

Abstract
Background
Patients with systemic sclerosis-associated pulmonary arterial hypertension (SSc-PAH) have a far worse prognosis than those with idiopathic PAH (IPAH). In the intact heart, SSc-PAH exhibits depressed rest and reserve right ventricular (RV) contractility as compared to IPAH. We tested whether this disparity involves underlying differences in myofilament function.
Methods
Cardiac myocytes were isolated from RV septal endomyocardial biopsies from patients with SSc-PAH, IPAH, or SSc with exertional dyspnea but no resting PAH (SSc-d); control RV septal tissue was obtained from non-diseased donor hearts (6–7/group). Isolated myocyte passive length-tension and developed tension-calcium relationships were determined and correlated with in vivo RV function and reserve. RV septal fibrosis was also examined.
Results
Myocyte passive stiffness from length-tension relations was similarly increased in IPAH and SSc-PAH over control, although SSc-PAH biopsies had more interstitial fibrosis. More striking disparities were found between active force-calcium relations. Compared to controls, maximal calcium-activated force (Fmax) was 28% higher in IPAH but 37% lower in SSc-PAH. Fmax in SSc-d was intermediate between control and SSc-PAH. The calcium concentration required for half-maximal force (EC50) was similar between control, IPAH, and SSc-d, but lower in SSc-PAH. This disparity disappeared in myocytes incubated with the active catalytic subunit of protein kinase A. Myocyte Fmax directly correlated with in vivo RV contractility assessed by end-systolic elastance (Ees, R2=0.46, P=0.002) and change in Ees with exercise (R2=0.49, P=0.008), and was inversely related with exercise-induced chamber dilation (R2=0.63, P<0.002), the latter also a marker of depressed contractile reserve.
Conclusions
A primary defect in human SSc-PAH resides in depressed sarcomere function, whereas this is enhanced in IPAH. These disparities correlate with in vivo RV contractility and contractile reserve, and are consistent with worse clinical outcomes in SSc-PAH. The existence of sarcomere disease prior to developing resting PAH in SSc-d patients suggests that earlier identification and intervention may prove useful.
Keywords: RV, pulmonary arterial hypertension, scleroderma, myofilament, PKA, tension
BACKGROUND
Pulmonary arterial hypertension (PAH) is a progressive disorder that imposes high resistive and pulsatile loads on the right ventricle (RV). The extent of RV compensation is a major determinant of PAH clinical outcome, and its decompensation is a primary cause of death.1,2 Among World Health Organization Group I PAH subtypes, systemic sclerosis (SSc)-associated PAH carries a particularly poor prognosis.3 Systemic sclerosis, or scleroderma (SSc), is an autoimmune inflammatory disorder characterized by excess fibrosis and depressed microvascular perfusion and angiogenesis, and affects multiple organs including the ventricular myocardium.4 As compared to patients with idiopathic PAH (IPAH), SSc-PAH subjects are less responsive to therapy3,5 and display higher pro-brain natriuretic peptide levels, indicative of increased myocardial stress.6
Conventional measures of RV function, including echocardiographic indices, invasive hemodynamic assessments of afterload and compliance, ejection fraction, RV dimensions and mass do not distinguish between SSc-PAH and IPAH.4,7–9 However, in 2013 we reported that SSc-PAH patients have an intrinsic defect in RV chamber contractility as compared to IPAH, and that this results in adverse RV-pulmonary artery coupling at rest.7 In a subsequent study, we showed SSc-PAH subjects also have depressed RV force-frequency responses and RV contractile reserve with exercise versus IPAH.8 A consequence of this disparate reserve is that the RV dilates during exercise in SSc-PAH but not IPAH patients.8 In contrast to systolic measures, diastolic chamber abnormalities, including relaxation and end-diastolic pressure were similarly altered in both groups.
These studies turned a spotlight away from the pulmonary vasculature and more on the RV as a critical pathophysiologic feature of SSc-PAH. Many factors could underlie the contractile differences, though the presence of systolic depression without corresponding relaxation defects in SSc-PAH7 led us to hypothesize that the sarcomere may itself be prominently involved. The dependence of myofilament force on calcium has only been reported in a few PAH studies, with only one conducted with human tissue, and the results have varied.10–12 Pulmonary artery constriction in the rat lowered maximal Ca2+-activated tension,11 whereas this was unaltered in hypoxia-induced PAH induced in calves,10 and was increased in human PAH.12 The human study mostly examined tissue from patients with PAH from congenital heart disease, and did not include any with SSc-PAH.
The current study tested the hypothesis that RV sarcomere defects differentiate SSc-PAH from IPAH, and predict intact RV contractility and reserve function. As echo-Doppler studies have reported RV defects in SSc patients without PAH,13 we also examined myocardium from SSc patients who presented with exertional dyspnea but no resting PAH (SSc-d). Control heart RV septal tissue was provided from explanted but unused donor hearts. We find a striking depression of RV maximal Ca2+-dependent tension in SSc-PAH, directionally opposite to increased tension in IPAH. SSc-d had intermediate abnormalities. Maximal sarcomere tension corresponds with intact RV rest and reserve function measured in the same patients. Thus, defects of the RV sarcomere are a core feature differentiating two of the most common forms of PAH, and pose a potential therapy target for patients with SSc-PAH and SSc-d.
METHODS
Study Subjects
We prospectively enrolled 24 patients clinically referred for right heart catheterization (RHC) to evaluate pulmonary arterial hypertension (PAH) or exercise-induced PH at the Johns Hopkins Hospital (JHH) from November 2012 to March 2017. SSc-PAH patients and SSc patients with exertional dyspnea but without resting PAH (SSc-d) were enrolled, as were subjects with idiopathic PAH (IPAH). Diagnostic and exclusionary criteria for these groups have been previously described.8 All patients consented to the procurement of endomyocardial biopsies from the RV septum, obtained under echocardiographic guidance. A subset of patients separately consented to invasive right ventricular pressure-volume (PV) relation analysis, with data measured both at rest and during supine bicycle exercise. Details of the PV methods and intact patient results have been previously reported.8 Endomyocardial tissue was obtained in 7 IPAH patients, 11 SSc-PAH patients, 6 SSc-d patients, and all compared to 7 RV septal samples obtained from age-matched control donor hearts. The protocols were approved by the Johns Hopkins University Institutional Review Board (IRB), and informed consent obtained in all patients. Not all procedures and assays could be performed in each subject (details in Supplemental Table 1). Donor RV septum was obtained using cold-cardioplegia and open chest surgical excision, and tissue rapidly dissected and frozen. The University of Pennsylvania IRB and the Gift-of-Life donor program in Philadelphia, PA approved the donor tissue protocol, with consent for research use of donated heart tissue obtained from the families of the deceased. The data and study materials will not be available to other researchers as they are sensitive human subjects data.
Study Design
Demographic information and clinical characteristics of the study group were obtained prospectively by PH-specialists employing standardized assessments. Six-minute walk tests and other laboratory data were measured either at or within six months prior to RHC. After routine catheterization study, 3–4 endomyocardial biopsies (~2–3 mg/biopsy) were obtained from the RV septum using a 7 Fr endomyocardial bioptome (Argon Medical, Plano, TX). Each piece was rapidly frozen in liquid nitrogen immediately upon removal, with one first placed in OCT (optimal cutting temperature) solution prior to freezing to improve tissue preservation. This piece was used for the myofilament studies. In a subset of patients another piece was placed in formalin for histopathology. Flash frozen donor RV septal tissue was shipped by overnight express and analyzed in our laboratory.
