

Abstract
Non-steroidal anti-inflammatory drugs (NSAIDs) are common analgesic drugs that also cause well-known, negative gastrointestinal (GI) side effects. The physiological mechanism(s) of NSAID-induced GI damage are unclear and are likely due to multiple causes. The most studied contributing mechanisms are increased gastric acid secretion and increased gastric motility. The present study was designed to determine which ulcerogenic effects of the NSAID diclofenac sodium are reversed by blocking the endocannabinoid catabolic enzyme monoacylglycerol lipase (MAGL). Both male and female mice were used to identify possible sex differences. We hypothesized that the MAGL inhibitor JZL184 would attenuate diclofenac-induced increases in both gastric acid secretion and gastric motility. Diclofenac dose-dependently induced gastric hemorrhages to a similar extent in both male and female mice. Gastric hemorrhage severity significantly correlated with gastric levels of myeloperoxidase, an objective measure of neutrophil infiltration. Similarly, JZL184 reduced gastric acidity, in controls as well as mice treated with pentagastrin, which stimulates gastric acid release. As hypothesized, JZL184 decreased gastric motility. Surprisingly, diclofenac also slowed gastric emptying, indicating that diclofenac-induced ulcers most likely occur through decreased gastric acid secretion, and not increased gastric motility, as measured in the present study. Thus, MAGL inhibition may proffer gastroprotection through modulating the secretory pathway of gastric hemorrhage. These data underscore the importance of sampling multiple time points and using both sexes in research, in addition to multiple mechanistic targets, and contribute to the basic understanding of NSAID-induced gastric inflammation.
Keywords: cannabinoid, monoacylglycerol lipase, gastric hemorrhage, dyspepsia, gastric motility, acid secretion
1. Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) are the most commonly used pharmaceutical therapies for pain (Lanas, 2009). Approximately 20% of chronic NSAID users develop gastric hemorrhages in the stomach or duodenum that require medical intervention (Wallace, 2001). The physiological mechanisms through which NSAIDs induce gastric hemorrhages are not clear (Schubert, 2008). The most characterized mechanisms include NSAID-induced increases in gastric acid secretion and stomach contractions (Takeuchi, 2012; Wallace, 2008, 2001; Yano et al., 1978). To prevent NSAID-induced hemorrhages, proton pump inhibitors (PPI) are regularly co-prescribed to reduce gastric acid secretion (Mössner, 2016). Chronic PPI use is also associated with negative side effects including gastric polyps, tumors, infection, vitamin deficiencies, bone fractures, cancer, and dementia (Kuller, 2016; Lodato et al., 2010; Mössner, 2016). The negative side effects of both NSAIDs and PPIs emphasize the need for new gastroprotective therapeutics.
The psychoactive component of Cannabis, i.e., Δ9-tetrahydrocannabinol (THC), strongly inhibits NSAID-induced gastric hemorrhages in mice (Kinsey et al., 2011; Kinsey and Cole, 2013). Similarly, synthetic cannabinoid receptor agonists suppress gastric acid secretion, in pentagastrin- and histamine-stimulated animal models via the CB1 cannabinoid receptor (Adami et al., 2002; Coruzzi et al., 2006, 1999; Pazos et al., 2008). The effects of cannabinoids on gastric ulceration and acid production suggest that the endogenous cannabinoid (endocannabinoid) system modulates gastric physiology. Inhibiting the catabolism of endocannabinoid agonists blocks gastric hemorrhage development (Kinsey et al., 2011; Naidu et al., 2009). For example, inhibiting the catabolic enzyme monoacylglycerol lipase (MAGL) blocks the formation of NSAID-induced ulcers (Ignatowska-Jankowska et al., 2014; Kinsey et al., 2011). Furthermore, the anti-ulcerogenic effects of the MAGL inhibitor, JZL184, occur through CB1, indicating that endocannabinoids modulate ulcer formation (Kinsey et al., 2011). JZL184 also increases stomach levels of the endocannabinoid 2-arachidonylglycerol (2-AG) (Kinsey et al., 2011). Thus, the endocannabinoid influences pathogenic effects of NSAID-induced gastric hemorrhages, albeit through unknown mechanisms.
While synthetic cannabinoid receptor agonists decrease gastric acid secretions (Adami et al., 2002; Coruzzi et al., 2006, 1999), it is unknown whether MAGL inhibition, per se, modulates gastric acid output. 2-AG decreases gastric motility through a CB1 mechanism of action (Aviello et al., 2008). However, cannabinoids have not been evaluated in the context of altered gastric motility following NSAID exposure. Therefore, the primary goal of this study was to evaluate the role of MAGL inhibition on gastric acid production and gastric motility. The present study also evaluated myeloperoxidase (MPO) as a potential biomarker for gastric hemorrhage severity. After initial damage, epithelial cells recruit and activate white blood cells, including neutrophils (Wallace, 2008; Wallace et al., 1990). MPO is an enzyme that is expressed within circulating neutrophils and is commonly used to indirectly quantify neutrophil activity in tissue (Liu et al., 1998; Loria et al., 2008). Thus, MPO level for each mouse was correlated with an established visual measure of hemorrhage severity.
Finally, published reports of cannabinoid modulation of gastric pathology have used only male animals. Sex differences have been noted in multiple cannabinoid studies (Craft, 2005; Craft et al., 2013a, 2013b). Thus, a goal of the present study was to establish the NSAID-induced gastric hemorrhage model, as well as its attenuation by MAGL inhibition, in female mice. The ultimate goal of this research is to improve the lives of patients taking NSAIDs by informing the development of new, cannabinoid-based gastroprotective therapeutics.
2. Material and Methods
2.1 Animals
Subjects consisted of male and female ICR mice (Envigo, Indianapolis, IN), aged approximately 8 weeks. Mice were housed 3-5 per cage in a temperature (20 ± 2°C) and humidity (50 ± 20%) controlled environment with ad libitum access to food and water, in an AAALAC-accredited facility at West Virginia University. All studies were approved by the Institutional Animal Care and Use Committee at West Virginia University prior to the start of any experiments. Mice were randomly assigned to treatment groups, and experiments and analyses were carried out by researchers who were unaware of the treatment groups.
