

Abstract
Congenital disorders of manganese metabolism are rare occurrences in children, and medical management of these disorders is complex and challenging. Homozygous exonic mutations in the manganese transporter SLC39A14 have recently been associated with a pediatric-onset neurodegenerative disorder characterized by brain manganese accumulation and clinical signs of manganese neurotoxicity, including parkinsonism-dystonia. We performed whole exome sequencing on DNA samples from two unrelated female children from the United Arab Emirates with progressive movement disorder and brain mineralization, identified a novel homozygous intronic mutation in SLC39A14 in both children, and demonstrated that the mutation leads to aberrant splicing. Both children had consistently elevated serum manganese levels and were diagnosed with SLC39A14-associated manganism. Over a four-year period, we utilized a multidisciplinary management approach for Patient 1 combining decreased manganese dietary intake and chelation with symptomatic management of dystonia. Our treatment strategy appeared to slow disease progression, but did not lead to a cure or reversal of already established deficits. Clinicians should consider testing for noncoding mutations in the diagnosis of congenital disorders of manganese metabolism and utilizing multidisciplinary approaches in the management of these disorders.
Keywords: Congenital manganism, SLC39A14, Manganese toxicity
1. Introduction
Congenital disorders of manganese metabolism in children are rare occurrences. Manganese is an essential trace metal that is crucial for numerous metabolic pathways and normal cell function. However, in chronic overdose states, the metal can cause clinical toxicity by a variety of upstream subcellular-level mechanisms. Manganese toxicity is associated with a parkinsonism-dystonia syndrome termed manganism, the neuroanatomic substrate for which is manganese deposition in the globus pallidus and substantia nigra of the basal ganglia [1, 2]. The neuroimaging correlate for manganese deposition is progressive T1 shortening, in the absence of significant abnormalities on susceptibility weight sequencing [1, 3].
There are currently 3 known genetic transportopathies that impair manganese homeostasis (Table 1). SLC39A8, an intestinal manganese transporter, is associated with an autosomal recessive disorder of manganese uptake that presents with short limb dwarfism, neurodevelopmental impairment, epilepsy, deafness, and altered glycosylation related to intracellular manganese deficiency [4–6]. This disorder may be amenable to supplementation with manganese and galactose. SLC30A10 is a transporter that serves to export hepatocellular manganese into bile. Biallelic mutations in this gene are associated with a recessive disorder that presents from childhood to early adulthood with progressive manganism, polycythemia, liver disease, and skin hyperpigmentation [7–12]. SLC39A14-related manganism is the most recent of these disorders to be reported in 2016 [13]. This disorder presents with childhood onset dystonia and parkinsonism, in the absence of systemic involvement, such as the polycythemia or liver disease seen in SLC30A10-related manganism. Biallelic exonic mutations in eleven patients from seven families have been reported to date [8, 13–15]. Here, we report 2 additional unrelated patients from the United Arab Emirates (UAE) presenting with this disorder in association with a novel homozygous intronic variant that causes aberrant splicing. We describe the natural history of the disorder, and additionally detail our treatment strategy over a four-year period.
Table 1.
Genetic transportopathies that impair manganese homeostasis
| SLC39A14 gene disorder | SLC30A10 gene disorder | SLC39A8 gene disorder | |
|---|---|---|---|
| Inheritance | Autosomal recessive | Autosomal recessive | Autosomal recessive |
| Serum manganese levels | Elevated | Elevated | Low |
| Cognition | Relatively spared | Relatively spared | Developmental delay, intellectual disability |
| Extrapyramidal symptoms | Parkinsonism, dystonia | Parkinsonism, dystonia | Dystonia |
| Seizures | None | None | Yes |
| Systemic symptoms | None | Liver disease, polycythemia | Skeletal dysplasia, deafness, liver disease |
| Treatment | Chelation | Chelation, iron supplementation | Oral galactose |
2. Materials and Methods
2.1 Patients and Sequencing
Patient 1
The proband and parents were enrolled in a research study at the Manton Center for Orphan Disease Research at Boston Children’s Hospital, approved by the Boston Children’s Hospital Institutional Review Board. Whole exome sequencing (WES) was performed using methods that have been previously described [16]. FASTQ files were filtered, aligned, and variants were filtered and annotated by Codified Genomics (proprietary algorithm, Houston, TX). Candidate variants were confirmed by dideoxy (Sanger) sequencing using standard methods. Likely pathogenic variants were selected to include nonsynonymous, splice site, and indel variants with an allele frequency <1% in the National Heart, Lung, and Blood Institute exome variant server database (http://evs.gs.washington.edu/EVS/) or 1000 genomes project (http://www.1000genomes.org) and were evaluated in the Exome Aggregation Consortium database (http://www.exac.broadinstitute.org). The pathogenicity of the variants was evaluated in silico using Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/), Sorting Intolerant From Tolerant (http://sift.bii.a-star.edu.sg/), and MutationTaster (http://www.mutationtaster.org/).
Patient 2
Trio WES of the proband and parents, and analysis to identify pathogenic variants was performed by GeneDx (Gaithersburg, MD). The Neurodegenerative Brain Iron Accumulation (NBIA) sequencing panel was performed by the Knight Diagnostic Laboratory at Oregon Health Sciences University (OHSU).