Invasive PV assessments at rest and during supine bicycle ergometry were performed as previously described.8 PV analyses include data from these as well as additional patients, the latter obtained using the Inca System (Millar, Houston, TX) and CD Leycom PV catheters (RP-CA-41103-PN, Millar, Houston, TX), as the previously employed catheters were discontinued.8 All PV data were analyzed with custom WinPVAN software.
Myofilament Function
Myocardium was frozen at −140°C until studied. Methods for force-calcium analysis of biopsy-derived skinned myocytes have been previously reported.14 Briefly, biopsies are incubated with 0.3% Triton X-100 in isolation buffer with protease inhibitors (Sigma-Aldrich, St. Louis, MO) and phosphatase inhibitors (PhosSTOP, Roche Applied Science, Penzberg, Germany) and then homogenized by low-speed pulverization to generate a skinned cellular preparation. After washing in isolation buffer, myocytes are affixed to a force transducer-length controller (Aurora Scientific, Ontario, Canada) using ultraviolet-activated adhesive (Norland Products, Cranbury, NJ) (Supplemental Figure 1). Sarcomere length (SL) measured by Fourier transform of digital images (IPX-VGA210, Imperx, Boca Raton, FL) is adjusted by micro-manipulators (Newport, Corporation, Irvine, CA). Force was normalized to myocyte cross sectional area (CSA) to derive tension expressed as mN/mm2.
Passive tension-SL relations were measured in relaxing solution (0 μM Ca2+) over a range of SL. SL was then set to 2.1 μm, and active tension-Ca2+ relations generated by varying Ca2+ concentration from 0–46.8 μM (latter being saturating conditions). Steady-state force versus log[Ca2+] (F-Ca2+) plots were fit to the Hill equation: F = Fmax × Cah/(EC50h + Cah) yielding maximal force (Fmax, μN), calcium sensitivity (EC50, i.e. [Ca2+] required to achieve 50% maximal force, μM), and cooperativity (Hill) coefficient (h). In a subset of myocytes, we exposed cells to the active catalytic subunit of Protein Kinase A (0.125 units/ml, 20 minutes; Sigma-Aldrich) and repeated measurements of the F-Ca2+ dependence.
Histology
Heart biopsies fixed in 10% formalin were embedded in paraffin, and 10 μm sections cut and stained with Masson Trichrome. Histology was analyzed blinded to group using Aperio Imagescope (Leica Biosystems, Nussloch, Germany) and percent fibrosis determined by calculating the total area of blue-hued staining as a percentage of total area. Standardized settings (hue 0.66 and threshold 0.33) were used to define positive fibrosis staining.
Statistical Analysis
Data are presented as mean ± standard deviation in the tables and text, and mean ± standard error of the mean in figures, unless otherwise indicated. Parametric tests were used when possible; non-parametric tests were employed when normality criteria was not met. Fibrosis, as defined by an excess of 2.5% of the biopsy area, was compared between groups using Fisher’s exact and Kruskal-Wallis analysis of variance (ANOVA). Passive tension at each sarcomere length was compared between groups using one-way ANOVA with post-hoc testing using the Tukey test. For cell mechanics studies, 2–3 myocytes were tested from each biopsy (majority was 3 per biopsy) yielding a total of 68 myocytes. Hill equation parameters (Fmax, EC50, h) and myocyte cross-sectional area (CSA) were compared by a mixed-model ANOVA to account for the random effects of multiple myocyte measurements per individual biopsy. Post-hoc comparisons were done using the Tukey test. Group mean comparisons were made using Wilcoxon rank sum testing, while proportions were compared with Fisher exact tests. Within disease groups, the effect on measurements pre- and post-protein-kinase A (PKA) was tested using paired student T-test; group comparisons and differential PKA effects between disease groups were tested using two-way ANOVA and an interaction term between disease and PKA. Myocyte Fmax was compared to in vivo pressure-volume parameters were made using Pearson correlation coefficients and linear regression. Statistical analysis and graphical depiction were performed using commercial software (Stata-15, StataCorp, College Station, TX; Prism 7, Graphpad, La Jolla, CA).
RESULTS
Study Population
Demographic and clinical data for each patient group are provided in Supplemental Table 2. The groups were matched in nearly all parameters, with somewhat fewer female controls and somewhat greater body surface area in IPAH subjects. Clinical and cardiac magnetic resonance-based features of IPAH and SSc-PAH subjects studied in the current cohort were very similar to that previously reported.7,8 Resting RV contractility as assessed by ventricular end-systolic elastance (Ees), and right ventricular-pulmonary arterial (RV-PA) coupling assessed by the ratio of Ees to arterial elastance (Ees/Ea), were both lower in SSc-PAH compared to IPAH (Table 1). SSc-d subjects had significantly lower right atrial and pulmonary artery pressures and pulmonary afterload (resistance and Ea) as compared to SSc-PAH and IPAH, and their Ees and Ees/Ea were similar to that in IPAH (Table 1). Measures of in vivo diastolic function (time constant of relaxation and peak flow rate normalized to EDV) were similar between groups. When available, the hemodynamic response to exercise is also provided in Table 1. Both PAH groups displayed abnormal pulmonary artery pressure rise. SSc-d patients did not have resting PH, but on average displayed exercise-induced PH, as defined by total pulmonary resistance >3 Wood Units and mean pulmonary artery pressure >30 mmHg during exercise.15,16
Table 1.