2.2 Drugs
The MAGL inhibitor JZL184 and the NSAID diclofenac sodium salt were purchased from Cayman Chemical (Ann Arbor, MI). The gastric acid secretagogue pentagastrin and the proton pump inhibitor omeprazole were purchased from Sigma Aldrich (St. Louis, MO). All compounds were dissolved in a vehicle consisting of ethanol, Cremophor (Sigma-Aldrich), and saline in a ratio of 1:1:18 parts, as described previously (Crowe et al., 2015; Kinsey and Cole, 2013). All solutions were warmed to room temperature and administered per oral (p.o.), intraperitoneally (i.p.), or subcutaneously (s.c.) at a volume of 10 μl/g body mass. Diclofenac was administered p.o., consistent with clinical use in that the NSAID comes into direct contact with the lining of the stomach. Omeprazole and JZL184 were administered i.p., and pentagastrin was administered s.c.
2.3 Induction of gastric hemorrhage
Gastric hemorrhages were induced per previously published methods (Kinsey et al., 2011; Naidu et al., 2009). Mice were weighed, then food-deprived for 24 h with free access to water. A wire mesh barrier was placed on the bottom of each cage to prevent coprophagia, with the goal of minimizing stomach contents. On the day of the hemorrhage induction, mice were weighed, administered diclofenac or vehicle, and returned to the home cage. After 6 h, mice were euthanized via CO2 asphyxiation and their stomachs were harvested, photographed, and the mucosal layer was isolated by scraping with a metal spatula. The mucus and stomach were separately snap-frozen in liquid nitrogen and were stored at -80°C.
2.4 Gastric hemorrhage quantification
Gastric hemorrhages were quantified per previously reported methods (Ignatowska-Jankowska et al., 2014; Kinsey et al., 2011). Six h after diclofenac treatment, stomachs were harvested, cut along the greater curvature, and rinsed with distilled water. The tissue was then gently flattened and placed on a lighted stage and the exposed mucosa was photographed, along with a 1-mm reference, using a Canon EOS Rebel XS digital camera with a Canon 250D 10× close-up lens (Adorama Inc., New York, NY). The images were pseudo randomized, renamed, and analyzed using Photoshop (Adobe Systems, Inc., San Jose, CA). The length of each hemorrhage and the reference were each marked with a 1-pixel-wide line and compared, such that the total hemorrhage score in millimeters for each stomach was calculated by the formula:
2.5 Biomarker quantification
Myeloperoxidase (MPO) is commonly used to indirectly quantify neutrophils in tissue (Loria et al., 2008). Gastric hemorrhages were induced with diclofenac (0, 11.11, 33.33, or 100 mg/kg), as detailed in section 2.3, and mucosal scrapings were isolated, using a metal spatula, and snap frozen in liquid nitrogen. Samples were homogenized in 5 ml ice cold phosphate-buffered saline using a Tissue Tearor (Bartlesville, OK), and MPO activity was quantified by sandwich enzyme-linked immunosorbent assay per manufacturer's instructions (R&D Systems, Minneapolis, MN). All samples were run in duplicate.
2.6 Gastric acid secretion
Mice were subjected to the gastric hemorrhage procedure, and gastric secretions were collected 6 h after diclofenac administration (i.e., at the same time as hemorrhages were photographed). For evaluation of basal vs. pentagastrin-stimulated gastric acid secretions, mice were administered JZL184 (40 mg/kg; (Kinsey et al., 2011)), omeprazole (20 mg/kg; positive control; (Astudillo et al., 2002; Kinsey et al., 2011)), or vehicle (negative control) 2 h prior to vehicle (basal condition) or pentagastrin (0.25 mg/kg; stimulated condition). Mice were euthanized via CO2 asphyxiation 1 h after pentagastrin administration, then gastric secretions were collected using gastric lavage, as described previously (Ferrero et al., 2000; Martinez et al., 1998), with minor modification. After euthanasia, the stomach was clamped at the lower esophageal sphincter and the pyloric region to block stomach emptying, and 200 μl of normal saline was injected into the stomach. The stomach was then removed and its contents were collected in a test tube. Samples were centrifuged for 7 min at 13000 × g and the supernatant was aliquoted and assessed for pH using a pH microelectrode (Thermo Scientific, PerpHecT ROSS microelectrode 8220BNWP).
It is noteworthy that the current study measured the pH of gastric secretions mixed with saline, and therefore did not directly measure acid secretion, but rather the amount of acidity in the collected stomach contents. The changes in gastric pH were inferred as changes in gastric secretions. However, gastric acid secretion is the primary determinant of gastric pH (Fuchs et al., 1991). For simplicity, the alterations in pH samples are hereafter referred to as alterations in gastric acid secretion.
2.7 Gastric motility
Gastric emptying was performed as described previously (Di Marzo et al., 2008), with minor modifications. Mice were weighed, placed on a wire barrier, and fasted for 24 h. Mice were administered JZL184 (40 mg/kg; i.p.; (Kinsey et al., 2011)) or vehicle. Then, 2 h later, diclofenac sodium (100 mg/kg, p.o.) or vehicle was administered 1 h before oral administration of 0.3 ml solution of phenol red. The phenol red solution as 0.5 mg/ml Phenolsulfonphthalein (phenol red; Sigma-Aldrich, St. Louis, MO) dissolved in a vehicle of distilled water and 1.5% volume of Carboxymethylcellulose (CMC) (Di Marzo et al., 2008). At 20 min post phenol red administration, each mouse was euthanized and its stomach was quickly clamped at the lower esophageal sphincter and pyloric region. Mice in the “baseline” group were administered phenol red and immediately euthanized. Samples recovered from the “baseline” mice were used as a standard baseline (i.e., 0% emptying). Each stomach was removed, opened along the greater curvature, and rinsed with 4 ml Nanopure filtered (Barnstead NANOpure Water Purification System), deionized water and contents collected into a polypropylene test tube. Prior to assay, 2 ml of 1 M sodium hydroxide (NaOH) was added to each tube to develop the maximum intensity of the phenol red color. The optical density of each sample was read at 560 nm on a microplate reader (Vmax kinetic microplate reader, Molecular Devices, Sunnyvale, CA). As the stomach empties, the phenol red exits the stomach. Thus, a saturated color indicates slower gastric motility, whereas unsaturated color indicates hypermotility. The percentage of gastric emptying was calculated according to the following formula:
2.8 Statistical analyses
Diclofenac dose-response data were analyzed using one-way analysis of variance (ANOVA), followed by Dunnett's post hoc comparisons. To evaluate sex differences in gastric hemorrhages, diclofenac dose-response data were analyzed using a two-way ANOVA with sex and drug treatment as the independent variables and hemorrhage length or MPO level as the dependent variable. As a higher body weight may indicate a larger stomach, thus affecting gastric hemorrhages, analysis of covariance (ANCOVA) was also used to determine the influence of sex and drug treatment after controlling for body weight. Correlations between gastric hemorrhage length and MPO were analyzed using the Pearson's correlation coefficient. Gastric acid secretion with diclofenac and gastric motility data were analyzed using a two-way ANOVA with pretreatment (vehicle, JZL184) and drug treatment (vehicle, diclofenac). Whereas gastric acid secretion with pentagastrin data were analyzed using a two-way ANOVA with pretreatment (vehicle, JZL184, omeprazole) and drug treatment (vehicle, pentagastrin) as independent variables. Two-way ANOVA was followed by Tukey's post hoc comparisons. Differences were considered statistically significant if α < .05.
