

Abstract
Ovarian cancer is a disease with a poor prognosis and little progress has been made to improve treatment. It is now recognized that there are several histotypes of ovarian cancer, each with distinct epidemiologic and genomic characteristics. Cancer therapy is moving beyond classical chemotherapy to include epigenetic approaches. Epigenetics is the dynamic regulation of gene expression by DNA methylation and histone post translational modification in response to environmental cues. Improvement in technology to study DNA methylation has enabled a more agnostic approach and, with larger samples sets, has begun to unravel how epigenetics contributes to the etiology, response to chemotherapy and prognosis in of ovarian cancer. Investigations into histone modifications in ovarian cancer are more nascent. Much more is needed to be done to fully realize the potential that epigenetics holds for ovarian cancer clinical care.
Keywords: Ovarian cancer, DNA methylation, Chromatin
1. Introduction
1.1. Ovarian cancer
Epithelial ovarian cancer (EOC) continues to confer a poor prognosis; diagnosis at a late stage and the high rate of resistance to the standard chemotherapy regimens contributing greatly to the mortality. EOC consists of several distinct diseases defined based on district histotypes with distinct genomic and epigenomic characteristics. High grade serous ovarian cancer (HGSOC) is the most common and one of the most aggressive histotypes, comprising some 70% of newly diagnosed cases. Endometrioid (ENDOC, 15%), clear cell carcinoma (CCOC, 12%), mucinous (MOC, 3%), low grade serous (LGSOC, < 10%) and are less frequently diagnosed histotypes. There are two histotypes of low malignant potential borderline disease (serous and mucinous) [1]. In addition to genomic characteristics, these EOC histotypes have different epidemiologic risk factors [2–4], expression signatures [5–7], clinical responses to therapy [8] and distinct precursor sites. HGSOC appears to originate from fallopian tube or ovarian surface epithelium [9], shows few mutational events compared with other histotypes [10] and generally have gross chromosomal instability exhibiting copy number variations [11,12]. LGSOC are considered to arise from borderline tumors [13] and commonly possess mutations in the Ras family of genes (KRAS, BRAF, NRAS, ERBB2, and PTEN) and are chromosomally stable [14]. EOC and CCOC are thought to emanate from endometrium or endometriosis and MOC from endocervix or intestinal mucosa [15]. The rarer histotypes, CCOC and MOC, also have distinct clinical, pathological and genetic characteristics [3,16–20]. ENDOC bears some similarity to CCOC, but unlike CCOC, can be high grade or low grade. High grade ENDOC shares some clinical features of HGSOC, while the low grade ENDOC are similar to CCOC. The work of many has led to identification of genetic differences between the different histotypes [21–27], and epigenetic changes are beginning to be characterized.
1.2. Epigenetics
Epigenetic regulatory elements are comprised of post-translationally modified cytosines, non-coding RNAs, and histone modifications [28]. These effects do not change the underlying DNA sequence, but are heritable and regulate the means by which the genome and environment interact [29]. DNA methylation (methyl CpG) is the most commonly studied epigenetic modification, whereby a methyl group is attached in the C5 position of cytosines (5mC), usually in a cytosine followed by a guanine (CpG) context. One percent of human genome is methylated and 5mC are spread throughout the genome with some aggregating into longer stretches of CpGs identified as CpG islands [30]. DNA methylation is necessary to silence retroviral elements that make up 5–8% of the human genome [31]. DNA methylation also plays a critical role in imprinting, cell differentiation during development and resultant phenotypic variability [32–34]. With advent of new sequencing methods, methylation of other nucleotide dyads are being described in mammalian genomes [35,36], widening the scope of epigenetic knowledge. DNA methylation may be associated with decreased expression of a gene; however, this does not directly imply a causal relationship. Showing an association between gene expression and DNA methylation is one means of inferring a possible functional consequence, however the application of methods such as CRISPR-based approaches in epigenome-editing offer a means to establish a causal function [37].
DNA methylation may be increased (hypermethylated) or decreased (hypomethylation) in disease settings compared to normal tissue. Hypermethylation of tumor suppressor gene promoters may lead to gene silencing and inactivation of pathways such as DNA mismatch repair in cancers with hypermethylation of MLH1 [38]. Extensive hypermethylation, a CpG island methylator phenotype (CIMP), has been documented in multiple cancers [39]. Alternatively, hypomethylation may lead to expression of normally silenced oncogenes. There is also evidence that methylation within the gene body, counter to previous thought, may also regulate gene expression [40]; gene body hypomethylation also exists [41]. In EOC, as in cancer generally, global hypomethylation is seen across all histotypes and is associated with increasing stage, grade and mortality [33,42,43]. Hypermethylation on the other hand differs across the EOC histotypes. Aberrant expression of repeat elements, as well as oncogenes, is the result of hypomethylation, while hypermethylation silences genes that may regulate critical functions. Heterogeneity in DNA methylation underscores the complexity of genomic underpinnings in cancer development in which mutations, structural aberrations and epigenetic dysfunction all play a role.
Histones play a fundamental role in chromatin organization, and along with DNA methylation, are an active area of research in cancer [44]. Histones are the most abundant of proteins bound to DNA, regulating gene expression and influencing how DNA is packaged around the nucleosome. Histone post-translational modifications include acetylation, methylation, phosphorylation, sumoylation, deamination, ADP ribosylation, proline isomerization, ubiquination all playing key roles in regulation of chromatin function. Histone acetylation of lysines is generally associated with euchromatin, a gene transcription-ready state, as are H3K4 mono-di and tri-methylation, mono methylation in H3K27, H3K79, H4K20 and H3BK5. Heterochromatin, on the other hand, is generally not permissive and contains repressive histone marks (H3K27me2, me3; H3K9me2, me3). Bivalent marks, “poised”, contain both repressive (e.g. H3K27me3) and active (e.g. H3K4me3) marks, are commonly encountered in embryonic stem cells [45].
Histone acetylation is dynamic with histoacetyltranferases (HATs) and deacetyltransferases (HDACs) playing a role [46]. HDACs, which are aberrantly expressed in a number of cancers including ovarian cancer [47], are comprised of eighteen proteins grouped into four classes; classes I, II and IV HDACs are zinc dependent enzymes, while class III are NAD+ dependent [48]. Class III HDACs include the sirtuin enzymes, products of the SIRT1-SIRT7 gene family [49]. Class I HDAC enzymes are expressed in all cell types, while class II enzymes show tissue-specific expression. Bromodomain and extra-terminal domain (BET) proteins (BRD2, BRD3, BRD4 and testis-specific BRDT proteins) act as “readers” of the histone marks by binding to the acetylated lysine on histone tails to promote transcription [50]. This review will examine DNA methylation changes in EOC considering the different histotypes, histone modifications and application of epigenetics in the clinical setting.
2. DNA methylation analyses in EOC
DNA methylation analyses have either focused on the methylation state of specific sets of genes or have assessed genome-wide DNA methylation comparing EOC histotypes or cancer and normal ovarian tissue. These studies vary widely in the number of samples analyzed, consideration of histotypes, and analysis methods.
2.1. Targeted analyses
Historically, early efforts to analyze DNA methylation in EOC were targeted to genes commonly mutated in cancer such as genes involved in DNA repair, cell cycle and growth regulation (BRCA1, PTEN, CDKN2A, MLH1, RASSF1A, CDH1 and others) [51–64]. Fig. 1 shows the current estimates for frequency of histotype specific DNA methylation for the above set of genes. DNA methylation of candidate genes is most commonly studied and reported in HGSOC; whereas the small number of studies including rarer histotypes highlights the difficulty in acquiring large enough sample numbers. The majority of analyses used methylation-specific PCR (MSP), which evaluates the presence of a PCR fragment amplified with primers that target the preserved methylated cytosines in bisulfite modified DNA. The presence of a methylated fragment is typically reported as a frequency of methylated samples per study. Frequencies of methylation vary widely between studies, due in part to small numbers of samples when considering grade or histotypes. Thus, determining whether DNA methylation of tumor suppressor genes is common in EOC from these studies is quite difficult.
Fig. 1.
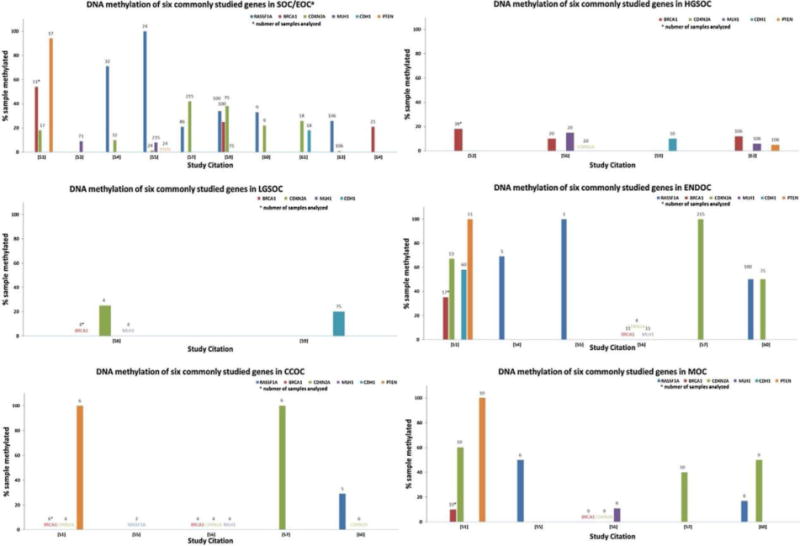
DNA methylation of selected genes in histotype specific epithelial ovarian cancer reported in targeted analysis studies.
