

Abstract
Group V phospholipase A2 (Pla2g5) is a lipid-generating enzyme necessary for macrophage effector functions in pulmonary inflammation. However, the lipid mediators involved and their cellular targets have not been identified. Mice lacking Pla2g5 showed markedly reduced lung ILC2 activation and eosinophilia following repetitive Alternaria Alternata inhalation. While Pla2g5-null mice had Wt levels of immediate IL-33 release after one Alternaria dose, they failed to upregulate IL-33 in macrophages following repeated Alternaria administration. Unexpectedly, while adoptive transfer of bone marrow-derived (BM)-macrophages restored ILC2 activation and eosinophilia in Alternaria-exposed Pla2g5-null mice, exogenous IL-33 did not. Conversely, transfers of Pla2g5-null BM-macrophages reduced inflammation in Alternaria-exposed Wt mice. Mass spectrometry analysis of free fatty acids (FFAs) demonstrated significantly reduced FFAs (including linoleic acid (LA) and oleic acid (OA)) in lung and BM-macrophages lacking Pla2g5. Exogenous administration of LA or LA+OA to Wt mice sharply potentiated IL-33-induced lung eosinophilia and ILC2 expansion in-vitro and in-vivo. In contrast, OA potentiated IL-33-induced inflammation and ILC2 expansion in Pla2g5-null mice, but LA was inactive both in-vivo and in-vitro. Notably, Pla2g5-null ILC2s showed significantly reduced expression of the FFA-receptor-1 compared to Wt ILC2s. Thus, macrophage-associated Pla2g5 contributes significantly to type-2 immunity through regulation of IL-33 induction and FFA-driven ILC2 activation.
Introduction
Alternaria Alternata is a common fungus that is a source of allergens associated with the development of asthma and asthma exacerbations. In mice, Alternaria allergens trigger the accumulation of eosinophils and the development of airway hyperreactivity1, 2, each of which prominently involves effectors of the innate immune system1, 3, 4. Acute exposure of the airways of naive mice to Alternaria causes the rapid release of IL-33 by epithelial cells, followed by the activation of group 2 innate lymphoid cells (ILC2s)3, 5. Long-term repetitive administration of Alternaria upregulates whole lung expression of IL-33, and promotes incremental ILC2-dependent lung eosinophilic inflammation1. ILC2s lack cell surface markers associated with major hematopoietic lineages (Lin−)6–8. In the lung they express Thy1.2 (CD45+ Lin− Thy1.2+)9 and inducible molecules including ST2 (IL1R1), Sca-1, CD278 (ICOS), CD25 (IL-2Rα), CD127 (IL-7Rα), CD117 (c-Kit), and IL-17RB (IL-25R)1, 10–12. Following activation, ILC2s produce IL-5 and IL-13 (as well as other cytokines), which mediate pulmonary eosinophilia, airway hyperreactivity1, 12, 13 and macrophage activation14. Although IL-33 in naive mouse lung is principally derived from structural cells15, hematopoietic cells (including macrophages) can express IL-33 inducibly16, 17. Macrophages can activate ILC2s through an IL-33-dependent mechanism in a model of influenza-induced airway hyperreactivity10. Whether macrophages contribute to activating ILC2s in allergic inflammation in general, or in Alternaria-induced pulmonary inflammation in particular is not known.
Phospholipases A2 (PLA2) are a family of enzymes that release lysophospholipids and free fatty acids (FFAs) from membrane glycerophospholipids18, 19. While FFAs such as arachidonic acid (AA) can be converted to receptor-active eicosanoids (including prostaglandins and leukotrienes), other FFAs can act directly at cognate receptors to regulate metabolic processes and inflammatory responses20. PLA2s may have substrate preferences and specific cell and tissue expression, therefore serving context-specific functions. Group V PLA2 (Pla2g5) preferentially releases lysophosphatidylcholine (LPC) and the FFAs linoleic acid (LA) and oleic acid (OA)21–23, and is prevalently expressed by innate immune cells, including dendritic cells and macrophages24–26, as well as epithelial cells25–27. Using a mouse model of allergic lung inflammation induced by the allergens of house dust mite Dermatophagoides farinae, we found that Pla2g5 was necessary for the effector functions of both dendritic cells and macrophages24, 25. Adoptive transfer studies showed that Pla2g5 expression by macrophages was required for their generation of CCL22 and recruitment of T cells into the lungs25. While the defects in cellular functions resulting from cell intrinsic absence of Pla2g5 suggest critical roles for endogenous lipids, neither the identity of the lipids nor their potential paracrine cellular targets are known.
Because eicosanoids may contribute to ILC2-mediated pulmonary inflammation9, 28, we hypothesized that Pla2g5-derived lipids generated from macrophages may contribute to ILC2 activation and subsequent pulmonary inflammation. Here we demonstrate that ILC2 activation is impaired in Pla2g5-null mice exposed to Alternaria. Moreover, adoptive transfers of macrophages restored ILC2 activation by a mechanism that is at least in part dependent on Pla2g5-dependent production of IL-33 and releases of LA, OA and AA by macrophages, which sustain ILC2 activation, and on Pla2g5-dependent expression of the LA-preferring FFA-receptor-1 (FFAR1) by ILC2s.
Results
Pulmonary inflammatory response to Alternaria requires Pla2g5
To investigate the role of Pla2g5 in activation of ILC2s, we used a model of allergic pulmonary inflammation induced by Alternaria, which relies on ILC2 activation to cause eosinophilic inflammation. We administered Alternaria (25μg/dose) every two days for four doses and lungs were collected 18h after the last dose1. Wt mice treated with Alternaria had significantly increased total lung cell numbers compared to Alternaria-treated Pla2g5-null mice (Figure 1A). The number of eosinophils (identified as CD45+/CD11c−/SiglecF+ cells)29 in Alternaria-treated Wt lungs was significantly higher than in Alternaria-treated Pla2g5-null lungs (Figure 1B). Alternaria-treated Wt mice had a significantly higher number of ILC2s, identified as CD45+, Lin−, Thy1.2+ cells9 (see Supplementary Figure 1A for staining controls), than equivalently treated Pla2g5-null mice (Figure 1C), although the percentages were similar (Figure 1C and data not shown). The expression of the inducible markers Sca-1, ST2, CD25, ICOS by lung ILC2s was drastically reduced in Alternaria-treated Pla2g5-null mice compared to ILC2s isolated from equally treated Wt mice (Figure 1D and Supplementary Figure 1B). The numbers (Figure 1E) and percentages (Supplementary Figure 1B and data not shown) of ILC2s expressing IL-5 or IL-13 were also significantly reduced in Pla2g5-null mice treated with Alternaria compared to Wt mice. Similar results were obtained by gating ILC2s as CD45+ Lin− Thy1.2+ Sca-1+ cells to exclude at least contaminating ILC330, 31 (Supplementary Figure 1C and data not shown). These data suggest that the absence of Pla2g5 affects both numbers and activation of lung ILC2s.
Figure 1. Alternaria-induced pulmonary inflammation requires Pla2g5.
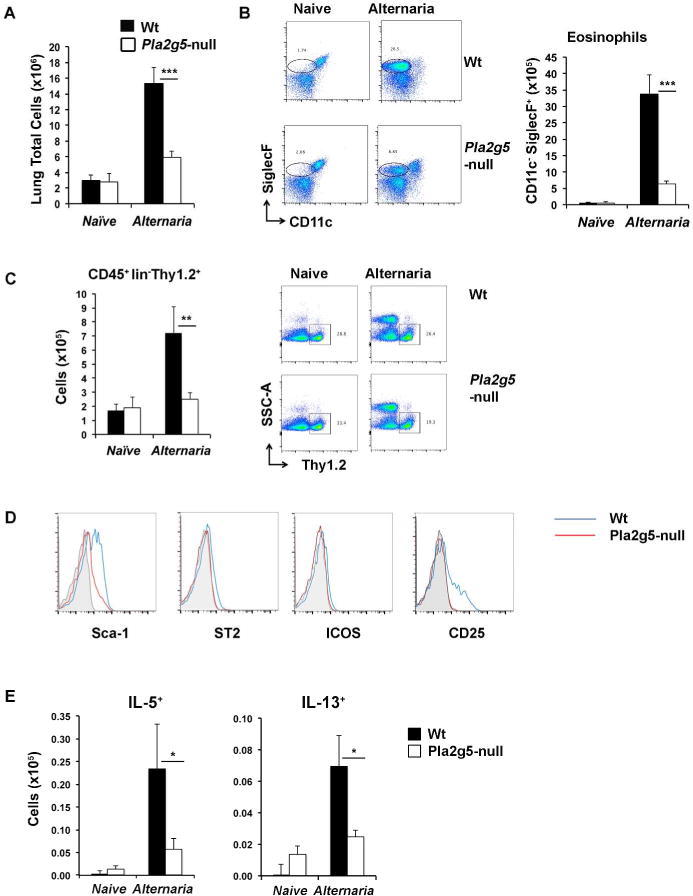
(A) Total cells counts from homogenate lungs of naive and Alternaria-treated Wt and Pla2g5-null mice. Analysis by flow cytometry of lung cell from naive and Alternaria-treated Wt and Pla2g5-null lung homogenates of (B) eosinophils gated as CD45+ CD11c− SiglecF+; (C) ILC2s gated as CD45+ Lin− Thy1.2+; (D) histograms of Sca-1, ST2, ICOS, CD25 gated on Thy1.2+ cells (isotype in gray, Wt in blue, Pla2g5-null in red), and (E) expression of intracellular IL-5 and IL-13 by Thy1.2+ ILC2s. Values are mean ± SEM of at least three independent experiments with 5–9 (naïve) or 10–21 (Alternaria-treated) mice per group. Images are from one representative mouse per group. *** P< 0.0005, **P< 0.005, *P< 0.05.
