

Abstract
Rationale:
Infantile-onset hypertrophic cardiomyopathy (HCMP) should be considered a largely genetic condition, although its onset is most often triggered by infection. Very long chain acyl-CoA dehydrogenase (VLCAD) deficiency is a rare autosomal recessive inborn error of mitochondrial fatty acid β-oxidation that often causes severe cardiomyopathy and/or sudden death during the neonatal period.
Patient concerns:
Herein, we report an infant with VLCAD deficiency who presented with severe cardiac manifestations, including massive pericardial effusion and HCMP. The subject's older sister died of unknown causes at three days of age; however, the subject exhibited a normal tandem mass-spectrometry profile during the neonatal period.
Diagnoses:
During her later cardiac presentation, the subject's C-14 and C-18 levels became elevated, and she was determined, via the conducted molecular analysis, to harbor a novel homozygous frameshift mutation (c.103_112dup) in ACADVL.
Interventions:
After VLCAD deficiency diagnosis, the subject was treated with the administration of a medium chain triglyceride formula and fluid therapy.
Outcomes:
The subject's cardiac status was markedly improved by the dietary intervention and fluid therapy.
Lessons:
This report highlights that genetic mutations should be investigated as possible causes of infantile-onset HCMP, and that early diagnosis and intervention can prevent mortality for patients with VLCAD deficiency.
Keywords: cardiomyopathy, long chain acyl-CoA dehydrogenase, medium chain triglyceride, mitochondrial β-oxidation, tandem mass screening
1. Introduction
Very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency (OMIM #201475) is a rare autosomal recessive disease characterized by an inborn error of mitochondrial fatty acid β-oxidation.[1] VLCAD, which is encoded by ACADVL located on chromosome 17p13.1, catalyzes the first step of mitochondrial β-oxidation for long-chain fatty acids consisting of 14 to 20 carbon molecules. Although some patients with VLCAD deficiency remain asymptomatic, most are triggered by fasting, infection, and/or excessive activity to present with either severe early onset cardiomyopathy, hepatic encephalopathy/severe hypoketotic hypoglycemia, or late onset episodic myopathy.[1,2] Early diagnosis and treatment of VLCAD deficiency has been shown to reduce patient mortality and morbidity; however, the introduction of newborn VLCAD deficiency screening has been significantly complicated by false positive diagnoses.[1,3]
Herein, we report an infant with VLCAD deficiency that presented with massive pericardial effusion and left ventricular hypertrophy (LVH). Despite knowledge of the unexplained sudden neonatal death of her older sibling, a normal tandem mass spectrometry result yielded during her stay in a neonatal intensive care unit delayed her diagnosis.
2. Case report
The subject was a 5-month-old girl, who presented with massive cardiomegaly and pericardial effusion. She was born by cesarian section at 29 weeks and 2 days of gestation, at a birth weight of 980 g. As a neonate, she received surfactant therapy and ventilator support for respiratory distress syndrome, as well as a single dose of ibuprofen for patent ductus arteriosus. The results of her initial tandem mass screening, (which was delayed until 20 days after birth due to her extreme prematurity), revealed a mild elevation of both C14:1 (1.083, reference < 0.599 μmol) and C14:2 (0.289, reference < 0.166 μmol). A repeat test conducted at 26 days of age yielded normal results; therefore, she subsequently received total parenteral nutrition with intravenous medium chain triglyceride (MCT) lipids during her neonatal intensive care. She was managed in a neonatal intensive unit for 3 months, before being discharged without medication or oxygen. Only a small atrial septal defect was observed during her discharge echocardiogram; however, by 5 months of age, the subject presented with poor oral intake, dyspnea, and lethargy following an upper respiratory infection. Severe cardiomegaly (Fig. 1), as well as both massive pericardial effusion and LVH were revealed by a chest radiograph and an echocardiogram, respectively. A pericardiocentesis identified the pericardial effusion as exudative, and the subject was administered ventilator support and intravenous inotropic therapy. At her initial presentation, the subject exhibited a plasma creatinine kinase (CK) level of 245 U/L (reference < 150 U/L), a ventricular ejection fraction of 48%, a left ventricular mass of 41.7 g (Z-score = 4.69), and a pericardial effusion depth of 12 mm. As several pericardiocenteses did not improve the subject's pericardial effusion, a pericardiostomy was performed, and a pericardial tube inserted. The subject's ventilator support was gradually reduced; however, her pericardial effusion persisted, and her plasma CK levels increased to 710 U/L. A second metabolic study that included an analysis of the subject's acylcarnitine, plasma carnitine, and urine organic acid profiles was conducted during her cardiac presentation. The results of this study showed elevated C-14 to C-18 levels (as identified via tandem mass spectrometry), and markedly reduced free and total carnitine levels. Several excreted C6–C10 dicarboxylic acids were identified during the conducted urine organic acid analysis. The subject also underwent various additional evaluations to identify other possible causes for infantile-onset LVH. For example, her urinary glycosaminoglycan and acid α-glucosidase levels were analyzed to screen for mucopolysaccharidosis and Pompe disease, respectively; however, all the additional tests yielded normal results.