3. Results
3.1 Diclofenac induces gastric hemorrhages and increases MPO activity in both male and female mice
In both male and female mice, gastric hemorrhages were induced using diclofenac (0, 11.11, 33.33, or 100 mg/kg), as detailed above in section 2.3. Diclofenac sodium caused a significant increase in gastric hemorrhages in male [F(3, 28) = 10.72, P < .01; Fig. 1A] and female [F(3, 29) = 8.45, P < .01; Fig. 1C] mice, compared to vehicle treated mice. Post hoc analyses revealed that diclofenac induced gastric hemorrhages at doses of ≥ 33.33 mg/kg in male mice and 100 mg/kg in female mice. Sex differences of gastric hemorrhage development were also evaluated. There was no interaction between sex and drug treatment [F(3, 57) = .917, P = .44; data not shown]. There was a main effect of treatment [F(3, 57) = 18.36, P < .01], such that diclofenac increased gastric hemorrhage formation. There was a main effect of sex [F(1, 57) = 4.27, P < .05], such that male mice had increased gastric hemorrhage length. However, the sex effect was lost after controlling for body weight (P = .45), indicating that diclofenac induced gastric hemorrhages to a similar extent regardless of sex.
Fig. 1.
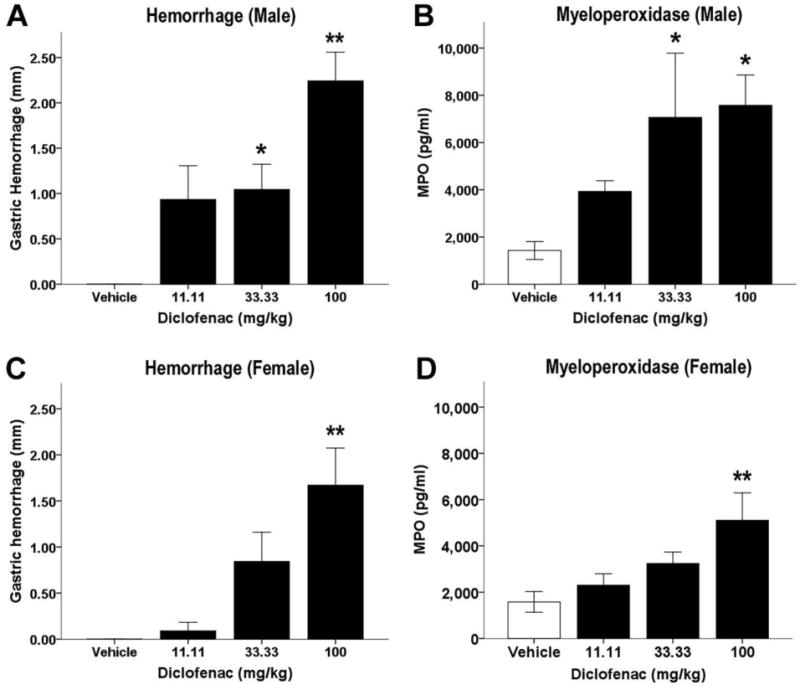
Diclofenac increases gastric hemorrhages and MPO levels. Mice were fasted for 24 h, administered diclofenac (100 mg/kg, p.o.) or vehicle, and stomachs were harvested and mucus collected 6 h later. In mucus samples, diclofenac dose-dependently increases gastric hemorrhage length and gastric mucus myeloperoxidase (MPO) levels in male (n = 8) (A, B) and female (n = 8 or 9) (C, D) mice. Data presented as mean ± S.E.M.; *P < 0.05 vs. vehicle; ** P < 0.01 vs. vehicle
MPO activity, an indirect measure of neutrophil activity, was quantified by ELISA. Diclofenac significantly increased MPO level in male [F(3, 28) = 3.53, P < .05; Fig. 1B] and female [F(3, 29) = 4.19, P < .05; Fig. 1D] mice. Post hoc analyses indicated that diclofenac increased MPO at ≥33.33 mg/kg in male mice and 100 mg/kg in female mice. Sex differences of gastric MPO levels were also evaluated. There was no interaction between sex and drug treatment [F(3, 57) = .95, P = .42]. There was a main effect of treatment [F(3, 57) = 6.48, P < .05], in that diclofenac increased gastric MPO concentration. As with hemorrhage length, there was a main effect of sex [F(1, 57) = 5.25, P < .05], indicating that male mice had higher levels of MPO. However, after controlling for body weight, there was no effect of sex (P = .08).
Because neutrophil infiltration contributes directly to gastric tissue damage, MPO could be used as an objective biomarker for gastric hemorrhage severity. Therefore, a Pearson's correlation coefficient was computed to elucidate the relationship between blind-scored total gastric hemorrhage length and gastric MPO level. There was a positive correlation between gastric hemorrhage length and gastric MPO level in male mice [r = 0.37, N = 32, P < .05]. The data between hemorrhage length and gastric MPO level in female mice was also positive, but did not reach statistical significance [r = 0.33, N = 33, P = .059]. These data indicate that MPO may be used as an objective biomarker for gastric damage.
