figs. 2–4). Immunocytochemistry staining confirmed CD42b (GPIbα) and GPVI expression on PNV-CSCs (Fig. 2f, upper panels) but not on control CSCs (Fig. 2f, lower panels). These data suggest successful PNV fusion with CSCs. Expression of primary platelet membrane proteins or protein subunits on PNV-CSCs revealed that the engineered PNV-CSC inherited the binding motifs of platelets. Flow cytometry analyses confirmed the expressions of platelet and exosomal markers on PNV-CSCs, but not on CSCs (Fig. 2g,h and Supplementary Fig. 5). In addition, we employed a fluorescence-based assay and estimated the numbers of PNVs coating each CSC (Supplementary Fig. 6), which decreased as expected with increasing passages.
Fig. 2. Generation and characterization of PNV-CSCs.
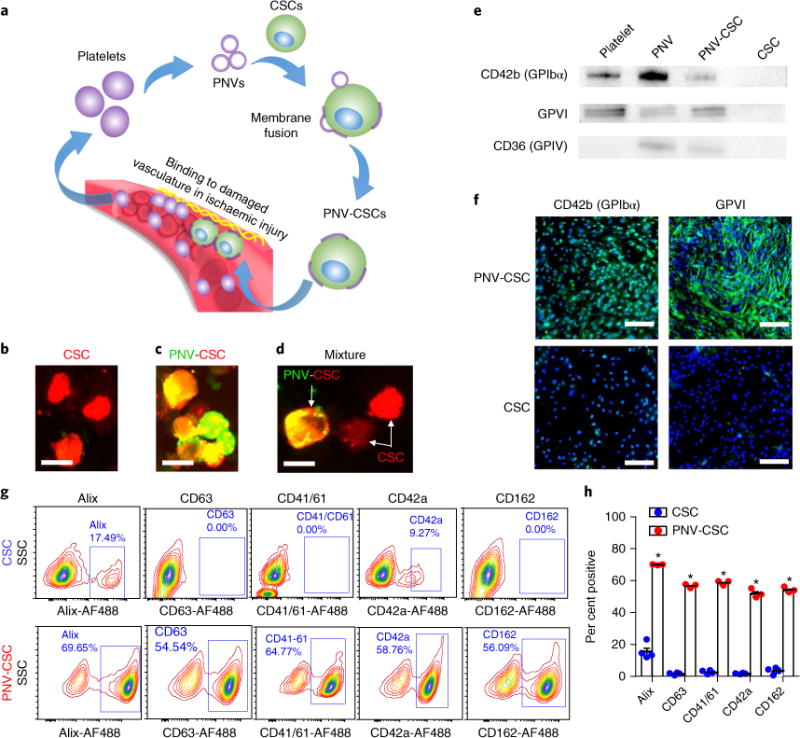
a, A schematic showing the overview of PNV decoration and PNV-CSC therapy. b,c, Red fluorescent DiI-labelled CSCs (b) were fused with green fluorescent DiO-labelled PNVs to form PNV-CSCs (c). d, Co-incubation of CSCs (red) with PNV-CSCs (yellow). Scale bar, 20 μm. e, Western blot analysis revealed the expressions of platelet-specific markers including CD42b (GPIbα), GPVI and CD36 (GPIV) in platelets, PNVs and PNV-CSCs, but not in CSCs. Original western blot images can be found in Supplementary Figs. 2–4. f, Immunocytochemistry staining confirmed CD42b (GPIbα) and GPVI expression in PNV-CSCs (top), but not in CSCs (bottom). Scale bars, 200 μm. g,h, Flow cytometric analysis of platelet and exosome surface marker expressions on PNV-CSCs (n = 3) and CSCs (n = 4). *P< 0.05. All values are mean ± s.d. Two-tailed t-test for comparison.
PNV decoration does not affect PNV-CSC viability and function
To further explore if PNV decoration affects the viability and function of PNV-CSCs, a live/dead assay was performed on PNV-CSCs and CSCs cultured on tissue culture plates for 7 d (Fig. 3a; green = Calcein-AM=live, red = EthD = dead). Cumulative data indicates comparable cell viabilities between PNV-CSCs and CSCs (Fig. 3b). A CCK-8 assay indicated the proliferation rates of PNV-CSCs and CSCs were indistinguishable (Fig. 3c). A trans-well migration assay showed that PNV-CSCs and CSCs had similar migration potencies at various time points (Fig. 3d). As the release of growth factors such as insulin-like growth factor (IGF)-1, stromal-cell-derived factor (SDF)-1, vascular endothelial growth factor (VEGF) and hepatocyte growth factor (HGF) is a major mode of action of injected stem cells, it was confirmed by ELISA that their release from PNV-CSCs was not undermined by PNV decoration (Fig. 3e). These data suggest that PNV decoration is nontoxic to CSCs, and that PNV-CSCs can retain the regenerative potency of the original CSCs.
Fig. 3. The effects of PNV decoration on CSC viability and functions.
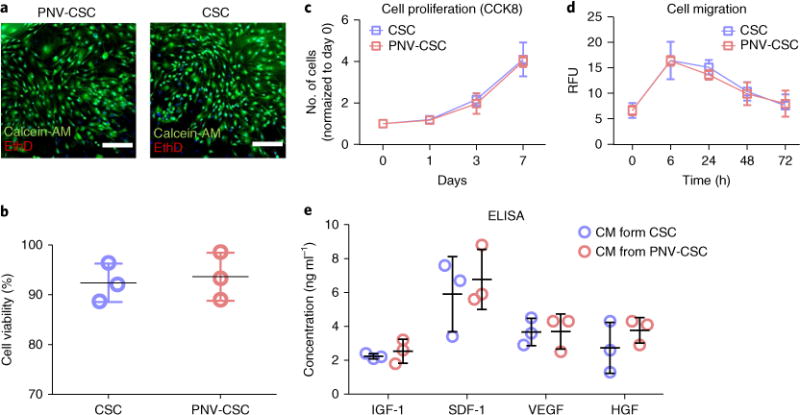
a, Representative fluorescent micrographs showing live (calcein-AM, green) and dead (EthD, red) staining of PNV-CSCs and CSCs cultured on tissue culture plates for 7 d. Scale bars, 200 μm. b, Pooled data of cell viability (n = 3 per group). c, CCK8 assay measurement of proliferation of PNV-CSCs and CSCs cultured on tissue culture plates (n = 3 per group at each time point). d, Trans-well migration assay showing the migration potencies of PNV-CSCs or CSCs (n = 3 per group at each time point). e, Determination of the release of growth factors IGF-1, SDF-1, VEGF and HGF in the conditioned media (CM) from CSCs and PNV-CSCs (n = 3 per group) by ELISA. Values are mean ± s.d. Two-tailed t-test for comparison.
