

Summary
Nitric oxide (NO) plays an established role in numerous physiological and pathological processes, but the specific cellular sources of NO in disease pathogenesis remain unclear, preventing the implementation of NO-related therapy. Argininosuccinate lyase (ASL) is the only enzyme able to produce arginine, the substrate for NO generation by nitric oxide synthase (NOS) isoforms. Here, we generated cell-specific conditional ASL knockout mice in combination with genetic and chemical colitis models. We demonstrate that NO derived from enterocytes alleviates colitis by decreasing macrophage infiltration and tissue damage, whereas immune cell-derived NO is associated with macrophage activation, resulting in increased severity of inflammation. We find that induction of endogenous NO production by enterocytes with supplements that upregulate ASL expression and complement its substrates results in improved epithelial integrity and alleviation of colitis and of inflammation-associated colon cancer.
Keywords: IBD, inflammation-associated colon cancer, nitric oxide metabolism, neutraceutical supplements
Graphical Abstract
Highlights
-
•
ASL CKO enables dissection of NO contribution to disease in a cell-specific manner
-
•
NO has different roles in the specific cell types relevant to colitis
-
•
NO synthesis in enterocytes is a native defense mechanism against colitis
-
•
Metabolic modulation of NO levels is beneficial for colitis and associated colon cancer
ASL levels metabolically regulate NO synthesis in a cell-specific manner. Here, we find that cell-autonomous production of NO by enterocytes can be protective as part of the innate immune response against colitis. Finally, we demonstrate the superior advantage of metabolic modulation as a therapy for colitis and inflammation-associated colon cancer.
Introduction
Inflammatory bowel disease (IBD), comprised of Crohn’s disease and ulcerative colitis, is a chronic inflammation of the digestive tract. Multiple factors, including genetic predisposition, environment, gut microbiota, altered barrier function of the intestinal lining, and dysregulated immune response, are key elements in IBD pathogenesis (Atreya and Neurath, 2015, de Souza and Fiocchi, 2016). Nitric oxide (NO), an important signaling molecule, is a homeostatic regulator of gastrointestinal integrity by maintaining perfusion, epithelial and vascular permeability, and gut motility (Alican and Kubes, 1996, Blaise et al., 2005, Moncada, 1992, Shah et al., 2004). In addition, NO signaling is important for the host immune response and tissue repair (Brown et al., 1993, Witthöft et al., 1998). Although a substantial increase in NO signaling has been implicated in the pathogenesis of human IBD, the specific cellular origin of NO and the exact roles of epithelial and immune cell-derived NO in intestinal inflammation remain unclear (Boughton-Smith et al., 1993, Kolios et al., 2004, Middleton et al., 1993, Suschek et al., 2004). A primary reason for the limited understanding of the role of cell- and context-dependent NO signaling in IBD is the redundancy of the three isoforms of nitric oxide synthase (NOS): endothelial (eNOS), neuronal (nNOS), and inducible (iNOS). The NOS isoforms are often co-expressed in multiple cell types, which prevents dissection of cell-specific NO contributions to disease pathogenesis in both in vitro and in vivo models (Knowles and Moncada, 1994, Soufli et al., 2016).These obstacles have precluded implementation of NO-related therapies, necessitating a translational model system that overcomes these limitations.
All three NOS isoforms use arginine as a substrate for NO synthesis. Argininosuccinate lyase (ASL), a urea cycle enzyme, is the only mammalian enzyme that can endogenously generate arginine. Outside of the liver, ASL, together with another urea cycle enzyme, argininosuccinate synthase (ASS1), participates in the citrulline-arginine cycle, in which arginine is recycled back to citrulline by NOS, which also generates NO in this reaction (Erez et al., 2011a, Nagamani et al., 2012). Because arginine is a semi-essential amino acid, ASL is likely to play a key role in maintaining arginine homeostasis at the tissue level in arginine-deficient states, such as intestinal inflammation (Erez et al., 2011a). We have previously shown that loss of ASL leads to metabolic restriction of arginine for all NOS-derived NOs (Erez et al., 2011b). Here we use models with cell-specific loss of ASL to better understand the cell-specific contributions of NO in the causation of IBD.
Results
Generating Cell-Specific Impairment of Arginine Production in Epithelial and Immune Cells and Induction of Colitis
To generate cell-specific conditional ASL knockout (CKO) mice, we crossed Aslf/f animals (Erez et al., 2011b) to three different transgenic mice expressing Cre recombinase under the enterocyte-specific Villin promoter (Madison et al., 2002), the hematopoietic Vav1 promoter (Ogilvy et al., 1998), and the macrophage/dendritic cell (DC)-specific CD11c promoter (Caton et al., 2007, Vander Lugt et al., 2014; Figures S1A–S1F). At baseline, all CKO mice were indistinguishable from their wild-type (WT) littermates and showed no observable phenotype; in particular, there were no differences in blood counts, body weight, arginine levels, and intestinal histology (data not shown; Figures S1E, S1G, and S2A). The severity of intestinal inflammation after induction of colitis was assessed comprehensively by endoscopy, histology, and MRI as well as by evaluating clinical parameters such as weight and survival. In all experiments, Cre−/−Aslf/f littermate mice (labeled Aslf/f) were used as controls.
Loss of ASL in Enterocytes Is Detrimental in Arginine-Deficient States
In human and murine enterocytes, expression of ASL is maximal in the second week of life, when the intestine is the principal site of arginine synthesis (De Jonge et al., 1998). In contrast, enterocyte expression of ASL in adult mice is rather limited and thus, expectedly, was not different compared with ASL expression in VillinCre:Aslf/f mice (Figure S2A). In agreement with ASL expression levels, plasma arginine levels were similar between VillinCre:Aslf/f adult mice and controls (Figure S1G). Colitis was induced in the CKO models chemically by using dextran sulfate sodium (DSS) (Whittem et al., 2010, Cooper et al., 1993). Following acute colitis induction, ASL levels in control enterocytes were not elevated significantly, and the severity of DSS-induced colonic inflammation was similar in controls and VillinCre:Aslf/f mice (Figure 1A; Figure S2B). These results are consistent with our previous work showing increased incidence of necrotizing enterocolitis in VillinCre:Aslf/f mice only in the neonatal period, when there is a significant expression of ASL in enterocytes (Premkumar et al., 2014). In an attempt to generate significant differential ASL expression in adult enterocytes of VillinCre:Aslf/f mice and control littermates, mice were maintained on an arginine-free diet. As previously described, the weight of these mice was 20% lower than that of the respective genotypes fed an arginine-sufficient diet (Marini et al., 2015). Importantly, dietary arginine restriction resulted in expression of ASL in the intestines of control mice but not in the VillinCre:Aslf/f animals, although no differences in the animal growth curves or immune infiltrates to the intestine were observed between CKO mice and controls (Figure S1G). Collectively, these results suggest that ASL induction in the adult mouse gut is a physiological mechanism to restore arginine homeostasis during arginine-deficient states.