Figure 1.
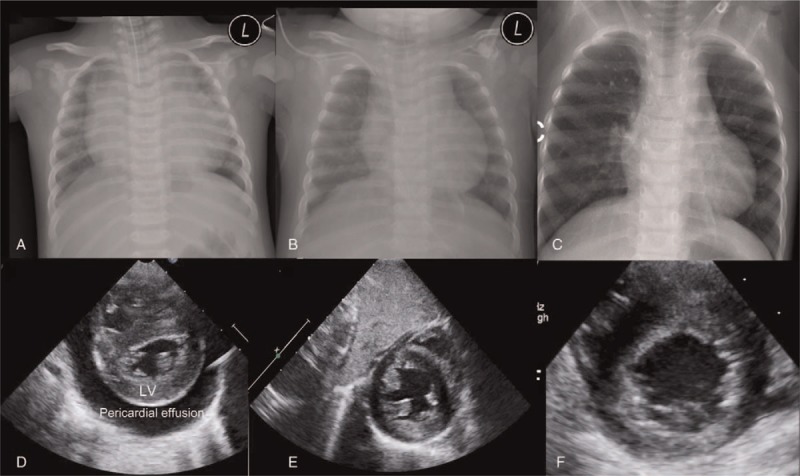
Patient chest radiographs and echocardiographs. (A–C) Chest x-rays showing severe cardiomegaly (A) that was markedly improved after 2 months (B) and 1 year (C) of dietary intervention. (D–F) A parasternal short axis view of the conducted echocardiograph, showing severe pericardial effusion and left ventricular hypertrophy (D) that was completely resolved after 2 months (E) and 1 year (F) of dietary intervention. L = patient's left side, LV = left ventricle.
A molecular analysis of the subject's ACADVL sequence revealed a homozygous novel frameshift mutation, comprising c.103_112dup (p.Arg38Profs∗24) (Fig. 2). Both of the subject's parents were identified to carry the mutation, despite being nonconsanguineous. The subject's family history included 2 older female siblings, the elder of which was confirmed to also be a mutation carrier. The mutation status of the middle sibling could not be determined as she died from unknown causes 3 days after her birth in a standard nursery; however, her newborn screening results (which were reported after death) revealed elevated C-14 (4.958, reference; 0.065–0.920 μmol), C-16 (10.352, reference: 0.555–5.891 μmol), and C-18 (2.733, reference: 0.213–1.688 μmol) levels. After VLCAD-deficiency diagnosis, the subject was administered a high concentration dextrose fluid, 50 mg riboflavin per day, and an MCT formula. Removal of the subject's pericardial catheter resulted in a poor overall condition and increased CK levels of 8000 U/L. After 2 months of dietary intervention (using only the MCT formula), an echocardiogram confirmed that the subject's cardiac function was normal (ejection fraction = 78%), comprising only mild LVH without pericardial effusion. The subject later experienced several acute illnesses with fever, including both pneumonia and gastroenteritis; however, her resulting metabolic crises were rapidly resolved by a combination of fluid therapy, and MCT formula (but not any other long-chain triglyceride) supplementation (Fig. 3). At 2 years of age, the subject's height, body weight, and head circumference were 83.7 cm (−0.79 SD), 9.7 kg (−1.96 SD), and 47.3 cm (0.3 SD), respectively. Her most recent echocardiogram showed normal cardiac function and an improving LV mass (51.3 g, Z-score = 1.81).