PNV decoration does not promote blood clotting or cell aggregation
We employed a pro-thrombotic assay to evaluate whether PNV decoration promotes blood clotting, which is a risk factor in cell infusion (Supplementary Fig. 7). As expected, platelet-poor plasma (PPP) alone did not clot. The addition of 2 × 103 cells ml−1 of either CSCs or PNV-CSCs also did not induce clotting. Increasing concentrations of CSCs or PNV-CSCs did induce some clotting, and the rate of clotting increased in a dose-dependent manner. However, it should be noted that the rates of clot polymerization in the presence of 2 × 104 or 6 × 104 cells ml−1 were minimal (0.002–0.004 nm min−1) compared to PPP polymerized in the presence of thrombin (0.024 nm min−1). No differences in polymerization rates between CSCs and PNV-CSCs were observed at any cell dosage, indicating that PNV decoration does not promote the thrombogenicity of CSCs. In addition, a cell aggregation assay (Supplementary Fig. 8) showed that after 24 hrs of storage in heparin-free saline, cell aggregation occurred with both CSCs and PNV-CSCs, as indicated by increases in particle sizes. With the addition of heparin, cell aggregation was not evident in either group, and so PNV decoration did not promote cell aggregation. Furthermore, an aggregometry assay indicated that PNV-CSC did not promote platelet aggregation with or without the platelet agonist collagen (Supplementary Fig. 9).
Binding of PNV-CSCs to collagen surface and denuded aortas
We tested the binding potency of PNV-CSCs ex vivo in excised damaged vasculatures. A segment of rat aorta was obtained and surgically scraped to expose the subendothelial matrix (Fig. 4a). Microscopic imaging showed no binding of DiI-labelled PNV-CSCs or CSCs on control (non-denuded) aortas (Fig. 4b,c). Interestingly, PNV-CSCs had robust binding to the denuded aorta (Fig. 4d) while control CSCs did not (Fig. 4e). Quantitative analysis confirmed the superior binding of PNV-CSCs to denuded aortas with excellent specificity and sensitivity (Fig. 4i). In addition, DiI-labelled PNV-CSCs or CSCs were plated onto GFP-tagged human umbilical vein endothelial cells (HUVECs) cultured on collagen-coated surfaces (Fig. 4f) and augmented adherence of PNV-CSCs was confirmed (Fig. 4g,h,j).
Fig. 4. PNV decoration promotes CSC binding to damaged rodent vasculatures.
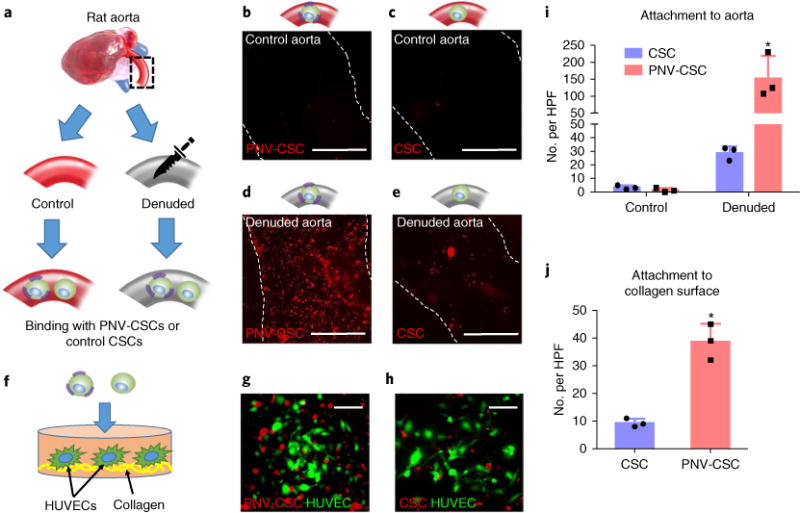
a, A schematic showing the experimental design for denuded rat aorta binding. b–e, Representative fluorescent micrographs showing the adherence of DiI-labelled PNV-CSCs and CSCs on control (b,c) or denuded (d,e) aortas. Scale bars, 1 mm. f, A schematic showing PNV-CSCs (left) or CSCs (right) seeded on collagen-coated tissue culture slides. HUVECs, human umbilical vein endothelial cells. g,h, Representative fluorescent images showing the binding of DiI-labelled PNV-CSCs (g) or control CSCs (h) on HUVECs cultured on collagen surfaces. Scale bars, 50 μm. i,j, Quantitative analysis of cell binding (n = 3 experiments per group). *P< 0.05 when compared to CSC group. All values are mean ± s.d. Two-tailed t-test for comparison between the two groups. HPF, high-power field.
PNV-CSCs exhibited superior retention and engraftment in rats with ischaemia/reperfusion injury
To test the therapeutic potential of PNV-CSCs, we employed a rat model of ischaemia/reperfusion by temporary left anterior descending coronary artery ligation for 1 hr (Fig. 5a) followed by reperfusion. After 20 mins of reperfusion, 500,000 PNV-CSCs or control CSCs were administered via an intracoronary injection. Cells were injected into the left ventricular cavity during temporary occlusions of the aorta, and so cells perfused into the myocardium through the coronary arteries in the closed circuit, which mimicked intracoronary injection. Ex vivo fluorescent imaging at 24 hrs revealed that PNV decoration boosted CSC retention in the heart (Fig. 5b). This was further confirmed by qPCR analysis. A cohort of female rats received PNV-CSCs or CSCs from male donors to quantify cell retention by SRY qPCR (Fig. 5c). Furthermore, immunohistochemistry staining on heart sections revealed superior engraftment of PNV-CSCs (red, Fig. 5e) over CSCs (red, Fig. 5d) in the post-MI heart. Quantitative analysis confirmed superior retention of PNV-CSCs in the heart (Fig. 5f). Thus, PNV-CSCs exhibited superior retention/engraftment in rats with ischaemia/reperfusion injury. Multiple-organ ex vivo fluorescent imaging revealed off-target expression in the liver and the kidney, which may represent cells that had escaped from the aorta and entered the main circulation after the temporary aortic occlusion procedure (Supplementary Fig. 10). Off-target expression could be mitigated by use of coronary catheters to carry out a true intracoronary injection. To rule out any immunogenicity that could have risen from the PNV decoration process, we examined CD3- and CD8-positive T cells in the hearts that had received PNV-CSCs or control CSCs. Only a negligible amount of CD3- and CD8-positive T cells were detected in CSC- or PNV-CSC-treated hearts, while the positive control group (hearts injected with human CSCs) exhibited severe T cell infiltration (Supplementary Fig. 11). Consistent with our previous work12,17, 4 weeks after injection, very few injected cells still persisted in the recipient heart, with even fewer cells exhibiting a cardiac phenotype. Nevertheless, the number of DiI/α-SA (α-sarcometric actin) double-positive cells was greater in the PNV-CSC group than that in the control CSC group (Supplementary Fig. 12).
Fig. 5. PNV decoration boosts CSC retention and therapeutic outcomes in rats with myocardial infarction.