Induced IL-33 expression requires Pla2g5
Whereas IL-33 is constitutively expressed by lung barrier cells, its expression can also be upregulated during sustained inflammatory responses, in part reflecting the contributions from hematopoietic cells32. To investigate whether the reduced ILC2 activation in Pla2g5-null mice was associated with a lack of either constitutive or inducible pools of IL-33, we measured IL-33 release into the BAL fluids of naive mice after administration of a single Alternaria dose. We also monitored the content of IL-33 in the lung at baseline and after 4 doses of Alternaria using western blotting. We found that naive Wt and Pla2g5-null mice released similar amounts of IL-33 into BAL at 1 and 3h after Alternaria challenge (Figure 2A), and showed equivalent amounts of immunoreactive IL-33 in lung lysates (Figure 2B). Only the full-length IL-33 was detected in the naive mice. After ten days and four doses of Alternaria, Wt lungs had increased amounts of IL-33 protein compared to naive mice, and both the preformed full-length form (34 kDa) and the proteolytically processed short length form (18kDa) were present. Compared with the Alternaria-treated Wt controls, the lungs of Pla2g5-null mice showed sharply diminished induction of both the 18 and 34 kDa forms of IL-33 (Figure 2B).
Figure 2. Induced IL-33 generation in the lung requires Pla2g5.
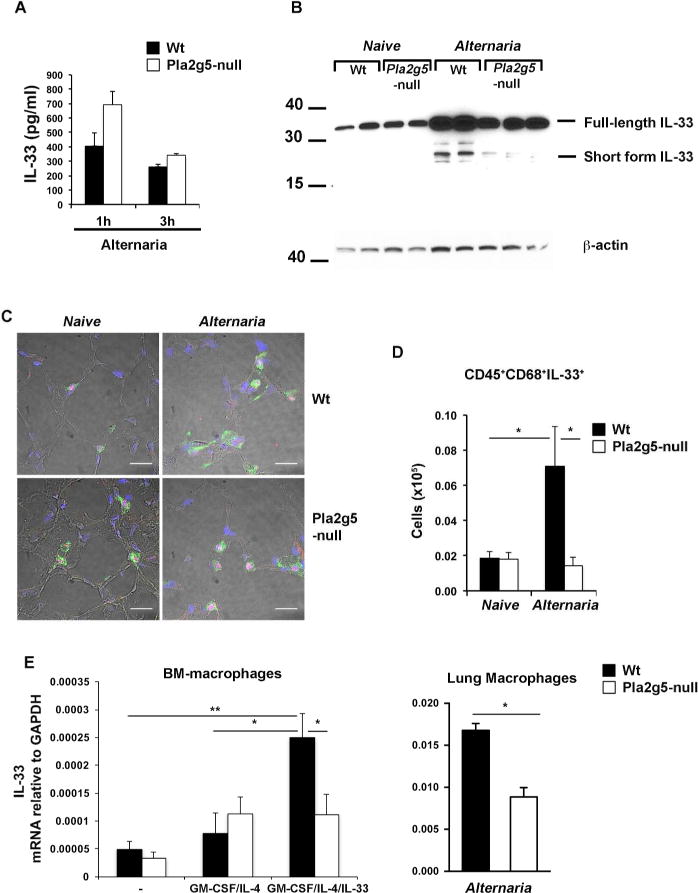
(A) IL-33 levels determined by ELISA of BAL 1 or 3 h after Alternaria administration in Wt and Pla2g5-null mice. (B) Expression of IL-33 protein by Western blot in homogenized lungs of naive and Alternaria-treated Wt and Pla2g5-null mice treated with Alternaria for 10 days (4 doses). Equivalent loading was confirmed by immunoblot analysis for β-actin. (C) Frozen sections from the lungs of Wt and Pla2g5-null mice naive or treated with Alternaria for 10 days, were stained for IL-33 (red), SPC (green) and nuclei (blue). Original magnification ×40. Size bar 50 μm. (D) Expression of IL-33 on gated CD68+ lung cells of naive and Alternaria-treated Wt and Pla2g5-null mice evaluated by flow cytometry. (E) Expression of IL-33 mRNA relative to GAPDH measured by qPCR in BM-macrophages unstimulated or stimulated with GM-CSF/IL-33/IL-4 or lung macrophages enriched by Percoll density gradients from 5–8 pulled lung homogenates of Alternaria-treated mice. Values are mean ± SEM from two or three independent experiments assayed in duplicate (E) or with 5–8 mice per group (A and D). (B and C) Images and panels are from one experiment representative of two with similar results. **P< 0.005, *P< 0.05.
To identify the cellular source(s) responsible for the constitutive and inducible pools of IL-33, we stained frozen sections of Wt and Pla2g5-null lungs with anti-IL-33. Since alveolar type 2 pneumocytes (AT2) are one of the major sources of IL-33 in Alternaria challenged mice15, we counterstained the lung sections with Abs against the AT2 cell marker surfactant protein C (SPC). Lungs of both Wt and Pla2g5-null mice showed IL-33 in the nuclei of SPC+ AT2 cells at baseline, with no differences between Wt and Pla2g5-null Alternaria challenged mice (Figure 2C). Since lung macrophages can also express IL-33 in a model of prolonged exposure to viral allergens10 and in the recovery phase of IAV infection33, we used intracellular staining and flow cytometry to determine whether macrophages contributed to the Alternaria-inducible pool of IL-33. Intracellular staining showed that Alternaria increased the number of CD68+/IL-33+ macrophages in Wt mice (Figure 2D). The number of CD68+/IL-33+ cells was significantly reduced in Alternaria-treated Pla2g5-null mice. To determine whether cell-intrinsic Pla2g5 was involved in inducing IL-33 expression by macrophages, we examined IL-33 expression by Wt and Pla2g5-null BM-macrophages stimulated with GM-CSF, IL-4, and IL-3325, and also by lung macrophages enriched from Alternaria-treated Wt and Pla2g5-null mice using consecutive Percoll gradients25. Wt BM-macrophages activated to full potential by GM-CSF/IL-4/IL-33 displayed robust induced expression of IL-33 mRNA compared to macrophages unstimulated or activated more weakly with GM-CSF/IL-4. In contrast, Pla2g5-null GM-CSF/IL-4/IL-33 BM-macrophages showed significantly reduced induction compared to equally treated Wt controls (Figure 2E, left panel). Wt lung macrophages enriched from Alternaria-treated mice expressed significantly higher IL-33 mRNA compared to equally treated Pla2g5-null lung macrophages (Figure 2E, right panel).
Pla2g5-sufficient macrophages, but not IL-33 alone, can restore ILC2 activation and inflammation to Pla2g5-null mice
Next, we wanted to ascertain whether exogenous recombinant (r)-IL-33 would restore eosinophilia and ILC2 activation in Pla2g5-null mice. Administration of IL-33 over 10 days (100ng/dose, Figure 3, inset) robustly increased the numbers of eosinophils, ILC2s and Sca-1+ ILC2s in Wt mice3. Surprisingly, Pla2g5-null mice showed markedly diminished numbers of eosinophils, total ILC2s, and Sca-1+ ILC2s after treatment with IL-33 compared with Wt controls (Figure 3A). Exogenous IL-33 also induced substantial macrophage activation in Wt mice, as determined by the detection of resistin-like molecule alpha (RELMα) in macrophages. In contrast, macrophage activation was markedly impaired in IL-33-treated Pla2g5-null animals (Figure 3B). To determine whether the defect in ILC2 function reflected the effects of ILC2-intrinsic Pla2g5, we sorted ILC2s from the lungs of Wt mice and performed qPCR. Pla2g5 transcripts were not detected in ILC2s (data not shown).
Figure 3. r-IL-33 induces pulmonary inflammation in Wt but not in Pla2g5-null mice.