2.2 Real-time Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR) and Sanger Sequencing
RNA was extracted from Patient 1 and control fibroblast cell lines and cDNA was synthesized using the Superscript IV kit (ThermoFisher, Waltham, MA) according to the manufacturer’s protocol. Complementary DNA (cDNA) was amplified using polymerase chain reaction (PCR) with three sets of primers and sent for Sanger sequencing. Set 1 was proximal to the mutation (forward primer in exon 3: GAGAATGAGCAGACGGAGGA, reverse primer in exon 4: CAATGGACAGGGCGATGAAG). Set 2 surrounded the mutation (forward primer in exon 5: TCCAAGTCTGCAGTGGTGTT, reverse primer in exon 6: GGAAGCGACTCAGAGGCATA). Set 3 also surrounded the mutation (forward primer in exon 5: ATTATGTCTCCAAGTCTGCAGT, reverse primer in exon 7: AGAGTAGCGGACACCTTTCA). qRTPCR was performed using the Fast SYBR Green Master Mix (ThermoFisher, Waltham, MA) with the three sets of primers according to the manufacturer’s protocol. Delta cycle threshold (dCT) values were calculated by normalizing the cycle threshold (CT) value of each sample to the loading control (GAPDH). Double delta cycle threshold (ddCT) values were calculated by normalizing the dCT value of each patient sample to the corresponding control sample. Relative expression levels were then calculated for each primer set and compared using a one-way Analysis of Variance and post-hoc Tukey honest significant difference Test.
2.3 Trace element analysis
Fibroblasts from controls and Patient 1 were grown to confluence in 10 cm2 tissue culture dishes in Dulbecco’s Modified Eagle Medium (ThermoFisher, Waltham, MA) with penicillin/streptomycin and 10% Fetal Bovine Serum (Sigma-Aldrich, St. Louis, MO) and analyzed for intracellular metals by inductively coupled plasma mass spectrometry (Trace Metals Laboratory, Harvard TH Chan School of Public Health, Boston, MA) as described previously [17]. The internal standard was 50 ppb indium. Briefly, cell samples were digested with 2 mL/g total wet weight nitric acid for 24 hours at room temperature. Ultrapure water was used for final sample dilution.
3. Results
3.1 Patient 1
Patient 1 is a 9-year old female from the UAE with progressive generalized dystonia and brain mineralization. She was the product of an uncomplicated term pregnancy. Her initial motor development was delayed: she sat at 8 months, and never stood. She said her first words around 1 year of age. At 2 years of age, she developed progressive generalized dystonia. She also displayed failure to thrive and required a G-tube for supplemental nutrition. Her parents are first cousins, and she has 3 healthy brothers, ages 3, 13, and 14 years (Figure 1A). There are no additional affected family members. There were no significant environmental exposures to heavy metals.
Figure 1.
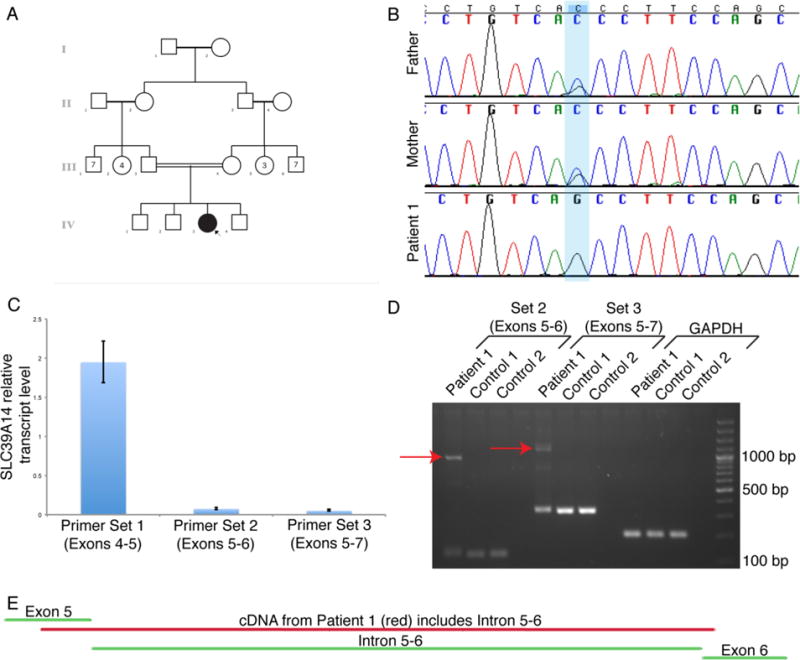
Identification of a homozygous intronic variant in SLC39A14 that leads to aberrant splicing (2-column figure)
A) Pedigree of Patient 1’s family. The square represents a male individual, the circle represents a female individual, the filled in circle designates Patient 1, and the double horizontal line between Patient 1’s parents indicates consanguinity.
B) Sanger sequencing chromatograms of genomic DNA demonstrate the homozygous intronic variant in SLC39A14 in Patient 1 and that both of her parents are heterozygous carriers of the variant.
C) qRT-PCR using RNA extracted from patient fibroblasts demonstrates significantly decreased SLC39A14 transcript levels surrounding the mutation (Sets 2 and 3) compared to proximal to the mutation (Set 1) (one-way ANOVA p<0.01; Tukey HSD p<0.01 for Set 1 versus Set 2, p<0.01 for Set 1 versus Set 3, p>0.05 for Set 2 versus Set 3).