The histogram indicates the percentage of samples showing methylation of the target genes (by colour for each gene) on the Y axis, with the number of samples analyzed above each bar, and the reference number for the studies on the X axis for each of the EOC histotypes, when reported: SOC/EOC serous ovarian cancer, grade not specified or EOC, histotype not specified; HGSOC high grade serous ovarian cancer; LGSOC low grade serous ovarian cancer; ENDOC endometrioid ovarian cancer; CCOC clear cell ovarian cancer; MOC mucinous ovarian cancer.
2.2. Genome-wide analyses
Methods genome-wide DNA methylation assessment came into use over a decade ago in EOC studies. Using an early array-based method three groups profiled 1505 CpGs (Illumina GoldenGate) [65–67] considering tumor grade and histotype in their analyses (Table 1). Several studies used normal ovarian or fallopian tissue as the control [11,12,66,68–70] while the other studies used hierarchical clustering to discern the methylation patterns within EOC cases [20,65,67], making comparison between analyses difficult. However, significant histotypespecific methylation was noted both between histotypes and when compared to a normal tissue. HGSOC, while showing some hypermethylation, is more hypomethylated than ENDOC or CCOC. In addition, HGSOC appeared distinct from LGSOC based on methylation patterns [67]. Importantly, Houshdaran et al. reported that commonly used EOC cell lines formed a separate cluster from EOC tissue, and there was no association between cell lines and any reported histotype lineage. Notably, following gene expression analysis revealed only a minority of methylated genes to have decreased expression [65].
Table 1.
Genome-wide DNA methylation analyses in EOC
| Method | Histotype and number of samples used a | Analysis | Key findings | Ref |
|---|---|---|---|---|
| GoldenGate | 5 SOC/EOC 5 ENDOC 4 CCOC 3MOC |
Differential methylation normal vs cancer | Hypermethylated CpG: SOC 20; MOC 37; 43% CpG unique ENDOC 15; CCOC 5 | [68] b |
| GoldenGate | 1 SOC/EOC 14 HGSOC 9 ENDOC 3 CCOC EOC cell lines |
Differential methylation within histotype Association with gene expression |
90 CpG in 68 genes had significant histotype specific methylation CCOC more likely hypermethylated. 10% of 492 CpGs are hypermethylation and associated with decreased gene expression. EOC cell lines distinct from tumors. |
[65] |
| GoldenGate | 46 HGSOC 8 LGSOC |
Differential methylation Within histotype | HGSOC more hypomethylated Hypermethylated at GFAP, TAL1, IPF1, AREG, HOXA9 |
[67] |
| 27 K | 489 HGSOC | Differential methylation normal vs cancer Association with gene expression |
168 genes methylated with altered gene expression. AMT, CCL21, SPARCL1 methylated majority of sample RAB25 methylated in a subset; BRCA1 methylated in 11.5% of sample. Four methylation subtypes identified |
[12] |
| 27 K | 11 ENDOCb 34 HGSOC 4 LGSOC |
Methylation cluster analysis using multiple algorithms | Three clusters: normal, EODOC and HGSOC Serous generally hypomethylated ENDOC very hypermethylated, with hypomethylation of usually methylated sites |
[69] |
| 27 K | fallopian tube 53 ENDOC 83 HGSOC |
Differential methylation normal vs cancer within histotype Association with gene expression |
12 CpG loci: ZNF154*; ZNF11;TUBGCP2; C19orf19; BTNL2*;KRTAP11-1*; TMC6; TMC8;DEFB118*; FLJ44674*;SNTB1;VHL*; LMLN;IQCG;CASP8* * seen in TCGA multiple cancers data |
[70] |
| 450 K | 344 HGSOC 80 ENDOC 16 LGSOC 20 CCOC 12 MOC |
Methylation cluster analysis using Gaussian distributed recursive portioned mixture model | CCOC methylation profile identified including KCNH2, VWA1, NRDG2, SLC25A9 | [20] |
| 450 K | 80 HGSOC 7 fallopian tube |
Differential methylation normal vs cancer | 433 genes hypermethylated correlated with decreased expression. ALDH1A3, AMT, LONRF2, NPDC1, SLC16A5 also hypermethylated in TCGA dataset |
[11] |
HGSOC: High grade serous ovarian cancer; LGSOC: Low grade serous ovarian cancer; ENDOC: Endometrioid ovarian cancer; CCOC: Clear cell ovarian cancer; MOC: Mucinous ovarian cancer.
SOC/EOC: Serous ovarian cancer, grade not specified or ovarian cancer, histology not specified.
5 ovarian endometrioid and 6 endometrial endometrioid tumors.
Higher resolution methylation arrays: Illumina’s Human Methylation 27 K, Human Methylation 450 K, and custom microarrays, have been utilized in the past seven years in studies with larger sample numbers [11,12,20,69–71] (Table 1). The Cancer Genome Atlas (TCGA), collaboration between the National Cancer Institute and National Human Genome Research Institute, has profiled the genomes of 33 types of cancer, including ovarian cancer. TCGA EOC study included mostly HGSOC, while the other large studies included additional histotypes. EOC methylation was compared with normal fallopian tube DNA methylation in four studies [11,12,69,70] while one compared methylation differences between the histotypes [20]. TCGA identified 168 genes with altered gene expression in HGSOC compared to normal fallopian tube, with 11.5% hypermethylated at BRCA1, similar to frequencies noted in HGSOC in the targeted analyses [12]. Four methylation subtypes were revealed using clustering analysis, but were not as stable as gene expression clustering subtypes identified in EOC. In another analysis of HGSOC [11], 543 hypermethylated genes with decreased gene expression were reported, and five of those genes overlapped with those identified in the TCGA dataset. Differences in statistical analysis techniques may account for the relatively low consistency of results across studies [72]. Sánchez-Vega et al. conducted a study comparing DNA methylation between both HGSOC and ENDOC and normal fallopian tissue. Of the 12 CpG loci that were differentially methylated in both HGSOC and ENDOC compared to normal tissue all but one CpG had reduced methylation in tumors compared to fallopian tube DNA [70]. Hypermethylation was noted in a CpG island downstream of the transcription start site for ZNF154; however not all ENDOC in this analysis showed hypermethylation at ZNF154 with some ENDOC cases having a low-intensity methylator phenotype, as also shown in TCGA study. Seven of the 12 loci that were found to be differentially methylated in the majority of cancer types in the TCGA, where not found by Sánchez-Vega et al. possibly to the relatively small number of samples in the original study [70]. Overall, ENDOC appears to have a significantly different methylation profile compared to HGSOC [69,70], with most having a CIMP phenotype [73].
A CCOC-specific methylation profile was identified in one study [20]. What is notable in CCOC genome-wide studies is the paucity of tumor suppressor gene hypermethylation. CCOC gene expression studies support a specific dysregulation of genes that are involved in oxidative stress response, including HNF1B and VCAN [17], supporting a role for microenvironment in tumor development. Interestingly, while HNF1B is overexpressed in CCOC, it is methylated in ∼50% of HGSOC [12,19]. A variant in HNF1B was identified as an HGSOC susceptibility locus [74] and recently Ross–Adams et al. [75] reported that the susceptibility allele was associated with HNF1B promoter methylation. This methylation, upstream of the transcription start site, also bears hallmarks of poised and active enhancers (H3K27Ac, H3K4me1 and H3K3Me3 histone marks). HNF1B plays a critical role in the epithelialmesenchymal transition (EMT), in which cells lose cellular adhesion and polarity acquiring an invasive phenotype [76]. EMT properties are also seen in cells under stress imbued by tumor microenvironment resulting in methylation dysregulation potentially contributing to a tumor promoting role. Thus, unmethylated HNF1B appears to act as an oncogene in CCOC, but when hypermethylated, acts like a tumor suppressor in the more aggressive HGSOC histotype [75]. This study provides an excellent example the complexity of genomic and epigenetics in EOC.
These studies demonstrate the growing consensus that the epigenome is altered in EOC. Both CCOC and ENDOC are more likely to have a hypermethylated phenotype compared with HSGOC which are overall hypomethylated and has fewer hypermethylation events. CCOC and ENDOC are thought to arise from endometrial epithelium and endometriosis, suggesting that inflammation may play a role.
3. Chromatin alterations in ovarian cancer
Regulation of gene expression is closely related to the chromatin state. Mutations in genes involved in chromatin remodeling (ARID1A, SPOP, KMT2D) have been reported in CCOC[77,78], implicating aberrant chromatin remodeling in this histotype. Assessing the chromatin state is classically achieved through the histone marks described earlier. These histone post-translational modifications and HDAC expression have been evaluated in EOC using immunohistochemistry (IHC) and next generation sequencing (NGS) approaches (Table 2). SIRT1, a class II HADC, was found to have higher expression in invasive serous EOC, compared to benign or borderline cases, and was higher in serous compared with mucinous tumors[79]. IHC-evaluated reduced expression of the repressive histone mark H3K27me3 was noted in 55% of EOC cases, 33% of borderline OC cases and cystadenomas, and 16% of normal ovary tissue. A decrease in H3K27me3 expression was associated with increasing stage, but not grade, patient, age, or histotype [80].
Table 2.