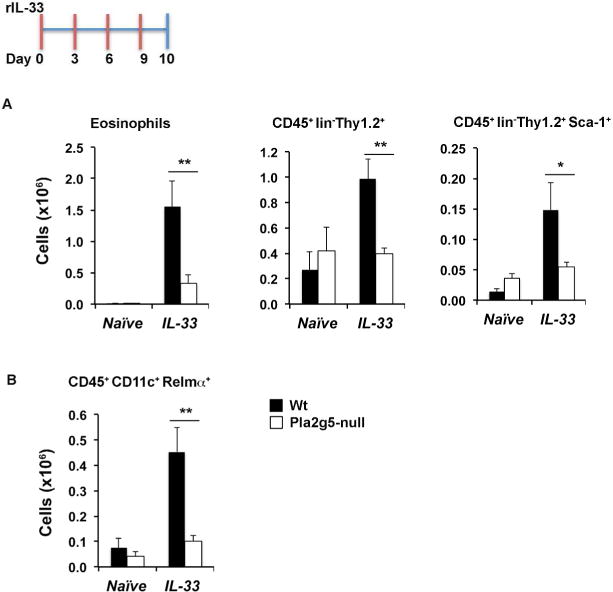
Flow cytometry analysis of (A) eosinophils, Thy1.2+ ILC2s and Thy1.2+ ILC2s expressing Sca-1, (B) Relm-α expression on gated CD45+CD11c+ cells from homogenate lungs of Wt and Pla2g5-null mice naive or administered r-IL-33 for 10 days. Values are mean ± SEM of two (B) or three (A) independent experiments with 7–15 mice per group. **P< 0.005, *P< 0.05.
Because macrophages require endogenous Pla2g5 for their functions in pulmonary inflammation25, we wanted to investigate whether ILC2 activation and downstream lung inflammation could be restored to Pla2g5-null mice by reconstituting Pla2g5 function in macrophages. We adoptively transferred unstimulated Wt BM-macrophages into Wt and Pla2g5-null recipient mice 24h before the second dose of Alternaria, then administered 3 more doses and analyzed eosinophil numbers and ILC2 activation (Figure 4, inset). Compared to Pla2g5-null mice receiving Alternaria without macrophage transfer, Pla2g5-null mice receiving Wt BM-macrophages plus Alternaria had significantly higher numbers of eosinophils and significantly higher numbers of ILC2s expressing Sca-1, CD25 or intracellular IL-5 (Figure 4A). In contrast, the transfer of Wt BM-macrophages into Alternaria-treated Wt mice did not significantly increase the recruitment of eosinophils or activation of ILC2s compared to Alternaria-treated Wt mice. Accordingly, transfers of Pla2g5-null macrophages into Pla2g5-null mice were ineffective (Figure 4A). However, transfers of Pla2g5-null macrophages in Wt mice (Figure 4A) significantly reduced the numbers of activated ILC2s expressing Sca-1, CD25, and IL-5 and eosinophil numbers, suggesting that Pla2g5-null macrophages could downregulate ILC2 activation when exposed to Th2 inflammatory environment. To further prove this point, we fully activated Pla2g5-null macrophages, and Wt macrophages as controls, with GM-CSF/IL-4/IL-33 and transferred them into Alternaria-treated Wt mice. As shown in Supplementary Figure 2, activated Pla2g5-null macrophages significantly reduced the numbers of IL-5+ILC2s and eosinophils in the lungs of Alternaria-challenged Wt mice. Transfers of Wt macrophages activated with GM-CSF/IL-4/IL-33 were ineffective.
Figure 4. Transfers of Wt and Pla2g5-null BM-macrophages regulate Alternaria-induced pulmonary inflammation.
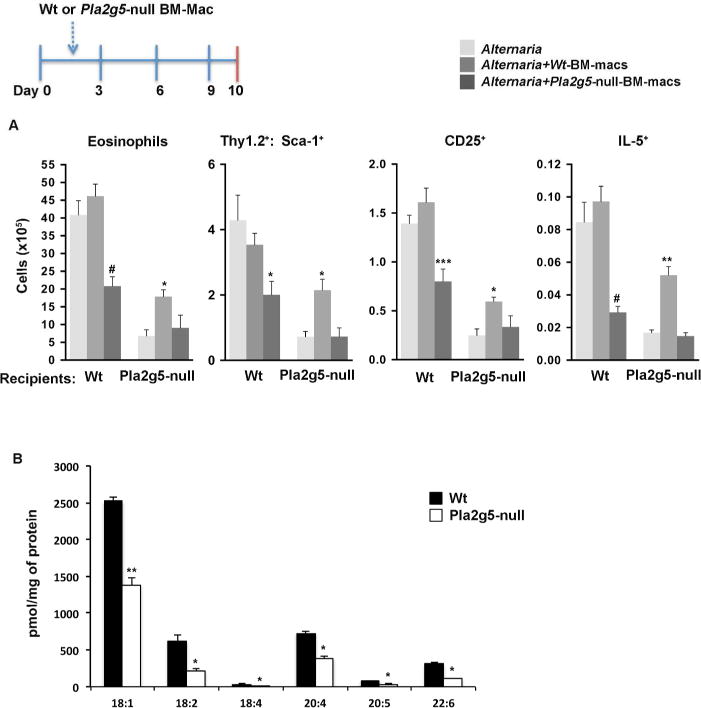
A) Pla2g5-null and Wt recipient mice received Wt (medium grey bars) or Pla2g5-null (dark gray bars) BM-macrophages intratracheally at day 2, followed by Alternaria i.n. at day 3, 6 and 9 or only Alternaria (light grey bars). Mice were euthanized 18 h after the last dose. Analysis by flow cytometry of eosinophils gated as CD45+ CD11c− SiglecF+ lung cells and expression of CD25, Sca-1 and intracellular IL-5 on Thy1.2+ ILC2s. B) Production of FFAs measured by mass spectrometry in Wt and Pla2g5-null BM-Macrophages. (A) Values are mean ± SEM of 2 independent experiments with 10–12 mice per group. (B) Data are from 3 independent experiments. # P<0.0001, *** P<0.0005, **P< 0.005, *P< 0.05.
Pla2g5-dependent generation of linoleic acid and oleic acid contribute to ILC2 activation and pulmonary inflammation
To identify candidate Pla2g5-derived mediators generated by macrophages that could contribute to ILC2 activation, we performed an unbiased assessment of lipids constitutively released by Wt and Pla2g5-null BM-macrophages, using mass spectrometry34. Compared to Wt BM-macrophages, Pla2g5-null BM-macrophages produced significantly lower quantities of medium- and long-chain FFAs, mostly represented by oleic acid (OA, 18:1), LA (18:2), and AA (20:4) (Figure 4B). Short chain FFAs were not different (data not shown). We also examined FFAs produced by lung macrophages enriched from Alternaria exposed mice by Percoll gradients, a technique previously shown to enrich lung macrophages >80%25. We did not sort CD68+ cells because staining for CD68 requires fixation with paraformaldehyde and permeabilization with saponin (as in Figure 2D) which could alter the lipid composition of the cells. Compared to Wt lung macrophages, Pla2g5-null macrophages had reduced quantities of OA, and LA (Figure 5A). AA was similar in both genotypes.
Figure 5. Intranasal administration of LA and OA in combination with r-IL-33 increased eosinophilia and ILC2 activation in Wt and Pla2g5-null mice.
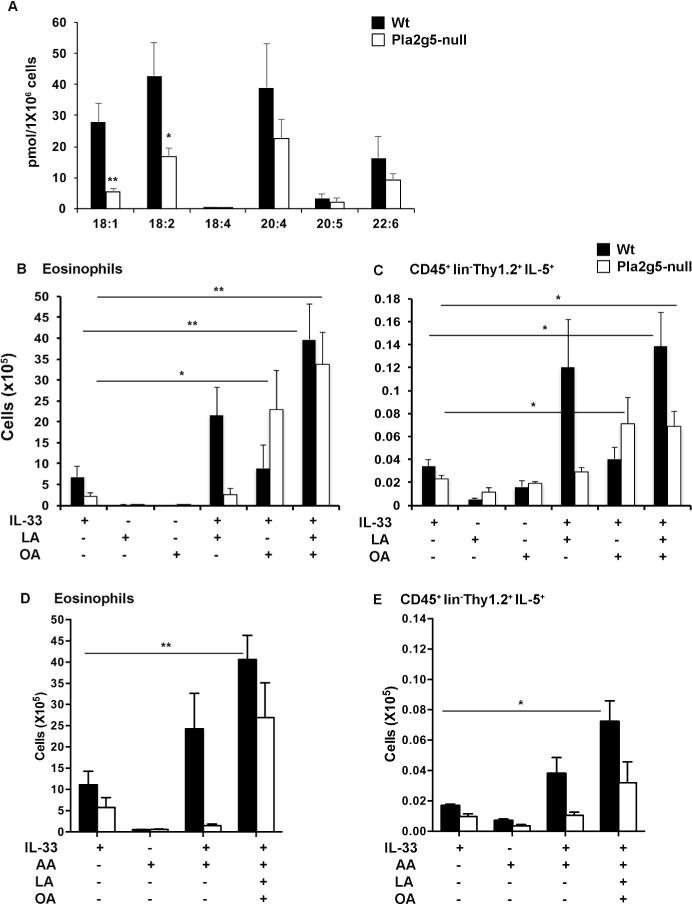
(A) Production of FFAs measured by mass spectrometry in Wt and Pla2g5-null lung macrophages enriched by Percoll gradients of lung homogenates pulled from 5–8 mice. Flow cytometry analysis of (B, D) numbers of eosinophils and (C, E) Thy1.2+ ILC2s expressing intracellular IL-5 in homogenate lungs of Wt (black bars) and Pla2g5-null mice (white bars) treated i.n. with IL-33, LA or OA (B, C), or IL-33, LA, OA and AA (D, E) as indicated. Values are mean ± SEM of two independent experiments with 5 samples (A), two-four independent experiments (B, C, D) or one representative experiment (E) with 4–12 mice per group, and were compared by t-test (A) or One-way ANOVA with Sidak’s correction for multiple comparisons. **P< 0.005, *P< 0.05.