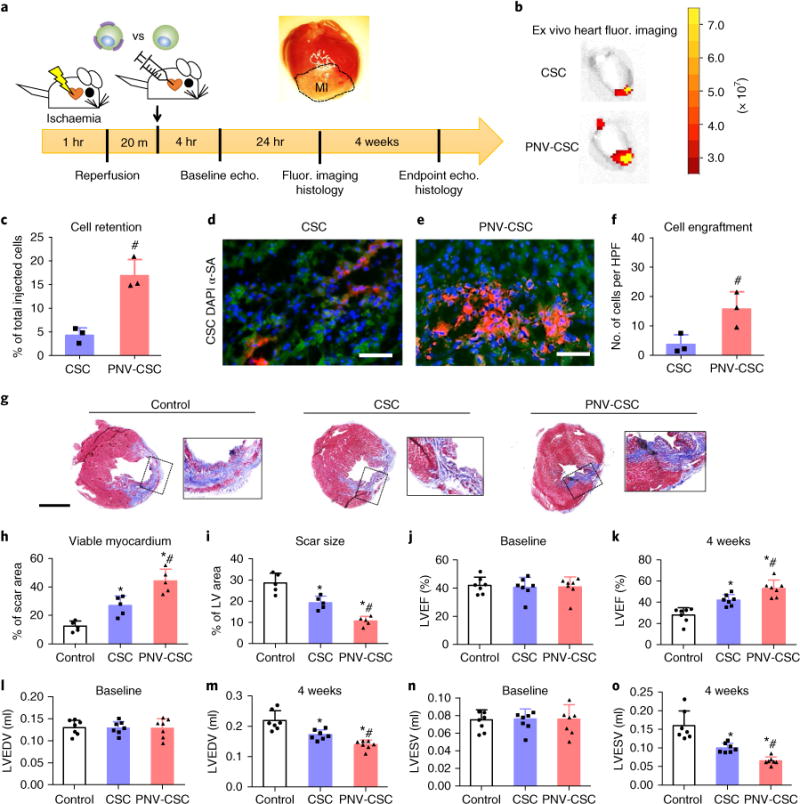
a, A schematic showing the animal study design. b, Representative ex vivo fluorescent imaging of ischaemia/reperfusion rat hearts 24 hrs after intracoronary infusion of DiI-labelled PNV-CSCs or CSCs. c, qPCR analysis revealed higher retention rates of PNV-CSCs as compared to those of CSCs (n = 3 animals/hearts per group). d,e, Representative fluorescent micrographs showing engrafted CSCs (d) or PNV-CSCs (e) in the post-MI hearts (green, α-SA antibody). Scale bars, 50 μm. f, Quantitative analysis of cell engraftment by histology (n = 3 animals/hearts per group). HPF, high-power field. g, Representative Masson’s trichrome-stained myocardial sections 4 weeks after treatment (blue, scar tissue; red, viable myocardium). Scale bar, 2 mm. h,i, Quantitative analyses of viable myocardium and scar size from the Masson’s trichrome images (n = 5 animals per group). LV, left ventricle. j,k, Left ventricular ejection fractions (LVEFs) measured by echocardiography at baseline (4 hrs post-MI) and 4 weeks later (n = 7 animals per group). l–o, Measurement of LV end-systolic (LVESV) and end-diastolic (LVEDV) volumes. *P<0.05 when compared to the control group; #P<0.05 when compared to the CSC groups. All values are mean ± s.d. Two-tailed t-test for comparison between two groups. One-way ANOVA with post-hoc Bonferroni test for comparisons that involve three or more groups.
Functional benefits of PNV-CSC therapy
To investigate whether increased cell retention translated into additional therapeutic benefits, we evaluated cardiac morphology, fibrosis and pump function. Masson’s trichrome staining 4 weeks after treatment (Fig. 5g; blue, scar tissue; red, viable myocardium) revealed apparent protection of heart morphology by CSCs compared to control-injected hearts, consistent with previous findings17,18. Interestingly, the best protective effects were observed in the PNV-CSC-treated hearts, which showed the highest amount of viable myocardium (Fig. 5h) with the smallest scar size (Fig. 5i). Left ventricular ejection fractions (LVEFs) were similar at baseline for all three groups (Fig. 5j). After 4 weeks, the LVEFs in control-treated hearts were reduced (white bar, Fig. 5k) while LVEF was retained in the CSC-treated animals (blue bar, Fig. 5k). The highest LVEFs were observed in the PNV-CSC group (red bar, Fig. 5k). In addition, LV end-systolic and end-diastolic volumes were well preserved in the PNV-CSC treatment group (Fig. 5l–o).
To investigate the mechanisms underlying the therapeutic benefits of PNV-CSCs, we performed a series of immunohistochemistry staining on the treated hearts. The results suggested that the PNV-CSC treatment led to increased cardiomyocyte cycling (Fig. 6a) and better retained relative blood flow, as assessed by lectin angiogram (Fig. 6b). Furthermore, PNV-CSC and CSC treatment increased vessel density compared to the control group (Fig. 6c).
Fig. 6. PNV-CSC therapy promotes myocyte proliferation and angiogenesis.
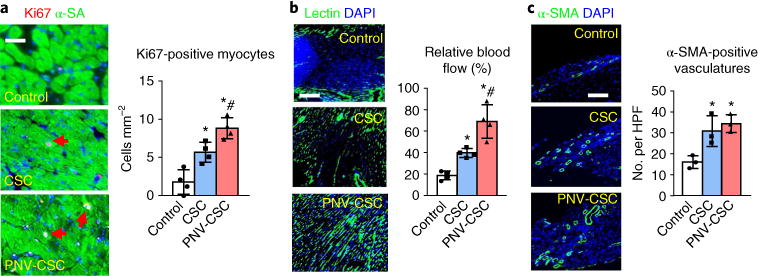
a, Representative images showing Ki67-positive cardiomyocyte nuclei (red, with red arrows) in control PBS-, CSC- or PNV-CSC-treated hearts at 4 weeks. Right: quantitative analysis of the numbers of Ki67-positive nuclei. n = 4 animals per group. Scale bar, 20 μm. b, Representative images indicating lectin-labelled blood vessels (green) in control PBS-, CSC- or PNV-CSC-treated hearts at 4 weeks. Right: quantification analysis of the lectin fluorescent intensities. n = 4 animals per group. Scale bar, 100 μm. c, Representative micrographs showing arterioles stained with antibody against α-smooth muscle actin (α-SMA, red) in PBS-, CSC- or PNV-CSC-treated hearts at 4 weeks. HPF, high-power field. The numbers of α-SMA-positive vasculatures were compared. n = 3 animals per group. Scale bar, 50 μm. *P< 0.05 when compared to the control group; #P< 0.05 when compared to the CSC group. All data are mean ± s.d. Comparisons were performed using one-way ANOVA followed by post-hoc Bonferroni test.
To further explore the adhesion molecules involved in the targeting of PNV-CSCs to the MI region, we used anti-CD42b neutralizing antibodies or isotype control antibodies to pretreat PNV-CSCs before the experiments. CD42b inhibition blunted the ability of PNV-CSCs to bind the denuded rat aorta (Fig. 7a,b), reduced the retention of PNV-CSCs in the heart (Fig. 7c,d) and ultimately diminished the therapeutic potency of PNV-CSCs in the same rat model of ischaemia/reperfusion injury (Fig. 7e). CD42b blocking had no effects on the retention of non-decorated CSCs in the heart (Supplementary Fig. 13). These results suggest CD42b plays an essential role in homing PNV-CSCs to MI injury. In addition, we screened other platelet receptors for their roles in mediating the binding potency of PNV-CSCs, using in vitro endothelial assays (Supplementary Fig. 14). Inhibition of CD42b and CD41/61, but not CD42a and CD162, reduced the ability of PNV-CSCs to bind endothelial cells.