HDACs and histone alterations in EOC.
| Targets | Samples useda | Analyses | Key findings | Ref |
|---|---|---|---|---|
| SIRT1 (HDAC) | TMA of 113 SOC 21 MOC 34 EOC 12 normal ovaries 31 borderline 26 cystadenomas |
IHC composite score of intensity × area | Higher SIRT1 expression in malignant SOC compared with benign or borderline cases and MOC | [79] |
| H3K27me3 | 164 EOC/SOC 21 MOC 34 ENDOC |
IHC composite score of intensity × area | Decreased expression in 55.3%, 38% borderline, 39% cystadenomas and 16% normal ovaries. | [80,78] |
| H3K4 | HGSOC Cell lines: A4-P;A4-T | MeDIPb | 76 genes hypomethylated and 31 genes hypermethylated with associated gene expression change | [82] |
| H3K9 | ChIP-on-chipc | 5 genes hypomethylated and 2 hypermethylated seen TCGA, in the following comparisons: A4-P with LGSOC and A4-T with HGSOC. | ||
| H3K27 | Gene expression | Histone changes: bivalent histone marks noted | ||
| H3K4me3 | 1 HGSOC | MeDIPb | Gene set 1: n = 580, bivalent signature (M3K37me3 and H3K4me3) | [83] |
| H3K27me3 | 8 fallopian tube 499 HGSOC (TCGA) |
ChIP-on-chipc Gene expression |
Gene set 2: n = 913 H3K27me3 Significant decrease in expression of genes with bivalent or repressive Histone PTM |
|
| H3K27 | CP70 cell line | H3K27R mutant Methylation |
Global methylation decrease Upregulation of tumor suppressor genes Resensitizes CP70 to cisplatin |
[85] |
HGSOC: High grade serous ovarian cancer; LGSOC: Low grade serous ovarian cancer; ENDOC: Endometrioid ovarian cancer; CCOC: Clear cell ovarian cancer; MOC: Mucinous ovarian cancer.
SOC/EOC: Serous ovarian cancer, grade not specified or ovarian cancer, histology not specified.
Three studies used NGS approches to examine histone marks. Two EOC cell lines developed from an HGSOC patient tumor ascites, defined as pre-transformed (A4 clone) and transformed (A4T, demonstrating expression of markers associated with a progenitor state) [81], were profiled for DNA methylation, histone marks, and gene expression [82]. Comparing A4 with LGSOC and A4T with TCGA HGSOC samples, a modest overlap of shared genes was noted. This may be may be due to the small number of LGSOC cases in TCGA or the definition of A4 cell line as pre-transformed EOC. Chapman-Rothe et al. [83] profiled a single primary HGSOC case for H3K27me3 (active) and H3K4me3 (repressive) marks and examined the histone-marked gene sets in two gene expression profiles sets, eight benign ovarian lesions, and the TCGA dataset of eight fallopian tube samples and 499 HGSOC. Bivalent (presence of both active and repressive marks) and H3K27me3 marked genes were associated with significantly lower expression in the HGSOC compared to eight benign serous ovarian lesions. Of the 580 bivalent marks in the HGSOC, 215 are similarly bivalent in embryonic stem cells, but 365 appear to be tumor–specific. These bivalent genes were enriched for the PI3K and TGF-β pathways. Methylation of H3K27 is mediated by the polycomb repressor complex 2 (PRC2) [84] and expression of PRC2-complex genes (EZH2, SUZ12, EED, RBBP7) were negatively correlated with H3K27me3, supporting H3K27 methylation role in mediating gene silencing in HGSOC. Another study examined DNA methylation and gene expression in a cisplatin-resistant ovarian cancer cell line with an altered H3K27 protein, in which the H3 lysine was mutated to arginine and thus not modifiable by methylation [82,85]. Loss of DNA methylation and altered gene expression was observed in this cell line as well as a sensitization to cisplatin. Tumor suppressor genes, MLH1, ARH1 and RASSFIA were upregulated, while NKX2 was down regulated. The mechanism underlying the sensitization may be due increased tumor suppressor gene expression, accessibility to DNA, or an undefined role for H3K27 methylation in adduct repair. These studies indicate that chromatin state is altered in at least some EOC, and the presence of tumor-specific bivalent histone marks may play a role in tumor progression and chemoresistance. As these data are based on limited sample numbers, further work is need to determine whether these findings extend to other HGSOC cases and the other histotypes.
4. Clinical aspects of epigenetics in EOC
4.1. Chemoresistance
Acquisition of resistance to therapy is common in EOC and contributes to the high mortality from the disease. Alterations in DNA methylation have been hypothesized to play a role, because DNA methylation is one mechanism cells employ to respond to environmental stimuli [29]. Platinum-based chemotherapy is the primary line of drug therapy for all advanced stage EOC cases; carboplatin combined with paclitaxel is the standard of care, irrespective of histotype [86,87]. Early studies on breast and ovarian cancer cell lines reported sensitivity to platinum-based chemotherapy in cells with either mutant or hypermethylated BRCA1 [88–90]. More recently, several studies have examined DNA methylation and chemoresistance [11,91,92] or survival [93–95] using genome-wide approaches (Table 3). One study, using a custom microarray [92], identified 749 CpG loci that differed in methylation between resistant and sensitive HGSOC, 509 were within promoter regions. They used a short hairpin RNA screen (which silences target genes via RNA interference) to examine the effect on carboplatin sensitivity in ovarian cancer cell lines (a normal human surface epithelial line and two EOC lines resistant to carboplatin), and identified 19 genes associated with carboplatin resistance. De Leon et al. [91] examined methylation changes in A2780 (ovarian carcinoma) cell line xenografts, treated with carboplatin or control diluent. Six genes had altered methylation of which two hypomethylated genes, TMEM88 and DAXX, had an associated increase in gene expression in platinum resistant xenografts. TMEM88 protein expression was evaluated in an ovarian tissue microarray with ∼50% of the EOC showing decreased expression. TMEM88 is a Wnt signaling inhibitor, and was inversely correlated with c-MYC gene expression in the TCGA dataset. In a study of acquired resistance, Patch et al. [11] examined methylation changes in 13 paired resistant and sensitive HGSOC and noted 94 CpG probes with > 10% methylation difference between the sensitive and resistant HGSOC, affecting COLA1A1, SPTBN4 and MYOID genes. Longer survival, using a cutoff of > 28 months, was associated with hypomethylation in GREB1, TGIF, TOB1 and hypermethylation in TMCO5, PTPRN, GUCY2C in a study of 20 EOC patients [93]. Methylation in peripheral blood leukocytes of EOC patients was compared between cancer presentation and relapse in 146 patients [94]. Nine CpG sites were validated in an independent set of cancer presentation and relapse samples (n = 45 each). Eight CpG were significantly associated with survival and consensus clustering revealed two classes of relapse samples, class 1 with a poorer survival than class 2. Notably, methylation at presentation of the eight CpG did not predict time to recurrence or overall survival. Two reports used methylation-capture sequencing (MethylCap-seq) to assess DNA methylation and survival. Huang et al. [95] identified differential methylated regions (DMR) associated with progression-free survival (PFS), and validated the DMR in platinum resistant and sensitive cell lines. Hafner et al. [71] evaluated DNA methylation difference between pools of HGSOC DNA with poor or good survival followed by validation in an independent set of samples revealing two genes, RUNX3 and CAM21, to be associated with PFS. In cell lines, increased methylation of these two genes was associated with reduced gene expression.
Table 3.
DNA methylation: therapy and prognosis.
| Method | Samples useda | Analyses/Comparisons | Key findings | Ref | |
|---|---|---|---|---|---|
| Chemoresistance | |||||
| 280 K custom | 36 HGSOC | 15refractory/resistant; | 749 CpG (∼60% hypermethylated and ∼40% hypomethylated in resistant HGSOC | [92] | |
| EOC cell lines: HOSE 6-3, SKOV3, CAOV3 | 21 sensitive HGSOC; 9 normal ovaries shRNA screen of 296 candidate genes (within 2KB of TSS) |
19 genes: GSK3B, DOK2, APRT, OXSR1, CENBO, FZD1, ESRRA, HIRIP3, GTF2b, SGPL1, GABPA, TWIST1, MDH1, NR2E, NR3C2, SOX9, TOB1, UNG, ZIC1 | |||
| 27 K | 20 EOC (mostly HGSOC); | Survival: 28, 47 month cutoff (note, 6 month cutoff didn’t reveal any significant findings) | 82 genes with > 10% difference, including hypomethylated: GREB1, TGIF, TOB1 hypermethylated: TMCO5, PTPRN, GUCY2C | [93] | |
| 27 K | 3 normal ovarian tissue EOC cell lines: A2780 xenografts EOC cell lines: A2780, CP70, PE1, PEO3 |
Xenograft treated with carboplatin or diluent: resistant vs control, mRNA Resistant vs sensitive EOC cell lines |
Hypomethylated: SSH3, SLC2A4, TMEM88, DAXX, MEST mRNA changed: TMEM88, DAXX Cell line study platinum resistant vs sensitive: only TMEM88 TMEM88 expressed in 24/47 EOC |
[91] | |
| Prognosis | |||||
| 450 K | IHC EOC TMA Peripheral blood lymphocytes from 146 EOCb 45 EOC |
Presentation/relapse PFS median < 10.8 Presentation/relapse months (n = 54) PFS median 48.5 months (n = 54) |
333 CpG significantly changed between presentation and relapse. Nine selected to validate Eight of the 333 CpG were significantly associated with survival, but in cluster analysis did not predict time of progression or overall survival |
[94] | |
| MethylCap-seq | 50 SOC 20 EOC (benign) 6 normal ovaries Cell lines: SKOV3 sensitive CP70 resistant IOSE normal |
Differentially methylated regions and PFS Validation of 16 DMR in cell lines |
63 DMR associated with PFS Pathway analysis: 23/63 genes connected to mTOR, MAPK, VEGF, ErbB, NOTCH, Wnt, TGF-β, hedgehog, cell cycle, adherens junction, |
[95] | |
| MethylCap-seq | Two pools each with 3 HGSOC and 3 HGSOC C with good and poor survival respectively. | 106 hypomethylated and 114 hypermethylated genes. | [71] | ||
| MSP | Validation in 18 HGSOC good survival and 30 with poor survival | 37 CpG: RUNX3/CAMK21 associated with PFS | |||
| Cell lines: SKOV3 (sensitive) SKOV3-12.8 (resistant) 6 platinum resistant and 42 sensitive EOC |
CAMK21 methylation | Increased methylation in resistant cells, and lower gene expression 2/6 resistant and 3/42 sensitive with CAMK21 hypermethylation |
HGSOC: High grade serous ovarian cancer; LGSOC: Low grade serous ovarian cancer; ENDOC: Endometrioid ovarian cancer;, CCOC: Clear cell ovarian cancer; MOC: Mucinous ovarian cancer.