To determine whether LA and/or OA could restore the IL-33-mediated induction of eosinophilic inflammation and ILC2 expansion, we administered intranasal LA and/or OA, alone and in combination with IL-33 (4 doses in 10 days), to Wt and Pla2g5-null mice. Neither LA nor OA alone caused pulmonary inflammation in either genotype (Figure 5B). The combination of LA + IL-33 increased the numbers of eosinophils in the lungs of Wt mice by ~3-fold when compared to IL-33 alone, and the combination of LA and OA + IL-33 further increased the numbers of eosinophils in this genotype (Figure 5B). In contrast, LA failed to potentiate IL-33-induced eosinophilia in Pla2g5-null mice, although OA + IL-33 was markedly active and the combination of LA and OA + IL-33 induced a modest further increase over IL-33 + OA. The effects of FFAs on the numbers of lung ILC2s expressing IL-5 paralleled their effects on eosinophil numbers (Figure 5C and Supplementary Figure 3 for representative FACS plots), although the numbers of IL5+ILC2s in IL-33+LA+OA treated Pla2g5-null mice were slightly lower than in equally treated Wt mice.
Because AA-derived eicosanoids were previously shown to contribute to ILC2 activation9, 28, to understand whether AA could increase the numbers of IL5+ ILC2s in FFAs treated Pla2g5-null mice to the same levels as equally treated Wt mice, in another set of experiments, we administered AA alone or in combination with IL-33+LA+OA to Wt and Pla2g5-null mice. AA alone did not induce pulmonary inflammation in either genotype (Figure 5D and E). In combination with IL-33, AA induced in Wt mice a 2-fold increase in eosinophil numbers (Figure 5D) and IL5+ILC2s (Figure 5E), but was ineffective in Pla2g5-null mice. The combination of IL-33+LA+OA+AA induced in Wt mice a significant increase in numbers of eosinophils and IL-5+ILC2s compared to IL-33-esposed mice (Figure 5D and E) and a nearly significant increase in Pla2g5-null mice. However, in both genotypes the effects of IL-33+LA+OA+AA were similar to those of IL-33+LA+OA (Figure 5B–E).
To determine whether LA and/or OA directly activated ILC2s, we sorted ILC2s from lungs of Alternaria-treated Wt and Pla2g5-null mice, rested them for 40h, and stimulated with LA, OA, IL-33 or a combination for 8h. Then we assayed ILC2s for their expression of intracellular IL-5. Staining controls and representative FACS plots are shown in Supplementary Figure 4. IL-33 significantly increased the percentage of IL-5-expressing ILC2s isolated from both Pla2g5-null and Wt mouse lungs. Neither LA nor OA induced significant IL-5 expression by ILC2s of either genotype. LA, but not OA, significantly potentiated IL-33-induced expression of IL-5 by Wt ILC2s, and the combination of LA + OA did not differ from the effects of LA (Figure 6A). In contrast LA suppressed the IL-33-induced increase in percentages of IL-5+ Pla2g5-null ILC2s (Figure 6B). OA was inactive. To determine whether LA and OA amplified the release of IL-5 by Wt ILC2s, we measured the quantity of secreted IL-5 from sorted lung ILC2s activated ex vivo (Figure 6C). IL-33 induced the release of large quantities of IL-5 in supernatant of sorted Wt ILC2s and the combination of IL-33+LA+OA significantly potentiated this release. In contrast, Pla2g5-null ILC2s released significantly less amount of IL-5 and failed to exhibit potentiation in response to LA+OA (Figure 6C).
Figure 6. Differential LA- and OA-induced activation of sorted Wt and Pla2g5-null ILC2s and FFAR1 expression.
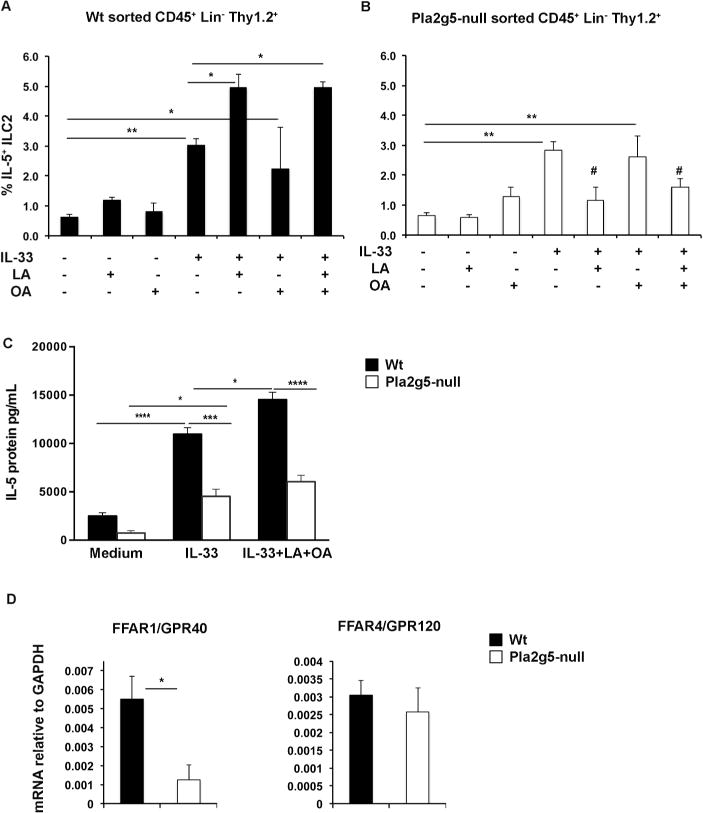
ILC2s were expanded in-vivo by 4 Alternaria challenges for 10 days. CD45+ lin-, Thy1.2+ cells were FACS sorted from 3–4 pulled lung homogenates, and rested for 40 hours prior to in-vitro stimulation with LA (200 μM), OA (200 μM), IL-33 (30ng/ml) or all together for 8 hours then analyzed by flow cytometry for percentage of IL-5 positive Wt (A) and Pla2g5-null (B) ILC2s. Unstimulated cells were used as controls. (C) IL-5 levels were measured by ELISA in the supernatants of sorted Wt and Pla2g5-null Thy1.2+ ILC2s stimulated as indicated. (D) Expression of FFAR1 and FFAR4 mRNA relative to GAPDH measured by qPCR in sorted ILC2s from Alternaria-treated Wt and Pla2g5-null mice. Data are from at least 3 independent experiments. Values are expressed as means ± SEM and were compared by One-way ANOVA (A–B), Two-way ANOVA (C) with Sidak’s correction for multiple comparisons, or t-test (D). ****P< 0.0001, *** P<0.0005, **P<0.005, *P< 0.05, # P<0.05 vs. IL-33 alone.
Medium and long chain FFAs signal through two G protein-coupled receptors, FFA receptor-1 (FFAR1) and FFA receptor-4 (FFAR4)35–39. To determine whether ILC2s expressed these receptors, and to determine the potential basis for the different responses of Wt and Pla2g5-null ILC2s to LA, we analyzed ILC2 expression of FFAR1 and FFAR4 in ILC2s sorted from Wt and Pla2g5-null Alternaria-treated mice. Wt ILC2s expressed FFAR1 mRNA and its expression was significantly higher compared to Pla2g5-null ILC2s (Figure 6D). Wt and Pla2g5-null ILC2s also expressed FFAR4 mRNA to similar extents (Figure 6D).
Discussion
It is now well established that ILC2s are key effectors of pulmonary inflammation. Their contribution is particularly evident in models triggered by the release of alarmins (IL-33, IL-25, TSLP) from epithelial cells2, 4, 9, 11, 40, 41 in response to environmental proteases11, many of which are relevant to asthma in humans13. IL-33, alone and in combination with IL-25, TSLP, and other cytokines can directly induce IL-5, IL-13, and IL-9 generation from ILC2s, promoting eosinophilic inflammation and goblet cell metaplasia that can occur independently of or in concert with adaptive immunity. The Alternaria model of pulmonary inflammation has been particularly useful to establish the contribution of innate, epithelial-derived alarmins and their downstream effects on ILC2 activation and subsequent development of airway inflammation1, 3. While macrophages can also express IL-3342, and other innate cell types have been proposed to interact with ILC2s43, no previous studies had established whether macrophages can activate ILC2s in Alternaria-induced pulmonary inflammation and which mediators might be involved. Pla2g5-null mice show markedly impaired type 2 pulmonary inflammation that reflects, at least in part, a requirement for cell-intrinsic Pla2g5 for macrophage effector functions25, 26. We therefore investigated the role of Pla2g5 in general and macrophage-associated Pla2g5 in particular, in lipid-generating function and its potential downstream effects on ILC2 activation in a model of pulmonary inflammation induced by Alternaria.