D) Gel electrophoresis demonstrates that cDNA from Patient 1 fibroblasts contains an additional, larger band representing the included intronic sequence compared to cDNA from control fibroblasts. The red arrows indicate the abnormal splicing products that contain the entire intron between exons 5 and 6.
E) Sanger sequencing of cDNA from Patient 1 fibroblasts confirms inclusion of the intronic sequence between exons 5 and 6 (image modified from Sequencher).
We first evaluated her at the age of 5 years. At that time, she could speak about 40-50 words, but no phrases or sentences. Her speech was dysarthric. She could W-sit and crawl but could not walk. She had a generalized dystonia and a dystonic smile. Ophthalmology evaluation was normal.
Her initial brain magnetic resonance imaging (MRI) was significant for T1 hyperintensity of bilateral globi pallidi and substantia nigra (Figure 2A–F). There was no abnormal T2 hyperintensity. Susceptibility weighted imaging disclosed no evidence of abnormal susceptibility artifact. Computed tomography scan of the brain did not demonstrate any calcifications.
Figure 2.
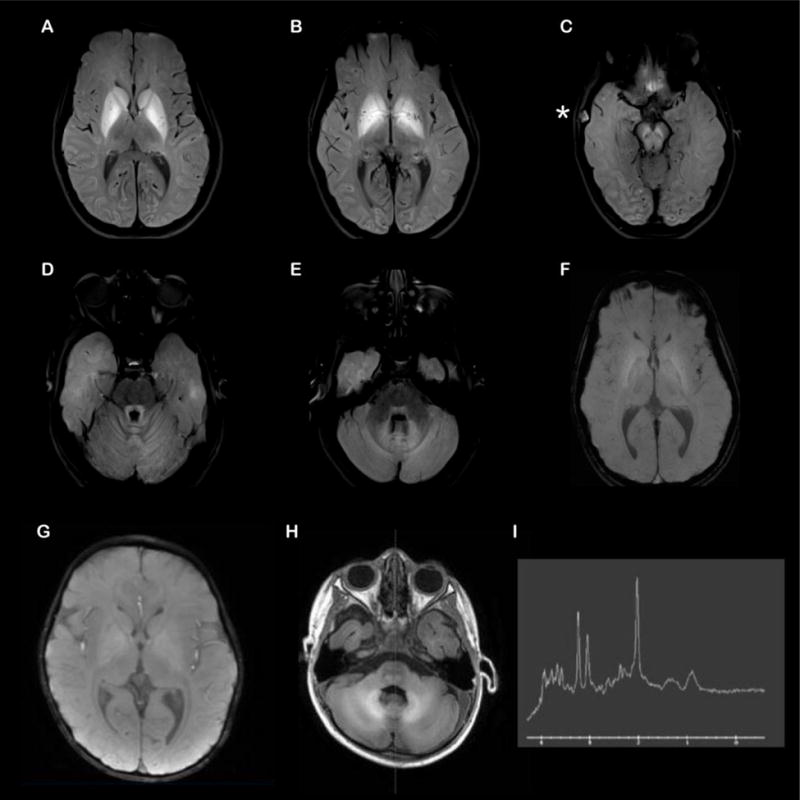
Brain magnetic resonance imaging (MRI) of Patients 1 and 2 (2-column figure) Axial T1 fat suppressed images of the brain from Patient 1 (A–F) demonstrate T1 shortening in the basal ganglia (A,B), subthalamic nuclei (B), midbrain including the substantia nigra (C), pontine tegmentum including the central tegmental tracts (D), superior/middle cerebellar peduncles (D,E), cerebellar folia (D,E), and the dentate nuclei (E). There are also questionable areas of T1 shortening in the perisylvian cortex (A) and occipital cortex (B,C). No pathologic susceptibility artifact was encountered in the areas of T1 shortening (representative slice in F). There was an incidental right temporal bone dermoid cyst (* in C). MRI of Patient 2 (G–H) shows abnormal T1 shortening within the globus pallidus, brainstem, cerebellum, and, to a lesser extent, within the surrounding cerebral white matter (G) and stable diffuse volume loss (H). The signal changes can be seen in the setting of hypermagnesemia or other metabolic disorders. MR spectroscopy (I) demonstrates normal metabolic peaks within the L basal ganglia. Technically limited left parietal white matter voxel demonstrates an abnormal decreased creatine peak but preserved choline and NAA peaks, which is of uncertain clinical significance.
Initial laboratory parameters demonstrated an elevated serum manganese (Mn) level of 64.2 μg/L (normal range 4-16.5 ug/L). Liver function tests, serum copper, ceruloplasmin, calcium, parathyroid hormone, ferritin, and urine oligosaccharides were normal. The complete blood count (CBC) demonstrated neutropenia. Serum iron was elevated at 207 mg/dL. Hair analysis corroborated elevated manganese storage, with a hair Mn concentration of 516 ppb. By comparison, the mother’s hair Mn was 147 ppb and her brother’s hair Mn concentration was 214 ppb. Serum Mn levels in her younger brother were initially elevated at 44.1 μg/L, but subsequently normalized. Her mother’s serum Mn level was mildly elevated at 28.7 μg/L. Her father’s serum Mn level was normal. Manganese excretion in urine was undetectable. Blood and urine levels of additional divalent heavy metals (arsenic, mercury, lead, copper, and zinc) were normal. Liver ultrasound was normal, with no evidence of portosystemic shunting or arteriovenous malformation.