Resting and Exercise Hemodynamics in IPAH, SSc-PAH, and SSc-d
| Hemodynamics | IPAH | SSc-PAH | P-value* | SSc-d | P-value† |
|---|---|---|---|---|---|
|
| |||||
| Resting | N=7 | N=11 | N=6 | ||
| RAP (mmHg) | 9 ± 7 | 8 ± 5 | 0.93 | 3 ± 1 | 0.02 |
| PASP (mmHg) | 70 ± 18 | 58 ± 20 | 0.17 | 33 ± 6 | 0.005 |
| PADP (mmHg) | 30 ± 8 | 23 ± 7 | 0.10 | 12 ± 4 | 0.003 |
| mPAP (mmHg) | 44 ± 10 | 36 ± 12 | 0.22 | 18 ± 5 | 0.002 |
| PAWP (mmHg) | 9 ± 4 | 10 ± 3 | 0.78 | 8 ± 2 | 0.08 |
| CO (L/min) | 4.2 ± 1.0 | 4.4 ± 1.0 | 0.59 | 4.2 ± 1.0 | 0.65 |
| PVR (Wood units) | 8.9 ± 4.3 | 7.3 ± 5.6 | 0.30 | 3.0 ± 1.9 | 0.04 |
| Ees (mmHg/ml) | 0.96 ± 0.32 | 0.44 ± 0.14 | 0.01 | 0.71 ± 0.29 | 0.08 |
| Ea (mmHg/ml) | 0.97 ± 0.60 | 1.06 ± 0.73 | 0.78 | 0.48 ± 0.17 | 0.02 |
| Ees/Ea | 1.39 ± 0.85 | 0.49 ± 0.16 | 0.08 | 1.53 ± 0.66 | 0.001 |
| τ(Suga) | 39.0 ± 7.9 | 32.2 ± 7.7 | 0.11 | 41.8 ± 5.3 | 0.11 |
| PFR/EDV (s−1)_ | 2.9 ± 1.2 | 3.3 ± 1.6 | 0.69 | 4.2 ± 1.9 | 0.27 |
|
| |||||
| Exercise | N=4 | N=10 | N=4 | ||
|
| |||||
| Peak VO2 (ml/kg/min) | 11.5 ± 2.4 | 9.9 ± 3.2 | 0.40 | 11.0 ± 4.2 | 0.74 |
| Peak CO (L/min) | 11.2 ± 3.3 | 10.1 ± 5.6 | 0.48 | 9.7 ± 4.3 | 0.89 |
| Peak mPAP (mmHg) | 64 ± 24 | 52 ±19 | 0.36 | 38 ± 8 | 0.20 |
| TPR (Wood Units) | 6.5 ± 4.1 | 7.7 ± 6.4 | 0.94 | 5.4 ± 4.6 | 0.78 |
Resting and exercise hemodynamics of IPAH, SSc-PAH, and SSc without resting PAH (SSc-d) patients. RAP, right atrial pressure; PASP, PA systolic pressure; PADP, PA diastolic pressure; mPAP, mean PA pressure; PAWP, PA wedge pressure, CO, cardiac output; PVR, pulmonary vascular resistance; Ees, end-systolic elastance; Ea, effective arterial elastance; Ees/Ea, RV-PA coupling ratio; τ(Suga), relaxation time constant; PFR/EDV, peak flow rate normalized to end-diastolic volume; TPR, total pulmonary resistance; peak VO2, peak oxygen consumption. Comparisons between groups made using Wilcoxon rank-sum test.
P-value comparing IPAH vs. SSc-PAH,
P-value comparing SSc-PAH vs SSc-d.
Passive myocyte stiffness is similarly increased in SSc-PAH and IPAH
Interstitial fibrosis is observed in patients with PAH, and is considered a particular feature of SSc. Interstitial fibrosis in formalin fixed endocardial biopsies exceeded a threshold of 2.5% in two-thirds of SSc-PAH subjects but in none of IPAH or SSc-d patients (P<0.01 by Fischer Exact Test; P<0.05 for Kruskal-Wallis test comparing the 3 groups) (Figure 1A-B). Tissue stiffness could not be assessed from the biopsy, but isolated myocyte stiffness was possible. Myocytes were stiffer compared to both forms of PAH versus controls (P<0.05 from sarcomere lengths of 2.2 to 2.6 μm), but this increase was virtually identical between SSc-PAH and IPAH (P>0.75 from sarcomere lengths of 2.2 to 2.6 μm) (Figure 1C).
Figure 1.
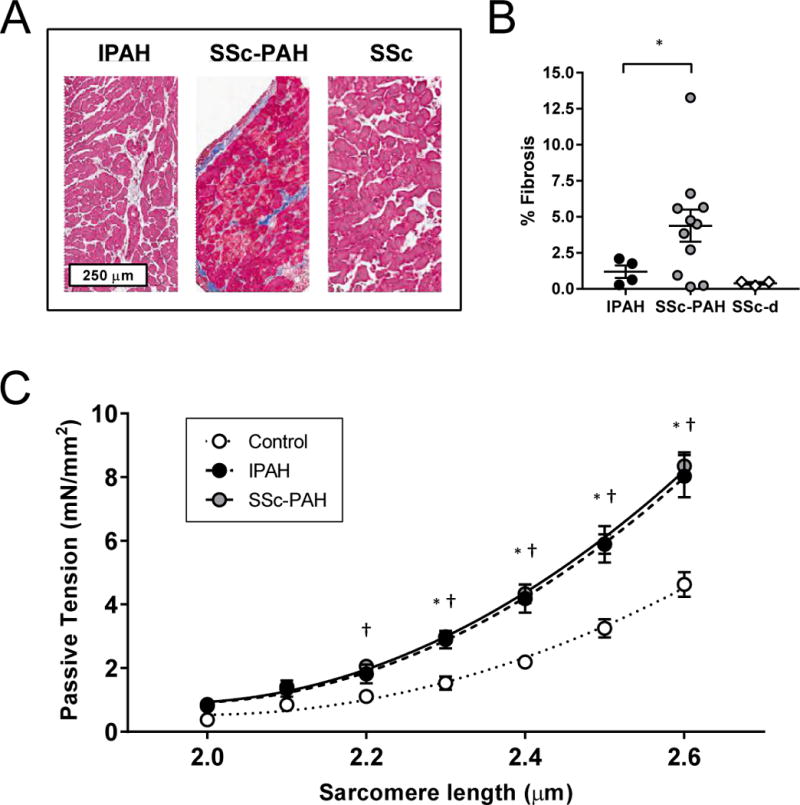
Effects of SSc-PAH versus IPAH on passive myocyte tension and fibrosis
(A) Representative Masson Trichrome histologic images from IPAH, SSc-PAH, and SSc. (B) Histologic fibrosis measured by Masson Trichrome as a percentage of total biopsy area was increased in SSc-PAH versus IPAH or SSc-d, based on a threshold of 2.5% fibrosis. (C) Passive tension as a function of escalating sarcomere length was measured in IPAH, SSc-PAH, and non-failing control myocytes (n=5 subjects/group). Passive tension between all three groups were compared using one-way analysis of variance (ANOVA) at each SL. * post-hoc P<0.05 between Control and IPAH; † post-hoc P<0.05 between Control and SSc-PAH.
Maximal Tension and Calcium Sensitivity Differs between SSc-PAH, IPAH, and SSc
Unlike passive myocyte stiffness, we found marked differences in active F-Ca2+ dependence between the PAH groups, each being different from controls (Fig. 2A). Maximal Ca2+-activated tension (e.g. normalized maximal force, or Fmax) was 28% higher in IPAH versus controls (23.4±2.7 vs. 18.2±2.7 mN/mm2, P<0.05), whereas in SSc-PAH, Fmax was depressed by 37% (11.4±2.6 mN/mm2, P<0.05 versus both other groups). The calcium required for half-maximal activation (EC50) was similar between IPAH, controls, and SSc-d (1.73±0.21, 1.85±0.25, and 1.88±0.28 μM respectively, P=0.35); however SSc-PAH had a lower EC50 (1.35±0.26 μM, P<0.05), shown by the left-shifted F-Ca2+ relation when data are normalized to their respective Fmax (Figure 2B). Figure 2C compares SSc-d to SSc-PAH and controls. SSc-d displayed an intermediate Fmax (15.1±3.5 mN/mm2, P<0.05 vs. control and SSc-PAH); other parameters were similar to control. Summary Hill equation parameters for all groups are provided in Figure 2D. Myocyte cross-sectional area is also shown and was similar among the four groups (Figure 2D). Human myocyte resting length in our experimental setup was generally 1.70 to 1.80 μm.