We subjected Wt and Pla2g5-null mice to a protocol involving the administration of Alternaria four times over a 10-day period, which elicits prominent contributions from IL-33 and ILC2s. The marked pulmonary eosinophilia and increases in the numbers of total and activated ILC2s observed in Wt mice (Figure 1) were all sharply reduced in Pla2g5-null mice. The reduced levels of both eosinophils and ILC2s were paralleled by reduced levels of IL-33 induction (Figure 2B), but not constitutively levels of IL-33 (Figure 2B), or by release of IL-33 in response to a single Alternaria dose (Figure 2A). AT2 cells are the dominant source of pre-formed IL-33 in the mouse lung, as well as of the pre-formed IL-33 in response to a single dose of Alternaria15. In our study, AT2 cells showed equivalent staining for IL-33 in Wt and Pla2g5-null mice (Figure 2C), suggesting that Pla2g5 functions are not required by AT2 to store or release IL-33. In marked contrast, IL-33 expression by lung macrophages was substantially induced in Alternaria-treated Wt mice but not in Pla2g5-null mice (Figure 2D), suggesting that macrophages may be one of the cell types accounting for the impaired induction of IL-33 in Pla2g5-null lungs. Our previous studies demonstrated that macrophage-intrinsic Pla2g5 was necessary for inducible expression of Th2 cell-active chemokines25. Our current results, supported by our ex vivo data (Figure 2E), suggest that this may also be the case for IL-33 induction.
When administered exogenously to naive Wt mice, r-IL-33 is sufficient alone to drive a robust type 2 inflammatory response that depends on ILC2s3, 40. Despite the evident role of Pla2g5 in IL-33 induction by macrophages, the direct administration of IL-33 to naive Pla2g5-null mice was insufficient to induce inflammation, ILC2 expansion, and macrophage activation (Figure 3). Combined with the fact that transfer of Wt macrophages almost fully restored these parameters in Pla2g5-null mice in response to Alternaria challenges (Figure 4A), we suspected the involvement of additional Pla2g5-dependent factors that could enable macrophages to activate ILC2s, alone or in concert with IL-33. We identified at least two candidate FFAs (LA and OA) as Pla2g5-dependent factors derived from BM and lung macrophages (Figure 4B and 5A). Both of these FFAs can signal to immune and non-immune cells through the GPCRs FFAR1 and FFAR4, although their potential roles as mediators of allergic inflammation in general and stimulants of ILC2 activation in particular had not been explored. The sharp potentiation of IL-33-driven eosinophilic inflammation and ILC2 expansion in Wt mice by LA, alone and in combination with OA (Figure 5B and C), was parallel by its effects on IL-33-induced IL-5 generation (Figure 6A) and secretion (Figure 6C) by ILC2s ex vivo. Interestingly in Pla2g5-null mice this LA-induced potentiation of inflammation in vivo (Figure 5A and B) and ex vivo (Figure 6B and C) was absent. The lack of LA responsiveness by Pla2g5-null ILC2s is consistent with the lack of ILC2-intrinsic expression of FFAR1 (Figure 6D), which exhibits a preference for LA over to OA44. Notably, although unable to directly activate Wt or Pla2g5-null ILC2s, OA did substantially enhance IL-33-induced eosinophilic inflammation and expand lung ILC2s in Pla2g5-null mice, reflecting a potential compensatory mechanism involving ILC2 activation by a yet to-be-determined OA-responsive cell. Since FFAR1 and FFAR4 are broadly expressed by immune and non-immune cell types45, it is likely that Pla2g5-derived FFAs potentiate innate type 2 immune responses and ILC2 activation by both direct and indirect pathways. We speculate that ILC2s require conditioning in vivo by one or more inductive factors that are deficient in Pla2g5-null mice in order to express FFAR1 and respond to LA. Moreover, the fact that FFAs including AA could not restore ILC2 activation in Pla2g5-null mice to the levels of equally treated Wt mice (Figure 5C and E), suggests that the presence of Pla2g5-null macrophages (Figure 4A and Supplementary Figure 2) through a yet unknown mechanism might limit ILC2 activation in Pla2g5-null mice.
Our data clearly identify a role for macrophages, and Pla2g5-derived FFAs, as activators of ILC2s, acting in concert with IL-33. It is likely that the coordinate action of ILC2s, macrophages and epithelial cells induces pulmonary inflammation, highlighting a complex interplay of innate cells in the lung4, 12, 46. These data also suggest that FFAs directly activate ILC2s through FFAR1, which is expressed on ILC2s in a yet to be identified Pla2g5-dependent fashion. However, it is likely that additional Pla2g5-generated factors might regulate type 2 immunity. Thus, our observations suggest that macrophage-derived FFAs amplify innate, IL-33-triggered type 2 immunopathology in diseases such as asthma. We speculate that LA, derived at least in part from Pla2g5-expressing macrophages, may contribute to the function of ILC2s in other circumstances, such as homeostasis of adipose tissue and glucose metabolism where macrophages, Pla2g5, IL-33, and ILC2s have all been implicated22, 47.
Methods
Lung inflammation
C57/BL6 Wt and Pla2g5-null mice48, 49 (9–12 wk-old males) received 25 μg of Alternaria alternata extract (Greer Laboratories, Lenoir, NC) in 20 uL of PBS or PBS alone intranasally (i.n.) on days 0,3,6 and 9 and euthanized 18h later1 or a single dose of 100 μg and were euthanized after 1h or 3h2. Alternatively, Wt and Pla2g5-null naïve mice were given mouse rIL-33 (R&D Systems, Minneapolis, MN) i.n. 100 ng/dose on days 0,3,6 and 9 with or without LA (132 nM)28 OA (106 nM) or AA (99 nM), and mice were euthanized 18h after the last dose. All animal experiments were approved by the Animal Care and Use Committee of the Dana-Farber Cancer Institute (Boston, MA).
Flow cytometry
Lungs were manually chopped to approximately 10mm pieces, then digested in RPMI containing 428 U/ml Collagenase IV (Worthington, Lakewood, NJ) and 20 mg/ml DNAse I (Roche, Mannheim, Germany) (30min, 37°C). After red cell lysis, the obtained cell suspension from single mouse was washed and counted. Cells were blocked (1h, 4 °C) with 1% of rat anti mouse CD16/CD32 (BD Biosciences, San Jose, CA) and 10% donkey serum and then stained (1h, 4 °C) with appropriate Abs: CD45 PercPCy5 (clone 30-F11, BioLegend, San Diego, CA), CD19 FITC (6D5, Biolegend), CD3 FITC (145-2C11, BioLegend), CD11b FITC (M1/70, BioLegend), CD11c PE-Cy7 FITC (N418, BioLegend), Ly6G/C FITC (RB6-8C5, eBiosciences), Nk1.1 FITC (PK136, Biolegend), FcεR1 FITC (MAR-1, Biolegend), Siglec-F PE (E50-2440, BD Bioscience), Thy 1.2 APC (53–2.1, eBioscience, San Diego, Ca), ICOS (C398.4A, eBiosciences), Sca-1 (D7, eBioscieces), CD25 (PC61, eBiosciences), ST2 biotin (clone DJ8, MD Bioscience) followed by PE streptavidin (eBiosciences). In selected experiments cells were fixed with 4% paraformaldehyde (7 min, 21°C), washed, permeabilized with 0.1% saponin (SigmaAldrich, St Louis, Ca) (7 min, 21°C) and stained with CD68 APC (FA-11, AbD Serotec, Raleigh, Nc), IL-5 PE (TRFK5, Biolegend), IL-13 (eBio13A eBiosciences), IL-33 PE (396118, R&D Systems, Minneapolis, MN), rabbit polyclonal anti murine RELM-α (Peprotech, Rocky Hill NJ)) and corresponding isotypes as controls. Alternatively, cells were permeabilized with BD Cytofix/Cytoperm kit (BD Biosciences, San Jose, CA). The acquisition was performed on a FACSCanto flow cytometer with FACSDiva software (BD Biosciences), and data were analyzed with FlowJo (Tree Star, Ashland, OR).
Airways analysis and lung cell processing
Bronchoalveolar lavage (BAL) was performed with 0.7 mL PBS (Sigma-Aldrich) containing 0.5 mM EDTA (three times). The BAL fluid was collected, and cell-free supernatant was aliquoted and frozen. ELISA was used to measure IL-33 (R&D Systems). In selected experiments lung macrophages were enriched by Percoll gradients25 of lung homogenates pulled from 3–4 Alternaria-treated Wt or Pla2g5-null mice. Cells were then counted and assayed by qPCR or Mass Spectrometry.