Figure 1.

Increased Colitis Severity in ASL CKO Enterocytes
(A) In mice fed an arginine-sufficient diet, there were no noticeable differences in body weight (left) or colitis score (right) between Aslf/f and VillinCre:Aslf/f mice (n ≥ 5 in each group, experiments were repeated at least three times).
(B–D) Colitis was induced by DSS in VillinCre:Aslf/f and control Aslf/f mice fed an arginine-free diet. VillinCre:Aslf/f mice had increased colitis severity compared with control mice as demonstrated by (B) a significantly weight loss (n = 18 in each group), (C) a higher endoscopic colitis score (the right shows a representative colonoscopy image for an experiment with n ≥ 8 in each group), and (D) shorter colons.
(E) A T2 map of colon section MRI showing increased relaxation time, a marker of inflammation in VillinCre:Aslf/f compared with control mice. The color gradient ranges from blue, which represents short relaxation, to red, which represents a longer relaxation time, correlating with more severe colon damage. The quantification graph of the images on the right shows a statistically significant increase in relaxation time in CKO enterocytes (n = 11 in each group).
(F) Left: a higher histological score in the VillinCre:Aslf/f mice (n ≥ 6 in each group). Right: a representative colon cross-section stained with H&E, showing severe edema and destruction of colon crypts in VillinCre:Aslf/f mice. The asterisk indicates an area of severe edema. Arrows indicate crypts present in the colon of Aslf/f mice only.
(G) Kaplan-Meier curve showing a higher mortality rate in VillinCre:Aslf/f mice compared with controls.
Error bars represent SEM.
With an arginine-free diet, induction of colitis with the standard dose of 1.5% DSS was lethal in the VillinCre:Aslf/f mice (data not shown), necessitating that a lower dose of DSS (0.8%) be used for all experiments. Even with such a reduced DSS challenge, the increased severity of colitis in the VillinCre:Aslf/f mice compared with controls was evidenced by greater weight loss, increased colonic inflammation, decreased colon length, decreased intestinal relaxation time in MRI (a surrogate marker of inflammation; Martin et al., 2005), a higher histology score, and decreased survival (Figures 1B–1G). To confirm that the enterocyte-specific loss of ASL also increases colitis severity in an independent IBD model, we induced colitis in Aslf/f and VillinCre:Aslf/f mice by irradiation and engraftment with CX3CR1Cre:Il10raf/f donor bone marrow (BM). Specifically, we had previously shown that macrophage-specific deletion of the receptor for the anti-inflammatory cytokine interleukin-10 (IL-10) results in spontaneous and chronic gut inflammation (Glocker et al., 2009, Zigmond et al., 2014). In an extension of this model, lethally irradiated WT animals that received a BM graft from CX3CR1Cre:Il10ra f/f mice developed spontaneous colitis; hence, this procedure was used by us to generate a genetic model for IBD (Figure S2C). Similarly to DSS-induced colitis, on an arginine-free diet, colitis driven by pro-inflammatory IL-10R-deficient macrophages in VillinCre:Aslf/f mice was exacerbated compared with controls, as evident by higher colitis and histology scores and decreased colon length (Figure S2D). These results, using two distinct models, suggest that colitis severity is at least in part governed by arginine availability, specifically in enterocytes during arginine-deficient states.
The Increased Colitis Severity Associated with ASL Loss in Enterocytes Is Mediated by NO
Although plasma arginine levels were similar, the combination of an arginine-restricted diet and colitis induction resulted in significantly reduced arginine levels in enterocytes of VillinCre:Aslf/f mice (Figure S2E). This suggested that cell-specific deficiency of arginine, and potentially one of its downstream metabolites, could be enhancing inflammation severity. Because arginine is also the substrate for polyamine synthesis via ornithine, and because polyamines have been shown to play a role in colitis (Coëffier et al., 2010, Rath et al., 2014), we measured the intestinal levels of these metabolites as well and found no significant difference between VillinCre:Aslf/f and control mice in the levels of spermidine and spermine or in the spermidine/spermine ratio, which was found to be critical for normal growth and development (Pegg, 2016; Figure S2F). Importantly, sequence analysis confirmed that, on an arginine-free diet without colitis induction, there were no significant differences in microbiome composition between VillinCre:Aslf/f mice and controls (Figure S3). Because we had previously shown a critical role for ASL in NO metabolism (Erez et al., 2011b), we next investigated whether enterocyte-specific arginine deficiency leads to decreased NO synthesis by measuring nitrite, an established NO biomarker (Mian et al., 2013), 10 days after colitis induction. This time point was chosen because, with DSS-induced colitis, the intestine is in the post-inflammatory regenerative phase (Whittem et al., 2010), and, thus, nitrite measurements from intestinal homogenates are expected to be more representative of NO production from enterocytes rather than immune cell infiltrates. We found, in enterocytes of VillinCre:Aslf/f mice, significantly decreased NO levels as well as reduced nitrosylation and decreased iNOS levels, likely because of the specific decrease in arginine levels (Figures 2A–2C). Simultaneously, during colitis, there was increased recruitment of CD45+immune cells to the intestine of VillinCre:Aslf/f animals (Figure 2D). Of note, recruited monocytes/macrophages in VillinCre:Aslf/f mice were phenotypically similar to cells infiltrating the intestine of controls because they were able to upregulate iNOS expression and generate NO in response to DSS at comparable levels (Figures 2E and 2F). These results highlight the requirement of arginine by enterocytes during colitis for NO synthesis and allude to the possibility that enterocyte-restricted NO deficiency might contribute to immune cell recruitment to the gut and, hence, exacerbate inflammation.
Figure 2.

Decreased NO Levels in Enterocytes Increases Colitis Severity
Shown is cell-specific analysis of colon cells from VillinCre:Aslf/f and control Aslf/f mice before and 10 days after colitis induction by DSS.