Figure 2.
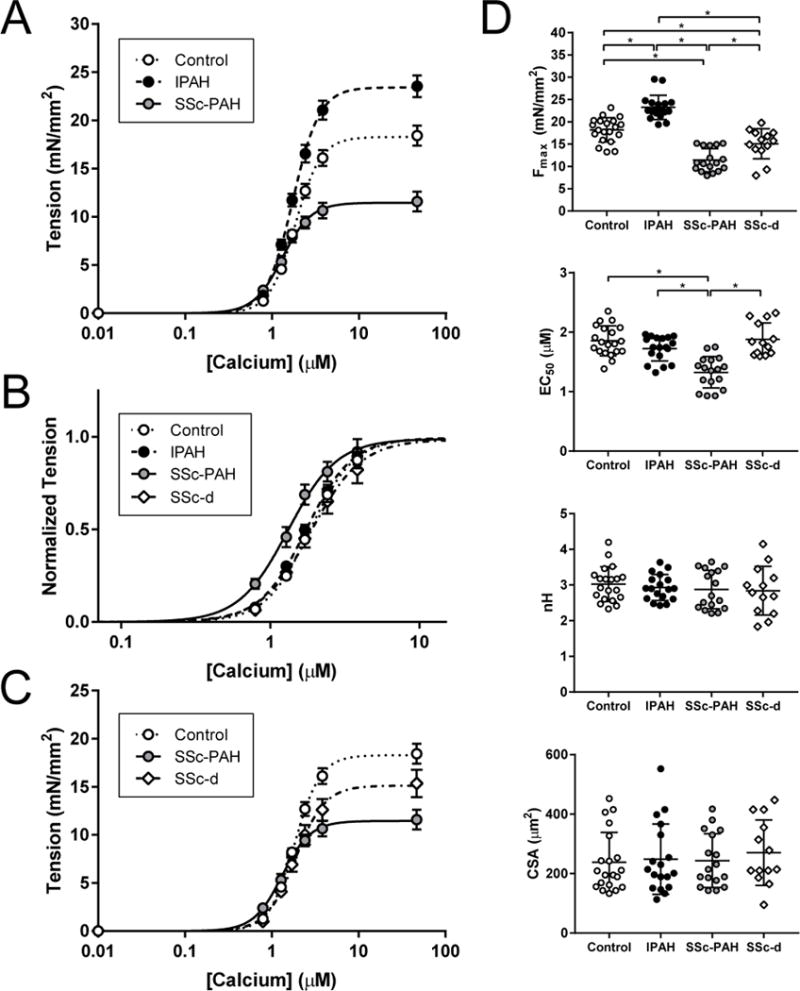
Force-Calcium curves in Control, IPAH, and SSc myofilaments
(A) Force-calcium data were obtained in non-failing control, IPAH, and SSc-PAH myocytes to obtain maximal calcium-activated tension (Fmax) and calcium sensitivity (EC50) (n=7 subjects/group). (B) Data were normalized to maximal tension to illustrate shifts in calcium sensitivity (measured as EC50, or the calcium concentration necessary for 50% maximal activation). (C) Myofilament force-calcium data of control, SSc-PAH, and SSc subjects free of PAH (SSc-d). (D) Fmax, EC50, Hill coefficient (h), and myocyte cross-sectional area (CSA) data; data presented as mean ± SD. Fmax was increased in IPAH versus controls, whereas it was decreased in SSc-PAH versus both control and IPAH; in SSc-d, Fmax was intermediate between control and SSc-PAH. EC50 was decreased in SSc-PAH versus control, IPAH, and SSc-d. There was no significant difference between groups in Hill coefficient or myocyte cross-sectional area (CSA). * significant difference in post-hoc comparison.
Protein Kinase A Exposure Equalizes Calcium Sensitivity Among Patient Groups
In both LV and RV failure, increased Ca2+ sensitivity (reduced EC50) has been reported and linked to depressed protein kinase A (PKA) phosphorylation of myofilament proteins, notably of troponin I.10,17,18 Only SSc-PAH patients displayed this, and we hypothesized this too might reflect depressed PKA activation. Myocytes from both PAH groups and controls were therefore further exposed to the catalytically active PKA subunit, and data before and after exposure compared. In IPAH and controls, PKA increased EC50 slightly (P<0.05 versus non-PKA); however this rise was significantly greater in SSc-PAH myocytes, resulting in near-identical EC50 values after PKA exposure (P-value for interaction between PAH group and PKA treatment = 0.002; Figure 3A-B). Fmax was unaltered by PKA (pre- vs. post PKA Fmax in controls, 17.3±1.0 vs. 17.2±1.0 mN/mm2, P=0.8; IPAH, 22.4±0.7 vs. 22.3±0.7 mN/mm2, P=0.8; SSc-PAH, 10.0±1.1 vs. 11.7±1.7 mN/mm2; P=0.4).
Figure 3.
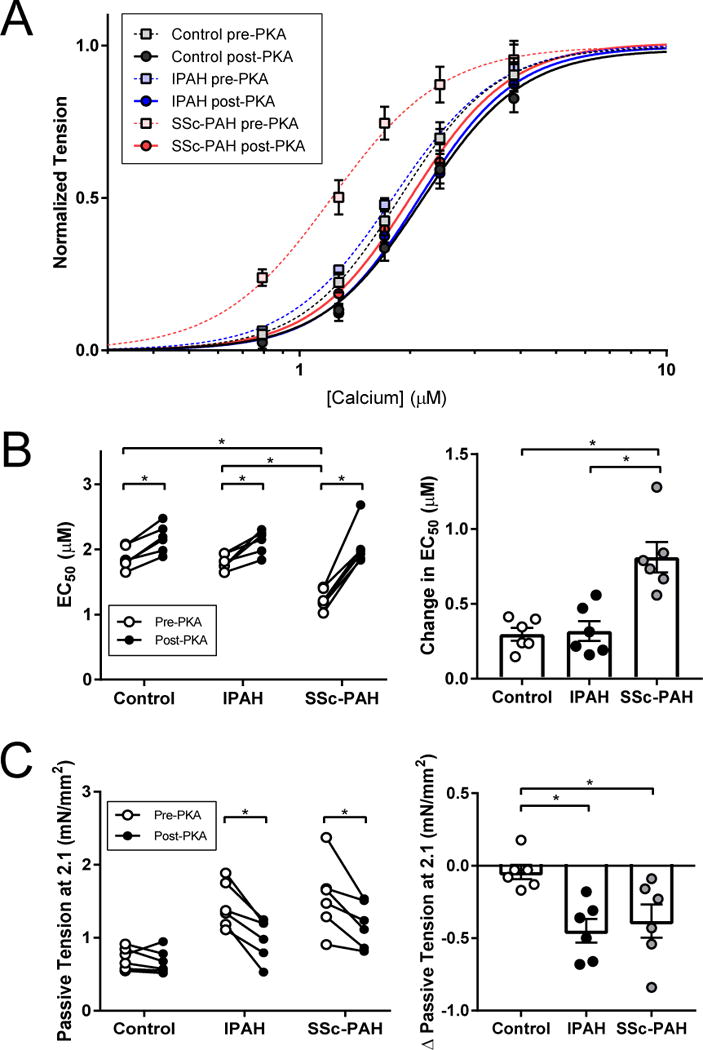
Effect of PKA treatment on myofilament function and passive myocyte stiffness
(A) Myofilaments from a subset of control, IPAH, and SSc-PAH subjects (3 subjects/group) were directly incubated with protein kinase A (PKA). (B) Baseline calcium sensitivity (EC50) in all three groups mirrored trends of the larger cohort. With PKA, there was a slight increase in EC50 in control and IPAH. On the other hand, PKA completely markedly increased EC50 in the SSc-PAH group to control levels. Change in EC50 was significantly greater in SSc-PAH than in both control and IPAH (Disease × group interaction P=0.002). (C) PKA treatment led to a decrease in passive tension, at a sarcomere length (SL) of 2.1 μm, in both IPAH and SSc-PAH, but no change in controls. There was no significant difference in PKA-mediated passive tension change between IPAH and SSc-PAH. * P<0.05; post-hoc P-value applied where applicable.