Western Blot
Right lungs were collected at the time of euthanasia and snap frozen. Proteins were isolated from tissue homogenates in RIPA buffer (Boston Bioproducts, Ashland, MA, USA) with protease inhibitors26. The protein concentration in cell lysates was measured using the BCA Assay (Pierce, Thermo Scientific). 20 μg of proteins were separated on a 10–20% Tris-Glycine gel (Novex, Life Technologies) and transferred to a PVDF membrane. After blocking overnight at 4°C in 5% milk, blots were incubated with a goat polyclonal IL-33 (1:500, R&D Systems) or mouse monoclonal β-actin (1:1000, Cell Signaling, Danvers, MA) antibodies diluted in TBST at RT for 2h, followed by a rabbit anti-goat or goat anti-mouse secondary antibody (1:3000, BioRad) diluted in TBST for 1h at RT. The blots were visualized using the Supersignal West Femto Chemiluminescent substrate (Thermo Scientific) and imaged by a KODAK M35A X-OMAT processor.
Frozen sections
Lungs of Wt and Pla2g5-null mice were excised and immersed in RPMI. Within 1h of surgery, the tissue was removed from RPMI and fixed in 4% paraformaldehyde, then embedded in Tissue-Tek® O.C.T.™ Compound (Sakura Finetek), and kept at −80°C until sectioning. Sections of 5-μm thickness were freshly cut, thaw-mounted onto slides, and stained for confocal microscopy. Frozen sections were rehydrated for 1h at RT then blocked with 10% donkey serum, followed by incubation with goat polyclonal IL-33 (AF3626, R&D Systems) and rabbit polyclonal proSPC (AB3786, Millipore, Temecula, CA) antibodies or appropriate isotypes controls at 4°C, overnight. Samples were washed, incubated at RT for 1h with appropriate secondary antibodies, washed and covered with Fluoroshield mounting media (Electron Microscopy Sciences, Hatfield, PA). Sections were imaged using a Nikon C1 plus laser scanner confocal system with a 40× oil Plan-Fluor NA1.3 objective lens. 8–10 Z-stack images of 0.5 μm were acquired through a small pinhole using Nikon EZ-C1 software. Images were analyzed using Image J (U.S. National Institute of Health, Bethesda, MD).
BM macrophage transfer
Wt or Pla2g5-null bone marrow (BM) cells were collected from femurs and tibiae of mice. The disaggregated cells were counted and suspended in complete medium (DMEM F12, 5%FBS, 100U/ml penicillin, 100ug/ml streptomycin, 0.1mM nonessential amino acids, 2mM L-glutamine and 0.05 μM 2-ME) containing 50ng/ml murine r-MCSF (PeproTech) at a concentration of 4.0 ×106 cells/ml in a 10ml/Petri dish. On day 3, 10 ml of medium containing r-MCSF were added to each dish. On day 7, cells were harvested with PBS containing Lidocaine (4mg/ml, 15min, 37C) and resuspended at concentration of 5×106 cells/ml in PBS. For adoptive transfer, 1 ×105 Wt or Pla2g5-null BM-macrophages were transferred i.t. into Wt and Pla2g5-null mice two days after the first dose of Alternaria followed by 3 more doses of Alternaria (25 μg in 20 μl PBS) i.n. on day 3, 6 and 9. Mice were euthanized 18h after the last dose.
Mass Spectrometry of lipids
Wt and Pla2g5-null BM-macrophages were cultured for 7 days in r-MCSF. Adherent cells were collected, frozen and shipped for analysis by mass spectrometry. In another set of experiments lung macrophages were enriched by multiple Percoll gradients25. Free fatty acid analysis was performed according to a previously published method34, 50. Briefly, the cell pellet was homogenized in 500 ul of PBS/10% methanol. An aliquot of 200 μl corresponding to about 0.5×106 cells was withdrawn and a cocktail of internal standards consisting of 15 deuterated fatty acids was added. The extraction was initiated with 500 μl of methanol and 25 μl of 1N HCl and a bi-phasic solution is formed by addition of 1.5 ml of isooctane. The phases are separated by centrifugation and the isooctane phase containing the free fatty acids FFA fraction was removed. The extraction is repeated once and the combined extracts are evaporated to dryness. The free fatty acids were derivatized with pentafluorobenzyl (PFB) bromide and the resulting fatty acid PFB esters were analyzed by gas chromatography/mass spectrometry using a negative chemical ionization mode (Agilent 6890N gas chromatograph equipped with an Agilent 5973 mass selective detector; Agilent, Santa Clara, CA). Standard curves for each of the fatty acids were acquired in parallel using identical conditions. The quantitative assessment of fatty acids in a sample was achieved by comparison of the mass spectrometric ion signal of the target molecule normalized to the internal standard with the matching standard curve according to the isotope dilution method and by protein content34.
ILC2 cells sorting and culture
Wt and Pla2g5-null mice received four doses of 25ug of Alternaria in 20ul of PBS i.n. on day 0, 3, 6 and 9 and euthanized 18h later in order to expand ILC2s prior to FACs sorting. Sorting of ILC2s (CD45+ Lin− (CD3, CD19, Ly6g, CD11c, CD11b, Nk1.1, FcεR1−), Thy1.2+) was performed using a FACSDiva 8.0.1 cell sorter (BD Bioscience). Purified CD45+ lin-Thy1.2+ cells (>98%) were rested for 40h with 10ng/mL rIL-2 and rIL-7 (R&D Systems, Minneapolis, MN) in 96 well around bottom plates (20000 cells per well). Prior to stimulation, the medium was changed to fresh medium. ILC2s were cultured with 30ng/mL rIL-33 (R&D Systems), 200 μM Linoleic Acid (Cayman Chemical) or 200 μM Oleic Acid (Cayman Chemical)22 or all together for 8h. For intracellular cytokine staining, 1 μl/mL of Golgi Plug (BD Bioscience) was added to ILC2s 6h before collection for FACs analysis.
ELISA
Wt and Pla2g5-null mice sorted ILC2s were obtained as described above and rested for 40h with 10ng/mL rIL-2 and rIL-7 in a 96 well round bottom plates (40000 cells per well). After changing to fresh media, ILC2s were stimulated with 30ng/mL rIL-33 or 30ng/mL rIL-33, 200 μM Linoleic Acid, 200 μM Oleic Acid for 8h. Supernatants were then collected. IL-5 ELISA (R&D Systems, M5000) was performed as per manufacturer protocol. For these experiments, during lung homogenization, Dispase (Gibco, Life Technologies, NY) was added (2U/ml) to increase the yield for ILC2s.
Real-time PCR
Total RNA was isolated from lysate with the RNeasy Micro Kit (Qiagen, Louisville, KY, USA), reverse transcribed into cDNA (High-Capacity cDNA Reverse Transcription Kit; Thermo science-Applied Biosystems, Foster City, CA, USA) and measured by real-time PCR with the use of SYBR Green/ROX master mix (SABiosciences, Frederick, MD, USA) on an Mx3005P thermal cycler (Stratagene, Santa Clara, CA, USA). The ratio of each mRNA relative to the GAPDH mRNA was calculated with the Ct threshold cycle method. The mouse primers used were GAPDH F: TCAACAGCAACTCCCACTCTTCCA; R: ACCCTGTTGCTGTAGCCGTATTCA. Pla2g5 F: TGGTTCCTGGCTTGCAGTGTG; R: TTCGCAGATGACTAGGCCATT. IL-33 F: TCCCAACAGAAGACCAAAG; R: 5′-GATACTGCCAAGCAAGGAT. FFAR1/GPR40 and FFAR4/GPR120 were from Qiagen. Real-time PCR products were run on a 1.5% agarose gel and visualized using chemilmager 4400 fluorscience system (Alpha Innotech, Missouri, TEX, USA)
Statistical analysis
Comparisons between 2 groups were made by using unpaired Student’s t-test. To compare three or more groups, we performed One-way ANOVA or Two-Way ANOVA with Sidak’s correction for multiple comparisons. Comparisons were performed with Prism software (GraphPad, La Jolla, CA). Data are expressed as mean ± SEM, and P<0.05 was considered significant.
Supplementary Material
Supplementary Figure 1. (A) Representative FACS plots showing gating strategy for ILC2s. Alternaria-treated Wt lung homogenates were stained for ILC2s gated as CD45+ Lin-Thy1.2+ and forward and side scatter representation of the lineage positive population (depicted in blue) with low side scatter, and lineage positive population (depicted in red) with high side scatter. Percentage expression of Sca-1, ST2, ICOS, CD25 and intracellular IL-5 on Thy1.2+ (B) or Thy1.2+ Sca-1+ (C) ILC2s from homogenate lungs of Alternaria-treated Wt and Pla2g5-null mice. Images are from one representative mouse per group from three experiments with 13–19 (A, B) or 4–10 (C) Alternaria-treated mice per group.
Supplementary Figure 2. Transfer of Wt and Pla2g5-null BM-macrophages activated for 24h with GM-CSF/IL-4/IL-33. Wt mice received Wt or Pla2g5-null BM-macrophages intratracheally at day 2 after the first dose of Alternaria, followed by Alternaria at day 3, 6 and 9 or only Alternaria. Mice were euthanized 18 h after the last dose for flow cytometric analysis of eosinophils (CD45+ CD11c− SiglecF+) and Thy1.2+ ILC2s expressing intracellular IL-5. Values are mean ± SEM of 3 independent experiments with 6–22 mice per group and were compared by t-test. *P< 0.05.