Fig. 7. The role of CD42b in targeting PNV-CSCs to Mi injury.
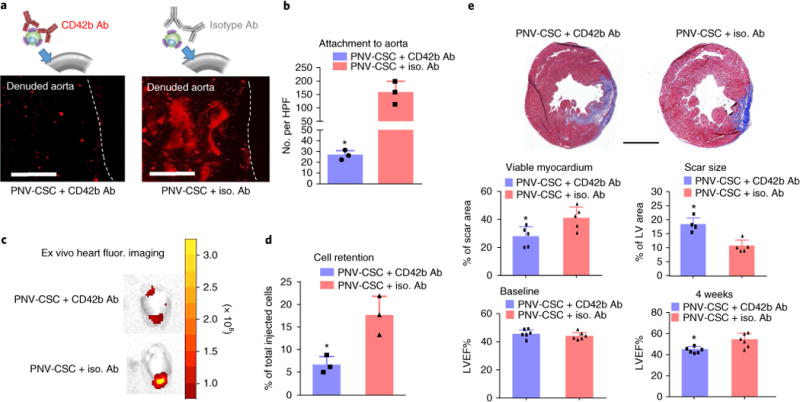
a, Representative fluorescent micrographs showing the adherence of anti-CD42b or isotype antibody pre-treated PNV-CSCs on denuded rat aortas. b, Quantitation of binding (n = 3 samples per group). HPF, high-power field. c, Representative ex vivo fluorescent imaging of ischaemia/reperfusion rat hearts 24 hrs after intracoronary infusion of anti-CD42b or isotype antibody pre-treated PNV-CSCs. d, Quantitation of cell retention by qPCR (n = 3 animals per group). e, Representative Masson’s trichrome-stained myocardial sections 4 weeks after treatment (blue, scar tissue; red, viable myocardium). Scale bar, 2 mm. Quantitative analyses of viable myocardium and scar size from the Masson’s trichrome images (n = 5 animals per group). Left ventricular ejection fractions (LVEFs) measured by echocardiography at baseline (4 hrs post-MI) and 4 weeks later (n = 6 animals per group). *P< 0.05 when compared to the PNV-CSC + iso. Ab group. All values are mean ± s.d. Two-tailed t-test for comparison.
We then translated the findings from the rodent study to a clinically relevant porcine model of acute MI injury. MIs were created in pigs with ischaemia/reperfusion (Fig. 8a). Blockage of blood flow in the left anterior descending coronary artery was evident in the angiogram when the balloon was inflated, and reperfusion was confirmed after the ischaemic period (Fig. 8b and Supplementary Videos 1–3). MI was further confirmed by an abnormal electrocardiogram reading after balloon occlusion (Fig. 8c). After 24 hrs the hearts were excised and sliced for cell retention and infarct size studies (Fig. 8d). Ex vivo fluorescent imaging revealed a stronger fluorescent signal (representing DiI-labelled cells) in the PNV-CSC-treated hearts (Fig. 8f), suggesting that PNV decoration enhanced cell retention in the injured heart. Triphenyl tetrazolium chloride (TTC) staining indicated that infusion of PNV-CSC therapy did not increase infarct size, suggesting the PNV modification on the stem cells did not exacerbate ischaemia/reperfusion injury (Fig. 8g,h).
Fig. 8. PNV decoration augments CSC retention in a porcine model of ischaemia/reperfusion.
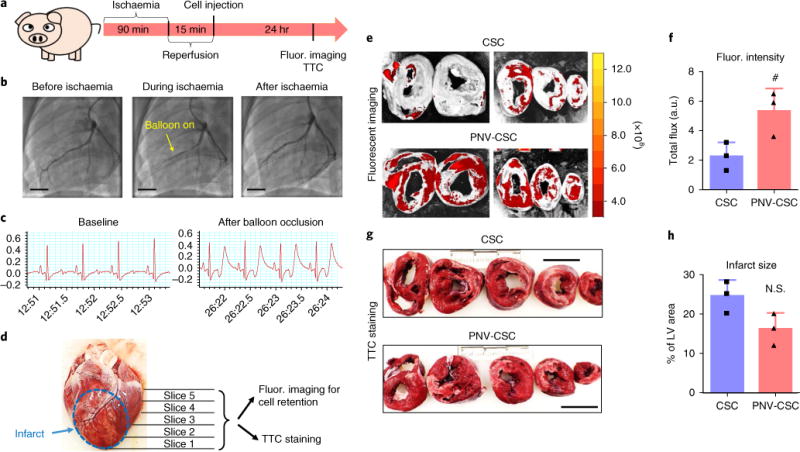
a, A schematic showing the pig study design. b, Angiograms showing coronary flow and placement of balloon before, during and after ischaemia. Scale bars, 15 mm. c, Representative electrocardiograms at baseline and after balloon occlusion. d, A representative excised pig heart image showing the preparation of myocardium slices for ex vivo fluorescent imaging and TTC staining. e, Representative ex vivo fluorescent imaging of ischaemia/reperfusion pig hearts 24 hrs after intracoronary infusion of CM-DiI-labelled CSCs or PNV-CSCs. f, Quantitation of fluorescent signals in CSC- or PNV-CSC-treated hearts (n = 3 pigs per group). g, Representative TTC staining images showing infarct region (white). Scale bars, 4 cm. h, Quantitation of infarct size (n = 3 pigs per group). #P< 0.05 when compared to the CSC group. All values are mean ± s.d. Two-tailed t-test for comparison.
Discussion
In the present study, we applied the natural injury-targeting power of platelets to enhance the vascular delivery and therapeutic outcome of CSCs. Our data showed that: (1) PNV decoration was effective and did not affect the viability and functions of CSCs in vitro; (2) PNV decoration enhanced CSC binding to denuded (injured) blood vessel ex vivo; (3) in a rat model of ischaemia/reperfusion, PNV decoration enhanced the targeting of CSCs to the MI injury site and augmented their ability to preserve cardiac pump functions and reduce infarct sizes; and (4) the targeting effects of PNV-CSCs were further confirmed in a porcine model of ischaemia/reperfusion.
To demonstrate this strategy, we first isolated platelets and confirmed their ability to target MI tissues (Fig. 1). PNVs were then generated from platelets and fused to CSCs to form PNV-CSCs, which expressed primary platelet membrane protein(s) subunits while retaining the regenerative potency of the CSCs (Figs. 2 and 3). Furthermore, PNV-CSCs exhibited superior binding to collagen surfaces in vitro and vascular lesions on denuded aortas ex vivo (Fig. 4). These data indicate that engineered PNV-CSCs inherited the binding ability and therapeutic potential of the parental platelets and CSCs, suggesting that they can lead to improved cell therapy in ischaemic heart diseases. In a rat ischaemia/reperfusion model, PNV-CSCs had higher cell retention, which translated into greater therapeutic benefits as compared to control CSCs (Fig. 5). Heart histology suggested that PNV-CSC treatment enhanced angiomyogenesis along with increased relative blood flow in the post-MI heart (Fig. 6). We also performed a mechanistic study to demonstrate that the platelet adhesion molecule CD42b played an important role in targeting PNV-CSCs to MI injury sites (Fig. 7). Finally, we translated the findings into a clinically relevant large animal (porcine) model of acute MI injury and confirmed the repeatability and robustness of our strategy (Fig. 8).