3.2 MAGL inhibition attenuates gastric hemorrhages in male and female mice
Consistent with previously reported data, the MAGL inhibitor JZL184 significantly attenuated gastric hemorrhages in male mice, the interaction of the pretreatment and drug treatment was significant [F(1, 27) = 22.69, P < .01; Fig. 2A]. There was a main effect of pretreatment, such that JZL184 pretreatment significantly decreased gastric hemorrhages [F(1, 27) = 27.42, P < .01]. There was a main effect of drug, in that diclofenac significantly increased gastric hemorrhage length [F(1, 27) = 26.82, P < .01]. Post hoc analyses revealed that JZL184 significantly decreased ulcer formation (P < .01). We extended these data by challenging diclofenac-induced gastric hemorrhage formation by inhibiting MAGL in female mice. As in male mice, JZL184 significantly attenuated gastric hemorrhages in female mice, the interaction between pretreatment and drug treatment was significant [F(1, 28) = 31.58, P < .01; Fig. 2C]. Post hoc analyses revealed that pretreatment of JZL184 prior to diclofenac significantly reduced gastric hemorrhages (P < .01).
Fig. 2.
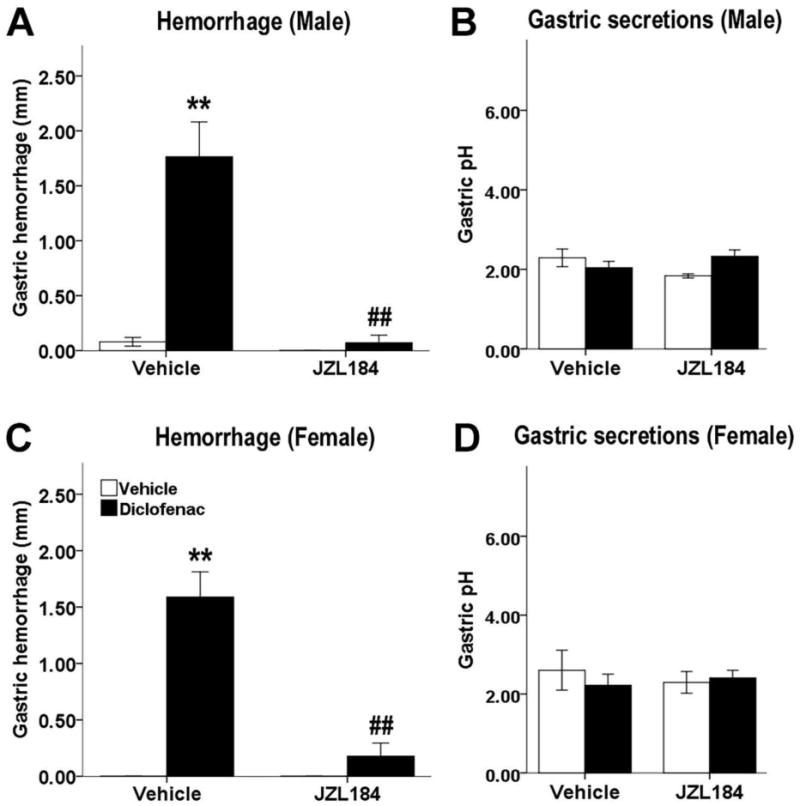
MAGL inhibition prevents the development of gastric hemorrhages induced by diclofenac in male (A) and female (C) mice. Mice were fasted for 22 h, administered JZL184 (40 mg/kg, i.p.) or vehicle, then diclofenac (100 mg/kg, p.o., black bars) or vehicle (white bars) 2 h later, and stomachs and gastric contents were harvested 6 h later. Diclofenac and JZL184 do not alter gastric acid secretions in either male (B) or female (D) mice 6 h or 8 h after administration, respectively. Data presented as mean ± S.E.M. (n = 7-10); **P < 0.01 vs. vehicle/vehicle; ## P < 0.01 vs. vehicle/diclofenac
A second goal of this experiment was to determine the efficacy of endocannabinoid attenuation of induced gastric acid secretion. Mice were subjected to the gastric hemorrhage procedure, with JZL184 (8 mg/kg) administered 2 h prior to diclofenac then acid was collected 6h later, as detailed above in section 2.6. In male mice, there was a significant interaction between JZL184 pretreatment and diclofenac treatment [F(1, 34) = 5.51, P < .05; Fig. 2B]. However, the post hoc analyses revealed no statistically significant differences of JZL184 pretreatment (P = 0.60) or drug treatment (P = .46). Similarly, in female mice, there was no effect of JZL184 pretreatment (P = .86; Fig. 2D) or diclofenac (P = .70) on acid secretions.
3.3 MAGL inhibition reduces pentagastrin-stimulated gastric acid secretion
Given that gastric acid secretion did not differ 6 h after ulcer induction, we next probed gastric acid secretion earlier, at 3 h after JZL184 treatment, under basal and pentagastrin-stimulated acid secretion conditions. There was a significant interaction between pretreatment (JZL184, omeprazole, vehicle) and drug treatment (pentagastrin, vehicle) [F(2, 42) = 9.46, P < .01; Fig. 3]. There was a main effect of pretreatment, indicating that JZL184 (40 mg/kg) and omeprazole (20 mg/kg) pretreatments significantly increased gastric pH [F(2, 42) = 21.07, P < .01], which indicates that either drug decreased gastric acid secretion. There was also a main effect of drug, such that pentagastrin (0.25 mg/kg) significantly decreased gastric pH [F(1, 42) = 13.18, P < .01]. Post hoc analyses revealed JZL184 (P < .05) and omeprazole (P < .01) each significantly decreased pentagastrin-stimulated gastric acid secretions. Further, JZL184, but not omeprazole, decreased basal (i.e., unstimulated) gastric acid secretion (P < .01).
Fig. 3.