Supplementary Figure 3. Percentage expression of intracellular IL-5 on Thy1.2+ ILC2s in homogenate lungs of Wt and Pla2g5-null mice treated with IL-33, OA, or LA in various combinations. Analysis by flow cytometry of Thy1.2+ ILC2s expressing intracellular IL-5 obtained from Wt and Pla2g5-null homogenate lungs treated as indicated. Images are from one representative mouse per group from two-four independent experiments with 4–12 mice per group.
Supplementary Figure 4. Percentage expression of intracellular IL-5 on Thy1.2+ ILC2s sorted from homogenate lungs of Wt and Pla2g5-null mice treated with Alternaria. Analysis by flow cytometry of ILC2s sorted from Wt and Pla2g5-null homogenate lungs, rested for 40 hours prior to in-vitro stimulation with LA (200 μM), OA (200 μM), IL-33 (30ng/ml) or all together for 8 hours, then analyzed by flow cytometry for percentages of IL-5 positive ILC2s. Images are from one representative sample per group from three independent experiments.
Acknowledgments
We thank Jennifer Zacharia and Jannatul Firdous for technical assistance.
Footnotes
The authors declare no conflict of interest.
Supplementary material is linked to the online version of this paper.
Author contributions
Acquisition of data: MY, SS, OQ, BB. Analysis and interpretation: MY, SS, OQ, JB, BB. Drafting or revising manuscript: MY, SS, OQ, JB, BB. Conception and design: BB.
Disclosure
The authors declare no conflicting financial interests. Dr. Balestrieri received a patent relating to the subject matter discussed in this manuscript.
References
- 1.Doherty TA, Khorram N, Chang JE, Kim HK, Rosenthal P, Croft M, et al. STAT6 regulates natural helper cell proliferation during lung inflammation initiated by Alternaria. American journal of physiology Lung cellular and molecular physiology. 2012;303(7):L577–588. doi: 10.1152/ajplung.00174.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kouzaki H, Iijima K, Kobayashi T, O’Grady SM, Kita H. The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. Journal of immunology. 2011;186(7):4375–4387. doi: 10.4049/jimmunol.1003020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL-33-responsive lineage-CD25+ CD44(hi) lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. Journal of immunology. 2012;188(3):1503–1513. doi: 10.4049/jimmunol.1102832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Halim TY, Steer CA, Matha L, Gold MJ, Martinez-Gonzalez I, McNagny KM, et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity. 2014;40(3):425–435. doi: 10.1016/j.immuni.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doherty TA, Khorram N, Sugimoto K, Sheppard D, Rosenthal P, Cho JY, et al. Alternaria induces STAT6-dependent acute airway eosinophilia and epithelial FIZZ1 expression that promotes airway fibrosis and epithelial thickness. Journal of immunology. 2012;188(6):2622–2629. doi: 10.4049/jimmunol.1101632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. 2010;464(7293):1367–1370. doi: 10.1038/nature08900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature. 2010;463(7280):540–544. doi: 10.1038/nature08636. [DOI] [PubMed] [Google Scholar]
- 8.Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(25):11489–11494. doi: 10.1073/pnas.1003988107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doherty TA, Khorram N, Lund S, Mehta AK, Croft M, Broide DH. Lung type 2 innate lymphoid cells express cysteinyl leukotriene receptor 1, which regulates TH2 cytokine production. The Journal of allergy and clinical immunology. 2013;132(1):205–213. doi: 10.1016/j.jaci.2013.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nature immunology. 2011;12(7):631–638. doi: 10.1038/ni.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Halim TY, Krauss RH, Sun AC, Takei F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity. 2012;36(3):451–463. doi: 10.1016/j.immuni.2011.12.020. [DOI] [PubMed] [Google Scholar]
- 12.Barlow JL, Bellosi A, Hardman CS, Drynan LF, Wong SH, Cruickshank JP, et al. Innate IL-13-producing nuocytes arise during allergic lung inflammation and contribute to airways hyperreactivity. The Journal of allergy and clinical immunology. 2012;129(1):191–198. e191–194. doi: 10.1016/j.jaci.2011.09.041. [DOI] [PubMed] [Google Scholar]
- 13.Licona-Limon P, Kim LK, Palm NW, Flavell RA. TH2, allergy and group 2 innate lymphoid cells. Nature immunology. 2013;14(6):536–542. doi: 10.1038/ni.2617. [DOI] [PubMed] [Google Scholar]
- 14.Molofsky AB, Nussbaum JC, Liang HE, Van Dyken SJ, Cheng LE, Mohapatra A, et al. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. The Journal of experimental medicine. 2013;210(3):535–549. doi: 10.1084/jem.20121964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hardman CS, Panova V, McKenzie AN. IL-33 citrine reporter mice reveal the temporal and spatial expression of IL-33 during allergic lung inflammation. European journal of immunology. 2013;43(2):488–498. doi: 10.1002/eji.201242863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim HY, Chang YJ, Subramanian S, Lee HH, Albacker LA, Matangkasombut P, et al. Innate lymphoid cells responding to IL-33 mediate airway hyperreactivity independently of adaptive immunity. The Journal of allergy and clinical immunology. 2012;129(1):216–227. e211–216. doi: 10.1016/j.jaci.2011.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Polumuri SK, Jayakar GG, Shirey KA, Roberts ZJ, Perkins DJ, Pitha PM, et al. Transcriptional regulation of murine IL-33 by TLR and non-TLR agonists. Journal of immunology. 2012;189(1):50–60. doi: 10.4049/jimmunol.1003554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dennis EA, Cao J, Hsu YH, Magrioti V, Kokotos G. Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chemical reviews. 2011;111(10):6130–6185. doi: 10.1021/cr200085w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murakami M, Taketomi Y, Miki Y, Sato H, Yamamoto K, Lambeau G. Emerging roles of secreted phospholipase A enzymes: The 3rd edition. Biochimie. 2014;107PA:105–113. doi: 10.1016/j.biochi.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 20.Alvarez-Curto E, Milligan G. Metabolism meets immunity: The role of free fatty acid receptors in the immune system. Biochemical pharmacology. 2016;114:3–13. doi: 10.1016/j.bcp.2016.03.017. [DOI] [PubMed] [Google Scholar]
- 21.Murakami M, Koduri RS, Enomoto A, Shimbara S, Seki M, Yoshihara K, et al. Distinct arachidonate-releasing functions of mammalian secreted phospholipase A2s in human embryonic kidney 293 and rat mastocytoma RBL-2H3 cells through heparan sulfate shuttling and external plasma membrane mechanisms. The Journal of biological chemistry. 2001;276(13):10083–10096. doi: 10.1074/jbc.M007877200. [DOI] [PubMed] [Google Scholar]
- 22.Sato H, Taketomi Y, Ushida A, Isogai Y, Kojima T, Hirabayashi T, et al. The adipocyte-inducible secreted phospholipases PLA2G5 and PLA2G2E play distinct roles in obesity. Cell metabolism. 2014;20(1):119–132. doi: 10.1016/j.cmet.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singer AG, Ghomashchi F, Le Calvez C, Bollinger J, Bezzine S, Rouault M, et al. Interfacial kinetic and binding properties of the complete set of human and mouse groups I, II, V, X, and XII secreted phospholipases A2. The Journal of biological chemistry. 2002;277(50):48535–48549. doi: 10.1074/jbc.M205855200. [DOI] [PubMed] [Google Scholar]
- 24.Giannattasio G, Fujioka D, Xing W, Katz HR, Boyce JA, Balestrieri B. Group V secretory phospholipase A2 reveals its role in house dust mite-induced allergic pulmonary inflammation by regulation of dendritic cell function. Journal of immunology. 2010;185(7):4430–4438. doi: 10.4049/jimmunol.1001384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ohta S, Imamura M, Xing W, Boyce JA, Balestrieri B. Group V secretory phospholipase A2 is involved in macrophage activation and is sufficient for macrophage effector functions in allergic pulmonary inflammation. Journal of immunology. 2013;190(12):5927–5938. doi: 10.4049/jimmunol.1203202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamaguchi M, Zacharia J, Laidlaw TM, Balestrieri B. PLA2G5 regulates transglutaminase activity of human IL-4-activated M2 macrophages through PGE2 generation. Journal of leukocyte biology. 2016 doi: 10.1189/jlb.3A0815-372R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Munoz NM, Meliton AY, Arm JP, Bonventre JV, Cho W, Leff AR. Deletion of secretory group V phospholipase A2 attenuates cell migration and airway hyperresponsiveness in immunosensitized mice. Journal of immunology. 2007;179(7):4800–4807. doi: 10.4049/jimmunol.179.7.4800. [DOI] [PubMed] [Google Scholar]