PKA also reduces passive myocyte tension by phosphorylating the sarcomeric protein titin,19 and reduced titin phosphorylation has been implicated as a mechanism of increased passive myocyte stiffness in the RV of patients with PAH.12,18 We therefore assessed the impact of PKA in both PAH groups by change in passive tension at a fixed SL of 2.1 μm. This similarly declined after PKA in both PAH groups, but was unchanged in controls (P<0.05 for both, Figure 3C).
Fmax Predicts RV Rest Contractility and Exercise-Reserve Response
To test if the lower Fmax in SSc-PAH, partial decline in SSc-d, and rise in IPAH correlated with in vivo RV systolic function, we regressed Fmax and resting chamber contractility (Ees), the latter derived from invasive RV pressure-volume analysis in the same patients. Figure 4A shows these variables directly correlated (R2 =0.46, P=0.002). We also tested if Fmax correlated with RV exercise performance. As previously reported,8 RV contractile reserve (change in Ees) is depressed in SSc-PAH subjects. This was associated with chamber dilation (higher RV end-diastolic volumes, RV EDV) at submaximal exercise whereas IPAH patients showed no dilation at the same exercise level (Stage 2, 25 Watts). Fmax positively correlated with Ees change during exercise (R2=0.49, P=0.008), and inversely correlated with chamber dilation (%Δ RV EDV, R2=0.63, P<0.002).
Figure 4.
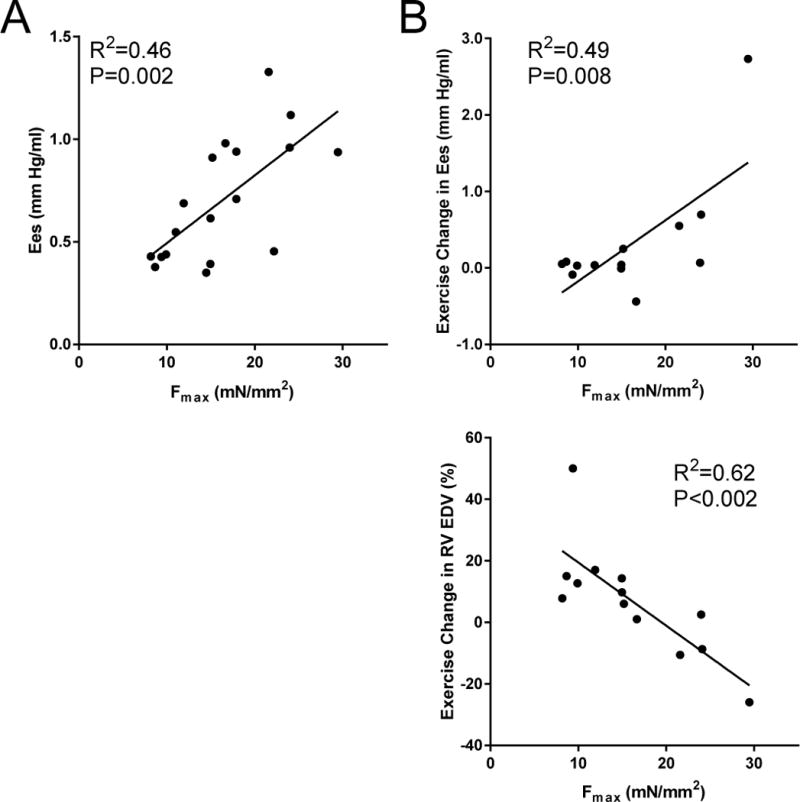
Myofilament Fmax correlates with in vivo RV function and functional reserve during exercise
(A) Myofilament Fmax correlated strongly with resting RV chamber-level contractility (Ees) (R2=0.46, P=0.002). (B) Fmax also correlated strongly with contractile reserve, or change in Ees with exercise (R2=0.49, P=0.008), as well as exercise RV chamber dilation, as measured by percent change in RV end-diastolic volume (RV EDV) after reaching a 25-Watt workload (R2=0.63, P<0.002).
DISCUSSION
The current study reveals that patients with SSc-PAH have depressed sarcomeric function manifest prominently by a substantial decline in maximal force-calcium dependence. The exact opposite, an increase in this dependence, is observed in IPAH. This disparity parallels and predicts corresponding differences in global RV chamber systolic function at rest and upon exertional demand. As the latter has been found to correlate with clinical outcome, the new results surprisingly focus the spotlight on the RV sarcomere rather than pulmonary vasculature in explaining SSc-PAH versus IPAH differences. This is further strengthened by the finding that SSc-d patients who do not yet have resting PAH also exhibit abnormal sarcomere function. While prior efforts to treat both SSc-PAH and IPAH have focused on pulmonary load, our results suggest tailored treatment focusing on RV sarcomeric disease maybe particularly useful for SSc-PAH.
To our knowledge, only one prior study by Rain and colleagues has reported on human RV force-calcium relations in PAH.12 In this report, the authors found myocytes were passively stiffer, much as we observed. From their F-Ca2+ data, they found Fmax was ~40% above that in non-failing controls, and EC50 was slightly lower. This study primarily examined tissue from congenital heart disease-PAH patients; with only 3/11 having IPAH and none with SSc-PAH. Congenital heart disease involves complex mechanical loads and chronic disease history, so comparisons with the current IPAH and SSc-PAH groups are difficult. The prior study was also based on tissue obtained at time of heart-lung transplantation, so patients all had end-stage disease.12 The current results are the first to include both SSc-PAH and SSc-d, and perform analysis from heart biopsies reflecting an earlier disease stage. Our findings in IPAH are similar to those of Rain et al.,12 suggesting this form of PAH retains RV sarcomere compensation even at late stages.
In any form of heart disease involving marked increases in chamber load, the net result depends on the interaction between load severity and the myocardium. As pulmonary arterial load was similar in both PAH groups, unique RV features linked to SSc would appear more important for the specific maladaptive response and disease severity. Whether SSc patients without any abnormal pulmonary vascular load have similar sarcomere defects remains unknown, as these patients are not referred for RHC or biopsy. However, non-invasive echo Doppler data such patients have found sub-clinical systolic and diastolic defects based on strain-based measurements.13 The intermediate defect in SSc-d may indicate that even intermittent PAH as induced during exercise is sufficient to manifest a sarcomere defect. While the underlying cause remains unknown, SSc-related immune activation with vasculopathy and inflammation could be factors leading to damaged myofibrillar proteins.