SOC/EOC: Serous ovarian cancer, grade not specified or ovarian cancer, histology not specified.
DNA form Peripheral blood leukocytes from EOC patients.
These studies reveal little overlap in the genes identified for chemoresistance or survival in part because of the use of cell line xenografts which may not be reflective of the original lineage, and the small number of patient derived xenografts or samples used. Only one study, from those reviewed, had > 100 patients and assessed blood leukocytes methylation rather than tumor methylation. Most studies evaluated HGSOC histotype, so there are no data available on rarer histotypes with a more methylated genome. As expressed in Patch et al. [11], development of chemoresistance is likely a complex process and involves more than a single gene. Much more needs to be done to understand how epigenetics, DNA methylation in particular, influences chemoresistance.
4.2. Epigenetic inhibitors in EOC
DNA demethylation agents such as 5-aza-cytidine (AZA) and decitabine have been used to express genes silenced by DNA methylation [96], and have been used with success in patients with acute myeloid leukemia (AML) and other malignancies [97]. These agents inhibit DNA methylation by binding covalently to DNA methyltransferase 1 (DNMT1) [98]. The current histone deacetylase (HDACi) inhibitors used clinically target zinc ion dependency of HDAC’s catalytic activity, which affects HDACS I, II and IV but not III. The proposed mechanism for HDACi in cancer treatment is based on histone deacetylases allowing for expression of tumor suppressor genes, however these agents also acetylate non-histone proteins which not part of epigenetic regulation [99]. These inhibitors have been shown to be effective cancer treatments in a variety of cancer types, and have been associated with tumor growth inhibition, apoptosis and cell differentiation [100].
The potential for epigenetic therapy for EOC was first elucidated by examining the effect of demethylating agents on platinum resistant ovarian cancer cell lines. AZA enhanced the sensitivity of platinum resistant ovarian cancer cell lines and [101,102]. Similarly, HDACi and BET inhibitors which work to alter histone tail state have been evaluated in cell lines [11,103–108], and have generally shown efficacy alone or when used in combination with platinum chemotherapy [103,109]. HDACs may also promote DNMT1 protein degradation, thereby inhibiting DNA methylation and allowing re-expression of repressed genes. HDACs are expressed at higher levels in all EOC histotypes compared to normal ovarian tissues [110,111].
Decitabine has been used in studies of DNA methylation and gene expression [112]. In EOC patients with previous chemotherapy and either resistant or refractory disease, treatment with decitabine has had varied efficacy, producing antitumor responses in 18–30% of cases [113,114], or having no effect [115]. Several phase I clinical trials using AZA or decitabine with paclitaxel/carboplatin in several cancers, including EOC patients, have been reported [113,115–118]. It should also be noted that these phase I–II studies had small sample numbers and a mix of EOC histotypes.
BET inhibitors, such as JQ1 and I-BET inhibit the binding of histone’s acetylated lysines, displacing BET proteins from chromatin and preventing gene transcription, have been shown to be variably effective in a number of cancers [119,120]. JQI represses the growth of cisplatin treated ovarian cancer cell lines and xenografts via suppression of BRD4-mediated ALDH1A1 expression, a gene associated with chemoresistance in ovarian cancer [107]. However, resistance to BET inhibitors has been reported [121].
HDACis have been reported to sensitize previously platinum resistant EOC cell lines [103,104], noting that different doses were required for at least two cell lines[103], suggesting histotype specific differences[122]. A response to an HDACi, suberoylamilide hydroxamic acid (SAHA), in combination with paclitaxel was reported in ascitesderived HGSOC cells from four patients initially resistant to paclitaxel [123]. Several clinical trials involving HDACi are ongoing or due are pending. A pan-HDACi, Quisinostat, in combination with paclitaxel and carboplatin is currently being evaluated for safety and efficacy in advanced platinum therapy resistant EOC [124]. A phase 1b/2 study of avelumab with and without entinostat (class I HDACi) in advanced EOC is planned [125]. This is an emerging therapy in EOC, and with development of more selective HDACi and perhaps targeting histotypes most likely to respond, this approach may find a way into clinical care.
4.3. Cell free DNA
Liquid biopsies, intended to detect circulating cell–free tumor DNA (ctDNA) and circulating tumor cells found in most cases of advanced malignancy [126,127]. though less frequently in patients with localized disease [128–130], are an active area of development [128]. DNA methylation based ctDNA liquid biopsies may be used to detect aberrant levels of methylation in genes or DNA regulatory elements previously detected in patient’s primary cancer or developed due to chemoresistance. Alternatively, liquid biopsies may be used to screen high risk patients for the presence of cancer. In EOC, most reports have focused on specific gene targets, such as tumor suppressor genes, evaluating methylation in matched EOC tumor tissues and plasma [131–133]. Notably, assessment of methylation in healthy populations, vital to improvement in sensitivity and specificity of liquid biopsies, has not been adequate. The presence of methylated RASSF1A and BRCA1 has been reported in plasma of EOC patients [54,132,134] and also in healthy controls [135–138], highlighting the need for larger sample numbers of healthy tissue. DNA methylation for seven genes (APC, RASSF1A, CDH1, RUNX3, TFP12, SRFP5, OPCML) was evaluated in 202 plasma samples from healthy individuals and patients with benign, early or late stage EOC. Including health controls in the study was reported to improve sensitivity and specificity of early stage EOC [138], however, no validation of this has been published. A phase III clinical trial using carboplatin/taxoid in EOC evaluated MLH1 methylation in plasma and found increased methylation at relapse which was associated with poor overall survival [131]. This non-invasive holds great promise and as methods for genome-wide methylation profiling of small amounts of degraded DNA are developed, and as we will learn more about the role of DNA methylation in EOC, the hope is to apply this to clinical care.
4.4. Molecular methylation profiling in EOC
Molecular methylation profiling can be helpful in understanding underlying differences in survival and response to therapy of previously homogenous ovarian cancer histotypes. Zhang et al. evaluated whether genome-wide DNA methylation profiles are associated with differential survival within HGSOC cases by clustering CpGs in 568 of the 583 TCGA HGSOC cases with both available methylation and survival data [139]. With 201 CpGs, those significantly associated with patient survival out of the 14,877 available, Zhang et al. was able to identify four methylation profiles with significantly different survival times. Two of the four TCGA methylation profiles (C1 and C3) showed higher global methylation and longer survival. The importance of this type of analysis is that it shows that histological subtyping of ovarian cancer can be made better defined with the addition of epigenetic information. In the clinic, molecular methylation profiling can eventually be used to build predictive models to communicate personalized response to therapy.
Summary.
Epigenetics is making its way into the clinic, thus understanding the role it plays in ovarian cancer is highly relevant. Whereas early studies of DNA methylation treated all ovarian cancer as one group, later studies have built histotype classification although the rarer ones have made it difficult to generate conclusive histotype-specific data. Nonetheless, the development of genome-wide approaches, and collaborative efforts to study the rarer histotypes, will yield more comprehensive data. Studies of histone modification proteins are more nascent, yet clinical trials in inhibitors of these proteins are underway. Much more needed to be done to fully realize the potential that epigenetics holds for ovarian cancer clinical care.