(A) CD45− cells fluorescence-activated cell sorting (FACS) analysis shows a significant increase in NO levels in the control group after colitis induction compared with VillinCre:Aslf/f mice using 4-amino-5-methylamino-2',7'-difluorofluorescein (DAF-FM) (n = 3 in each group).
(B) ELISA of S-nitrosocysteine expression by CD45– cells (i.e., non-hematopoietic cells), showing no significant increase in nitrosylation after colitis induction in VillinCre:Aslf/f mice.
(C–F) FACS analysis.
(C) A significant increase in iNOS expression in CD45− CD326+ (i.e., colonic enterocytes) in the control group but not in the VillinCre:Aslf/f mice after colitis induction.
(D) Increased recruitment of CD45+ immune cells to the colon of VillinCre:Aslf/f mice compared with control.
(E and F) A similar upregulation of NO (E) and iNOS (F) in CD64+; CD11c+ colon macrophages in VillinCre:Aslf/f mice and in the control group.
Error bars represent SEM.
Reduced NO Production and Colitis Severity in Mice with Immune Cell-Specific ASL Deficiency
Because immune cells were found to have detectable levels of ASL even during basal conditions, no dietary intervention was necessary to reveal differential ASL expression between Vav1Cre: Aslf/f mice and controls; thus, mice were given a regular diet (Figure S1D). Following colitis induction by a standard dose of DSS (1.5%–2.5%), the degree of colonic inflammation was significantly milder in Vav1Cre: Aslf/f mice compared with control littermates (Figures 3A–3E). In addition, although control mice had decreased hemoglobin levels following colitis, Vav1Cre: Aslf/f mice displayed normal hemoglobin levels, likely reflecting decreased blood loss to the stool (Figure 3F). Hence, modulation of ASL levels in a cell type-specific manner differentially affects colitis severity; although ASL produced by enterocytes appears to have a protective role, ASL expression in immune cells exacerbates colonic injury.
Figure 3.

Reduced Colitis Severity in ASL CKO Immune Cells
Colitis was induced by DSS in Vav1Cre: Aslf/f and in control Aslf/f mice.
(A) A representative graph of endoscopic evaluation scores on day 12 after colitis induction, showing a significant improvement in the colitis score of the Vav1Cre: Aslf/f group compared with the control group. The experiment was performed 3 times with a total of n ≥ 20 mice in each group with 3 repetitions. Right: a representative image of the colon taken by colonoscopy.
(B) A representative experiment showing a significant increase in colon length in Vav1Cre: Aslf/f mice compared with the control group. The experiment was performed three times with a total of n ≥ 19 mice in each group.
(C) A T2 map of representative colon sections, demonstrating increased relaxation time in the control group compared with Vav1Cre: Aslf/f mice. Right: quantification of the images using Image-pro Pulse software. The experiment was performed on a total of n = 5 mice in each group.
(D and E) A significant reduction in histological score (D) and a lower mortality rate (E) in Vav1Cre: Aslf/f mice compared with controls.
(F) Anemia developed only in the control group after colitis induction, as shown by decreased red blood cells (RBCs), hemoglobin (HgB), and hematocrit (HCT) levels. No differences between groups were detected in the white blood cell (WBC) count (n = 3 in each group).
Error bars represent SEM.
Increased NO production by immune cells is a well-known phenomenon associated with inflammation (Eiserich et al., 1998, Guzik et al., 2003, Kolios et al., 2004). In addition, it was recently shown that red blood cells (RBCs) control systemic NO bioavailability by synthesizing, transporting, and releasing NO metabolic products (Cortese-Krott and Kelm, 2014). Indeed, following colitis induction, we found a robust increase in nitrite production by RBCs only in control mice and not in Vav1Cre: Aslf/f animals (Figure 4A). To confirm that the effect on the inflammatory cascade is caused specifically by decreased NO production by the immune cells of the mutant mice, we conducted ex vivo studies with peritoneal macrophages isolated from the Vav1Cre: Aslf/f mice, the major immune cell type producing NO (Lorsbach et al., 1993), and measured their response to lipopolysaccharide (LPS). Similar to the finding at the whole-organism level, Vav1Cre:Aslf/f macrophages produced less nitrite in response to LPS (Figure 4B). Lately, several studies have shown that, in addition to the paracrine effects of macrophage-derived NO on other cells, NO has an autocrine effect in polarizing macrophages toward a pro-inflammatory (or “M1”) state. The metabolic mechanism behind this phenomenon was proposed to be a prominent induction of glycolysis (Kelly and O’Neill, 2015, Vatassery et al., 2004). We hence measured lactate and glucose levels in the medium of cultured peritoneal macrophages as a readout for macrophage activation (Bustos and Sobrino, 1992). Although macrophages of control mice showed decreased glucose levels and increased lactate production, consistent with increased glycolysis, no such changes were observed in Vav1Cre: Aslf/f macrophages, suggesting their decreased activation (Figures 4C and 4D). Furthermore, colon histology after induction of colitis revealed decreased recruitment of activated monocytes/macrophages to the intestine in immune Asl CKO mice, as demonstrated by a decreased ratio between the general number of macrophages (marked with F-4/80) and the number of activated macrophages (marked with MAC-2) (Rosenberg et al., 1991; Figure 4E). To specifically investigate intestinal macrophages, we analyzed the levels of iNOS and NO in gut hematopoietic cells (CD45+) and enterocytes (CD326+) (King et al., 2012). As expected, we found ASL levels to be significantly lower in intestinal CD45+ cells of Vav1Cre:Aslf/f mice, correlating with decreased iNOS expression and decreased NO production; in contrast, there were no significant differences in these parameters in enterocytes of the same animals (Figures S4A–S4F). To corroborate these results, we generated CD11cCre:Aslf/f mice and induced DSS colitis in these animals. Challenged CD11cCre:Aslf/f mice displayed a decrease in weight loss, colitis scores, and pro-inflammatory cytokines and an increase in colon length, all supporting ameliorated colitis (Figures 4F–4H). Collectively, our data suggest that loss of ASL in enterocytes has a significant effect on increasing the severity of inflammation, whereas loss of ASL in immune cells, and in particular CD11c+ mononuclear phagocytes, alleviates colitis severity, likely because of alterations in NO levels. Interestingly, analysis of the GEO database revealed that ASL expression is increased in chronic IBD patients with Crohn’s disease or ulcerative colitis compared with controls. Moreover, in contrast to the expression of the rate-limiting enzyme in polyamine synthesis (i.e., ornithine decarboxylase [ODC]), ASL expression correlates with iNOS expression (Figure 4I; Figure S4J). These data provide further support for our assumption of the importance of ASL re-expression in enterocytes during colitis for NO synthesis and for the potential relevance of our findings to human IBD.