The PNV decoration strategy is not immunogenic. There were no increases in CD3- or CD8-positive cell numbers in hearts treated with PNV-CSCs as compared to CSCs (Supplementary Fig. 11). It has been reported that HLA antigens (class I and II) are lost from platelets during lysis and centrifugation19. In addition, previous work used human platelet membrane coated particles in immunocompetent rodents and reported no inappropriate immunoresponse20. We previously described the sequence of non-PNV modified CSC adhesion and extravasation after vascular delivery in MI hearts. Our data indicate that by 24 hrs post-injection CSCs are found adhering to the blood vessels and surrounded by endothelial projections21. By 72 hrs, the infused cells have extravasated into the extravascular space via a process we recently termed angiopellosis22. With the PNV modification, we speculate that the extravasation process can be expedited or amplified. Myocardial ischaemia leads to endothelial denudation and exposure of the thrombogenic subendothelial matrix to circulating platelets, and initiates platelet recruitment to the injured vessel wall23. Moreover, activated platelets can also bind to injured endothelial cells. It has been reported that platelet activation starts as early as 2 mins post-reperfusion after 45 mins of ischaemia24, and activation and recruitment of platelets can persist for days to weeks after MI injury9. Our data suggest that PNV decoration resulted in a boost of CSC retention in the heart. In vitro assays confirmed that PNVs help CSCs to adhere to endothelial cells and denuded blood vessels. Future studies should focus on whether PNV decoration also enhances the extravasation of CSCs.
The cell delivery method in the rat studies was a surrogate of intracoronary infusion as the cells were injected into the left ventricular cavity during a temporary aortic occlusion. Unavoidably, some cells may have escaped the cavity and entered the main circulation, causing off-target cell retention in the kidney and liver (Supplementary Fig. 10). The intracoronary infusion in our pig studies did not have this problem. Cells were directly injected while the catheter remained in the coronary artery. This mode of delivery should have reduced the incidence of off-target cell retention. Additionally, PNVs are not live platelets, and so lack the intracellular machinery to promote coagulation. Our pro-coagulation and prothrombosis assays (Supplementary Figs. 7–9) indicated that PNV decoration does not exacerbate the thrombotic risk of CSC infusion. In fact, if there are any pro-thrombotic risks, they would come from the CSCs rather than the PNVs. Nevertheless, we did detect some dose-dependent effects on the fibrin polymerization kinetics of CSCs as well as PNV-CSCs (Supplementary Fig. 7). However, there were no differences between CSCs and PNV-CSCs. Moreover, the safety of intracoronary CSC infusion has been demonstrated in human patients15,16.
Platelets have an innate ability to home to the vascular wall and specifically target vascular injury sites. To that end, platelet-like nanoparticles have been designed for haemostatic functions and targeted drug delivery25. Here, we have showed that platelet membranes can be employed to target stem cells to the injury sites. Nevertheless, our study has several limitations. First, to mimic intracoronary injection in rats, we temporarily occluded the aorta and injected the cells into the left ventricular cavity during this brief time period. Although the duration of aortic occlusion was standardized, the exact number of cells that actually reached the target myocardium may have varied among different animals, thereby introducing a bias in the interpretation of retention and engraftment rates and functional outcomes. Second, although the platelet membrane cloaking approach represents a straightforward and robust method to enhance injury targeting without genetic alternation of the cells, patients who have a prolonged history of taking anti-platelet medications may not be able to benefit from this targeting therapy. As another limitation, it should be recognized that because of the short-term nature of the pig study, it was impossible to demonstrate whether the observed cell retention enhancement translated into a better recovery of function in those pigs.
In summary, we provide evidence that platelet membranes can be employed to quickly modify stem cells before injection. Such modification is nontoxic and can increase the disease-targeting ability and functional outcomes of stem cell therapy.
Methods
Isolation of platelets and generation of platelet nanovesicles
Isolation of platelets and generation of platelet nanovesicles were performed as previously described20. Briefly, blood from WKY male rats was collected into an EDTA tube and then centrifuged at 100g for 20 mins at room temperature to separate red blood cells and white blood cells. The supernatant was collected containing platelets (namely platelet-rich plasma, or PRP) and further centrifuged at 100g for 20 mins to remove remaining blood cells. PBS with 1 mM of EDTA and 2 mM of prostaglandin E1 (PGE1, Sigma Aldrich, MO, USA) was added to the purified PRP to keep platelets inactivated. The isolated platelets were then pelleted by centrifugation at 800 g for 20 mins at RT. After removing the supernatant, the platelets were re-suspended in PBS containing 1 mM of EDTA and mixed with protease inhibitor (Thermo Fisher Scientific, MA, USA). Platelets were aliquoted into 1 ml samples and placed in −80 °C for storage before use. To generate PNVs, platelet samples were thawed to RT and centrifuged. The samples were then washed with PBS and mixed with protease inhibitors, followed by sonication. The size distribution and morphology of PNVs were examined using NanoSight (Malvern) and transmission electron microscopy, respectively.
Derivation and culture of rat CSCs
We obtained rat CSCs from the hearts of WKY rats as described in previously published papers12,26,27. Briefly, hearts were minced into fragments less than 2 mm3, followed by digestion with collagenase and then seeded onto fibronectin-coated plates to create explant-derived cells. In about 14 days, explant-derived cells were harvested and seeded in ultralow attachment flasks (Corning Life Sciences, Durham, NC) and grown in suspended culture for cardiosphere formation. CSCs were obtained by replating cardiospheres on fibronectin-coated plates.
Generation and characterization of PNV-CSCs
PNV decoration of CSCs was performed by mixing cells in the presence of polyethylene glycol (PEG), which is widely used to aid cell fusion28–31. Briefly, 1 × 106 DiI-labelled CSCs and 1 × 1010 DiO-labelled PNVs were mixed in 50 μl PEG for 5 mins. The suspension was then diluted by 10 ml warm serum-free medium and treated cells were retrieved by centrifugation at 410g for 5 mins. A fluorescence-based assay was performed to estimate the numbers of PNVs on each CSC after various passages. Cell proliferation, viability and migration of PNV-CSCs were characterized and compared to control CSCs. Cell Counting Kit-8 (Dojindo Molecular Technologies, MD, USA) was used to quantify cellular proliferation at day 0, 1, 3 and 7. The absorbance rate was read by a microplate reader (Tecan Sunrise, Switzerland). For cell viability, PNV-CSCs or CSCs were cultured on tissue culture plates for 7 days and then stained with a LIVE/DEAD Viability/Cytotoxicity Kit (Thermo Fisher Scientific). Live cell numbers in three randomized selected microscopic fields were counted. A trans-well plate setup allowed for cell migration through pores into the lower chamber where they could be detected. Fluorescently labelled PNV-CSCs or CSCs were incorporated and FBS served as a chemoattractant in the lower chamber. When the PNV-CSCs or CSCs migrated from the upper to the lower chamber, fluorescence (relative fluorescent unit; RFU) increased. Secretion of growth factors including IGF-1 (IGF-1 Quantikine ELISA Kit, R&D Systems, MN, USA), SDF-1 (CXCL12/SDF-1 alpha Quantikine ELISA Kit, R&D Systems), VEGF (VEGF Quantikine ELISA kit, R&D Systems), and HGF (HGF Quantikine ELISA Kit, R&D Systems) by PNV-CSCs and CSCs was determined by ELISA kits.