Analysis of cultured skin fibroblasts for heavy metal content in Patient 1 compared to 2 healthy controls demonstrated similar levels of manganese and iron, but significantly increased levels of zinc (p < 0.05, Student’s t-test) (Table 2). Sequencing and deletion/duplication analysis of the SLC30A10 gene was negative [7, 8, 10, 12]. Whole exome sequencing demonstrated a homozygous intronic variant in the SLC39A14 gene, c.751-9C>G (NM_001128431, [Hg19] chr8: 22273273 C>G) (Figure 1B), which was not found in the gnomAD database. Both parents and two of the three unaffected siblings were heterozygous carriers, and the third unaffected sibling was homozygous for the wild type allele. Gel electrophoresis suggested both normal and abnormal splicing in Patient 1 fibroblast cells, and sequencing of cDNA demonstrated inclusion of the entire intronic sequence between exons 5 and 6. qRT-PCR analysis on fibroblast cells confirmed aberrant splicing, demonstrating significantly decreased SLC39A14 transcript levels around the mutation compared to proximal to the mutation (Figure 1C–E).
Table 2.
Heavy metal content in cultured skin fibroblasts.
| Control Fibroblasts #1 | Control Fibroblasts #2 | Patient Fibroblasts | |
|---|---|---|---|
| Manganese (ng/mg) | 510.3 ± 0.40 | 509.8 ± 0.65 | 519.5 ± 1.20 |
| Iron (μg/mg) | 2.48 ± 0.27 | 1.85 ± 0.03 | 2.42 ± 0.28 |
| Zinc (ug/mg) | 1.84 ± 0.05 | 1.58 ± 0.04 | 5.09 ± 0.18* |
Fibroblasts were collected from 10 cm2 dish and intracellular metal levels were measured by ICP-MS. Data are means ± SD from 10 measurements.
P < 0.05 between control and patient fibroblasts; Student’s t-test.
Patient 1 has been followed intermittently for 4 years at our institution, although she and her family returned to the UAE for long periods between hospitalizations. During this time, her blood manganese levels have ranged between 49.3-79.6 μg/L (Figure 3). Beginning at the age of 5 years, we treated her with repeated 5-day cycles of chelation with parenteral edetate calcium disodium (CaNa2EDTA) (40 mg/kg/day) over a 4-year period. The CaNa2EDTA was well tolerated, except for a couple of occasions when it had to be held because of worsening neutropenia. The CaNa2EDTA effected a urine Mn diuresis from negligible concentrations pretreatment to >25 μg/L; however, serum Mn was not significantly altered. We attempted to coordinate monthly courses of chelation, but because of practical limitations with an international patient, she would often go several months or more between courses. Each course of chelation was associated with a small window of subjective improvement in dystonia and motor functioning, typically lasting weeks.
Figure 3.
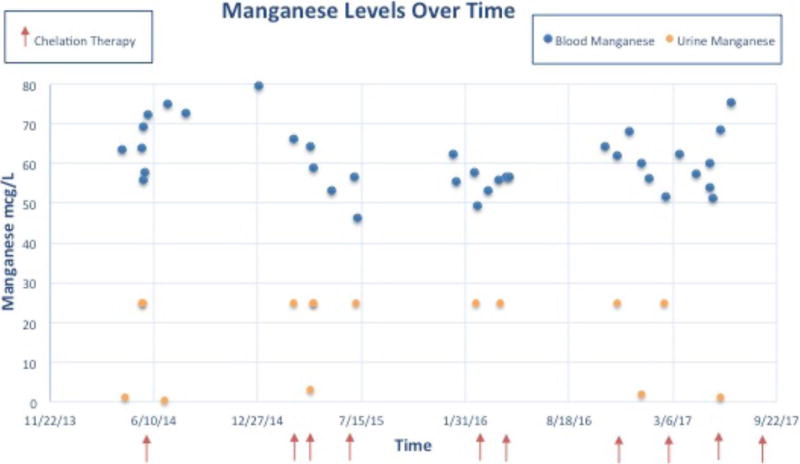
Treatment outcome over a four-year period for Patient 1 (2-column figure) Blue dots indicate blood manganese levels, orange dots indicate urine manganese levels, and red arrows indicate when chelation therapy occurred.
Two oral chelants, dimercaptosuccinic acid (DMSA, 200 mgs PO BID for 28 days) and d-penicillamine (125 mg PO BID for 30 days), were trialed and proved ineffectual in increasing urinary Mn excretion or lowering blood Mn levels. The two oral chelants were given in separate trials, with a substantial wash-out period in-between their use. We attempted a treatment trial with para-amino salicylic acid (PAS), a drug historically used for tuberculosis with some evidence of benefit in occupational studies of Mn toxicity [18, 19] and animal studies [20, 21]. The patient was not able to swallow the medication, and it could not be administered through her G-tube or effectively dissolved because of its enteric coating.
In addition, since the age of 7 years, the patient has been maintained on a reduced manganese diet using modular formulas. At the age of 8 years, we introduced days with a “Mn free diet”, or days of the week when she receives negligible Mn from oral food. To achieve this, she is entirely fed via manganese-depleted formula for these days, and is supplemented with a manganese free multivitamin. Her macronutrient and micronutrient profile on this diet is summarized in Supplementary Tables 1–3. We began with 2 days a week of “Mn free diet”, and then ultimately increased to 3 days a week. We considered supplementation with iron to decrease gastrointestinal manganese absorption, but opted not to because of baseline high-normal serum iron levels.