Figure 4.

Decreased NO Levels in ASL CKO Immune Cells
(A) Nitrite (NO2−) levels were measured by dedicated high-performance liquid chromatography (HPLC) in blood samples before and after colitis induction. Results show a significant elevation in NO levels after colitis induction in RBCs of the control group but not in Vav1Cre: Aslf/f mice (n ≥ 3 in each group).
(B) A significant elevation in NO2− levels is seen in peritoneal macrophages growth medium 18 hr following LPS administration in the control group compared with the Vav1Cre: Aslf/f group (n = 6 in each group).
(C and D) A significant reduction in glucose levels (C) and elevation in lactate levels (D) in response to LPS administration is demonstrated in control but not in Vav1Cre: Aslf/f macrophages. Measurements were performed using a NOVA measuring instrument, (n ≥ 4 in each group).
(E) Immunohistochemistry staining for the total number of macrophages (using the F4-80 marker, top) as well as for activated macrophages (using the MAC2 marker of activation, bottom), in distal colon sections after colitis induction. The ratio between F4-80- and MAC2-positive cells shows a significant reduction in the number of activated macrophages in Vav1Cre: Aslf/f mice compared with controls. Cells were counted from at least 8 microscopic fields taken from 3 different animals. Right: quantification analysis.
(F and G) Colitis was induced by DSS in CD11cCre:Aslf/f and in control Aslf/f mice.
(F) Right: a representative graph showing that CD11cCre:Aslf/f mice have decreased colitis severity compared with control mice, as demonstrated by endoscopic evaluation on day 12 after colitis induction. The experiment was performed on a total of n ≥ 13 mice in each group. Left: a representative image of the colon taken by colonoscopy.
(G) A representative graph showing a significant increase in colon length in CD11cCre:Aslf/f mice compared with the control group. The experiment was performed on a total of n ≥ 10 mice in each group.
(H) ELISA measurement of colon tissues extracted from mice on day12 following colitis induction, showing significant elevation of pro-inflammatory cytokine levels in the control group compared with CD11cCre:Aslf/f mice.
(I) Human data taken from a GEO dataset analysis (GEO: GSE57945; ID:200057945), showing significant upregulation of ASL and iNOS RNA expression in patients suffering from Crohn’s disease (CD) and ulcerative colitis (UC) compared with controls, with no significant change in ornithine decarboxylase (ODC) expression.
Error bars represent SEM.
Inducing Endogenous NO Production by Enterocytes Alleviates Colitis and Inflammation-Associated Colon Cancer
To investigate whether enhancing NO production can alleviate colitis severity, we induced colitis in WT mice and, 6 days later, examined the response to different NO-related drug interventions. Mice were supplemented with either arginine or citrulline to support NOS-dependent NO generation or with sodium nitrite, an NOS-independent NO supplement. All three interventions led to significant colitis amelioration, as evaluated by change in weight, endoscopy score, colon length, and histology score (Figures 5A–5D). Among these interventions, citrulline induced ASL expression the most (Figure 5E), likely because it metabolically drives ASL activity by increasing its substrate (Wijnands et al., 2012). Furthermore, intestines of mice treated with citrulline showed increased NO generation by enterocytes and decreased apoptosis, correlating with decreased colitis severity (Figures 5F and 5G). Thus, we sought to further investigate the enterocyte-specific induction of ASL and NO using citrulline.
Figure 5.

NO-Related Metabolites Alleviate Colitis Severity
Treatment with arginine pathway-related metabolites leads to a significant improvement in colitis severity. Colitis was induced in wild-type C57BL/6J.OlaHsd mice. Mice were treated with DSS for 6 days, followed by administration of either arginine solution in drinking water, citrulline, or NaNO2 or with water as a control for an additional 6 days.
(A) Significant weight loss was documented in the control group compared with the three treatments (n ≥ 25 in each group, experiments were repeated at least three times).
(B) Endoscopic evaluation shows a significant improvement in colitis score on day 12 for all 3 treatments. In comparison with arginine, citrulline was more beneficial (n ≥ 25 in each group). Bottom: a representative image of the colon taken by colonoscopy.
(C) A representative plot showing a significant reduction in colon length in the control group compared with the treated colons (n ≥ 15 in each group).
(D) The histological score for colitis was significantly lower in the treated colons compared with controls.
(E) Immunohistochemistry staining of colons for ASL shows higher expression after citrulline treatment compared with other treatments.
(F) FACS analysis showing that citrulline treatment is more effective than arginine in inducing NO synthesis in enterocytes.
(G) Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining showing increased apoptosis in control colons compared with colons taken from mice treated with citrulline. Bottom: a quantification graph generated using Image-Pro Pulse software.
Error bars represent SEM.
To boost ASL expression directly, we performed a screen for Food and Drug Administration (FDA)-approved small molecules that increase ASL transcription using the non-biased computational method Mantra, which has been previously developed by us (Iorio et al., 2010). In several cancer cell lines, the flavanol molecule fisetin (3,7,3′,4′-tetrahydroxyflavone) was found to significantly upregulate the RNA expression levels of ASS1 and ASL, which are both required for arginine synthesis from citrulline (Table S1). Fisetin is commonly found in many fruits and vegetables and has been shown to have an anti-inflammatory property (Sahu et al., 2016). We hence aimed to understand its specific effect on enterocytes. Encouragingly, in vivo supplementation with fisetin upregulated ASL and ASS1 expression in enterocytes (Figure 6A). Moreover, ASL induction correlated with increased iNOS expression and NO production levels specifically in enterocytes (Figures 6B and 6C; Figures S5A–S5F). Importantly, fisetin therapy alleviated colitis severity in WT mice with respect to all parameters compared with NO donors; however, the compound had no effect in VillinCre:Aslf/f mice harboring the enterocyte-specific ASL deficiency. This establishes that it is the specific upregulation of ASL in the intestinal epithelium that is ameliorating colitis (Figures 6D–6F).