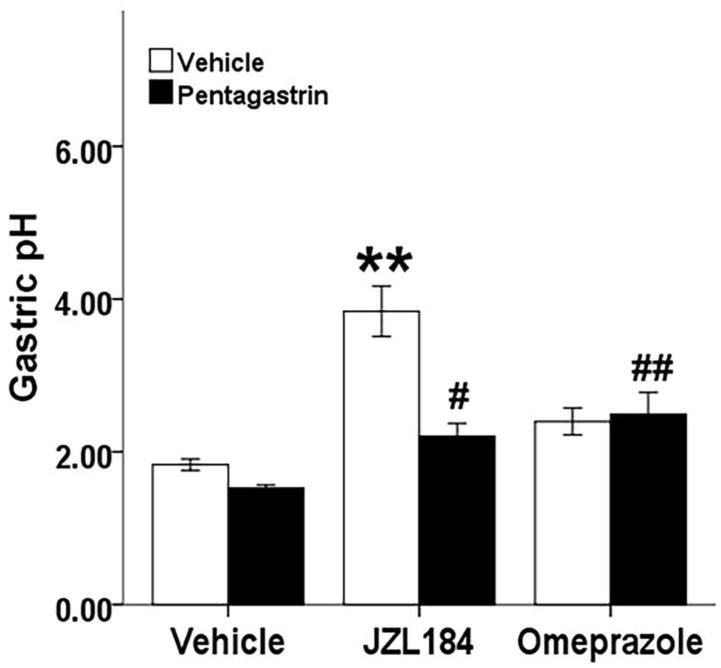
MAGL inhibition reduces pentagastrin-induced and basal gastric acid secretion. Mice were fasted for 22 h, administered JZL184 (40 mg/kg, i.p.), omeprazole (20 mg/kg, i.p.) or vehicle (i.p.), acid secretion was stimulated 2 h later with pentagastrin (0.25 mg/kg, s.c., black bars) or vehicle (s.c., white bars), and gastric contents were collected 1 h later. Data presented as mean ± S.E.M. (n = 8); ** P < .01 vs vehicle/vehicle, # P < .05, ## P < .01 vs vehicle/pentagastrin
3.4 MAGL inhibition decreases gastric motility
Gastric motility was assessed in fasted mice, using phenol red to quantify percent gastric emptying. There was a significant interaction between JZL184 and diclofenac treatments [F(1, 28) = 6.11, P < .05; Fig. 4]. Post hoc analyses revealed that JZL184 decreased gastric motility in the absence (P < .01) and presence (P < .01) of diclofenac, as compared to controls. Similarly, diclofenac administered prior to vehicle decreased gastric motility (P < .01).
Fig. 4.
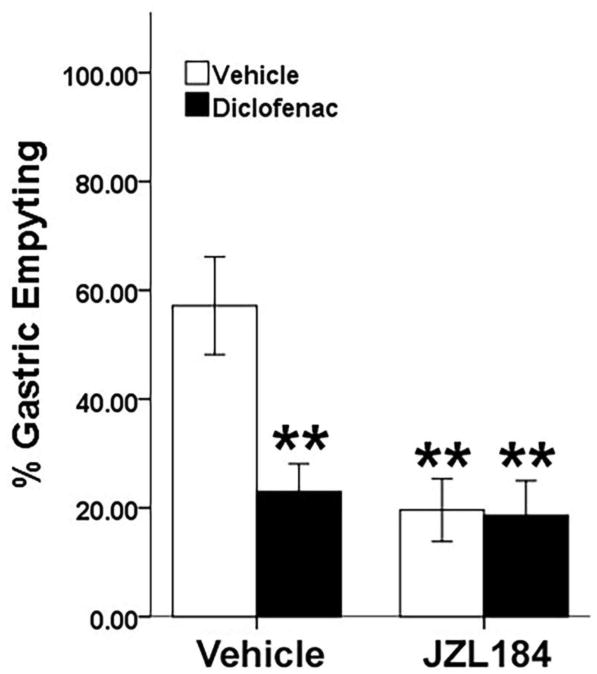
MAGL inhibition decreases gastric emptying. Mice were fasted for 24 h. The mice were administered JZL184 (40 mg/kg) or vehicle, then 2 h later, diclofenac sodium (100 mg/kg, p.o., black bars) or vehicle (p.o., white bars) was administered 1 h before oral administration of phenol red (0.5 mg/ml). JZL184 and diclofenac significantly reduce gastric emptying of phenol red as compared to control mice. Data presented as mean ± S.E.M. (n = 8); * P < .05 vs vehicle treatment.
4. Discussion
The results of the present study align with previous reports that the NSAID, diclofenac sodium, induces gastric hemorrhages, whereas the MAGL inhibitor, JZL184, curtails the formation of these hemorrhages. We included female mice and determined that diclofenac induces gastric hemorrhages to a similar degree in both sexes, once body mass is controlled. Similarly, the MAGL inhibitor JZL184 effectively blocked the formation of diclofenac-induced gastric hemorrhages in both males and females, opening future studies to include females in their designs. In addition, the present data found significant correlations between published hemorrhage quantification methods and neutrophil levels, as assessed by MPO activity. These data support the use of MPO as an objective measure of the severity of NSAID-induced gastric pathology.
The current study also explored the primary mechanisms through which NSAID-induced gastric hemorrhaging is suspected to occur, i.e., increased gastric acid secretion and motility. The MAGL inhibitor JZL184 had a robust antacid effect, both in mice induced by pentagastrin to release acid and under basal conditions. Increased gastric acid secretion is proposed as a primary, probable pathogenic mechanism of NSAID-induced gastric hemorrhages (Wallace, 2008). NSAIDs in vivo increase baseline and stimulated gastric acid secretion, and support the idea that gastric acid is involved with ulcer formation (Salvatella et al., 2004). In the present study, JZL184 significantly decreased gastric acid secretion in both basal and pentagastrin-stimulated conditions 3 h after JZL184 administration. However, JZL184 did not alter gastric acid in the absence or presence of diclofenac following gastric hemorrhage induction, i.e., 8 h after JZL184 administration, indicating that while JZL184 may initially reduce acid secretions, it does not lead to a prolonged reduction in gastric acidity.
In addition to gastric acid secretions, one of the primary, suspected mechanisms through which NSAIDs cause gastric hemorrhages is by increasing mechanical compressions of the stomach (i.e., gastric motility) (Takeuchi, 2012; Wallace, 2008). Gastric motility was assessed in the present study, using a functional model of gastric emptying. The MAGL inhibitor JZL184 significantly attenuated gastric emptying. The present data are in concordance with published reports that anandamide and exogenous cannabinoids receptor agonists slow gastric motility (Aviello et al., 2008; Di Marzo et al., 2008). Due to rapid degradation of 2-AG by MAGL, in vivo, pharmacological inhibition of MAGL via JZL184 is used to indirectly increase levels of 2-AG (Crowe et al., 2014; Kinsey et al., 2009; Long et al., 2009).