There were 2 longer periods of time when the patient went off diet and parenteral chelation therapy while temporarily returning home to the UAE due to logistical hurdles in securing the customized Mn depleted formula. There was a 6-month period between ages 6-7 years, and another 6-month period between ages 7-8 years. Both of these treatment hiatuses were associated with a marked clinical deterioration from which she did not entirely recover following subsequent chelation treatments.
She has also been managed symptomatically for her dystonia with oral trihexyphenidyl and botulinum toxin injections. In addition, she underwent tendon lengthening surgery. She was previously trialed on baclofen, clonazepam, and L-dopa/carbidopa with little clinical benefit. She is being considered for deep brain stimulation.
Despite these interventions, her serum manganese levels have remained essentially unchanged (Figure 3) and she has demonstrated a gradual disease progression. She currently has more difficulty W-sitting and crawling. Her dysarthria has progressed. She reaches with a significant action tremor. Her intelligence has scored within the average range on formal neuropsychological evaluations, within the limits imposed by her motor impairment. She receives some of her nutrition though a G-tube because of feeding difficulties.
Serial brain MRIs, the most recent of which was at the age of 7 years, have demonstrated stable mineralization of brain parenchyma evidenced on her most recent imaging as T1 hyperintensity of basal ganglia, hypothalamus, anterior commissure, amygdala, cerebral peduncles (including the substantia nigra), dorsal pons, superior cerebellar peduncles, dentate nuclei, cerebellar folia, and pyramids. Abdominal MRI did not demonstrate hepatic manganese accumulation, and liver function tests in blood have remained normal.
3.2 Patient 2
Patient 2 presented at 2 years old for evaluation of a progressive movement disorder and loss of ambulation skills. She was born full term via non-spontaneous vaginal delivery, weighing 3.1 kg. There were no post-birth complications. Her mother received prenatal care. There were no maternal or fetal infections noted. The pregnancy was complicated by gestational diabetes, which was well controlled throughout the pregnancy.
Her early development was normal. At the age of 18 months, she first started having trouble walking. The parents first noticed that she started to limp and then started falling frequently. This progressed to her walking on the tips of her toes and then over a few months, she became unable to walk at all. A few weeks later, she was no longer able to sit independently.
Along with this regression in motor milestones, her parents also noticed increased stiffness in all of her extremities and odd positioning of her upper extremities and fingers. Symptoms have continued to progress in a caudal to rostral direction—she subsequently began to develop problems with chewing food and sticking out her tongue. She also lost the ability to say a few words, such as “dada” and “mama”, which she was able to do before the regression started. She is still able to swallow but the difficulty in chewing has resulted in poor weight gain. There is no history of seizures.
There is an older sister (10 years old) and younger brother (6 months old) who are unaffected. The parents are healthy and deny consanguinity. Two maternal uncles died at 18 and 19 years of age of an unspecified liver condition. There are no other early deaths in the family. There is a paternal niece with dystonia diagnosed by another provider at the same hospital, but no genetic testing was pursued.
Patient 2 was seen by specialists in her home country (UAE) and baseline investigation included a gene panel for pantothenate kinase-associated neurodegeneration (PKAN), mitochondrial DNA sequencing, full gene sequencing of SLC30A10, and whole exome sequencing, which were all negative. Her MRI showed abnormal T1 shortening within the globus pallidus, brainstem, cerebellum, and, to a lesser extent, within the surrounding cerebral white matter (Figure 2G–I).
Additional testing included normal electroencephalography and normal biochemical labs, including plasma amino acids, urine organic acids, serum lactate, plasma carnitine, and plasma acylcarnitines. Initial testing demonstrated an elevated serum manganese level of 78 μg/L. Liver function tests, serum copper, ceruloplasmin, calcium and ferritin were normal. Zinc level was elevated at 123 mcg/dL (normal range: 29-115 mcg/dL). CBC and serum iron were normal. The NBIA sequencing panel completed by the Knight Diagnostic Laboratory at OHSU identified the same homozygous variant in intron 5 of SLC39A14, c.751-9C>G.
Feeding and speech evaluations performed in the United States demonstrated decreased oral motor skills, using suckling, munching, and suction to eat. She did not exhibit tongue lateralization and mastication to manipulate food. A soft pureed diet was recommended as well as a swallow study. Patient 2 returned to the UAE and received chelation therapy for a year but parents decided not to continue with it.
4. Discussion
4.1 SLC39A14-associated manganism
Biallelic exonic mutations in the manganese transporter SLC39A14 have recently been implicated in a pediatric-onset neurodegenerative disorder associated with brain manganese accumulation and clinical signs of manganese neurotoxicity, including parkinsonism-dystonia. Previously, 11 patients from 7 families had been reported with exonic missense, nonsense, and frameshift mutations[13–15]. In this article, we report 2 additional unrelated patients with a novel homozygous intronic variant that leads to aberrant splicing. Inclusion of the intron between exons 5 and 6, confirmed by cDNA sequencing, results in a stop codon at the beginning of the intron and suggests there is thus less functional protein. All 13 patients presented in infancy or early childhood with rapidly progressive dystonia, loss of developmental milestones, and/or bulbar dysfunction, and all had elevated levels of Mn in blood or serum.