Figure 6.

Induction of Endogenous NO Production by Enterocytes Alleviates Colitis
(A) Immunohistochemistry staining of distal colon sections after colitis induction shows up-regulation of ASL and ASS1 expression following fisetin treatment.
(B and C) FACS analysis of colon cells from mice treated with fisetin after colitis induction shows a significant increase in NO levels (B) and in iNOS expression (C) compared with cells from untreated controls.
(D–F) Colitis was induced in enterocyte CKO mice and littermate Cre−/− controls. Mice were treated with DSS for 5 days, followed by administration of either NO donors (NaNO2) or fisetin (n ≥ 6 in each group). Although NO donors were beneficial to both groups, fisetin treatment was not effective in VillinCre: Aslf/f mice, as shown by (D) decreased survival, (E) decreased body weight gain, and (F) a higher colitis score compared with controls.
Error bars represent SEM.
Finally, because citrulline is the upstream metabolic substrate for ASS1, we expected that combining citrulline with fisetin would have a synergistic effect. Indeed, supplementing WT mice on a regular diet with fisetin together with citrulline was the most effective modality to generate NO in enterocytes in both the chemically and genetically induced colitis models (Figures 7A–7F). Improved epithelial integrity following increased NO production by fisetin and citrulline in the genetic model of colitis was corroborated by significantly decreased plasma levels of fluorescein isothiocyanate (FITC)-conjugated dextran (Figure 7G). To confirm the human relevance of our findings, we measured conductivity in Caco-2 human epithelial cells (Araki et al., 2006) following exposure to DSS and different drug regimens. In support of our conclusions from the mouse models, we found that the combined treatment improved conductivity, reflecting preservation of the epithelial barrier (Sambuy et al., 2005; Figure 7H).
Figure 7.

Enterocyte Self-Regulation of NO Levels Is the Most Beneficial Treatment for Colitis
Colitis was induced in C57BL/6J.OlaHsd mice with DSS for 6 days, followed by administration of either intraperitoneal (i.p.) fisetin, citrulline solution in drinking water, a combination of both treatments, or only DMSO as a control; the experiment was repeated 3 times.
(A) FACS analysis of colon epithelial cells from mice treated with the combined treatment shows a significant increase in NO levels compared with the control group (n = 4 in each group).
(B) A significant weight loss is documented in the control group compared with the treated mice (n ≥ 20 in each group).
(C) An endoscopic evaluation demonstrates a significant improvement on day 12 in the colitis score of the treated groups compared with the control. Among the treatments, the combined treatment was the most beneficial (n ≥ 20 in each group).
(D) A significant longer colon length is seen in the treated groups compared with the control. In addition, the colon is significantly longer with combined treatment compared with each treatment separately (n = 15 in both groups).
(E) A significant reduction in histological score in all treatment groups compared with the control.
(F and G) Colitis was induced by bone marrow (BM) implantation from either WT or CX3CR1Cre:Il10rafl/fl donors to lethally irradiated Aslf/f mice. Two weeks after transplantation, mice were treated with a combination of citrulline and fisetin and compared with control animals treated with DMSO only. Colitis severity was evaluated 6 weeks after the implantation.
(F) No colitis signs were observed in animals implanted with WT donor BM. In animals implanted with CX3CR1Cre:Il10rafl/fl BM, a significant reduction in colitis severity was seen in treated animals compared with control mice, as shown by reduced weight loss (top left), a reduced colitis score (top right), increased colon length (bottom left), and a reduced histology score (bottom right) (n > 5 in each group, experiments were repeated twice).
(G) In vivo intestinal permeability assay to assess epithelial barrier function, performed using FITC-labeled dextran, showing significantly decreased permeability in treated mice compared with controls.
(H) Caco-2 cells plotted for resistance percentage over time, showing a significant decrease in conductivity of the control group compared with the group treated with citrulline and fisetin (the experiment included at least 5 biological repetitions).
Error bars represent SEM.
Because colitis severity has been shown to predispose to colon cancer development (Barral et al., 2016), we used the DSS-azoxymethane (AOM) model for colitis-associated cancer (Thaker et al., 2012). Although there was no significant difference in cancer severity between Aslf/f and Vav1Cre: Aslf/f (data not shown), VillinCre: Aslf/f mice, even when given a regular diet, displayed a higher tumor burden, reflected by increased numbers and sizes of tumors, again supporting the protective effect of ASL upregulation in chronic inflammation (Figures 8A and 8B). Importantly, treatment of WT mice following the AOM-DSS protocol with our combined citrulline/fisetin treatment reduced colon cancer severity in all parameters evaluated (Figures 8C and 8D).
Figure 8.

Metabolic Treatment Decreases Colitis Severity and Inflammation-Associated Colon Cancer
(A and B) The inflammation-associated cancer model was generated twice using the AOM-DSS model.
(A) A significant elevation in tumor score was seen in the VillinCre:Aslf/f group compared with the control. Right: a representative image of the colon taken by colonoscopy.
(B) A significant increase in tumor number was seen in the VillinCre:Aslf/f group compared with control mice.
(C and D) C57BL/6J.OlaHsd mice were treated with the AOM-DSS protocol. In addition, mice were treated for 15 days with either fisetin i.p., citrulline in drinking water, a combination of both treatments, or DMSO as a control. A significant elevation in tumor score (C) and tumor number (D) was seen in the treated groups compared with the control. The combined treatment was more beneficial compared with each treatment by itself (n ≥ 6 in each group).