The EC50 is actively regulated by post-translational changes in regulatory thin filament proteins including troponins I, C, and T (TnI, TnC, TnT), myosin binding protein C, myosin light chain, and tropomyosin.20 It is controlled by multiple kinases, including phosphorylation by PKA,17 PKG,21 PKC,22 PKD,23 and AMPK24 (particularly targeting TnI and TnT), and by oxidation,25 nitrosylation, and acetylation.26 Many of these factors lead to Ca2+-desensitization (increase EC50), although some (e.g. TnI phosphorylation by AMPK and PKCβ2) result in sensitization. EC50 often declines with LV failure, and this has been attributed to reduced TnI phosphorylation by PKA due to down-regulation of β-adrenergic signaling, since PKA activation restores it to control levels.17 Such sensitization may in part compensate for depressed calcium transients in heart failure, but it may also worsen diastolic dysfunction.27 Evidence that RV PKA deficiency occurs with PAH has been reported.10,12,18 We found evidence for PKA deficiency in active force-Ca2+ dependency in SSc-PAH, as observed in forms of LV failure. This suggests another component of abnormal systolic reserve in these patients versus IPAH, perhaps related to greater inhibition of sympathetic stimulation. This remains to be tested in vivo. PKA-effects on passive myocyte stiffness were similar between groups, though our capacity to detect differences may have been limited by having restricted the analysis to a SL of 2.1 μm, where stiffness differences are slight.
Unlike EC50 or passive stiffness, Fmax is not modified by PKA, and indeed most kinases that alter sarcomere function leave Fmax intact. Prior studies have found that Fmax is depressed by proteolysis of regulatory thin filament proteins and perturbations in structural sarcomere integrity.28 For example, Fmax declines after ischemic injury in conjunction with troponin I proteolysis by calpain-1 at the C-terminus.29,30 Mice expressing a mutant TnI with this truncation exhibit a depressed Fmax and develop heart failure31 Another protein that can undergo destructive proteolysis is myosin binding protein C, and in this case, the cleaved by-product serves as a poison peptide to depress Fmax.32 Fmax also declines with oxidative stress,33 and in a canine tachypacing heart failure model, reduced Fmax was correlated with structural disruption of the sarcomere with reduced force generation by ~40% of myofibrils.34 At present, we do not know what modifies Fmax in SSc-PAH and to a smaller extent in SSc-d, but ongoing studies in additional patients hope to reveal factors to further guide precision therapy. However, even without a specific molecular explanation for the Fmax defect in SSc-PAH, its presence and correlation to intact RV physiology suggests that small molecule inotropes designed to enhance sarcomere function20 may be particularly useful.
We previously described that the RV contractile response to faster heart rates (force-frequency response, FFR) and calcium cycling also predicted to be diminished in SSc-PAH versus IPAH.8 However, as calcium transients could not be measured in vivo, we were unable to test if these defects were due to calcium transient disparities or to abnormal sarcomere responses to calcium. The Fmax and EC50 disparities between IPAH and SSc-PAH shown here would be adequate to explain FFR differences as well, since the latter depends on levels of stimulating Ca2+. This was shown in a study employing a calcium sensitizer that modifies Fmax but not Ca2+ cycling to improve FFR.35 Thus, improving sarcomere dysfunction in SSc-PAH should also help frequency dependent systolic reserve.
Diastolic stiffening of PAH RV myocytes has been described,12,18 and our results are concordant with such data. Chamber stiffness combines properties of myocytes, the interstitium, and extrinsic factors (e.g. RV-LV interaction, pericardium). In this regard, our in vivo analysis has not revealed significant disparities between IPAH and SSc-PAH. While SSc-PAH had more endocardial fibrosis, this could be consistent with an autopsy study which found SSc fibrosis to be disproportionately reflected in the endocardium and interventricular septum.36 By contrast, we found T2-weighted cardiac magnetic resonance imaging analysis of fibrosis was similar between IPAH and SSc-PAH.8 Myocyte stiffness is also similarly increased in both diseases, supporting the net results in intact patients. Thus, while subtle differences may still exist, it is unlikely that diastolic properties are a primary contributor to worsened RV function or clinical course in SSc-PAH.
This study has several limitations. Given its invasive nature, endomyocardial biopsies from healthy controls could not be obtained. The RV septal tissue from non-failing donor hearts can be impacted by the patient history, anesthesia, neurohormonal activation, and other conditions at time of organ procurement. We did not assess the RV free wall given the higher risk of myocardial perforation, but prior studies suggest homogeneity of myofilament function from multiple anatomic sites within a given RV.10 While the individual sample sizes (number of hearts) were relatively small for each group, we analyzed many cells, which confer their own variability given the extraction procedure. Our analysis was adjusted for the random effects of multiple measurements per biopsy piece. Our ability to correlate the individual results to the same subject’s intact heart data strengthens the conclusions.
Conclusion
We demonstrate that in contrast to the hyper-contractile response of IPAH myofilaments, RV myofilaments from SSc-PAH exhibit a marked decrease in maximal calcium-activated force and abnormal increase in calcium sensitivity. These findings are strengthened by their correlation to measures of in vivo RV contractility and reserve. Abnormalities in calcium sensitivity, but not maximal calcium-activated tension, were restored by PKA. The presence of intermediate defects in SSc-d further supports a primary influence of SSc on the RV. Together, these data highlight the critical role of RV sarcomere dysfunction in SSc-PAH and suggest that the SSc-PAH RV is characterized by a phenotype of RV failure, not compensation. Targeted therapies to enhance RV myofilament function in this PAH subgroup, with more vigilant surveillance and perhaps early intervention in borderline SSc patients, should be explored.
Supplementary Material
CLINICAL PERSPECTIVE.
What is new?
Right ventricular (RV) myofilaments isolated from human subjects with systemic sclerosis-associated pulmonary arterial hypertension (SSc-PAH) exhibit diminished contractile force and abnormal calcium sensitivity versus control myofilaments, in contrast to hyper-contractile compensation in idiopathic pulmonary arterial hypertension.
SSc patients with dyspnea and only exercise-induced pulmonary hypertension (PH) exhibited an intermediate RV myofilament phenotype.
These myofilament contractile abnormalities correlate strongly with in vivo right ventricular (RV) function at rest and RV contractile reserve during exercise, suggesting a central role of RV myofilament dysfunction in SSc-PAH.
What are the clinical implications?
Among patients with PAH, those with SSc-PAH suffer disproportionately poor outcomes, even with treatment using standard PAH-directed therapies.
These findings uncover key deficiencies in the SSc-PAH RV and suggest that therapies targeted at RV myofilament contractile dysfunction may prove particularly useful for this vulnerable PAH sub-population.
Furthermore, SSc patients with only exercise-induced PH would appear to comprise an at-risk population that may warrant improved screening and treatment paradigms.