Examination of platelet-specific surface markers on PNV-CSCs
To further confirm successful membrane fusion, we performed western blot analysis on PNV- CSCs using antibodies against major platelet surface markers including rabbit anti-rat GPVI (Novus Bio, NBP1-76941), rabbit anti-CD42b (Santa Cruz, sc-292722) and rabbit anti-CD36 (Santa Cruz, sc-9154), followed by a 1 hr incubation with a goat anti-rabbit HRP-conjugated secondary antibody. For immunocytochemistry staining, PNV-CSCs or CSCs were plated on 4-well culture chamber slides (EMD Millipore, PEZGS0416). Slides were fixed with 4% PFA for 30 mins at room temperature followed by permeabilization and blocking with Dako Protein block containing 0.1% saponin for 1 hr at room temperature. Rabbit anti-rat GPVI (Novus Bio, NBP1-76941) and rabbit anti-CD42b (Santa Cruz, sc-292722) antibodies were incubated overnight at 4 °C followed by a 90 min incubation with a goat anti-rabbit Alexa fluor-488 secondary antibody (Abcam, ab150077). Nuclei were stained with DAPI for 10 mins at room temperature (Life Technologies, R37606). Slides were imaged on a fluorescent microscope (Olympus; Olympus IX81, Center Valley PA). Flow cytometry was performed on the CytoFlex (Beckman Coulter, Indianapolis, IN, http://www.beckman.com) and analysed using FCS Express (De Novo Software, Glendale, CA, https://www.denovosoftware.com). PNV-CSCs and CSCs were labelled with the following antibodies: mouse anti-CD162 (BD, 556053), mouse anti-CD41/61 (Novus, NBP1-97563), rabbit anti-Alix (Abcam, ab186429), mouse anti-CD42a (BD, 558816) and mouse anti-CD63 (Abcam, ab193349).
Pro-thrombotic and aggregation assay
To determine if PNV decoration induced any adverse pro-thrombotic effects, polymerization of human platelet-poor plasma (PPP) was analysed in the absence or presence of control (undecorated) CSCs and PNV-CSCs32,33. Briefly, samples were prepared to contain 85% PPP, 5% control CSCs, PNV-CSCs or ultrapure water, and 10% CaCl2 in ultrapure water. Three doses of CSCs or PNV-CSCs (2 × 103, 2 × 104 or 6 × 104 cells ml−1) were used. CaCl2 was used at a final concentration of 5 mM. We also included PPP in the presence of 0.5 U ml−1 human α-thrombin as a positive control of robust clotting. Immediately following mixing, samples were transferred to a 96-well plate and polymerization was monitored by measuring absorbance at 350 nm every minute for 2 hrs; as the clot polymerizes absorbance increases. The slope of the curve can be used to determine the rate of clot polymerization. Baseline absorbance readings were subtracted from the readings and the linear portion of the polymerization curves was fit using linear regression to determine the slope. The slope corresponds to the rate of polymerization and is expressed in nm per minute. Since CSCs will be stored in cold saline before administration to patients, we also performed a cell aggregation assay. PNV-CSCs and CSCs were stored in PBS with or without 100 U ml−1 heparin at 4 °C for 24 hrs. Cell clumping was analysed under a microscope. For aggregometry, PRP was harvested by collecting whole rat blood into 3.2% sodium citrated plastic tubes. The whole blood was centrifuged at 650g for 3 mins at room temperature. The PRP supernatant was then collected and 450 μl was added to a glass cuvette for aggregometry. Aggregometry was performed using a commercial optical (light transmission) aggregometer (Chrono-log 700 manual, Chrono-log Corp.) with or without the addition of collagen (at a final concentration of 10 μg ml−1) as a platelet agonist. The following groups were analysed: (1) positive control (PRP with collagen); (2) negative control (PRP with PBS); (3) PNV-CSC or CSC with PRP alone; and (4) PNV-CSC or CSC with PRP and collagen. The output from the aggregometer reported maximal percentage aggregation.
Collagen surface binding assay
GFP-tagged HUVECs (Angio-Proteomie, cAP-0001GFP, Boston, MA) were seeded on collagen-coated (Sigma Aldrich) 4-well culture chamber slides (Thermo Fisher Scientific) and cultured in vascular cell basal medium (ATCC PCS-100-030) supplemented with endothelial cell growth kit-VEGF (ATCC PCS-100-041). The cells were then incubated with DiI-loaded PNV-CSCs in PBS at 4 °C for 30 s. Then the cells were washed with PBS twice and imaged on a fluorescent microscope (Olympus IX81).
Denuded rat aorta binding assay
To examine PNV-CSCs binding on injured (denuded) vascular walls, aortas from WKY rats were dissected and denuded on their lumen walls with a pair of fine forceps to scrape the endothelial layer. Successful denudation was confirmed by microscopic examination. Both denuded or control aortas were incubated with DiI-labelled PNV-CSCs or CSCs for 30 s. Following PBS rinses, the samples were examined for cell binding on a fluorescent microscope (Olympus IX81).
Rat ischaemia/reperfusion model
Animal care was in accordance with the Institutional Animal Care and Use Committee (IACUC) guidelines. The ischaemia/reperfusion procedure was performed as described in a previous publication17. Briefly, we generated the ischaemia/reperfusion model using female Wistar-Kyoto (WKY) rats (6–8 weeks old, Charles River Laboratories). A left thoracotomy was created, followed by 60 min left anterior descending coronary artery ligation. Intracoronary injection was achieved by injection into the left ventricle cavity during a 25 s temporary aorta occlusion with a looped suture. Animals were randomized into three treatment groups: (1) control, intracoronary injection of PBS; (2) CSCs, intracoronary injection of 500,000 CSCs in PBS; and (3) PNV-CSCs, intracoronary injection of 500,000 PNV-CSCs in PBS. CSCs and PNV-CSCs were pre-labelled with CM-DiI (Vybrant CM-DiI Cell-Labeling Solution, Thermo Fisher Scientific). A cohort of animals was euthanized 24 hrs after injection for ex vivo fluorescent imaging, qPCR and histological analysis of PNV-CSCs or CSCs retention while the rest of the animals were followed for another 4 weeks.