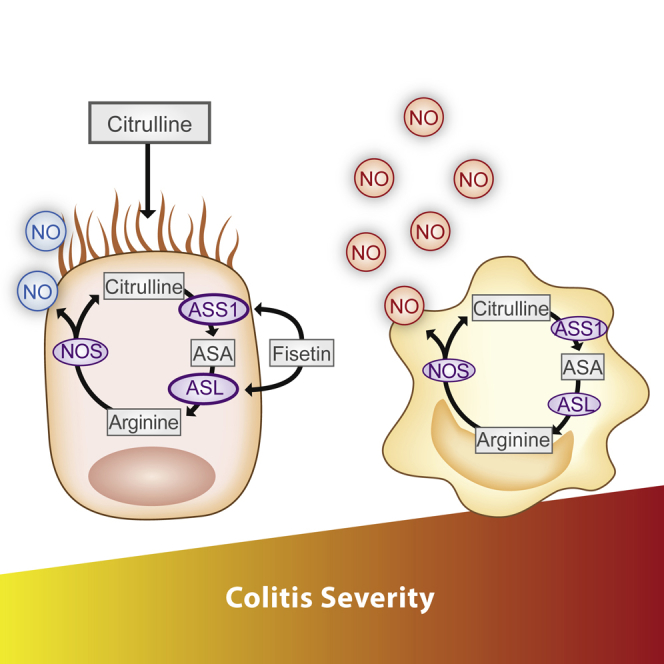
(E) A summary illustration demonstrating the distinct role of NO in each cell type. Although NO produced by macrophages is harmful, NO generated by enterocytes is protective. Colitis severity is reduced maximally by metabolically activating ASL in enterocytes using citrulline and fisetin supplementation.
Error bars represent SEM.
Collectively, our results show that NO levels can be metabolically modulated specifically in enterocytes by regulation of ASL function, which increases endogenous NO production at the precise amount and dosages needed to strengthen the epithelial barrier and, hence, mitigate colitis and decrease inflammation-associated colon cancer (Figure 8E).
Discussion
IBD causes significant disease burden on affected individuals and their families and on the health care system (Limsrivilai et al., 2017). Treatment is typically long-term or requires surgery, and, hence, adjuvant therapy that specifically targets key pathogenic mechanisms would be of significant value. NO has been known to play important but ambivalent roles in IBD (Cross and Wilson, 2003, Kolios et al., 2004). Indeed, in different models of colitis, inhibition of NO increased tissue injury, whereas, in other models, inhibition of NO provided benefits (Kolios et al., 2004). Similarly, administration of NOS inhibitors in general and of iNOS specifically for colitis treatment gave inconsistent results (Kubes, 2000, Miller et al., 1993, Miller et al., 1994). To add to this complexity, NO has been shown to play different roles along the colitis course, both in the inflammatory phase (Oshima et al., 2001) as well as in the regenerative phase (Aoi et al., 2008). These paradoxical and opposing results prevent implementation of NO-related drugs as a treatment regimen for colitis, and it is clear that there is a need to dissect the cell-specific contributions of NO in causation of IBD (Soliman and Mazzio, 1998). Because ASL is essential for NO production by all NOSs (Erez et al., 2011b), we abolished its expression in a cell-specific manner and defined a distinctive role for NO in each cell type.
Although the role of NO in promoting the immune response during colitis has been well established (Kolios et al., 2004), recent studies using different animal models propose that intestinal epithelial cells upregulate iNOS and NO levels against invading virulent pathogens as a host defense mechanism against intestinal infections (Meyerhoff et al., 2012, Vareille et al., 2008). Indeed, we find that promoting NO synthesis by elevating ASL expression with fisetin and citrulline was beneficial against colitis because it strengthened the epithelial barrier and prevented immune cell infiltration.
Emerging metabolic strategies have shown promise as treatments for inflammation and cancer by reducing the availability of glucose and glutamine levels, whereas therapies that increase inflammation and energy metabolites in the cancer microenvironment have been demonstrated to enhance tumor progression (Seyfried et al., 2015). We show here that boosting the metabolic ability of the enterocytes to synthesize and adjust the level and localization of NO to the exact amount and place where it is needed is the most beneficial treatment for reducing colitis severity. Because the duration, severity, and extent of colitis have all been shown to increase the risk for colon cancer development (Dyson and Rutter, 2012), not surprisingly, our combination of citrulline/fisetin also decreased inflammation-associated colon cancer.
Broadly, our study serves as a proof of concept for three main ideas. First, it confirms that regulating ASL allows metabolic regulation of NO in a cell-specific manner. Second, it shows that, during disease, cell-autonomous production of NO by iNOS can be protective as part of the intestinal innate immune response. Last, our study demonstrates the superior metabolic advantage of supplementing with substrates and upregulating the relevant metabolic enzymes over giving the deficient molecule directly as therapy. Because both citrulline and fisetin are nutraceutical agents that are available as supplements, the findings from this study could be translated rather straightforward into the human context.
Experimental Procedures
Animals
Mice were monitored daily by staff and veterinary personnel for health and activity. Mice were given ad libitum access to water and standard mouse chow with 12-hr light/dark cycles. Colonies were maintained in a specific pathogen-free (SPF) barrier facility, with quarterly testing of pathogens in sentinel animals housed in the same room. All laboratory staff wore personal protective clothing, and all manipulations were performed in biosafety cabinets in procedure rooms in the same animal suite. All animal procedures were approved by the Institutional Animal Care and Use Committee (applications 07291113-2, 08020114-2, 24330116-3, 15990115-1, and 24340116-2) and performed in strict adherence to Weizmann Institute Animal Care and Use guidelines, following the NIH, European Commission, and Israeli guidelines. Mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). B6.Cg-Tg(Vav1-icre)A2Kio/J and B6.SJL-Tg(Vil-cre0997 Gum/J. C57BL/6JOlaHsd mice were purchased from ENVIGO RMS (ISRAEL). The B6.Cg-Tg (Itgax-cre)1-1Reiz/J. Cre mice were received from Prof. Jung.
Colitis induction experiments were conducted on 8- to 10-week-old male mice. Colitis-associated cancer experiments were conducted on 10- to 12-week-old female mice. Histologic sections of pups were taken from 10-day-old pups. Generation of BM chimeric mice was conducted on 10- to 12-week-old male mice.
Statistics
All statistical analyses were performed using one-way ANOVA, two-way ANOVA, repeated measures ANOVA, or Student’s t test. Contrasts comparing groups with one specific treatment or control were tested with Dunnett’s test. Log-transformed data were used where differences in variance were significant, and variances were correlated with means. Sample sizes were chosen in advance on the basis of common practice of the described experiment and are mentioned for each experiment. No statistical methods were used to predetermine sample size. Each experiment was conducted with biological and technical replicates and with three replications of the entire experiment unless specified otherwise. Statistical tests were done using Statsoft’s STATISTICA, version 10. All error bars represent SE. p < 0.05 was considered significant in all analyses (∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.0005). A survival analysis, accounting for genotype and batch, was conducted using a Cox proportional hazards model (using the “survival” package in R).