Acknowledgments
SOURCES OF FUNDING
The study was supported by National Institutes of Health-National Heart, Lung, and Blood Institute (P50-HL084946, 5-R01-HL114910, R35-135827, T32-HL007227), the Scleroderma Research Foundation, and the Jerome L. Greene Scholarship.
Footnotes
Hsu, RV Myofilaments in Scleroderma vs. Idiopathic PAH
DISCLOSURES
- R.J.T. is a member of an Abbott Pulmonary Hypertension Study steering committee (modest) and is an Actelion core lab consultant (significant).
- All other authors have no relevant disclosures to report.
References
- 1.Ryan JJ, Archer SL. The right ventricle in pulmonary arterial hypertension: disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ Res. 2014;115:176–188. doi: 10.1161/CIRCRESAHA.113.301129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vonk Noordegraaf A, Haddad F, Chin KM, Forfia PR, Kawut SM, Lumens J, Naeije R, Newman J, Oudiz RJ, Provencher S, Torbicki A, Voelkel NF, Hassoun PM. Right heart adaptation to pulmonary arterial hypertension: physiology and pathobiology. J Am Coll Cardiol. 2013;62:D22–D33. doi: 10.1016/j.jacc.2013.10.027. [DOI] [PubMed] [Google Scholar]
- 3.Ruiz-Cano MJ, Escribano P, Alonso R, Delgado J, Carreira P, Velazquez T, Sanchez MAG, Sáenz de la Calzada C. Comparison of baseline characteristics and survival between patients with idiopathic and connective tissue disease-related pulmonary arterial hypertension. J Heart Lung Transplant. 2009;28:621–627. doi: 10.1016/j.healun.2009.02.016. [DOI] [PubMed] [Google Scholar]
- 4.Chaisson NF, Hassoun PM. Systemic sclerosis-associated pulmonary arterial hypertension. Chest. 2013;144:1346–1356. doi: 10.1378/chest.12-2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Argula RG, Karwa A, Lauer A, Gregg D, Silver RM, Feghali-Bostwick C, Schanpp LM, Egbert K, Usher BW, Ramakrishnan V, Hassoun PM, Strange C. Differences in Right Ventricular Functional Changes during Treatment between Systemic Sclerosis-associated Pulmonary Arterial Hypertension and Idiopathic Pulmonary Arterial Hypertension. Ann Am Thorac Soc. 2017;14:682–689. doi: 10.1513/AnnalsATS.201608-655OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mathai SC, Bueso M, Hummers LK, Boyce D, Lechtzin N, Le Pavec J, Campo A, Champion HC, Housten T, Forfia PR, Zaiman AL, Wigley FM, Girgis RE, Hassoun PM. Disproportionate elevation of N-terminal pro-brain natriuretic peptide in scleroderma-related pulmonary hypertension. Eur Respir J. 2010;35:95–104. doi: 10.1183/09031936.00074309. [DOI] [PubMed] [Google Scholar]
- 7.Tedford RJ, Mudd JO, Girgis RE, Mathai SC, Zaiman AL, Housten-Harris T, Boyce D, Kelemen BW, Bacher AC, Shah AA, Hummers LK, Wigley FM, Russell SD, Saggar R, Saggar R, Maughan WL, Hassoun PM, Kass DA. Right ventricular dysfunction in systemic sclerosis-associated pulmonary arterial hypertension. Circ Heart Fail. 2013;6:953–963. doi: 10.1161/CIRCHEARTFAILURE.112.000008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsu S, Houston BA, Tampakakis E, Bacher AC, Rhodes PS, Mathai SC, Damico RL, Kolb TM, Hummers LK, Shah AA, McMahan Z, Corona-Villalobos CP, Zimmerman SL, Wigley FM, Hassoun PM, Kass DA, Tedford RJ. Right Ventricular Functional Reserve in Pulmonary Arterial Hypertension. Circulation. 2016;133:2413–2422. doi: 10.1161/CIRCULATIONAHA.116.022082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramjug S, Hussain N, Hurdman J, Billings C, Charampopoulos A, Elliot CA, Kiely DG, Sabroe I, Rajaram S, Swift AJ, Condliffe R. Idiopathic and Systemic Sclerosis associated Pulmonary Arterial Hypertension: A Comparison of Demographic, Haemodynamic and Magnetic Resonance Imaging Characteristics and Outcomes. Chest. 2017;152:92–102. doi: 10.1016/j.chest.2017.02.010. [DOI] [PubMed] [Google Scholar]
- 10.Walker LA, Walker JS, Glazier A, Brown DR, Stenmark KR, Buttrick PM. Biochemical and myofilament responses of the right ventricle to severe pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2011;301:H832–H840. doi: 10.1152/ajpheart.00249.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fan D, Wannenburg T, de Tombe PP. Decreased myocyte tension development and calcium responsiveness in rat right ventricular pressure overload. Circulation. 1997;95:2312–2317. doi: 10.1161/01.CIR.95.9.2312. [DOI] [PubMed] [Google Scholar]
- 12.Rain S, Handoko ML, Trip P, Gan CT-J, Westerhof N, Stienen GJ, Paulus WJ, Ottenheijm CAC, Marcus JT, Dorfmüller P, Guignabert C, Humbert M, Macdonald P, Remedios Dos C, Postmus PE, Saripalli C, Hidalgo CG, Granzier HL, Vonk Noordegraaf A, van der Velden J, de Man FS. Right ventricular diastolic impairment in patients with pulmonary arterial hypertension. Circulation. 2013;128:2016–2025. doi: 10.1161/CIRCULATIONAHA.113.001873. [DOI] [PubMed] [Google Scholar]
- 13.Mukherjee M, Chung S-E, Ton VK, Tedford RJ, Hummers LK, Wigley FM, Abraham TP, Shah AA. Unique Abnormalities in Right Ventricular Longitudinal Strain in Systemic Sclerosis Patients. Circ Cardiovasc Imaging. 2016;9:e003792. doi: 10.1161/CIRCIMAGING.115.003792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kirk JA, Holewinski RJ, Kooij V, Agnetti G, Tunin RS, Witayavanitkul N, de Tombe PP, Gao WD, Van Eyk J, Kass DA. Cardiac resynchronization sensitizes the sarcomere to calcium by reactivating GSK-3β. J Clin Invest. 2014;124:129–138. doi: 10.1172/JCI69253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hervé P, Lau EM, Sitbon O, Savale L, Montani D, Godinas L, Lador F, Jaïs X, Parent F, Günther S, Humbert M, Simonneau G, Chemla D. Criteria for diagnosis of exercise pulmonary hypertension. Eur Respir J. 2015;46:728–737. doi: 10.1183/09031936.00021915. [DOI] [PubMed] [Google Scholar]
- 16.Kovacs G, Hervé P, Barbera JA, Chaouat A, Chemla D, Condliffe R, Garcia G, Grünig E, Howard L, Humbert M, Lau E, Laveneziana P, Lewis GD, Naeije R, Peacock A, Rosenkranz S, Saggar R, Ulrich S, Vizza D, Vonk Noordegraaf A, Olschewski H. An official European Respiratory Society statement: pulmonary haemodynamics during exercise. Eur Respir J. 2017;50:1700578. doi: 10.1183/13993003.00578-2017. [DOI] [PubMed] [Google Scholar]