Figure 2.
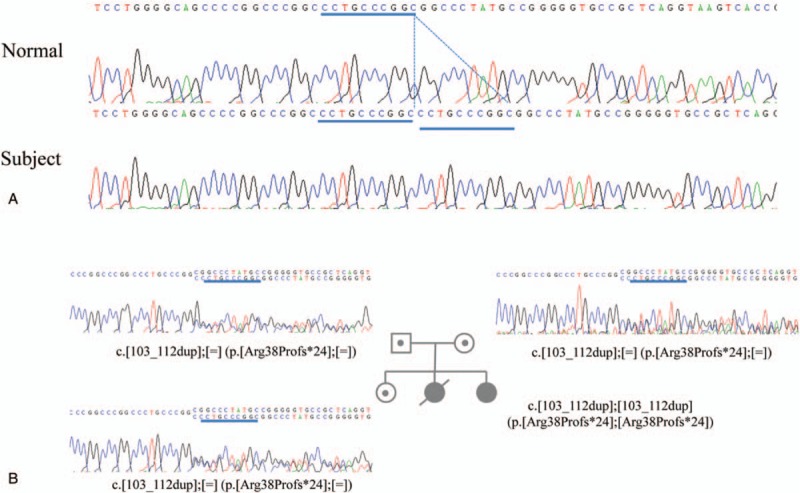
(A) The partial ACADVL sequence, and included novel c.4399_4400del (p.R38Pfs∗4) mutation, that was exhibited by the patient with very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency. (B) The conducted segregation analysis revealed that the patient inherited a copy of this mutant sequence from both her mother and her father.
Figure 3.
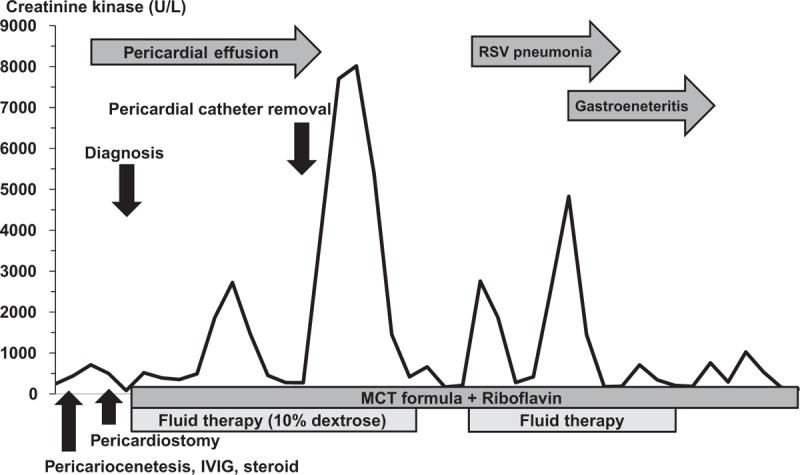
Variation in the patient's creatinine kinase levels, with respect to the onset of her infantile very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency, and the administration of various clinical interventions. IV = intravenous, IVIG = intravenous immunoglobulin, MCT = medium chain triglycerides, RSV = respiratory syncytial virus, VLCAD = very long-chain acyl-CoA dehydrogenase.
3. Discussion
The present report highlights the importance of the repetitive metabolic analyses conducted during the patient's catabolic state; in fact, these analyses yielded data critical for the clinical diagnosis of a fatty acid oxidation disorder (FAOD). The report also emphasizes the importance of considering treatable genetic conditions, including FAODs such as VLCAD deficiency, as potential causes of infantile-onset HCMP. Although FAOD is a rare inborn error of metabolism, it accounts for approximately 5% of sudden infantile deaths.[4] Since the introduction of newborn screening, the majority of patients with VLCAD deficiency can be diagnosed early, enabling them to avoid life-threatening metabolic crises;[1] however, the current screen is not sufficient to accurately confirm VLCAD deficiency. In fact, false positive results are estimated to occur at a rate of up to 69%, often identifying asymptomatic ACADVL mutation carriers, and thus incurring unnecessary costs and stress.[1,6] Conversely, false negative results impede VLCAD diagnosis, as shown here.[1,5] In this case, the initial positive newborn screen result was ignored due to the normal result obtained during the follow-up repeated test, despite the fact that the subject's family history included an unexplained neonatal death. The appearance of the subject's symptoms was likely delayed by the neonatal intensive care, high dextrose liquid, and MCT lipid supplement that she received as treatment for the respiratory distress syndrome induced by her extreme prematurity.