Acknowledgments
We acknowledge and thank the Weizmann Institute for providing financial and infrastructural support. We are very thankful for the intellectual input of Prof. Brendan Lee and Prof. Eran Elinav. We appreciate the support given by Prof. Shimon Harrus, the statistical analysis by R. Rotkopf, and the technical contributions of Yuval Moshe, Yulia Frug, Amir Marinov, Sivan Galai, Jung-Seok Kim, and Sivan Pinto. A.E. is the incumbent of the Leah Omenn Career Development Chair and is supported by research grants from the European Research Program (CIG618113 and ERC614204), the Israel Science Foundation (1343/13 and 1952/13), and a Minerva grant award (711730). A.E. received additional support from the Adelis Foundation, the Henry S. and Anne S. Reich Research Fund, the Dukler Fund for Cancer Research, the Paul Sparr Foundation, the Saul and Theresa Esman Foundation, Joseph Piko Baruch, and the estate of Fannie Sherr. A. Sarver was supported by the Israel Cancer Research Fund (ICRF 711982). N.B.-P was supported by the European Research Council (IEMTx), D.d.B. and N.B.-P. was supported by Fondazione Telethon, and S.C.S.N. was supported by Baylor College of Medicine Intellectual and Developmental Disabilities Research Center Grant 1 U54 HD083092 and by the Doris Duke Charitable Foundation (DDCF 2013095).
Author Contributions
N.S. conducted and was in charge of all experiments. C.R. and S.G.-C. conducted the FACS analysis. B.B. helped to generate the IL-10R-deficient colitis model. A. Sarver helped with NO analysis. J.F. preformed ELISA and cell culture experiments. A. Silberman and A.B. helped with the metabolic analysis. N.N.C.-N. analyzed human data. R.E. performed immunohistochemistry staining. I.B. performed MRI. M.P.-F. and N.Z. preformed 16S rRNA analysis. K.B.H. and S.I. performed the FISH experiments. R.M. and G.D. helped with the colonoscopy. N.N.C.-N. performed protein assays. D.d.B. and N.B.-P. performed the computational drug analysis. M.H.P. and S.C.S.N. gave scientific input. A.H. performed all pathological analyses and gave scientific input. S.J. helped design the studies and with writing the manuscript. A.E. was the leading scientist who initiated the study and wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: May 15, 2018
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, five figures, and one table and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.04.053.
Supplemental Information
References
- Alican I., Kubes P. A critical role for nitric oxide in intestinal barrier function and dysfunction. Am. J. Physiol. 1996;270:G225–G237. doi: 10.1152/ajpgi.1996.270.2.G225. [DOI] [PubMed] [Google Scholar]
- Aoi Y., Terashima S., Ogura M., Nishio H., Kato S., Takeuchi K. Roles of nitric oxide (NO) and NO synthases in healing of dextran sulfate sodium-induced rat colitis. J. Physiol. Pharmacol. 2008;59:315–336. [PubMed] [Google Scholar]
- Araki Y., Sugihara H., Hattori T. In vitro effects of dextran sulfate sodium on a Caco-2 cell line and plausible mechanisms for dextran sulfate sodium-induced colitis. Oncol. Rep. 2006;16:1357–1362. [PubMed] [Google Scholar]
- Atreya R., Neurath M.F. IBD pathogenesis in 2014: Molecular pathways controlling barrier function in IBD. Nat. Rev. Gastroenterol. Hepatol. 2015;12:67–68. doi: 10.1038/nrgastro.2014.201. [DOI] [PubMed] [Google Scholar]
- Barral M., Dohan A., Allez M., Boudiaf M., Camus M., Laurent V., Hoeffel C., Soyer P. Gastrointestinal cancers in inflammatory bowel disease: An update with emphasis on imaging findings. Crit. Rev. Oncol. Hematol. 2016;97:30–46. doi: 10.1016/j.critrevonc.2015.08.005. [DOI] [PubMed] [Google Scholar]
- Blaise G.A., Gauvin D., Gangal M., Authier S. Nitric oxide, cell signaling and cell death. Toxicology. 2005;208:177–192. doi: 10.1016/j.tox.2004.11.032. [DOI] [PubMed] [Google Scholar]
- Boughton-Smith N.K., Evans S.M., Hawkey C.J., Cole A.T., Balsitis M., Whittle B.J., Moncada S. Nitric oxide synthase activity in ulcerative colitis and Crohn’s disease. Lancet. 1993;342:338–340. doi: 10.1016/0140-6736(93)91476-3. [DOI] [PubMed] [Google Scholar]
- Brown J.F., Keates A.C., Hanson P.J., Whittle B.J. Nitric oxide generators and cGMP stimulate mucus secretion by rat gastric mucosal cells. Am. J. Physiol. 1993;265:G418–G422. doi: 10.1152/ajpgi.1993.265.3.G418. [DOI] [PubMed] [Google Scholar]
- Bustos R., Sobrino F. Stimulation of glycolysis as an activation signal in rat peritoneal macrophages. Effect of glucocorticoids on this process. Biochem. J. 1992;282:299–303. doi: 10.1042/bj2820299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caton M.L., Smith-Raska M.R., Reizis B. Notch-RBP-J signaling controls the homeostasis of CD8- dendritic cells in the spleen. J. Exp. Med. 2007;204:1653–1664. doi: 10.1084/jem.20062648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coëffier M., Marion-Letellier R., Déchelotte P. Potential for amino acids supplementation during inflammatory bowel diseases. Inflamm. Bowel Dis. 2010;16:518–524. doi: 10.1002/ibd.21017. [DOI] [PubMed] [Google Scholar]
- Cooper H.S., Murthy S.N., Shah R.S., Sedergran D.J. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab. Invest. 1993;69:238–249. [PubMed] [Google Scholar]
- Cortese-Krott M.M., Kelm M. Endothelial nitric oxide synthase in red blood cells: key to a new erythrocrine function? Redox Biol. 2014;2:251–258. doi: 10.1016/j.redox.2013.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross R.K., Wilson K.T. Nitric oxide in inflammatory bowel disease. Inflamm. Bowel Dis. 2003;9:179–189. doi: 10.1097/00054725-200305000-00006. [DOI] [PubMed] [Google Scholar]
- De Jonge W.J., Dingemanse M.A., de Boer P.A.J., Lamers W.H., Moorman A.F.M. Arginine-metabolizing enzymes in the developing rat small intestine. Pediatr. Res. 1998;43:442–451. doi: 10.1203/00006450-199804000-00002. [DOI] [PubMed] [Google Scholar]