Cell retention assay by fluorescence imaging and quantitative PCR
Animals were euthanized and hearts were harvested 24 hrs after cell infusion. We placed hearts in a Xenogen IVIS imaging system (Caliper Life Sciences, Mountain View, CA) to detect RFP fluorescence. Furthermore, we performed qPCR for precise measurement of the number of cells engrafted. As previously described26, we injected CSCs derived from male donor WKY rats into the myocardium of female recipients to utilize the detection of SRY gene located on the Y chromosome; the TaqMan assay was used to quantify the number of transplanted cells with the rat SRY gene as the template (see primer information in Supplementary Table 1). Real-time PCR was performed with a real-time PCR system. Cell numbers per mg of heart tissue and percentages of retained cells of the total injected cells were calculated.
Cardiac function assessment
The transthoracic echocardiogram procedure was performed by a cardiologist, who was blinded to the animal group allocation, using a Philips CX30 ultrasound system coupled with a L15 high-frequency probe34,35. Animals were anesthetized using a mixture of isoflurane and oxygen before undergoing the echo procedure at the 4 hr and 4 week time points. Hearts were imaged 2D in long-axis views at the level of the greatest left ventricle diameter. Left ventricular end-diastolic volume (LVEDV) and end-systolic volume (LVESV) were measured. Ejection fraction was determined by measurement from views taken from the infarcted area.
Heart morphometry
Hearts were harvested and cut into 10 μm-thick tissue sections. Masson’s trichrome staining was performed and images were acquired with a PathScan Enabler IV slide scanner (Advanced Imaging Concepts, Princeton, NJ). Image analyses related to viable myocardium and scar size were performed using ImageJ software. Three selected sections were quantified for each animal.
Histology
For immunohistochemistry staining, heart cryosections were fixed with 4% paraformaldehyde, permeabilized and blocked with Protein Block Solution (DAKO, Carpinteria, CA) containing 0.1% saponin (Sigma), and then incubated with the mouse anti-alpha sarcomeric actin (a7811, Sigma), rabbit anti-Ki67 (ab15580, Abcam), rabbit anti-alpha smooth muscle actin (ab5694, Abcam), rabbit anti-CD3 (ab16669, Abcam), and mouse anti-CD8 (MCA48R, Bio-Rad) antibodies overnight at 4 °C. For assessment of angiogenesis, heart cryosections were incubated with lectin (FL-1171, Vector Laboratories, Burlingame, CA, USA). Images were taken on a fluorescent microscope (Olympus IX81).
Platelet receptor blocking experiment
To explore which platelet adhesion molecules contributed to the targeting of PNV-CSCs, PNV-CSCs were pre-treated with anti-CD42b neutralizing antibodies (sc-292722, rabbit polyclonal, Santa Cruz) or isotype control antibodies (ab37415, rabbit polyclonal isotype control, Abcam) for 30 mins. The cells were then used in the ex vivo and in vivo experiments as previously described. The effects of CD42b blocking on control CSCs by pre-treating CSCs were evaluated using anti-CD42b neutralizing antibodies and the cell retention rate in the heart was measured. In addition, blocking assays on platelet markers CD42a, CD42b, CD162 and CD41/61 were performed using cultured endothelial cells.
Porcine model of ischaemia/reperfusion
Animal care was in accordance with the IACUC guidelines. MIs were created in adult farm pigs (male, 3.5-4 months old) by inflation using an angioplasty balloon (TREK OTW 3 mm, Abbott Vascular, Santa Clara, CA) in the mid-left anterior descending artery (distal to the second diagonal branch) for 1.5 hr. At the end of the ischaemic period, the vessel was reperfused. The vessel was allowed to reperfuse for 15 mins before cell infusion and remained perfused throughout the study. Animals were randomized to two treatment groups and 10 million DiI-labelled CSCs or PNV-CSCs were delivered intracoronarily using an over-the-wire catheter (without balloon inflations, to exclude possible confounding effects related to ischaemic post-conditioning). The cells were administered in 3 equally divided cycles with wash solution infusion in between. The animals were euthanized and the hearts were excised and sliced for fluorescent imaging (for cell retention) and triphenyl tetrazolium chloride (TTC) staining (for infarct size measurement) 24 hrs after the procedure.
Statistical analysis
All results are expressed as mean ± standard deviation (s.d.). Comparison between two groups were conducted by two-tailed Student’s t-test. One-way ANOVA test was used for comparison among three or more groups with Bonferroni post-hoc correction. Differences were considered statistically significant when P < 0.05. Our main efficacy endpoint is change in LVEF as measured by echocardiography. Based on previous rodent echo studies, experimental groups differed by 10% on average in left ventricular ejection fractions with standard deviations as large as 5.0%. We calculated that a sample size of 7 animals would be needed in each experimental group (assuming a 5% confidence level and a 90% power level; 10–15% animal loss due to the model). Kolmogorov-Smirnov test was performed for normal distribution.
Animal randomization method
Animal cages were housed in a random order on the shelves. Physical randomization was performed before animal experiments using a paper-drawing method. All measurements were done in random order, with the surgeon and echocardiographer being blind to the treatment groups.
Life Sciences Reporting Summary
Further information on experimental design is available in the Life Sciences Reporting Summary.
Data availability
The authors declare that all data supporting the findings of this study are available within the article and its Supplementary Information.
Supplementary Material
Acknowledgments
This work was supported by US National Institute of Health (HL123920 and HL137093), NC State University Chancellor’s Faculty Excellence Program, NC State Chancellor’s Innovation Fund, University of North Carolina General Assembly Research Opportunities Initiative grant and the National Natural Science Foundation of China (81370216, 81570274).
Footnotes
Author contributions
J.T., T.S., K.H., P.-U.D. and K.C. designed the research, performed biochemical, cellular and animal experiments, analysed the data and drafted the paper. P.-U.D., Z.W., A.V., M.T.H., T.A., J.C., T.L., E.S., E.M., L.L., L.R., A.L., A.B., T.G.C., D.S., Z.G. and G.A.S. performed cellular and in vitro experiments, and/or provided comments to improve the paper. K.C. and J.Z. provided financial support. K.C. directed the research.
Competing interests
The authors declare no competing interests.