Abbreviations
- 5mC
5hydroxymethylcytosine
- AML
acute myeloid leukemia
- BET
bromodomain inhibitors
- CCOC
clear cell ovarian cancer
- ctDNA
cell-free tumor DNA
- ChIP-onchip
chromatin immunoprecipitation with DNA microarray
- CIMP
CpG island methylator phenotype
- CpG
cytosine followed by guanine
- DMR
differentially methylated regions
- EMT
epithelial-mesenchymal transition
- ENDOC
endometrioid ovarian cancer
- EOC
epithelial ovarian cancer
- HGSOC
high grade serous ovarian cancer
- HDAC
histone deacetylase
- HDACi
histone deacetylase inhibitor
- IHC
immunohistochemistry
- LGSOC
low grade serous ovarian cancer
- MeDIP
Methylated DNA immunoprecipitation
- MethylCap-seq
methylation-capture sequencing
- MOC
mucinous ovarian cancer
- MSP
methylation-specific PCR
- NGS
next generation sequencing
- PBL
peripheral blood leukocytes
- PFS
progression free survival
- PTM
post-translation modification
- SAHA
suberoylamilide hydroxamic acid
- SOC
serous ovarian cancer
- TCGA
the cancer genome atlas
References
- 1.Devouassoux-Shisheboran M, Genestie C. Pathobiology of ovarian carcinomas. Chin J Cancer. 2015;34:50–55. doi: 10.5732/cjc.014.10273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Faber MT, Kjaer SK, Dehlendorff C, Chang-Claude J, Andersen KK, Hogdall E, et al. Cigarette smoking and risk of ovarian cancer: a pooled analysis of 21 case-control studies. Cancer Causes Control: CCC. 2013;24:989–1004. doi: 10.1007/s10552-013-0174-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pearce CL, Templeman C, Rossing MA, Lee A, Near AM, Webb PM, et al. Association between endometriosis and risk of histological subtypes of ovarian cancer: a pooled analysis of case-control studies. Lancet Oncol. 2012;13:385–394. doi: 10.1016/S1470-2045(11)70404-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pearce CL, Wu AH, Gayther SA, Bale AE, Beck PA, Beesley J, et al. Progesterone receptor variation and risk of ovarian cancer is limited to the invasive endometrioid subtype: results from the Ovarian Cancer Association Consortium pooled analysis. Br J Cancer. 2008;98:282–288. doi: 10.1038/sj.bjc.6604170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonome T, Lee JY, Park DC, Radonovich M, Pise-Masison C, Brady J, et al. Expression profiling of serous low malignant potential, low-grade, and high-grade tumors of the ovary. Cancer Res. 2005;65:10602–10612. doi: 10.1158/0008-5472.CAN-05-2240. [DOI] [PubMed] [Google Scholar]
- 6.Tothill RW, Tinker AV, George J, Brown R, Fox SB, Lade S, et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin Cancer Res. 2008;14:5198–5208. doi: 10.1158/1078-0432.CCR-08-0196. [DOI] [PubMed] [Google Scholar]
- 7.Zorn KK, Bonome T, Gangi L, Chandramouli GV, Awtrey CS, Gardner GJ, et al. Gene expression profiles of serous, endometrioid, and clear cell subtypes of ovarian and endometrial cancer. Clin Cancer Res. 2005;11:6422–6430. doi: 10.1158/1078-0432.CCR-05-0508. [DOI] [PubMed] [Google Scholar]
- 8.Vaughan S, Coward JI, Bast RC, Jr, Berchuck A, Berek JS, Brenton JD, et al. Rethinking ovarian cancer: recommendations for improving outcomes. Nat Rev Cancer. 2011;11:719–725. doi: 10.1038/nrc3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klinkebiel D, Zhang W, Akers SN, Odunsi K, Karpf AR. DNA methylome analyses implicate fallopian tube epithelia as the origin for high-Grade serous ovarian cancer. Mol Cancer Res. 2016;14:787–794. doi: 10.1158/1541-7786.MCR-16-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45:1127–1133. doi: 10.1038/ng.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patch AM, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015;521:489–494. doi: 10.1038/nature14410. [DOI] [PubMed] [Google Scholar]
- 12.Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vang R, Shih Ie M, Kurman RJ. Ovarian low-grade and high-grade serous carcinoma: pathogenesis, clinicopathologic and molecular biologic features, and diagnostic problems. Adv Anat Pathol. 2009;16:267–282. doi: 10.1097/PAP.0b013e3181b4fffa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen M, Jin Y, Bi Y, Yin J, Wang Y, Pan L. A survival analysis comparing women with ovarian low-grade serous carcinoma to those with high-grade histology. Onco Targets Ther. 2014;7:1891–1899. doi: 10.2147/OTT.S67812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marquez RT, Baggerly KA, Patterson AP, Liu J, Broaddus R, Frumovitz M, et al. Patterns of gene expression in different histotypes of epithelial ovarian cancer correlate with those in normal fallopian tube, endometrium, and colon. Clin Cancer Res. 2005;11:6116–6126. doi: 10.1158/1078-0432.CCR-04-2509. [DOI] [PubMed] [Google Scholar]
- 16.Yamaguchi K, Matsumura N, Mandai M, Baba T, Konishi I, Murphy SK. Epigenetic and genetic dispositions of ovarian carcinomas. Oncoscience. 2014;1:574–579. doi: 10.18632/oncoscience.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamaguchi K, Mandai M, Oura T, Matsumura N, Hamanishi J, Baba T, et al. Identification of an ovarian clear cell carcinoma gene signature that reflects inherent disease biology and the carcinogenic processes. Oncogene. 2010;29:1741–1752. doi: 10.1038/onc.2009.470. [DOI] [PubMed] [Google Scholar]
- 18.Shen J, Wei J, Wang H, Yang Y, Yue G, Wang L, et al. SULF2 methylation is associated with in vitro cisplatin sensitivity and clinical efficacy for gastric cancer patients treated with a modified FOLFOX regimen. PLoS One. 2013;8:e75564. doi: 10.1371/journal.pone.0075564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen H, Fridley BL, Song H, Lawrenson K, Cunningham JM, Ramus SJ, et al. Epigenetic analysis leads to identification of HNF1B as a subtype-specific susceptibility gene for ovarian cancer. Nat Commun. 2013;4:1628. doi: 10.1038/ncomms2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cicek MS, Koestler DC, Fridley BL, Kalli KR, Armasu SM, Larson MC, et al. Epigenome-wide ovarian cancer analysis identifies a methylation profile differentiating clear-cell histology with epigenetic silencing of the HERG K+ channel. Hum Mol Genet. 2013;22:3038–3047. doi: 10.1093/hmg/ddt160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bojesen SE, Pooley KA, Johnatty SE, Beesley J, Michailidou K, Tyrer JP, et al. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat Genet. 2013;45:371–384. 84e1–2. doi: 10.1038/ng.2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chornokur G, Lin HY, Tyrer JP, Lawrenson K, Dennis J, Amankwah EK, et al. Common genetic variation In cellular transport genes and epithelial ovarian cancer (EOC) risk. PLoS One. 2015;10:e0128106. doi: 10.1371/journal.pone.0128106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gayther SA, Pharoah PD. The inherited genetics of ovarian and endometrial cancer. Curr Opin Genet Dev. 2010;20:231–238. doi: 10.1016/j.gde.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuchenbaecker KB, Ramus SJ, Tyrer J, Lee A, Shen HC, Beesley J, et al. Identification of six new susceptibility loci for invasive epithelial ovarian cancer. Nat Genet. 2015;47:164–171. doi: 10.1038/ng.3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lynch HT, Conway T, Lynch J. Hereditary ovarian cancer pedigree studies, part II. Cancer Genet Cytogenet. 1991;53:161–183. doi: 10.1016/0165-4608(91)90094-b. [DOI] [PubMed] [Google Scholar]
- 26.Shulman LP. Hereditary breast and ovarian cancer (HBOC): clinical features and counseling for BRCA1 and BRCA2, Lynch syndrome, Cowden syndrome, and LiFraumeni syndrome. Obstet Gynecol Clin North Am. 2010;37:109–133. doi: 10.1016/j.ogc.2010.03.003. Table of Contents. [DOI] [PubMed] [Google Scholar]
- 27.Song H, Ramus SJ, Tyrer J, Bolton KL, Gentry-Maharaj A, Wozniak E, et al. A genome-wide association study identifies a new ovarian cancer susceptibility locus on 9p22.2. Nat Genet. 2009;41:996–1000. doi: 10.1038/ng.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, et al. Environmental and heritable factors in the causation of cancer–analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343:78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- 30.Bird A, Taggart M, Frommer M, Miller OJ, Macleod D. A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell. 1985;40:91–99. doi: 10.1016/0092-8674(85)90312-5. [DOI] [PubMed] [Google Scholar]
- 31.Belshaw R, Pereira V, Katzourakis A, Talbot G, Paces J, Burt A, et al. Longterm reinfection of the human genome by endogenous retroviruses. Proc Natl Acad Sci U S A. 2004;101:4894–4899. doi: 10.1073/pnas.0307800101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38:23–38. doi: 10.1038/npp.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Widschwendter M, Jiang G, Woods C, Muller HM, Fiegl H, Goebel G, et al. DNA hypomethylation and ovarian cancer biology. Cancer Res. 2004;64:4472–4480. doi: 10.1158/0008-5472.CAN-04-0238. [DOI] [PubMed] [Google Scholar]