In fact, the subject's family history was the most important diagnostic “clue” in the current case of VLCAD deficiency. The results of a previous report similarly emphasized the importance of a familial history of recurrent sudden-infant-deaths in diagnosing VLCAD deficiency. That study described a subject whose previous siblings had died within 36 hours after birth, but yielded retrospective acylcarnitine analysis results that were not significantly abnormal. That subject's family history prompted screening for VLCAD deficiency, which was subsequently confirmed via the identification of homozygous truncated ACADVL mutations.[7] The present study found that the subject instead harbored novel homozygous frameshift mutations in exon 1 of ACADVL. Though as yet uncharacterized, these mutations may have led to a protein truncation and/or loss of ACADVL function. Notably, ACADVL truncation mutations have been previously shown to be associated with severe infantile-onset cardiomyopathy, pericardial effusion, and very often (80% of ACADVL truncation mutations), with severe cardiomyopathy.[8]
The subject had a pericardiostomy performed, and an indwelling cardiac catheter inserted to address her persistent pericardial effusion prior to VLCAD diagnosis. Cardiac catheters have been previously shown to irritate the myocardium in some patients, leading to an aggravated cardiac manifestation and/or catabolic status. Thus, extracorporeal membrane oxygenation (ECMO) might also increase the severity of VLCAD pathogenesis by increasing the patient's catabolic rate, and/or causing persistent cardiac irritation.[9] This is supported by the fact that the subject in this report exhibited a markedly increased CK level, and an overall poor condition after pericardial catheter removal.
Previous studies have suggested various low-fat diets for patients with VLCAD deficiency, and similarly, have recommended that MCT oil or formula be administered at various dosages dependent upon the total patient caloric intake.[10] However, in the present study, administration of MCT formula alone during the subject's acute cardiac manifestation induced a rapid improvement of both the subject's pericardial effusion and LVH.
Similarly, a previous report showed early MCT formula administration to be effective in mitigating severe HCMP and pericardial effusion.[11–13] Nevertheless, while treatment with MCT formula/oil and riboflavin is well established to be effective against FAOD,[10] additional therapeutic targets are urgently required to address the metabolic crisis induced by fasting, infection, and/or exercise. Recently, a long-term study tested both the medium odd-chain (C7) triglyceride triheptanoin and its anaplerotic metabolites (UX 007) as potential therapeutic agents for patients with long-chain FAOD, and showed a resultant improvement in both patient cardiac function and quality of life.[14]
In conclusion, the present study reported a female born with extreme prematurity, who was determined to harbor a novel homozygous ACADVL mutation. She exhibited normal tandem mass spectrometry results during her stay in neonatal intensive care, but was subsequently diagnosed with delayed-onset VLCAD deficiency. Both her HCMP and massive pericardial effusion were successfully treated via MCT formula administration. Given that VLCAD deficiency is a relatively common and reversible infantile HCMP like infantile-onset Pompe disease (OMIM 232300), we suggest that patient acylcarnitine and urine organic acid profiles should be analyzed during any catabolic status assessment, irrespective of previous normal newborn screening results.
Author contributions
Conceptualization: Yoo-Mi Kim.
Data curation: Yoo-Mi Kim, Geena Kim.
Formal analysis: Han-Wook Yoo.
Investigation: Hoon Ko, Hyoung Doo Lee.
Resources: Hyoung Doo Lee.
Writing – original draft: Yoo-Mi Kim.
Writing – review & editing: Yoo-Mi Kim, Geena Kim, Han-Wook Yoo.