Considering established research showing that the administration of cannabinoid receptor agonists reduces gastrointestinal motility in mice, rats, and humans through the activation of CB1 receptors (Aviello et al., 2008; Esfandyari et al., 2006; Izzo and Sharkey, 2010; Maccarrone et al., 2015; McCallum et al., 1999), we hypothesized that JZL184 would attenuate NSAID-induced increases in gastric motility. As hypothesized, JZL184 decreased gastric motility. Surprisingly, diclofenac also slowed gastric emptying. Thus, it is tempting to conclude MAGL inhibition exerts its gastroprotective effects solely by decreasing gastric acid and not alterations in motility. But, neither of these mechanisms has been identified as the unequivocal mediating factor resulting in gastric hemorrhages. It is plausible that either mechanism is necessary, but not sufficient to induce gastric hemorrhaging in the presence of a high dose NSAID. In previous reports, NSAIDs increased gastric motility when measured continuously. Gastric motility is generally quantified via balloon manometry, in which a balloon is inserted into the stomach and changes in pressure are recorded to determine gastric contractions (Mersereau and Hinchey, 1982; Takeuchi, 2012; Takeuchi et al., 1986; Tanaka et al., 2001). The gastric motility assay used in the present study is a functional measure of gastric emptying that quantifies movement of a dye from the stomach. Conversely, balloon manometry measures fluctuations in frequency and amplitude of muscle contractions over time and may reveal a more subtle mucosal compression than can be determined in whole stomach movement, as measured by gastric emptying in the present study. In addition, motility is generally assessed in rats, as opposed to mice, due largely to the physiological size limitations in mice (Monroe et al., 2004).
More potential mechanisms involved in the formation of NSAID-induced ulcers exist than can be feasibly addressed in one study. One such alternative is deficient wound healing, caused by NSAID-induced hindering of microcirculation of the gastric mucosa (Ma et al., 2001; Wallace, 2008). Specifically, COX inhibition is proposed to alter neutrophil activation and gastrointestinal permeability, thereby increasing ulceration (Reuter et al., 1997; Rodriguez-Stanley et al., 2006). The present data indicate that diclofenac dose-dependently increases MPO, suggesting that the stomachs of mice treated with diclofenac had more neutrophil activation, thus supporting the idea that neutrophil infiltration contributes to gastric ulceration.
Neutrophils are also proposed to contribute to NSAID-induced gastric damage by adhering to the vascular endothelium, thereby inhibiting the ability of capillaries to maintain normal blood flow to the mucosa (i.e., by physically blocking the movement of red blood cells), and by producing oxygen-derived free radicals, which cause tissue damage (Beck et al., 2000; Wallace and Vong, 2008). Through the activation of neutrophils, NSAIDs foster oxygen metabolite free radical synthesis and also decrease the antioxidizing enzymes that defend against reactive oxygen metabolites (Pohle et al., 2001), thus further increasing free radical activity. Free radicals cause oxidative degradation of lipids, which damages endothelial cells, subsequently forming gaps in the cell membrane (Beck et al., 2000; Kwiecien et al., 2002). The free radical induced damage increases permeability of the stomach barrier, which reduces the protective lining of the gastrointestinal system (Kwiecien et al., 2002; Pohle et al., 2001). NSAIDs dose-dependently increase extravasation in the intestines (Beck et al., 2000; Reuter et al., 1997), indicating the intestinal barrier is compromised.. Protection against barrier deficiency and hemorrhagic effects of the mutant mice indicate the neutrophil-dependent release of oxides may play a bigger role in hemorrhage formation than neutrophil adhesion to the endothelium itself. Thus, an important future area of investigation would evaluate endocannabinoid modulation of free radical production in the gastric epithelium.
In conclusion, the present data indicate that the NSAID diclofenac induces gastric hemorrhaging and neutrophil infiltration in both male and female mice. These data support targeting MAGL as a therapeutic approach to protect against NSAID-induced gastric hemorrhages. As with previously published reports indicate, NSAIDs appear to cause gastric hemorrhages through multiple, simultaneous pathways that are necessary, but not individually sufficient to induce gastric ulceration. That is, one potential pathomechanism of NSAID-induced hemorrhages by itself will not likely result in gastric hemorrhages, but it is the combined physiological changes that is prompting ulcer development.
Acknowledgments
The authors thank Sara Nass and Kristen Trexler for technical and editorial assistance.
Funding: This research was supported by the National Institutes of Health [AR066806, DA038714, and GM081741].
Footnotes
Conflict of Interest: none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adami M, Frati P, Bertini S, Kulkarni-Narla A, Brown DR, de Caro G, Coruzzi G, Soldani G. Gastric antisecretory role and immunohistochemical localization of cannabinoid receptors in the rat stomach. Br J Pharmacol. 2002;135:1598–606. doi: 10.1038/sj.bjp.0704625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astudillo L, Rodriguez JA, Schmeda-Hirschmann G. Gastroprotective activity of oleanolic acid derivatives on experimentally induced gastric lesions in rats and mice. J Pharm Pharmacol. 2002;54:583–588. doi: 10.1211/0022357021778718. [DOI] [PubMed] [Google Scholar]
- Aviello G, Romano B, Izzo aa. Cannabinoids and gastrointestinal motility: animal and human studies. Eur Rev Med Pharmacol Sci. 2008;12(Suppl 1):81–93. [PubMed] [Google Scholar]