The function of the SLC39A14 transporter has not been fully elucidated; however, it is presumed to function in hepatic Mn uptake for biliary excretion through SLC30A10 [8]. Aberrant SLC39A14 function likely results in insufficient clearance of manganese from blood, and subsequent accumulation in brain. It is not known what additional transport roles it may have in other organs or cellular compartments. The SLC39A14 protein localizes to the plasma membrane, specifically to the basolateral domain in polarized hepaocytes where it likely functions in hepatic Mn uptake from blood, and also localizes intracellularly where it may play a role in intracellular trafficking [8, 22, 23]. In addition to manganese toxicity in brain, it is not yet clear if intracellular manganese deficiency in other cellular compartments may contribute to the pathogenesis of this disorder. Arguing against this, our patient did not have any abnormalities on transferrin mass spectrometry to suggest dysfunction of the manganese dependent enzyme β-1,4-galactosyltransferase that is seen in SLC39A8 gene related disorder.
4.2 Effectiveness of Mn Chelation with CaNaEDTA
There have been case reports that SLC30A10 can be effectively treated with IV chelation with calcium disodium edetate and iron supplementation, the latter to reduce intestinal manganese absorption through competitive inhibition across shared transporters. The previous report on this disorder suggested biochemical, clinical, and neuroimaging improvement with monthly chelation with intravenous calcium disodium edetate [10, 18, 24]. In terms of SLC39A14-associated maganism, the previous reports suggest variable clinical response with chelation with CaNaEDTA. In three patients, chelation led to a reduction in Mn levels; one patient started on chelation at 17 years old continued to deteriorate, a second patient had reported worsening of cervical dystonia and discontinued treatment, and a third patient had some clinical improvement in terms of dystonia and motor abilities [13–15]. A fourth patient started on IV chelation at five years old had major clinical improvement and was able to walk again [13]. Patient 1 in this article was also treated with IV chelation over a four year period, and although urine diuresis of Mn was observed, there was no change in serum Mn levels and her disease continued to progress. Thus, further investigation is needed to understand the variable clinical response, which may be influenced by the age at which treatment is started, disease severity, the specific mutation, or other factors. One limitation of parenteral CaNa2 EDTA is its inability to cross the blood brain barrier, and so its ability to remove brain manganese may be quite limited. Another practical limitation was that parenteral chelation in our patient required hospitalization, which made consistent monthly courses difficult to arrange. It is possible that this treatment may have been more effective if the frequency of administration was increased.
4.3 Other Chelants for Mn
PAS, an anti-tuberculous drug that has some chelating properties for manganese, has potential for treatment of this disorder since it can cross the blood brain barrier [18–21]. Further, it is more practical to administer since it is an oral medication and can be taken continuously. Unfortunately, our patient was not able to tolerate PAS because she could not swallow capsules and they could not be administered through her G-tube or dissolved given their enteric coating.
4.4 Clinical Outcomes
For Patient 1, despite significant dietary restriction of manganese and multiple courses of parenteral chelation, we were not able to lower serum manganese levels to an appreciable degree or effect any significant improvement in disease course. Further, brain imaging did not demonstrate any improvement in established mineralization. However, it is likely that this treatment has slowed the progression of her disease as evidenced by her dramatic clinical deterioration when stopping her therapies for 6 month intervals. We have lost contact with Patient 2 as she returned to the UAE and we have not been able to sustain communication with her family.
5. Conclusions
Chronic hypermanganesemia due to SLC39A14 transporter defect is a rare and disabling pediatric neuro-metabolic disorder. We report here two additional cases of SLC39A14 gene-related manganism in association with a common homozygous splice region variant. This is the first case series to describe a multidisciplinary approach to pediatric patient management, combining decreased manganese dietary intake and chelation with symptomatic dystonia treatment.
Our study had limitations, notably that logistical hurdles prevented us from optimally managing Patient 1 as she travelled back and forth between the United States and the UAE, and prevented us from starting any treatment for Patient 2 as she returned to the UAE and we lost contact with her family. In particular, treatment for Patient 1 may have been more effective if these logistical hurdles were not present. Given the rarity of this disorder and thus the small sample size, future studies of patients with this condition and treatment outcomes over long term periods will be critical to understand the most effective management approaches.
It appears based on our results that while parenteral chelation therapy with CaNa2EDTA and dietary manganese restriction may slow disease progression, it is not curative and does not appear to significantly reverse established deficits. This may in part relate to the inability of CaNa2EDTA to cross the blood brain barrier. It is possible that treatment would have been more effective if instituted at a younger age, and administered with a greater frequency and perhaps for a longer duration.
Acknowledgments
This work was supported by the American Academy of Pediatrics (AAP) and funded (in part) by the Agency for Toxic Substances and Disease Registry (ATSDR) (cooperative agreement FAIN: 1U61TS000237-02). The U.S. Environmental Protection Agency (EPA) supports the PEHSU by providing partial funding to the ATSDR (Inter-Agency Agreement number DW-75-95877701). Neither EPA nor ATSDR endorse the purchase of any commercial products or services mentioned in PEHSU publications. AMD was supported by the NIGMS (grant number T32GM007753). PBA was supported by the NIH/NIAMS (grant number 1R01AR068429-01) and the NICHD/NHGRI/NIH (grant number U19HD077671). M.W.-R. was supported by NIH/NIEHS (grant number 1R01ES014638) and metal analysis was supported by NIH/NIEHS (grant number P01 ES000002). We thank Jose Carlo Sosa for technical help with ICP-MS experiments. The funding sources did not play any role in the design of this study; the collection, analysis, and interpretation of data; the writing of the manuscript; or the decision to submit the manuscript for publication. The first draft of the manuscript was written by LHR; no honorarium, grant, or other form of payment was given to anyone to produce the manuscript. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work. The authors would like to acknowledge the outstanding health care providers caring for these patients and families, specifically, Dr. Elizabeth Barkoudah, Dr. Christine Lee, Dr. David Fogelman, Dr. Leslie Martell as well as these patients and families for their continued partnership.