- 34.Goodier JL, Kazazian HH., Jr Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell. 2008;135:23–35. doi: 10.1016/j.cell.2008.09.022. [DOI] [PubMed] [Google Scholar]
- 35.Ziller MJ, Muller F, Liao J, Zhang Y, Gu H, Bock C, et al. Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet. 2011;7:e1002389. doi: 10.1371/journal.pgen.1002389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu TP, Wang T, Seetin MG, Lai Y, Zhu S, Lin K, et al. DNA methylation on N (6)-adenine in mammalian embryonic stem cells. Nature. 2016;532:329–333. doi: 10.1038/nature17640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stricker SH, Koferle A, Beck S. From profiles to function in epigenomics. Nat Rev Genet. 2017;18:51–66. doi: 10.1038/nrg.2016.138. [DOI] [PubMed] [Google Scholar]
- 38.Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138:2073–2087 (e3). doi: 10.1053/j.gastro.2009.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller BF, Sanchez-Vega F, Elnitski L. The emergence of pan-cancer CIMP and its elusive interpretation. Biomolecules. 2016;6 doi: 10.3390/biom6040045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. 2014;26:577–590. doi: 10.1016/j.ccr.2014.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mendizabal I, Zeng J, Keller TE, Yi SV. Body-hypomethylated human genes harbor extensive intragenic transcriptional activity and are prone to cancer-associated dysregulation. Nucleic Acids Res. 2017 doi: 10.1093/nar/gkx020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeimet AG, Fiegl H, Goebel G, Kopp F, Allasia C, Reimer D, et al. DNA ploidy, nuclear size, proliferation index and DNA-hypomethylation in ovarian cancer. Gynecol Oncol. 2011;121:24–31. doi: 10.1016/j.ygyno.2010.12.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. 2009;1:239–259. doi: 10.2217/epi.09.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chi P, Allis CD, Wang GG. Covalent histone modifications–miswritten, mis-interpreted and mis-erased in human cancers. Nat Rev Cancer. 2010;10:457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li F, Wan M, Zhang B, Peng Y, Zhou Y, Pi C, et al. Bivalent histone modifications and development. Curr Stem Cell Res Ther. 2017;12:1–8. doi: 10.2174/1574888X12666170123144743. [DOI] [PubMed] [Google Scholar]
- 46.Verdin E, Dequiedt F, Kasler HG. Class II histone deacetylases: versatile regulators. Trends Genet. 2003;19:286–293. doi: 10.1016/S0168-9525(03)00073-8. [DOI] [PubMed] [Google Scholar]
- 47.Nakagawa M, Oda Y, Eguchi T, Aishima S, Yao T, Hosoi F, et al. Expression profile of class I histone deacetylases in human cancer tissues. Oncol Rep. 2007;18:769–774. [PubMed] [Google Scholar]
- 48.Pchejetski D, Alfraidi A, Sacco K, Alshaker H, Muhammad A, Monzon L. Histone deacetylases as new therapy targets for platinum-resistant epithelial ovarian cancer. J Cancer Res Clin Oncol. 2016;142:1659–1671. doi: 10.1007/s00432-015-2064-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ceccacci E, Minucci S. Inhibition of histone deacetylases in cancer therapy: lessons from leukaemia. Br J Cancer. 2016;114:605–611. doi: 10.1038/bjc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell. 2014;54:728–736. doi: 10.1016/j.molcel.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang HJ, Liu VW, Wang Y, Tsang PC, Ngan HY. Differential DNA methylation profiles in gynecological cancers and correlation with clinico-pathological data. BMC Cancer. 2006;6:212. doi: 10.1186/1471-2407-6-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Press JZ, De Luca A, Boyd N, Young S, Troussard A, Ridge Y, et al. Ovarian carcinomas with genetic and epigenetic BRCA1 loss have distinct molecular abnormalities. BMC Cancer. 2008;8:17. doi: 10.1186/1471-2407-8-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Helleman J, van Staveren IL, Dinjens WN, van Kuijk PF, Ritstier K, Ewing PC, et al. Mismatch repair and treatment resistance in ovarian cancer. BMC Cancer. 2006;6:201. doi: 10.1186/1471-2407-6-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ibanez de Caceres I, Battagli C, Esteller M, Herman JG, Dulaimi E, Edelson MI, et al. Tumor cell-specific BRCA1 and RASSF1A hypermethylation in serum, plasma, and peritoneal fluid from ovarian cancer patients. Cancer Res. 2004;64:6476–6481. doi: 10.1158/0008-5472.CAN-04-1529. [DOI] [PubMed] [Google Scholar]
- 55.Ozdemir F, Altinisik J, Karateke A, Coksuer H, Buyru N. Methylation of tumor suppressor genes in ovarian cancer. Exp Ther Med. 2012;4:1092–1096. doi: 10.3892/etm.2012.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Strathdee G, Appleton K, Illand M, Millan DW, Sargent J, Paul J, et al. Primary ovarian carcinomas display multiple methylator phenotypes involving known tumor suppressor genes. Am J Pathol. 2001;158:1121–1127. doi: 10.1016/S0002-9440(10)64059-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wiley A, Katsaros D, Chen H, Rigault de la Longrais IA, Beeghly A, Puopolo M, et al. Aberrant promoter methylation of multiple genes in malignant ovarian tumors and in ovarian tumors with low malignant potential. Cancer. 2006;107:299–308. doi: 10.1002/cncr.21992. [DOI] [PubMed] [Google Scholar]
- 58.Furlan D, Carnevali I, Marcomini B, Cerutti R, Dainese E, Capella C, et al. The high frequency of de novo promoter methylation in synchronous primary endometrial and ovarian carcinomas. Clin Cancer Res. 2006;12:3329–3336. doi: 10.1158/1078-0432.CCR-05-2679. [DOI] [PubMed] [Google Scholar]
- 59.Feng Q, Deftereos G, Hawes SE, Stern JE, Willner JB, Swisher EM, et al. DNA hypermethylation, Her-2/neu overexpression and p53 mutations in ovarian carcinoma. Gynecol Oncol. 2008;111:320–329. doi: 10.1016/j.ygyno.2008.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Makarla PB, Saboorian MH, Ashfaq R, Toyooka KO, Toyooka S, Minna JD, et al. Promoter hypermethylation profile of ovarian epithelial neoplasms. Clin Cancer Res. 2005;11:5365–5369. doi: 10.1158/1078-0432.CCR-04-2455. [DOI] [PubMed] [Google Scholar]
- 61.Moselhy SS, Kumosani TA, Kamal IH, Jalal JA, Jabaar HS Abdul, Dalol A. Hypermethylation of P15, P 16, and E-cadherin genes in ovarian cancer. Toxicol Ind Health. 2013;31:924–930. doi: 10.1177/0748233713484657. [DOI] [PubMed] [Google Scholar]
- 62.Niederacher D, Yan HY, An HX, Bender HG, Beckmann MW. CDKN2A gene inactivation in epithelial sporadic ovarian cancer. Br J Cancer. 1999;80:1920–1926. doi: 10.1038/sj.bjc.6690621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Teodoridis JM, Hall J, Marsh S, Kannall HD, Smyth C, Curto J, et al. CpG island methylation of DNA damage response genes in advanced ovarian cancer. Cancer Res. 2005;65:8961–8967. doi: 10.1158/0008-5472.CAN-05-1187. [DOI] [PubMed] [Google Scholar]
- 64.Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst. 2000;92:564–569. doi: 10.1093/jnci/92.7.564. [DOI] [PubMed] [Google Scholar]
- 65.Houshdaran S, Hawley S, Palmer C, Campan M, Olsen MN, Ventura AP, et al. DNA methylation profiles of ovarian epithelial carcinoma tumors and cell lines. PLoS One. 2010;5:e9359. doi: 10.1371/journal.pone.0009359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yoon MS, Suh DS, Choi KU, Sol MY, Shin DH, Park WY, et al. Highthroughput DNA hypermethylation profiling in different ovarian epithelial cancer subtypes using universal bead array. Oncol Rep. 2010;24:917–925. doi: 10.3892/or.2010.917. [DOI] [PubMed] [Google Scholar]
- 67.Shih Ie M, Chen L, Wang CC, Gu J, Davidson B, Cope L, et al. Distinct DNA methylation profiles in ovarian serous neoplasms and their implications in ovarian carcinogenesis. Am J Obstet Gynecol. 2010;203(584):e1–22. doi: 10.1016/j.ajog.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yoon JH, Dammann R, Pfeifer GP. Hypermethylation of the CpG island of the RASSF1A gene in ovarian and renal cell carcinomas. Int J Cancer. 2001;94:212–217. doi: 10.1002/ijc.1466. [DOI] [PubMed] [Google Scholar]
- 69.Kolbe DL, DeLoia JA, Porter-Gill P, Strange M, Petrykowska HM, Guirguis A, et al. Differential analysis of ovarian and endometrial cancers identifies a methylator phenotype. PLoS One. 2012;7:e32941. doi: 10.1371/journal.pone.0032941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sanchez-Vega F, Gotea V, Petrykowska HM, Margolin G, Krivak TC, DeLoia JA, et al. Recurrent patterns of DNA methylation in the ZNF154, CASP8, and VHL promoters across a wide spectrum of human solid epithelial tumors and cancer cell lines. Epigenetics. 2013;8:1355–1372. doi: 10.4161/epi.26701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hafner N, Steinbach D, Jansen L, Diebolder H, Durst M, Runnebaum IB. RUNX3 and CAMK2N1 hypermethylation as prognostic marker for epithelial ovarian cancer. Int J Cancer. 2015;138:217–228. doi: 10.1002/ijc.29690. [DOI] [PubMed] [Google Scholar]