Footnotes
Abbreviations: CK = creatinine kinase, FAOD = fatty acid oxidation disorder, HCMP = hypertrophic cardiomyopathy, LVH = left ventricular hypertrophy, MCT = medium chain triglyceride, VLCAD = very long chain acyl-CoA dehydrogenase.
Informed Consent: Informed consent was obtained from the patient's parents for the inclusion of her medical history in this case report. No additional ethics approval was required, since the study did not comprise a clinical trial, nor use any unapproved medications.
The authors have no conflicts of interest to disclose.
References
- [1].Miller MJ, Burrage LC, Gibson JB, et al. Recurrent ACADVL molecular findings in individuals with a positive newborn screen for very long chain acyl-coA dehydrogenase (VLCAD) deficiency in the United States. Mol Genet Metab 2015;116:139–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Saudubray JM, Martin D, De Lonlay P, et al. Recognition and management of fatty acid oxidation defects: a series of 107 patients. J Inherit Metab Dis 1999;22:488–502. [DOI] [PubMed] [Google Scholar]
- [3].Pena LD, van Calcar SC, Hansen J, et al. Outcomes and genotype-phenotype correlations in 52 individuals with VLCAD deficiency diagnosed by NBS and enrolled in the IBEM-IS database. Mol Genet Metab 2016;118:272–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Boles RG, Buck EA, Blitzer MG, et al. Retrospective biochemical screening of fatty acid oxidation disorders in postmortem livers of 418 cases of sudden death in the first year of life. J Pediatr 1998;132:924–33. [DOI] [PubMed] [Google Scholar]
- [5].Boneh A, Andresen BS, Gregersen N, et al. VLCAD deficiency: pitfalls in newborn screening and confirmation of diagnosis by mutation analysis. Mol Genet Metab 2006;88:166–70. [DOI] [PubMed] [Google Scholar]
- [6].Merritt JLII, Vedal S, Abdenur JE, et al. Infants suspected to have very-long chain acyl-CoA dehydrogenase deficiency from newborn screening. Mol Genet Metab 2014;111:484–92. [DOI] [PubMed] [Google Scholar]
- [7].Scalais E, Bottu J, Wanders RJ, et al. Familial very long chain acyl-CoA dehydrogenase deficiency as a cause of neonatal sudden infant death: improved survival by prompt diagnosis. Am J Med Genet A 2015;167A:211–4. [DOI] [PubMed] [Google Scholar]
- [8].Mathur A, Sims HF, Gopalakrishnan D, et al. Molecular heterogeneity in very-long-chain acyl-CoA dehydrogenase deficiency causing pediatric cardiomyopathy and sudden death. Circulation 1999;99:1337–43. [DOI] [PubMed] [Google Scholar]
- [9].Kajimoto M, O’Kelly Priddy CM, Ledee DR, et al. Extracorporeal membrane oxygenation promotes long chain fatty acid oxidation in the immature swine heart in vivo. J Mol Cell Cardiol 2013;62:144–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Arnold GL, Van Hove J, Freedenberg D, et al. A Delphi clinical practice protocol for the management of very long chain acyl-CoA dehydrogenase deficiency. Mol Genet Metab 2009;96:85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dereddy NR, Kronn D, Krishnan U. Defects in long chain fatty acid oxidation presenting as severe cardiomyopathy and cardiogenic shock in infancy. Cardiol Young 2009;19:540–2. [DOI] [PubMed] [Google Scholar]
- [12].Spiekerkoetter U, Tenenbaum T, Heusch A, et al. Cardiomyopathy and pericardial effusion in infancy point to a fatty acid b-oxidation defect after exclusion of an underlying infection. Pediatr Cardiol 2003;24:295–7. [DOI] [PubMed] [Google Scholar]
- [13].Sharef SW, Al-Senaidi K, Joshi SN. Successful treatment of cardiomyopathy due to very long-chain acyl-CoA dehydrogenase deficiency: first case report from Oman with literature review. Oman Med J 2013;28:354–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Vockley J, Marsden D, McCracken E, et al. Long-term major clinical outcomes in patients with long chain fatty acid oxidation disorders before and after transition to triheptanoin treatment–a retrospective chart review. Mol Genet Metab 2015;116:53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]