Abbreviations
- BID
twice daily, CaNa2EDTA, edetate calcium disodium
- CBC
complete blood count
- cDNA
complementary DNA
- dCT
delta cycle threshold
- ddCT
double delta cycle threshold
- DMSA
dimercaptosuccinic acid
- Mn
manganese
- MRI
magnetic resonance imaging
- NIDA
Neurodegenerative Brain Iron Accumulation
- PAS
para-amino salicylic acid
- OHSU
Oregon Health Sciences University
- PCR
polymerase chain reaction
- PKAN
pantothenate kinase-associated neurodegeneration
- PO
by mouth, qRT-PCR, real-time quantitative reverse transcription polymerase chain reaction
- UAE
United Arab Emirates
- WES
whole exome sequencing
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors have no potential, perceived, or real conflicts of interest to disclose.
References
- 1.Avelino MA, Fusao EF, Pedroso JL, Arita JH, Ribeiro RT, Pinho RS, Tuschl K, Barsottini OG, Masruha MR. Inherited manganism: the “cock-walk” gait and typical neuroimaging features. J Neurol Sci. 2014;341:150–152. doi: 10.1016/j.jns.2014.03.057. [DOI] [PubMed] [Google Scholar]
- 2.Peres TV, Schettinger MR, Chen P, Carvalho F, Avila DS, Bowman AB, Aschner M. Manganese-induced neurotoxicity: a review of its behavioral consequences and neuroprotective strategies. BMC Pharmacol Toxicol. 2016;17:57. doi: 10.1186/s40360-016-0099-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lakhan SE, Abboud H. Teaching neuroimages: manganese neurotoxicity of the basal ganglia and thalamus. Neurology. 2013;81:e111. doi: 10.1212/WNL.0b013e3182a6cb86. [DOI] [PubMed] [Google Scholar]
- 4.Boycott KM, Beaulieu CL, Kernohan KD, Gebril OH, Mhanni A, Chudley AE, Redl D, Qin W, Hampson S, Kury S, Tetreault M, Puffenberger EG, Scott JN, Bezieau S, Reis A, Uebe S, Schumacher J, Hegele RA, McLeod DR, Galvez-Peralta M, Majewski J, Ramaekers VT, C. Care4Rare Canada. Nebert DW, Innes AM, Parboosingh JS, Abou Jamra R. Autosomal-Recessive Intellectual Disability with Cerebellar Atrophy Syndrome Caused by Mutation of the Manganese and Zinc Transporter Gene SLC39A8. Am J Hum Genet. 2015;97:886–893. doi: 10.1016/j.ajhg.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park JH, Hogrebe M, Gruneberg M, DuChesne I, von der Heiden AL, Reunert J, Schlingmann KP, Boycott KM, Beaulieu CL, Mhanni AA, Innes AM, Hortnagel K, Biskup S, Gleixner EM, Kurlemann G, Fiedler B, Omran H, Rutsch F, Wada Y, Tsiakas K, Santer R, Nebert DW, Rust S, Marquardt T. SLC39A8 Deficiency: A Disorder of Manganese Transport and Glycosylation. Am J Hum Genet. 2015;97:894–903. doi: 10.1016/j.ajhg.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riley LG, Cowley MJ, Gayevskiy V, Roscioli T, Thorburn DR, Prelog K, Bahlo M, Sue CM, Balasubramaniam S, Christodoulou J. A SLC39A8 variant causes manganese deficiency, and glycosylation and mitochondrial disorders. J Inherit Metab Dis. 2017;40:261–269. doi: 10.1007/s10545-016-0010-6. [DOI] [PubMed] [Google Scholar]
- 7.Chen P, Parmalee N, Aschner M. Genetic factors and manganese-induced neurotoxicity. Front Genet. 2014;5:265. doi: 10.3389/fgene.2014.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mukhopadhyay S. Familial manganese-induced neurotoxicity due to mutations in SLC30A10 or SLC39A14. Neurotoxicology. 2017 doi: 10.1016/j.neuro.2017.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mukhtiar K, Ibrahim S, Tuschl K, Mills P. Hypermanganesemia with Dystonia, Polycythemia and Cirrhosis (HMDPC) due to mutation in the SLC30A10 gene. Brain Dev. 2016;38:862–865. doi: 10.1016/j.braindev.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 10.Quadri M, Federico A, Zhao T, Breedveld GJ, Battisti C, Delnooz C, Severijnen LA, Di Toro Mammarella L, Mignarri A, Monti L, Sanna A, Lu P, Punzo F, Cossu G, Willemsen R, Rasi F, Oostra BA, van de Warrenburg BP, Bonifati V. Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am J Hum Genet. 2012;90:467–477. doi: 10.1016/j.ajhg.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tuschl K, Clayton PT, Gospe SM, Jr, Gulab S, Ibrahim S, Singhi P, Aulakh R, Ribeiro RT, Barsottini OG, Zaki MS, Del Rosario ML, Dyack S, Price V, Rideout A, Gordon K, Wevers RA, Chong WK, Mills PB. Syndrome of Hepatic Cirrhosis, Dystonia, Polycythemia, and Hypermanganesemia Caused by Mutations in SLC30A10, a Manganese Transporter in Man. Am J Hum Genet. 2016;99:521. doi: 10.1016/j.ajhg.2016.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tuschl K, Clayton PT, Gospe SM, Jr, Mills PB. Dystonia/parkinsonism, hypermanganesemia, polycythemia, and chronic liver disease. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews(R) University of Washington; Seattle, WA: 1993. [PubMed] [Google Scholar]