- 72.Earp MA, Cunningham JM. DNA methylation changes in epithelial ovarian cancer histotypes. Genomics. 2015;106:311–321. doi: 10.1016/j.ygeno.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Whitcomb BP, Mutch DG, Herzog TJ, Rader JS, Gibb RK, Goodfellow PJ. Frequent HOXA11 and THBS2 promoter methylation, and a methylator phenotype in endometrial adenocarcinoma. Clin Cancer Res. 2003;9:2277–2287. [PubMed] [Google Scholar]
- 74.Pharoah PD, Tsai YY, Ramus SJ, Phelan CM, Goode EL, Lawrenson K, et al. GWAS meta-analysis and replication identifies three new susceptibility loci for ovarian cancer. Nat Genet. 2013;45:362–370. 70e1–2. doi: 10.1038/ng.2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ross-Adams H, Ball S, Lawrenson K, Halim S, Russell R, Wells C, et al. HNF1B variants associate with promoter methylation and regulate gene networks activated in prostate and ovarian cancer. Oncotarget. 2016;7:74734–74746. doi: 10.18632/oncotarget.12543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 77.Arildsen NS, Jonsson JM, Bartuma K, Ebbesson A, Westbom-Fremer S, Masback A, et al. Involvement of chromatin remodeling genes and the rho GTPases RhoB and CDC42 in ovarian clear cell carcinoma. Front Oncol. 2017;7:109. doi: 10.3389/fonc.2017.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jones S, Wang TL, Ie M Shih, Mao TL, Nakayama K, Roden R, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228–231. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jang KY, Kim KS, Hwang SH, Kwon KS, Kim KR, Park HS, et al. Expression and prognostic significance of SIRT1 in ovarian epithelial tumours, Pathology (Phila.) 2009;41:366–371. doi: 10.1080/00313020902884451. [DOI] [PubMed] [Google Scholar]
- 80.He WP, Li Q, J H Zhou, HF ZS Kung, Guan XY, et al. Decreased expression of H3K27me3 in human ovarian carcinomas correlates with more aggressive tumor behavior and poor patient survival. Neoplasma. 2015;62:932–937. doi: 10.4149/neo_2015_113. [DOI] [PubMed] [Google Scholar]
- 81.Bapat SA, Mali AM, Koppikar CB, Kurrey NK. Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res. 2005;65:3025–3029. doi: 10.1158/0008-5472.CAN-04-3931. [DOI] [PubMed] [Google Scholar]
- 82.Singh AK, Chandra N, Bapat SA. Evaluation of epigenetic drug targeting of heterogenous tumor cell fractions using potential biomarkers of response in ovarian cancer. Clin Cancer Res. 2015;21:5151–5163. doi: 10.1158/1078-0432.CCR-15-0505. [DOI] [PubMed] [Google Scholar]
- 83.Chapman-Rothe N, Curry E, Zeller C, Liber D, Stronach E, Gabra H, et al. Chromatin H3K27me3/H3K4me3 histone marks define gene sets in high-grade serous ovarian cancer that distinguish malignant, tumour-sustaining and chemoresistant ovarian tumour cells. Oncogene. 2013;32:4586–4592. doi: 10.1038/onc.2012.477. [DOI] [PubMed] [Google Scholar]
- 84.Widschwendter M, Jones A, Teschendorff AE. Epigenetics makes its mark on women-specific cancers–an opportunity to redefine oncological approaches? Gynecol Oncol. 2013;128:134–143. doi: 10.1016/j.ygyno.2012.09.027. [DOI] [PubMed] [Google Scholar]
- 85.Abbosh PH, Montgomery JS, Starkey JA, Novotny M, Zuhowski EG, Egorin MJ, et al. Dominant-negative histone H3 lysine 27 mutant derepresses silenced tumor suppressor genes and reverses the drug-resistant phenotype in cancer cells. Cancer Res. 2006;66:5582–5591. doi: 10.1158/0008-5472.CAN-05-3575. [DOI] [PubMed] [Google Scholar]
- 86.Vella N, Aiello M, Russo AE, Scalisi A, Spandidos DA, Toffoli G, et al. ‘Genetic profiling’ and ovarian cancer therapy (review) Mol Med Rep. 2011;4:771–777. doi: 10.3892/mmr.2011.512. [DOI] [PubMed] [Google Scholar]
- 87.Joerger M, Huitema AD, Richel DJ, Dittrich C, Pavlidis N, Briasoulis E, et al. Population pharmacokinetics and pharmacodynamics of doxorubicin and cyclophosphamide in breast cancer patients: a study by the EORTC-PAMM-NDDG. Clin Pharmacokinet. 2007;46:1051–1068. doi: 10.2165/00003088-200746120-00005. [DOI] [PubMed] [Google Scholar]
- 88.Bolton KL, Chenevix-Trench G, Goh C, Sadetzki S, Ramus SJ, Karlan BY, et al. Association between BRCA1 and BRCA2 mutations and survival in women with invasive epithelial ovarian cancer. JAMA. 2012;307:382–390. doi: 10.1001/jama.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Stefansson OA, Villanueva A, Vidal A, Marti L, Esteller M. BRCA1 epigenetic inactivation predicts sensitivity to platinum-based chemotherapy in breast and ovarian cancer. Epigenetics. 2012;7:1225–1229. doi: 10.4161/epi.22561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yang D, Khan S, Sun Y, Hess K, Shmulevich I, Sood AK, et al. Association of BRCA1 and BRCA2 mutations with survival, chemotherapy sensitivity, and gene mutator phenotype in patients with ovarian cancer. JAMA. 2011;306:1557–1565. doi: 10.1001/jama.2011.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.de Leon M, Cardenas H, Vieth E, Emerson R, Segar M, Liu Y, et al. Transmembrane protein 88 (TMEM88) promoter hypomethylation is associated with platinum resistance in ovarian cancer. Gynecol Oncol. 2016;142:539–547. doi: 10.1016/j.ygyno.2016.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lum E, Vigliotti M, Banerjee N, Cutter N, Wrzeszczynski KO, Khan S, et al. Loss of DOK2 induces carboplatin resistance in ovarian cancer via suppression of apoptosis. Gynecol Oncol. 2013;130:369–376. doi: 10.1016/j.ygyno.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 93.Bauerschlag DO, Ammerpohl O, Brautigam K, Schem C, Lin Q, Weigel MT, et al. Progression-free survival in ovarian cancer is reflected in epigenetic DNA methylation profiles. Oncology. 2011;80:12–20. doi: 10.1159/000327746. [DOI] [PubMed] [Google Scholar]
- 94.Flanagan JM, Wilson A, Koo C, Masrour N, Gallon J, Loomis E, et al. Platinum-based chemotherapy induces methylation changes in blood DNA associated with overall survival in ovarian cancer patients. Clin Cancer Res. 2016;23:2213–2222. doi: 10.1158/1078-0432.CCR-16-1754. [DOI] [PubMed] [Google Scholar]
- 95.Huang RL, Gu F, Kirma NB, Ruan J, Chen CL, Wang HC, et al. Comprehensive methylome analysis of ovarian tumors reveals hedgehog signaling pathway regulators as prognostic DNA methylation biomarkers. Epigenetics. 2013;8:624–634. doi: 10.4161/epi.24816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Momparler RL. Pharmacology of 5-Aza-2′-deoxycytidine (decitabine) Semin Hematol. 2005;42:S9–S16. doi: 10.1053/j.seminhematol.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 97.Malik P, Cashen AF. Decitabine in the treatment of acute myeloid leukemia in elderly patients. Cancer Manage Res. 2014;6:53–61. doi: 10.2147/CMAR.S40600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Smith HJ, Straughn JM, Buchsbaum DJ, Arend RC. Epigenetic therapy for the treatment of epithelial ovarian cancer: a clinical review. Gynecol Oncol Rep. 2017;20:16–81. doi: 10.1016/j.gore.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 100.Mottamal M, Zheng S, Huang TL, Wang G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules. 2015;20:3898–3941. doi: 10.3390/molecules20033898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Plumb JA, Strathdee G, Sludden J, Kaye SB, Brown R. Reversal of drug resistance in human tumor xenografts by 2′-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene promoter. Cancer Res. 2000;60:6039–6044. [PubMed] [Google Scholar]
- 102.Li Y, Hu W, Shen DY, Kavanagh JJ, Fu S. Azacitidine enhances sensitivity of platinum-resistant ovarian cancer cells to carboplatin through induction of apoptosis. Am J Obstet Gynecol. 2009;200(177):e1–e9. doi: 10.1016/j.ajog.2008.08.030. [DOI] [PubMed] [Google Scholar]
- 103.Lapinska K, Housman G, Byler S, Heerboth S, Willbanks A, Oza A, et al. The effects of histone deacetylase inhibitor and calpain inhibitor combination therapies on ovarian cancer cells. Anticancer Res. 2016;36:5731–5742. doi: 10.21873/anticanres.11156. [DOI] [PubMed] [Google Scholar]
- 104.Muscolini M, Cianfrocca R, Sajeva A, Mozzetti S, Ferrandina G, Costanzo A, et al. Trichostatin A up-regulates p73 and induces Bax-dependent apoptosis in cisplatin-resistant ovarian cancer cells. Mol Cancer Ther. 2008;7:1410–1419. doi: 10.1158/1535-7163.MCT-08-0299. [DOI] [PubMed] [Google Scholar]
- 105.Weberpals JI, O’Brien AM, Niknejad N, Garbuio KD, Clark-Knowles KV, Dimitroulakos J. The effect of the histone deacetylase inhibitor M344 on BRCA1 expression in breast and ovarian cancer cells. Cancer Cell Int. 2011;11:29. doi: 10.1186/1475-2867-11-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang Z, Ma P, Jing Y, Yan Y, Cai MC, Zhang M, et al. BET bromodomain inhibition as a therapeutic strategy in ovarian cancer by downregulating FoxM1. Theranostics. 2016;6:219–230. doi: 10.7150/thno.13178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yokoyama Y, Zhu H, Lee JH, Kossenkov AV, Wu SY, Wickramasinghe JM, et al. BET inhibitors suppress ALDH activity by targeting ALDH1A1 super-enhancer in ovarian cancer. Cancer Res. 2016;76:6320–6330. doi: 10.1158/0008-5472.CAN-16-0854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Garrido-Laguna I, Janku F, Vaklavas C, Falchook GS, Fu S, Hong DS, et al. Validation of the royal marsden hospital prognostic score in patients treated in the phase I clinical trials program at the MD anderson cancer center. Cancer. 2012;118:1422–1428. doi: 10.1002/cncr.26413. [DOI] [PubMed] [Google Scholar]