Supplementary information is available for this paper at https://doi.org/10.1038/s41551-017-0182-x.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Mozaffarian D, et al. Heart disease and stroke statistics—2015 update: a report from the American Heart Association. Circulation. 2015;131:29–322. doi: 10.1161/CIR.0000000000000152. [DOI] [PubMed] [Google Scholar]
- 2.Madonna R, et al. Cell-based therapies for myocardial repair and regeneration in ischemic heart disease and heart failure. Eur Heart J. 2016;37:1789–1798. doi: 10.1093/eurheartj/ehw113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sepantafar M, et al. Stem cells and injectable hydrogels: synergistic therapeutics in myocardial repair. Biotechnol Adv. 2016;34:362–379. doi: 10.1016/j.biotechadv.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Weissman IL. Translating stem and progenitor cell biology to the clinic: barriers and opportunities. Science. 2000;287:1442–1446. doi: 10.1126/science.287.5457.1442. [DOI] [PubMed] [Google Scholar]
- 5.Cheng K, et al. Magnetic antibody-linked nanomatchmakers for therapeutic cell targeting. Nat Commun. 2014;5:4880. doi: 10.1038/ncomms5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tongers J, et al. Stem and progenitor cell-based therapy in ischaemic heart disease: promise, uncertainties, and challenges. Eur Heart J. 2011;32:1197–1206. doi: 10.1093/eurheartj/ehr018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Spoel TI, et al. Human relevance of pre-clinical studies in stem cell therapy: systematic review and meta-analysis of large animal models of ischaemic heart disease. Cardiovasc Res. 2011;91:649–658. doi: 10.1093/cvr/cvr113. [DOI] [PubMed] [Google Scholar]
- 8.Lippi G, et al. Arterial thrombus formation in cardiovascular disease. Nat Rev Cardiol. 2011;8:502–512. doi: 10.1038/nrcardio.2011.91. [DOI] [PubMed] [Google Scholar]
- 9.Stellos K, et al. Circulating platelet-progenitor cell coaggregate formation is increased in patients with acute coronary syndromes and augments recruitment of CD34+ cells in the ischaemic microcirculation. Eur Heart J. 2013;34:2548–2556. doi: 10.1093/eurheartj/eht131. [DOI] [PubMed] [Google Scholar]
- 10.Li TS, et al. Direct comparison of different stem cell types and subpopulations reveals superior paracrine potency and myocardial repair efficacy with cardiosphere-derived cells. J Am Coll Cardiol. 2012;59:942–953. doi: 10.1016/j.jacc.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith RR, et al. Regenerative potential of cardiosphere-derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation. 2007;115:896–908. doi: 10.1161/CIRCULATIONAHA.106.655209. [DOI] [PubMed] [Google Scholar]
- 12.Cheng K, et al. Magnetic targeting enhances engraftment and functional benefit of iron-labeled cardiosphere-derived cells in myocardial infarction. Circ Res. 2010;106:1570–1581. doi: 10.1161/CIRCRESAHA.109.212589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee ST, et al. Intramyocardial injection of autologous cardiospheres or cardiosphere-derived cells preserves function and minimizes adverse ventricular remodeling in pigs with heart failure post-myocardial infarction. J Am Coll Cardiol. 2011;57:455–465. doi: 10.1016/j.jacc.2010.07.049. [DOI] [PubMed] [Google Scholar]
- 14.Cheng K, et al. Human cardiosphere-derived cells from advanced heart failure patients exhibit augmented functional potency in myocardial repair. JACC Heart Fail. 2014;2:49–61. doi: 10.1016/j.jchf.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malliaras K, et al. Intracoronary cardiosphere-derived cells after myocardial infarction: evidence of therapeutic regeneration in the final 1-year results of the CADUCEUS trial (cardiosphere-derived autologous stem cells to reverse ventricular dysfunction) J Am Coll Cardiol. 2014;63:110–22. doi: 10.1016/j.jacc.2013.08.724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Makkar RR, et al. Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): a prospective, randomised phase 1 trial. Lancet. 2012;379:895–904. doi: 10.1016/S0140-6736(12)60195-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng K, et al. Magnetic enhancement of cell retention, engraftment, and functional benefit after intracoronary delivery of cardiac-derived stem cells in a rat model of ischemia/reperfusion. Cell Transplant. 2012;21:1121–1135. doi: 10.3727/096368911X627381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanazawa H, et al. Cellular postconditioning: allogeneic cardiosphere-derived cells reduce infarct size and attenuate microvascular obstruction when administered after reperfusion in pigs with acute myocardial infarction. Circ Heart Fail. 2015;8:322–332. doi: 10.1161/CIRCHEARTFAILURE.114.001484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nasiri S. Infusible platelet membrane as a platelet substitute for transfusion: an overview. Blood Transfus. 2013;11:337–342. doi: 10.2450/2013.0209-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu CM, et al. Nanoparticle biointerfacing by platelet membrane cloaking. Nature. 2015;526:118–121. doi: 10.1038/nature15373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng K, et al. Brief report: mechanism of extravasation of infused stem cells. Stem Cells. 2012;30:2835–2842. doi: 10.1002/stem.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allen TA, et al. Angiopellosis as an alternative mechanism of cell extravasation. Stem Cells. 2017;35:170–180. doi: 10.1002/stem.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gawaz M. Role of platelets in coronary thrombosis and reperfusion of ischemic myocardium. Cardiovasc Res. 2004;61:498–511. doi: 10.1016/j.cardiores.2003.11.036. [DOI] [PubMed] [Google Scholar]
- 24.Xu Y, et al. Activated platelets contribute importantly to myocardial reperfusion injury. Am J Physiol Heart Circ Physiol. 2006;290:H692–699. doi: 10.1152/ajpheart.00634.2005. [DOI] [PubMed] [Google Scholar]
- 25.Anselmo AC, et al. Platelet-like nanoparticles: mimicking shape, flexibility, and surface biology of platelets to target vascular injuries. ACS Nano. 2014;8:11243–11253. doi: 10.1021/nn503732m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vandergriff AC, et al. Magnetic targeting of cardiosphere-derived stem cells with ferumoxytol nanoparticles for treating rats with myocardial infarction. Biomaterials. 2014;35:8528–8539. doi: 10.1016/j.biomaterials.2014.06.031. [DOI] [PubMed] [Google Scholar]
- 27.Li TS, et al. Cardiospheres recapitulate a niche-like microenvironment rich in stemness and cell-matrix interactions, rationalizing their enhanced functional potency for myocardial repair. Stem Cells. 2010;28:2088–2098. doi: 10.1002/stem.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li C, et al. Allogenic dendritic cell and tumor cell fused vaccine for targeted imaging and enhanced immunotherapeutic efficacy of gastric cancer. Biomaterials. 2015;54:177–187. doi: 10.1016/j.biomaterials.2015.03.024. [DOI] [PubMed] [Google Scholar]
- 29.Kawada M. Vaccination of fusion cells of rat dendritic and carcinoma cells prevents tumor growth in vivo. Int J Cancer. 2003;105:520–526. doi: 10.1002/ijc.11120. [DOI] [PubMed] [Google Scholar]
- 30.Lentz BR. Polymer-induced membrane fusion: potential mechanism and relation to cell fusion events. Chem Phys Lipids. 1994;73:91–106. doi: 10.1016/0009-3084(94)90176-7. [DOI] [PubMed] [Google Scholar]
- 31.Huang Y, et al. Fusions of tumor-derived endothelial cells with densritic cells induces antitumor immunity. Sci Rep. 2017;7:46544. doi: 10.1038/srep46544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown AC, et al. Molecular interference of fibrin’s divalent polymerization mechanism enables modulation of multiscale material properties. Biomaterials. 2015;49:27–36. doi: 10.1016/j.biomaterials.2015.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brown AC, et al. Ultrasoft microgels displaying emergent platelet-like behaviours. Nat Mater. 2014;13:1108–1114. doi: 10.1038/nmat4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang J, et al. Heart repair using nanogel-encapsulated human cardiac stem cells in mice and pigs with myocardial infarction. ACS Nano. 2017;11:9738–9749. doi: 10.1021/acsnano.7b01008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang J, et al. Therapeutic microparticles functionalized with biomimetic cardiac stem cell membranes and secretome. Nat Commun. 2017;8:13724. doi: 10.1038/ncomms13724. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all data supporting the findings of this study are available within the article and its Supplementary Information.