- 28.Wojno ED, Monticelli LA, Tran SV, Alenghat T, Osborne LC, Thome JJ, et al. The prostaglandin D(2) receptor CRTH2 regulates accumulation of group 2 innate lymphoid cells in the inflamed lung. Mucosal immunology. 2015;8(6):1313–1323. doi: 10.1038/mi.2015.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stevens WW, Kim TS, Pujanauski LM, Hao X, Braciale TJ. Detection and quantitation of eosinophils in the murine respiratory tract by flow cytometry. Journal of immunological methods. 2007;327(1–2):63–74. doi: 10.1016/j.jim.2007.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells–a proposal for uniform nomenclature. Nature reviews Immunology. 2013;13(2):145–149. doi: 10.1038/nri3365. [DOI] [PubMed] [Google Scholar]
- 31.Walker JA, Barlow JL, McKenzie AN. Innate lymphoid cells–how did we miss them? Nature reviews Immunology. 2013;13(2):75–87. doi: 10.1038/nri3349. [DOI] [PubMed] [Google Scholar]
- 32.Makrinioti H, Toussaint M, Jackson DJ, Walton RP, Johnston SL. Role of interleukin 33 in respiratory allergy and asthma. The Lancet Respiratory medicine. 2014;2(3):226–237. doi: 10.1016/S2213-2600(13)70261-3. [DOI] [PubMed] [Google Scholar]
- 33.Gorski SA, Hahn YS, Braciale TJ. Group 2 innate lymphoid cell production of IL-5 is regulated by NKT cells during influenza virus infection. PLoS pathogens. 2013;9(9):e1003615. doi: 10.1371/journal.ppat.1003615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quehenberger O, Armando AM, Dennis EA. High sensitivity quantitative lipidomics analysis of fatty acids in biological samples by gas chromatography-mass spectrometry. Biochimica et biophysica acta. 2011;1811(11):648–656. doi: 10.1016/j.bbalip.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Briscoe CP, Tadayyon M, Andrews JL, Benson WG, Chambers JK, Eilert MM, et al. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. The Journal of biological chemistry. 2003;278(13):11303–11311. doi: 10.1074/jbc.M211495200. [DOI] [PubMed] [Google Scholar]
- 36.Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, et al. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature. 2003;422(6928):173–176. doi: 10.1038/nature01478. [DOI] [PubMed] [Google Scholar]
- 37.Kotarsky K, Nilsson NE, Flodgren E, Owman C, Olde B. A human cell surface receptor activated by free fatty acids and thiazolidinedione drugs. Biochemical and biophysical research communications. 2003;301(2):406–410. doi: 10.1016/s0006-291x(02)03064-4. [DOI] [PubMed] [Google Scholar]
- 38.Hirasawa A, Tsumaya K, Awaji T, Katsuma S, Adachi T, Yamada M, et al. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nature medicine. 2005;11(1):90–94. doi: 10.1038/nm1168. [DOI] [PubMed] [Google Scholar]
- 39.Oh da Y, Walenta E, Akiyama TE, Lagakos WS, Lackey D, Pessentheiner AR, et al. A Gpr120-selective agonist improves insulin resistance and chronic inflammation in obese mice. Nature medicine. 2014;20(8):942–947. doi: 10.1038/nm.3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barlow JL, Peel S, Fox J, Panova V, Hardman CS, Camelo A, et al. IL-33 is more potent than IL-25 in provoking IL-13-producing nuocytes (type 2 innate lymphoid cells) and airway contraction. The Journal of allergy and clinical immunology. 2013;132(4):933–941. doi: 10.1016/j.jaci.2013.05.012. [DOI] [PubMed] [Google Scholar]
- 41.Halim TY, MacLaren A, Romanish MT, Gold MJ, McNagny KM, Takei F. Retinoic-acid-receptor-related orphan nuclear receptor alpha is required for natural helper cell development and allergic inflammation. Immunity. 2012;37(3):463–474. doi: 10.1016/j.immuni.2012.06.012. [DOI] [PubMed] [Google Scholar]
- 42.Wills-Karp M, Rani R, Dienger K, Lewkowich I, Fox JG, Perkins C, et al. Trefoil factor 2 rapidly induces interleukin 33 to promote type 2 immunity during allergic asthma and hookworm infection. The Journal of experimental medicine. 2012;209(3):607–622. doi: 10.1084/jem.20110079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu S, Kim HY, Chang YJ, DeKruyff RH, Umetsu DT. Innate lymphoid cells and asthma. The Journal of allergy and clinical immunology. 2014;133(4):943–950. doi: 10.1016/j.jaci.2014.02.015. quiz 951. [DOI] [PubMed] [Google Scholar]
- 44.Christiansen E, Watterson KR, Stocker CJ, Sokol E, Jenkins L, Simon K, et al. Activity of dietary fatty acids on FFA1 and FFA4 and characterisation of pinolenic acid as a dual FFA1/FFA4 agonist with potential effect against metabolic diseases. The British journal of nutrition. 2015;113(11):1677–1688. doi: 10.1017/S000711451500118X. [DOI] [PubMed] [Google Scholar]
- 45.Heng TS, Painter MW, Immunological Genome Project C The Immunological Genome Project: networks of gene expression in immune cells. Nature immunology. 2008;9(10):1091–1094. doi: 10.1038/ni1008-1091. [DOI] [PubMed] [Google Scholar]
- 46.Van Dyken SJ, Mohapatra A, Nussbaum JC, Molofsky AB, Thornton EE, Ziegler SF, et al. Chitin activates parallel immune modules that direct distinct inflammatory responses via innate lymphoid type 2 and gammadelta T cells. Immunity. 2014;40(3):414–424. doi: 10.1016/j.immuni.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee MW, Odegaard JI, Mukundan L, Qiu Y, Molofsky AB, Nussbaum JC, et al. Activated type 2 innate lymphoid cells regulate beige fat biogenesis. Cell. 2015;160(1–2):74–87. doi: 10.1016/j.cell.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Satake Y, Diaz BL, Balestrieri B, Lam BK, Kanaoka Y, Grusby MJ, et al. Role of group V phospholipase A2 in zymosan-induced eicosanoid generation and vascular permeability revealed by targeted gene disruption. The Journal of biological chemistry. 2004;279(16):16488–16494. doi: 10.1074/jbc.M313748200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Balestrieri B, Hsu VW, Gilbert H, Leslie CC, Han WK, Bonventre JV, et al. Group V secretory phospholipase A2 translocates to the phagosome after zymosan stimulation of mouse peritoneal macrophages and regulates phagocytosis. The Journal of biological chemistry. 2006;281(10):6691–6698. doi: 10.1074/jbc.M508314200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quehenberger O, Armando AM, Brown AH, Milne SB, Myers DS, Merrill AH, et al. Lipidomics reveals a remarkable diversity of lipids in human plasma. Journal of lipid research. 2010;51(11):3299–3305. doi: 10.1194/jlr.M009449. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. (A) Representative FACS plots showing gating strategy for ILC2s. Alternaria-treated Wt lung homogenates were stained for ILC2s gated as CD45+ Lin-Thy1.2+ and forward and side scatter representation of the lineage positive population (depicted in blue) with low side scatter, and lineage positive population (depicted in red) with high side scatter. Percentage expression of Sca-1, ST2, ICOS, CD25 and intracellular IL-5 on Thy1.2+ (B) or Thy1.2+ Sca-1+ (C) ILC2s from homogenate lungs of Alternaria-treated Wt and Pla2g5-null mice. Images are from one representative mouse per group from three experiments with 13–19 (A, B) or 4–10 (C) Alternaria-treated mice per group.
Supplementary Figure 2. Transfer of Wt and Pla2g5-null BM-macrophages activated for 24h with GM-CSF/IL-4/IL-33. Wt mice received Wt or Pla2g5-null BM-macrophages intratracheally at day 2 after the first dose of Alternaria, followed by Alternaria at day 3, 6 and 9 or only Alternaria. Mice were euthanized 18 h after the last dose for flow cytometric analysis of eosinophils (CD45+ CD11c− SiglecF+) and Thy1.2+ ILC2s expressing intracellular IL-5. Values are mean ± SEM of 3 independent experiments with 6–22 mice per group and were compared by t-test. *P< 0.05.
Supplementary Figure 3. Percentage expression of intracellular IL-5 on Thy1.2+ ILC2s in homogenate lungs of Wt and Pla2g5-null mice treated with IL-33, OA, or LA in various combinations. Analysis by flow cytometry of Thy1.2+ ILC2s expressing intracellular IL-5 obtained from Wt and Pla2g5-null homogenate lungs treated as indicated. Images are from one representative mouse per group from two-four independent experiments with 4–12 mice per group.
Supplementary Figure 4. Percentage expression of intracellular IL-5 on Thy1.2+ ILC2s sorted from homogenate lungs of Wt and Pla2g5-null mice treated with Alternaria. Analysis by flow cytometry of ILC2s sorted from Wt and Pla2g5-null homogenate lungs, rested for 40 hours prior to in-vitro stimulation with LA (200 μM), OA (200 μM), IL-33 (30ng/ml) or all together for 8 hours, then analyzed by flow cytometry for percentages of IL-5 positive ILC2s. Images are from one representative sample per group from three independent experiments.