- 109.Weberpals JI, O’Brien AM, Niknejad N, Garbuio KD, Clark-Knowles KV, Dimitroulakos J. The effect of the histone deacetylase inhibitor M344 on BRCA1 expression in breast and ovarian cancer cells. Cancer Cell Int. 2011;11:29. doi: 10.1186/1475-2867-11-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Weichert W, Denkert C, Noske A, Darb-Esfahani S, Dietel M, Kalloger SE, et al. Expression of class I histone deacetylases indicates poor prognosis in endometrioid subtypes of ovarian and endometrial carcinomas. Neoplasia. 2008;10:1021–1027. doi: 10.1593/neo.08474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Khabele D, Son DS, Parl AK, Goldberg GL, Augenlicht LH, Mariadason JM, et al. Drug-induced inactivation or gene silencing of class I histone deacetylases suppresses ovarian cancer cell growth: implications for therapy. Cancer Biol Ther. 2007;6:795–801. doi: 10.4161/cbt.6.5.4007. [DOI] [PubMed] [Google Scholar]
- 112.Tomar T, de Jong S, Alkema NG, Hoekman RL, Meersma GJ, Klip HG, et al. Genome-wide methylation profiling of ovarian cancer patient-derived xenografts treated with the demethylating agent decitabine identifies novel epigenetically regulated genes and pathways. Genome Med. 2016;8:107. doi: 10.1186/s13073-016-0361-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Falchook GS, Fu S, Naing A, Hong DS, Hu W, Moulder S, et al. Methylation and histone deacetylase inhibition in combination with platinum treatment in patients with advanced malignancies. Invest New Drugs. 2013;31:1192–1200. doi: 10.1007/s10637-013-0003-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fang F, Balch C, Schilder J, Breen T, Zhang S, Shen C, et al. A phase 1 and pharmacodynamic study of decitabine in combination with carboplatin in patients with recurrent, platinum-resistant, epithelial ovarian cancer. Cancer. 2010;116:4043–4053. doi: 10.1002/cncr.25204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Glasspool RM, Brown R, Gore ME, Rustin GJ, McNeish IA, Wilson RH, et al. A randomised, phase II trial of the DNA-hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in combination with carboplatin vs carboplatin alone in patients with recurrent, partially platinum-sensitive ovarian cancer. Br J Cancer. 2014;110:1923–1929. doi: 10.1038/bjc.2014.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fang F, Zuo Q, Pilrose J, Wang Y, Shen C, Li M, et al. Decitabine reactivated pathways in platinum resistant ovarian cancer. Oncotarget. 2014;5:3579–3589. doi: 10.18632/oncotarget.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fu Y, Nachtigal MW. Analysis of epigenetic alterations to proprotein convertase genes in disease. Methods Mol Biol. 2011;768:231–245. doi: 10.1007/978-1-61779-204-5_12. [DOI] [PubMed] [Google Scholar]
- 118.Cohen AL, Ray A, Van Brocklin M, Burnett DM, Bowen RC, Dyess DL, et al. A phase I trial of azacitidine and nanoparticle albumin bound paclitaxel in patients with advanced or metastatic solid tumors. Oncotarget. 2016 doi: 10.18632/oncotarget.14183. [DOI] [PMC free article] [PubMed]
- 119.Lockwood WW, Zejnullahu K, Bradner JE, Varmus H. Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proc Natl Acad Sci U S A. 2012;109:19408–19413. doi: 10.1073/pnas.1216363109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A. 2011;108:16669–16674. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kurimchak AM, Shelton C, Duncan KE, Johnson KJ, Brown J, O’Brien S, et al. Resistance to BET bromodomain inhibitors is mediated by kinome reprogramming in ovarian cancer. Cell Rep. 2016;16:1273–1286. doi: 10.1016/j.celrep.2016.06.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Beaufort CM, Helmijr JC, Piskorz AM, Hoogstraat M, Ruigrok-Ritstier K, Besselink N, et al. Ovarian cancer cell line panel (OCCP): clinical importance of in vitro morphological subtypes. PLoS One. 2014;9:e103988. doi: 10.1371/journal.pone.0103988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sonnemann J, Gange J, Pilz S, Stotzer C, Ohlinger R, Belau A, et al. Comparative evaluation of the treatment efficacy of suberoylanilide hydroxamic acid (SAHA) and paclitaxel in ovarian cancer cell lines and primary ovarian cancer cells from patients. BMC Cancer. 2006;6(183) doi: 10.1186/1471-2407-6-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tjulandin SA. Safety and Efficacy of Quisinostat, a Histone Deacetylase Inhibitor in Combination With Chemotherapy. 2017 [Google Scholar]
- 125.Matulonis U. In: Phase 1b/2 Study of Avelumab With or Without Entinostat in Patients With Advanced Epithelial Ovarian Cancer. Meyers M, editor. [Google Scholar]
- 126.Alix-Panabieres C, Schwarzenbach H, Pantel K. Circulating tumor cells and circulating tumor DNA. Annu Rev Med. 2012;63:199–215. doi: 10.1146/annurev-med-062310-094219. [DOI] [PubMed] [Google Scholar]
- 127.Stroun M, Lyautey J, Lederrey C, Olson-Sand A, Anker P. About the possible origin and mechanism of circulating DNA apoptosis and active DNA release. Clin Chim Acta: Int J Clin Chem. 2001;313:139–142. doi: 10.1016/s0009-8981(01)00665-9. [DOI] [PubMed] [Google Scholar]
- 128.Warton K, Samimi G. Methylation of cell-free circulating DNA in the diagnosis of cancer, Front. Mol Biosci. 2015;2:13. doi: 10.3389/fmolb.2015.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wittenberger T, Sleigh S, Reisel D, Zikan M, Wahl B, Alunni-Fabbroni M, et al. DNA methylation markers for early detection of women’s cancer: promise and challenges. Epigenomics. 2014;6:311–327. doi: 10.2217/epi.14.20. [DOI] [PubMed] [Google Scholar]
- 130.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in earlyand late-stage human malignancies. Sci Transl Med. 2014;6:224ra24. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gifford G, Paul J, Vasey PA, Kaye SB, Brown R. The acquisition of hMLH1 methylation in plasma DNA after chemotherapy predicts poor survival for ovarian cancer patients. Clin Cancer Res. 2004;10:4420–4426. doi: 10.1158/1078-0432.CCR-03-0732. [DOI] [PubMed] [Google Scholar]
- 132.Wu Y, Zhang X, Lin L, Ma XP, Ma YC, Liu PS. Aberrant methylation of RASSF2A in tumors and plasma of patients with epithelial ovarian cancer. Asian Pac J Cancer Prev. 2014;15:1171–1176. doi: 10.7314/apjcp.2014.15.3.1171. [DOI] [PubMed] [Google Scholar]
- 133.Giannopoulou L, Chebouti I, Pavlakis K, Kasimir-Bauer S, Lianidou ES. RASSF1A promoter methylation in high-grade serous ovarian cancer: a direct comparison study in primary tumors, adjacent morphologically tumor cell-free tissues and paired circulating tumor DNA. Oncotarget. 2017;8:21429–21443. doi: 10.18632/oncotarget.15249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Liggett TE, Melnikov A, Yi Q, Replogle C, Hu W, Rotmensch J, et al. Distinctive DNA methylation patterns of cell-free plasma DNA in women with malignant ovarian tumors. Gynecol Oncol. 2011;120:113–120. doi: 10.1016/j.ygyno.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Belinsky SA, Klinge DM, Dekker JD, Smith MW, Bocklage TJ, Gilliland FD, et al. Gene promoter methylation in plasma and sputum increases with lung cancer risk. Clin Cancer Res. 2005;11:6505–6511. doi: 10.1158/1078-0432.CCR-05-0625. [DOI] [PubMed] [Google Scholar]
- 136.Hsu HS, Chen TP, Hung CH, Wen CK, Lin RK, Lee HC, et al. Characterization of a multiple epigenetic marker panel for lung cancer detection and risk assessment in plasma. Cancer. 2007;110:2019–2026. doi: 10.1002/cncr.23001. [DOI] [PubMed] [Google Scholar]
- 137.Radpour R, Barekati Z, Kohler C, Lv Q, Burki N, Diesch C, et al. Hypermethylation of tumor suppressor genes involved in critical regulatory pathways for developing a blood-based test in breast cancer. PLoS One. 2011;6:e16080. doi: 10.1371/journal.pone.0016080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhang Q, Hu G, Yang Q, Dong R, Xie X, Ma D, et al. A multiplex methylationspecific PCR assay for the detection of early-stage ovarian cancer using cell-free serum DNA. Gynecol Oncol. 2013;130:132–139. doi: 10.1016/j.ygyno.2013.04.048. [DOI] [PubMed] [Google Scholar]
- 139.Zhang Z, Huang K, Gu C, Zhao L, Wang N, Wang X, et al. Molecular subtyping of serous ovarian cancer based on multi-omics data. Sci Rep. 2016;6:26001. doi: 10.1038/srep26001. [DOI] [PMC free article] [PubMed] [Google Scholar]