- Beck PL, Xavier R, Lu N, Nanda NN, Dinauer M, Podolsky DK, Seed B. Mechanisms of NSAID-induced gastrointestinal injury defined using mutant mice. Gastroenterology. 2000;119:699–705. doi: 10.1053/gast.2000.16497. doi:S0016508500973098. [pii] [DOI] [PubMed] [Google Scholar]
- Coruzzi G, Adami M, Coppelli G, Frati P, Soldani G. Inhibitory effect of the cannabinoid receptor agonist WIN 55,212-2 on pentagastrin-induced gastric acid secretion in the anaesthetized rat. Naunyn Schmiedebergs Arch Pharmacol. 1999;360:715–8. doi: 10.1007/s002109900135. [DOI] [PubMed] [Google Scholar]
- Coruzzi G, Adami M, Guaita E, Menozzi A, Bertini S, Giovannini E, Soldani G. Effects of cannabinoid receptor agonists on rat gastric acid secretion: discrepancy between in vitro and in vivo data. Dig Dis Sci. 2006;51:310–7. doi: 10.1007/s10620-006-3130-2. [DOI] [PubMed] [Google Scholar]
- Craft RM. Sex differences in behavioral effects of cannabinoids. Life Sci. 2005;77:2471–8. doi: 10.1016/j.lfs.2005.04.019. [DOI] [PubMed] [Google Scholar]
- Craft RM, Kandasamy R, Davis SM. Sex differences in anti-allodynic, anti-hyperalgesic and anti-edema effects of Δ9-tetrahydrocannabinol in the rat. Pain. 2013a;154:1709–1717. doi: 10.1016/j.pain.2013.05.017. [DOI] [PubMed] [Google Scholar]
- Craft RM, Marusich JA, Wiley JL. Sex differences in cannabinoid pharmacology: A reflection of differences in the endocannabinoid system? Life Sci. 2013b;92:476–481. doi: 10.1016/j.lfs.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe MS, Leishman E, Banks ML, Gujjar R, Mahadevan A, Bradshaw HB, Kinsey SG. Combined inhibition of monoacylglycerol lipase and cyclooxygenases synergistically reduces neuropathic pain in mice. Br J Pharmacol. 2015;172:1700–1712. doi: 10.1111/bph.13012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe MS, Nass SR, Gabella KM, Kinsey SG. The endocannabinoid system modulates stress, emotionality, and inflammation. Brain Behav Immun. 2014;42:1–5. doi: 10.1016/j.bbi.2014.06.007. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Capasso R, Matias I, Aviello G, Petrosino S, Borrelli F, Romano B, Orlando P, Capasso F, Izzo AA. The role of endocannabinoids in the regulation of gastric emptying: alterations in mice fed a high-fat diet. Br J Pharmacol. 2008;153:1272–1280. doi: 10.1038/sj.bjp.0707682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esfandyari T, Camilleri M, Ferber I, Burton D, Baxter K, Zinsmeister AR. Effect of a cannabinoid agonist on gastrointestinal transit and postprandial satiation in healthy human subjects: A randomized, placebo-controlled study. Neurogastroenterol Motil. 2006;18:831–838. doi: 10.1111/j.1365-2982.2006.00834.x. [DOI] [PubMed] [Google Scholar]
- Ferrero RL, Avé P, Radcliff FJ, Labigne A, Huerre MR. Outbred mice with long-term Helicobacter felis infection develop both gastric lymphoid tissue and glandular hyperplastic lesions. J Pathol. 2000;191:333–340. doi: 10.1002/1096-9896(2000)9999:9999<∷AID-PATH619>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Fuchs KH, DeMeester TR, Hinder RA, Stein HJ, Barlow AP, Gupta NC. Computerized identification of pathologic duodenogastric reflux using 24-hour gastric pH monitoring. Ann Surg. 1991;213:13–20. doi: 10.1097/00000658-199101000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatowska-Jankowska BM, Ghosh S, Crowe MS, Kinsey SG, Niphakis MJ, Abdullah RA, Tao Q, O' Neal ST, Walentiny DM, Wiley JL, Cravatt BF, Lichtman aH. In vivo characterization of the highly selective monoacylglycerol lipase inhibitor KML29: antinociceptive activity without cannabimimetic side effects. Br J Pharmacol. 2014;171:1392–407. doi: 10.1111/bph.12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo AA, Sharkey KA. Cannabinoids and the gut: new developments and emerging concepts. Pharmacol Ther. 2010;126:21–38. doi: 10.1016/j.pharmthera.2009.12.005. [DOI] [PubMed] [Google Scholar]
- Kinsey SG, Cole EC. Acute Δ(9)-tetrahydrocannabinol blocks gastric hemorrhages induced by the nonsteroidal anti-inflammatory drug diclofenac sodium in mice. Eur J Pharmacol. 2013;715:111–6. doi: 10.1016/j.ejphar.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Long JZ, O'Neal ST, Abdullah RA, Poklis JL, Boger DL, Cravatt BF, Lichtman AH. Blockade of endocannabinoid-degrading enzymes attenuates neuropathic pain. J Pharmacol Exp Ther. 2009;330:902–10. doi: 10.1124/jpet.109.155465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Nomura D, O'Neal S, Long JZ, Mahadevan A, Cravatt B, Grider J, Lichtman AH. Inhibition of monoacylglycerol lipase attenuates nonsteroidal anti-inflammatory drug-induced gastric hemorrhages in mice. J Pharmacol Exp Ther. 2011;338:795–802. doi: 10.1124/jpet.110.175778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogan NM, Mechoulam R. Cannabinoids in health and disease. Dialogues Clin Neurosci. 2007;9:413–430. doi: 10.31887/DCNS.2007.9.4/nkogan. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuller LH. Do Proton Pump Inhibitors Increase the Risk of Dementia? JAMA Neurol. 2016;73:379–381. doi: 10.1001/jamaneurol.2015.4931. [DOI] [PubMed] [Google Scholar]
- Kwiecien S, Brzozowski T, Konturek SJ. Effects of reactive oxygen species action on gastric mucosa in various models of mucosal injury. J Physiol Pharmacol. 2002;53:39–50. [PubMed] [Google Scholar]
- Lanas A. Nonsteroidal antiinflammatory drugs and cyclooxygenase inhibition in the gastrointestinal tract: a trip from peptic ulcer to colon cancer. Am J Med Sci. 2009;338:96–106. doi: 10.1097/MAJ.0b013e3181ad8cd3. [DOI] [PubMed] [Google Scholar]
- Liu W, Okajima K, Murakami K, Harada N, Isobe H, Irie T. Role of neutrophil elastase in stress-induced gastric mucosal injury in rats. J Lab Clin Med. 1998;132:432–439. doi: 10.1016/S0022-2143(98)90114-7. [DOI] [PubMed] [Google Scholar]
- Lodato F, Azzaroli F, Turco L, Mazzella N, Buonfiglioli F, Zoli M, Mazzella G. Adverse effects of proton pump inhibitors. Best Pract Res Clin Gastroenterol. 2010;24:193–201. doi: 10.1016/j.bpg.2009.11.004. [DOI] [PubMed] [Google Scholar]
- Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavon FJ, Serrano AM, Selley DE, Parsons LH, Lichtman AH, Cravatt BF. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol. 2009;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loria V, Dato I, Graziani F, Biasucci LM. Myeloperoxidase: A new biomarker of inflammation in ischemic heart disease and acute coronary syndromes. Mediators Inflamm. 2008;2008 doi: 10.1155/2008/135625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Elliott SN, Cirino G, Buret a, Ignarro LJ, Wallace JL. Platelets modulate gastric ulcer healing: role of endostatin and vascular endothelial growth factor release. Proc Natl Acad Sci U S A. 2001;98:6470–6475. doi: 10.1073/pnas.111150798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccarrone M, Bab I, Bíró T, Cabral GA, Dey SK, Di Marzo V, Konje JC, Kunos G, Mechoulam R, Pacher P, Sharkey KA, Zimmer A. Endocannabinoid signaling at the periphery: 50 years after THC. Trends Pharmacol Sci. 2015;36:277–296. doi: 10.1016/j.tips.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez V, Curi AP, Torkian B, Schaeffer JM, Wilkinson HA, Walsh JH, Tache Y. High Basal Gastric Acid Secretion in Somatostatin Receptor. Gastroenterology. 1998;114:1125–1132. doi: 10.1016/s0016-5085(98)70417-2. [DOI] [PubMed] [Google Scholar]
- McCallum RW, Soykan I, Sridhar KR, Ricci DA, Lange RC, Plankey MW. Delta-9-tetrahydrocannabinol delays the gastric emptying of solid food in humans: A double-blind, randomized study. Aliment Pharmacol Ther. 1999;13:77–80. doi: 10.1046/j.1365-2036.1999.00441.x. [DOI] [PubMed] [Google Scholar]
- Mersereau WA, Hinchey EJ. Prevention of phenylbutazone ulcer in the rat by glucose: Role of a glycoprivic receptor system. Am J Physiol Gastrointest Liver Physiol. 1982;242:G429–G432. doi: 10.1152/ajpgi.1982.242.4.G429. [DOI] [PubMed] [Google Scholar]
- Monroe MJ, Hornby PJ, Partosoedarso ER. Central vagal stimulation evokes gastric volume changes in mice: A novel technique using a miniaturized barostat. Neurogastroenterol Motil. 2004;16:5–11. doi: 10.1046/j.1365-2982.2003.00464.x. [DOI] [PubMed] [Google Scholar]
- Mössner J. The indications, applications, and risks of proton pump inhibitors - a review after 25 years. Dtsch Arztebl Int. 2016;113:477–483. doi: 10.3238/arztebl.2016.0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naidu PS, Booker L, Cravatt BF, Lichtman AH. Synergy between enzyme inhibitors of fatty acid amide hydrolase and cyclooxygenase in visceral nociception. J Pharmacol Exp Ther. 2009;329:48–56. doi: 10.1124/jpet.108.143487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazos MR, Tolón RM, Benito C, Rodríguez CF, Gorgojo JJ, Nevado M, Alvarez M, Arias F, Almodóvar F, Fernández MTP, Lledó JL, González S, Fernández-Ruiz JJ, Romero J. Cannabinoid CB1 receptors are expressed by parietal cells of the human gastric mucosa. J Histochem Cytochem. 2008;56:511–6. doi: 10.1369/jhc.2008.950741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohle T, Brzozowski T, Becker JC, Van Der Voort IR, Markmann A, Konturek SJ, Moniczewski A, Domschke W, Konturek JW. Role of reactive oxygen metabolites in aspirin-induced gastric damage in humans: Gastroprotection by vitamin C. Aliment. Pharmacol Ther. 2001;15:677–687. doi: 10.1046/j.1365-2036.2001.00975.x. [DOI] [PubMed] [Google Scholar]
- Reuter B, Davies N, Wallace JL. Nonsteroidal anti-inflammatory drug enteropathy in rats: Role of permeability, bacteria, and enterohepatic circulation. Gastroenterology. 1997;112:109–117. doi: 10.1016/S0016-5085(97)70225-7. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Stanley S, Redinger N, Miner PB. Effect of naproxen on gastric acid secretion and gastric pH. Aliment Pharmacol Ther. 2006;23:1719–1724. doi: 10.1111/j.1365-2036.2006.02897.x. [DOI] [PubMed] [Google Scholar]
- Salvatella MA, Rossi I, Del Valle JC, Gutierrez Y, Pereda C, Samper B, Feliu JE. Inhibition of acid secretion by the nonsteroidal anti-inflammatory drugs diclofenac and piroxicam in isolated gastric glands: analysis of a multifocal mechanism. Am J Physiol Liver Physiol. 2004;286:G711–G721. doi: 10.1152/ajpgi.00305.2003. [DOI] [PubMed] [Google Scholar]
- Schubert ML. Hormonal Regulation of Gastric Acid Secretion. Curr Gastroenterol Rep. 2008;10:523–527. doi: 10.1007/s11894-008-0097-5. [DOI] [PubMed] [Google Scholar]
- Takeuchi K. Pathogenesis of NSAID-induced gastric damage: importance of cyclooxygenase inhibition and gastric hypermotility. World J Gastroenterol. 2012;18:2147–2160. doi: 10.3748/wjg.v18.i18.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K, Ueki S, Okabe S. Importance of gastric motility in the pathogenesis of indomethacin-induced gastric lesions in rats. Dig Dis Sci. 1986;31:1114–1122. doi: 10.1007/BF01300266. [DOI] [PubMed] [Google Scholar]
- Tanaka A, Araki H, Komoike Y, Hase S, Takeuchi K. Inhibition of both COX-1 and COX-2 is required for development of gastric damage in response to nonsteroidal anti-inflammatory drugs. J Physiol Paris. 2001;95:21–27. doi: 10.1016/S0928-4257(01)00005-5. [DOI] [PubMed] [Google Scholar]
- Wallace JL. Prostaglandins, NSAIDs , and Gastric Mucosal Protection: Why Doesn't the Stomach Digest Itself? Physiol Rev. 2008;88:1547–1565. doi: 10.1152/physrev.00004.2008. [DOI] [PubMed] [Google Scholar]
- Wallace JL. Pathogenesis of NSAID-induced gastroduodenal mucosal injury. Best Pract Res Clin Gastroenterol. 2001;15:691–703. doi: 10.1053/bega.2001.0229. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Keenan CM, Granger DN. Gastric ulceration induced by nonsteroidal anti-inflammatory drugs is a neutrophil-dependent process. Am J Physiol. 1990;259:G462–7. doi: 10.1152/ajpgi.1990.259.3.G462. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Vong L. NSAID-induced gastrointestinal damage and the design of GI-sparing NSAIDs. Curr Opin Investig Drugs. 2008;9:1151–1156. [PubMed] [Google Scholar]
- Yano S, Akahane M, Harada MA. Role of gastric motility in development of stress-induced gastric lesions of rats. Jpn J Pharmacol. 1978;28:607–615. doi: 10.1254/jjp.28.607. [DOI] [PubMed] [Google Scholar]