- 13.Tuschl K, Meyer E, Valdivia LE, Zhao N, Dadswell C, Abdul-Sada A, Hung CY, Simpson MA, Chong WK, Jacques TS, Woltjer RL, Eaton S, Gregory A, Sanford L, Kara E, Houlden H, Cuno SM, Prokisch H, Valletta L, Tiranti V, Younis R, Maher ER, Spencer J, Straatman-Iwanowska A, Gissen P, Selim LA, Pintos-Morell G, Coroleu-Lletget W, Mohammad SS, Yoganathan S, Dale RC, Thomas M, Rihel J, Bodamer OA, Enns CA, Hayflick SJ, Clayton PT, Mills PB, Kurian MA, Wilson SW. Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism-dystonia. Nat Commun. 2016;7:11601. doi: 10.1038/ncomms11601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Juneja M, Shamim U, Joshi A, Mathur A, Uppili B, Sairam S, Ambawat S, Dixit R, Faruq M. A novel mutation in SLC39A14 causing hypermanganesemia associated with infantile onset dystonia. J Gene Med. 2018:e3012. doi: 10.1002/jgm.3012. [DOI] [PubMed] [Google Scholar]
- 15.Marti-Sanchez L, Ortigoza-Escobar JD, Darling A, Villaronga M, Baide H, Molero-Luis M, Batllori M, Vanegas MI, Muchart J, Aquino L, Artuch R, Macaya A, Kurian MA, Duenas P. Hypermanganesemia due to mutations in SLC39A14: further insights into Mn deposition in the central nervous system. Orphanet J Rare Dis. 2018;13:28. doi: 10.1186/s13023-018-0758-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joshi M, Anselm I, Shi J, Bale TA, Towne M, Schmitz-Abe K, Crowley L, Giani FC, Kazerounian S, Markianos K, Lidov HG, Folkerth R, Sankaran VG, Agrawal PB. Mutations in the substrate binding glycine-rich loop of the mitochondrial processing peptidase-alpha protein (PMPCA) cause a severe mitochondrial disease. Cold Spring Harb Mol Case Stud. 2016;2:a000786. doi: 10.1101/mcs.a000786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seo YA, Wessling-Resnick M. Ferroportin deficiency impairs manganese metabolism in flatiron mice. FASEB J. 2015;29:2726–2733. doi: 10.1096/fj.14-262592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang YM, Mo XA, Du FQ, Fu X, Zhu XY, Gao HY, Xie JL, Liao FL, Pira E, Zheng W. Effective treatment of manganese-induced occupational Parkinsonism with p-aminosalicylic acid: a case of 17-year follow-up study. J Occup Environ Med. 2006;48:644–649. doi: 10.1097/01.jom.0000204114.01893.3e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ky SQ, Deng HS, Xie PY, Hu W. A report of two cases of chronic serious manganese poisoning treated with sodium para-aminosalicylic acid. Br J Ind Med. 1992;49:66–69. doi: 10.1136/oem.49.1.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hong L, Jiang W, Pan H, Jiang Y, Zeng S, Zheng W. Brain regional pharmacokinetics of p-aminosalicylic acid and its N-acetylated metabolite: effectiveness in chelating brain manganese. Drug Metab Dispos. 2011;39:1904–1909. doi: 10.1124/dmd.111.040915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng W, Jiang YM, Zhang Y, Jiang W, Wang X, Cowan DM. Chelation therapy of manganese intoxication with para-aminosalicylic acid (PAS) in Sprague-Dawley rats. Neurotoxicology. 2009;30:240–248. doi: 10.1016/j.neuro.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu C, Hutchens S, Jursa T, Shawlot W, Polishchuk EV, Polishchuk RS, Dray BK, Gore AC, Aschner M, Smith DR, Mukhopadhyay S. Hypothyroidism induced by loss of the manganese efflux transporter SLC30A10 may be explained by reduced thyroxine production. J Biol Chem. 2017;292:16605–16615. doi: 10.1074/jbc.M117.804989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nam H, Wang CY, Zhang L, Zhang W, Hojyo S, Fukada T, Knutson MD. ZIP14 and DMT1 in the liver, pancreas, and heart are differentially regulated by iron deficiency and overload: implications for tissue iron uptake in iron-related disorders. Haematologica. 2013;98:1049–1057. doi: 10.3324/haematol.2012.072314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brazier MW, Volitakis I, Kvasnicka M, White AR, Underwood JR, Green JE, Han S, Hill AF, Masters CL, Collins SJ. Manganese chelation therapy extends survival in a mouse model of M1000 prion disease. J Neurochem. 2010;114:440–451. doi: 10.1111/j.1471-4159.2010.06771.x. [DOI] [PubMed] [Google Scholar]