- 17.van der Velden J, de Jong JW, Owen VJ, Burton PB, Stienen GJ. Effect of protein kinase A on calcium sensitivity of force and its sarcomere length dependence in human cardiomyocytes. Cardiovasc Res. 2000;46:487–495. doi: 10.1016/s0008-6363(00)00050-x. [DOI] [PubMed] [Google Scholar]
- 18.Rain S, Bos DDSG, Handoko ML, Westerhof N, Stienen G, Ottenheijm C, Goebel M, Dorfmüller P, Guignabert C, Humbert M, Bogaard H-J, Remedios CD, Saripalli C, Hidalgo CG, Granzier HL, Vonk Noordegraaf A, van der Velden J, de Man FS. Protein changes contributing to right ventricular cardiomyocyte diastolic dysfunction in pulmonary arterial hypertension. J Am Heart Assoc. 2014;3:e000716. doi: 10.1161/JAHA.113.000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Granzier H, Wu Y, Siegfried L, LeWinter M. Titin: physiological function and role in cardiomyopathy and failure. Heart Failure Reviews. 2005;10:211–223. doi: 10.1007/s10741-005-5251-7. [DOI] [PubMed] [Google Scholar]
- 20.Hwang PM, Sykes BD. Targeting the sarcomere to correct muscle function. Nat Rev Drug Discov. 2015;14:313–328. doi: 10.1038/nrd4554. [DOI] [PubMed] [Google Scholar]
- 21.Bonnevier J, Fässler R, Somlyo AP, Somlyo AV, Arner A. Modulation of Ca2+ sensitivity by cyclic nucleotides in smooth muscle from protein kinase G-deficient mice. J Biol Chem. 2004;279:5146–5151. doi: 10.1074/jbc.M306532200. [DOI] [PubMed] [Google Scholar]
- 22.Sumandea MP, Pyle WG, Kobayashi T, de Tombe PP, Solaro RJ. Identification of a functionally critical protein kinase C phosphorylation residue of cardiac troponin T. J Biol Chem. 2003;278:35135–35144. doi: 10.1074/jbc.M306325200. [DOI] [PubMed] [Google Scholar]
- 23.Cuello F, Bardswell SC, Haworth RS, Yin X, Lutz S, Wieland T, Mayr M, Kentish JC, Avkiran M. Protein kinase D selectively targets cardiac troponin I and regulates myofilament Ca2+ sensitivity in ventricular myocytes. Circ Res. 2007;100:864–873. doi: 10.1161/01.RES.0000260809.15393.fa. [DOI] [PubMed] [Google Scholar]
- 24.Oliveira SM, Zhang Y-H, Solis RS, Isackson H, Bellahcene M, Yavari A, Pinter K, Davies JK, Ge Y, Ashrafian H, Walker JW, Carling D, Watkins H, Casadei B, Redwood C. AMP-activated protein kinase phosphorylates cardiac troponin I and alters contractility of murine ventricular myocytes. Circ Res. 2012;110:1192–1201. doi: 10.1161/CIRCRESAHA.111.259952. [DOI] [PubMed] [Google Scholar]
- 25.Steinberg SF. Oxidative stress and sarcomeric proteins. Circ Res. 2013;112:393–405. doi: 10.1161/CIRCRESAHA.111.300496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gupta MP, Samant SA, Smith SH, Shroff SG. HDAC4 and PCAF bind to cardiac sarcomeres and play a role in regulating myofilament contractile activity. J Biol Chem. 2008;283:10135–10146. doi: 10.1074/jbc.M710277200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.LeWinter MM, Meyer M. Mechanisms of diastolic dysfunction in heart failure with a preserved ejection fraction: If it’s not one thing it’s another. Circ Heart Fail. 2013;6:1112–1115. doi: 10.1161/CIRCHEARTFAILURE.113.000825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Papp Z, van der Velden J, Stienen GJ. Calpain-I induced alterations in the cytoskeletal structure and impaired mechanical properties of single myocytes of rat heart. Cardiovasc Res. 2000;45:981–993. doi: 10.1016/s0008-6363(99)00374-0. [DOI] [PubMed] [Google Scholar]
- 29.Gao WD, Atar D, Liu Y, Perez NG, Murphy AM, Marbán E. Role of troponin I proteolysis in the pathogenesis of stunned myocardium. Circ Res. 1997;80:393–399. doi: 10.1161/01.res.0000435855.49359.47. [DOI] [PubMed] [Google Scholar]
- 30.van der Velden J, Merkus D, Klarenbeek BR, James AT, Boontje NM, Dekkers DHW, Stienen GJM, Lamers JMJ, Duncker DJ. Alterations in myofilament function contribute to left ventricular dysfunction in pigs early after myocardial infarction. Circ Res. 2004;95:e85–e95. doi: 10.1161/01.RES.0000149531.02904.09. [DOI] [PubMed] [Google Scholar]
- 31.Murphy AM, Kögler H, Georgakopoulos D, McDonough JL, Kass DA, Van Eyk JE, Marbán E. Transgenic mouse model of stunned myocardium. Science. 2000;287:488–491. doi: 10.1126/science.287.5452.488. [DOI] [PubMed] [Google Scholar]
- 32.Witayavanitkul N, Ait Mou Y, Kuster DWD, Khairallah RJ, Sarkey J, Govindan S, Chen X, Ge Y, Rajan S, Wieczorek DF, Irving T, Westfall MV, de Tombe PP, Sadayappan S. Myocardial infarction-induced N-terminal fragment of cardiac myosin-binding protein C (cMyBP-C) impairs myofilament function in human myocardium. J Biol Chem. 2014;289:8818–8827. doi: 10.1074/jbc.M113.541128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hertelendi Z, Tóth A, Borbély A, Galajda Z, van der Velden J, Stienen GJM, Edes I, Papp Z. Oxidation of myofilament protein sulfhydryl groups reduces the contractile force and its Ca2+ sensitivity in human cardiomyocytes. Antioxid Redox Signal. 2008;10:1175–1184. doi: 10.1089/ars.2007.2014. [DOI] [PubMed] [Google Scholar]
- 34.Kirk JA, Chakir K, Lee KH, Karst E, Holewinski RJ, Pironti G, Tunin RS, Pozios I, Abraham TP, de Tombe P, Rockman HA, Van Eyk JE, Craig R, Farazi TG, Kass DA. Pacemaker-induced transient asynchrony suppresses heart failure progression. Sci Transl Med. 2015;7:319ra207. doi: 10.1126/scitranslmed.aad2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Senzaki H, Isoda T, Paolocci N, Ekelund U, Hare JM, Kass DA. Improved mechanoenergetics and cardiac rest and reserve function of in vivo failing heart by calcium sensitizer EMD-57033. Circulation. 2000;101:1040–1048. doi: 10.1161/01.CIR.101.9.1040. [DOI] [PubMed] [Google Scholar]
- 36.Bulkley BH, Ridolfi RL, Salyer WR, Hutchins GM. Myocardial lesions of progressive systemic sclerosis. A cause of cardiac dysfunction. Circulation. 1976;53:483–490. doi: 10.1161/01.CIR.53.3.483. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.