- de Souza H.S.P., Fiocchi C. Immunopathogenesis of IBD: current state of the art. Nat. Rev. Gastroenterol. Hepatol. 2016;13:13–27. doi: 10.1038/nrgastro.2015.186. [DOI] [PubMed] [Google Scholar]
- Dyson J.K., Rutter M.D. Colorectal cancer in inflammatory bowel disease: what is the real magnitude of the risk? World J. Gastroenterol. 2012;18:3839–3848. doi: 10.3748/wjg.v18.i29.3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiserich J.P., Hristova M., Cross C.E., Jones A.D., Freeman B.A., Halliwell B., van der Vliet A. Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. Nature. 1998;391:393–397. doi: 10.1038/34923. [DOI] [PubMed] [Google Scholar]
- Erez A., Nagamani S.C., Lee B. Argininosuccinate lyase deficiency-argininosuccinic aciduria and beyond. Am. J. Med. Genet. C. Semin. Med. Genet. 2011;157C:45–53. doi: 10.1002/ajmg.c.30289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erez A., Nagamani S.C.S., Shchelochkov O.A., Premkumar M.H., Campeau P.M., Chen Y., Garg H.K., Li L., Mian A., Bertin T.K. Requirement of argininosuccinate lyase for systemic nitric oxide production. Nat. Med. 2011;17:1619–1626. doi: 10.1038/nm.2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glocker E.-O., Kotlarz D., Boztug K., Gertz E.M., Schäffer A.A., Noyan F., Perro M., Diestelhorst J., Allroth A., Murugan D. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N. Engl. J. Med. 2009;361:2033–2045. doi: 10.1056/NEJMoa0907206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzik T.J., Korbut R., Adamek-Guzik T. Nitric oxide and superoxide in inflammation and immune regulation. J. Physiol. Pharmacol. 2003;54:469–487. [PubMed] [Google Scholar]
- Iorio F., Bosotti R., Scacheri E., Belcastro V., Mithbaokar P., Ferriero R., Murino L., Tagliaferri R., Brunetti-Pierri N., Isacchi A., di Bernardo D. Discovery of drug mode of action and drug repositioning from transcriptional responses. Proc. Natl. Acad. Sci. USA. 2010;107:14621–14626. doi: 10.1073/pnas.1000138107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly B., O’Neill L.A.J. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015;25:771–784. doi: 10.1038/cr.2015.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King J.B., von Furstenberg R.J., Smith B.J., McNaughton K.K., Galanko J.A., Henning S.J. CD24 can be used to isolate Lgr5+ putative colonic epithelial stem cells in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012;303:G443–G452. doi: 10.1152/ajpgi.00087.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles R.G., Moncada S. Nitric oxide synthases in mammals. Biochem. J. 1994;298:249–258. doi: 10.1042/bj2980249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolios G., Valatas V., Ward S.G. Nitric oxide in inflammatory bowel disease: a universal messenger in an unsolved puzzle. Immunology. 2004;113:427–437. doi: 10.1111/j.1365-2567.2004.01984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubes P. Inducible nitric oxide synthase: a little bit of good in all of us. Gut. 2000;47:6–9. doi: 10.1136/gut.47.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limsrivilai J., Stidham R.W., Govani S.M., Waljee A.K., Huang W., Higgins P.D.R. Factors that predict high health care utilization and costs for patients with inflammatory bowel diseases. Clin. Gastroenterol. Hepatol. 2017;15:385–392.e2. doi: 10.1016/j.cgh.2016.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorsbach R.B., Murphy W.J., Lowenstein C.J., Snyder S.H., Russell S.W. Expression of the nitric oxide synthase gene in mouse macrophages activated for tumor cell killing. Molecular basis for the synergy between interferon-gamma and lipopolysaccharide. J. Biol. Chem. 1993;268:1908–1913. [PubMed] [Google Scholar]
- Madison B.B., Dunbar L., Qiao X.T., Braunstein K., Braunstein E., Gumucio D.L. Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J. Biol. Chem. 2002;277:33275–33283. doi: 10.1074/jbc.M204935200. [DOI] [PubMed] [Google Scholar]
- Marini J.C., Agarwal U., Didelija I.C. Dietary arginine requirements for growth are dependent on the rate of citrulline production in mice. J. Nutr. 2015;145:1227–1231. doi: 10.3945/jn.114.209668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D.R., Danrad R., Herrmann K., Semelka R.C., Hussain S.M. Magnetic resonance imaging of the gastrointestinal tract. Top. Magn. Reson. Imaging. 2005;16:77–98. doi: 10.1097/01.rmr.0000179461.55234.7d. [DOI] [PubMed] [Google Scholar]
- Meyerhoff R.R., Nighot P.K., Ali R.A., Blikslager A.T., Koci M.D. Characterization of turkey inducible nitric oxide synthase and identification of its expression in the intestinal epithelium following astrovirus infection. Comp. Immunol. Microbiol. Infect. Dis. 2012;35:63–69. doi: 10.1016/j.cimid.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mian A.I., Aranke M., Bryan N.S. Nitric oxide and its metabolites in the critical phase of illness: rapid biomarkers in the making. Open Biochem. J. 2013;7:24–32. doi: 10.2174/1874091X01307010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton S.J., Shorthouse M., Hunter J.O. Increased nitric oxide synthesis in ulcerative colitis. Lancet. 1993;341:465–466. doi: 10.1016/0140-6736(93)90211-x. [DOI] [PubMed] [Google Scholar]
- Miller M.J., Chotinaruemol S., Sadowska-Krowicka H., Kakkis J.L., Munshi U.K., Zhang X.J., Clark D.A. Nitric oxide: the Jekyll and Hyde of gut inflammation. Agents Actions. 1993;39:C180–C182. doi: 10.1007/BF01972759. [DOI] [PubMed] [Google Scholar]
- Miller M.J., Munshi U.K., Sadowska-Krowicka H., Kakkis J.L., Zhang X.J., Eloby-Childress S., Clark D.A. Inhibition of calcium-dependent nitric oxide synthase causes ileitis and leukocytosis in guinea pigs. Dig. Dis. Sci. 1994;39:1185–1192. doi: 10.1007/BF02093782. [DOI] [PubMed] [Google Scholar]
- Moncada S. The 1991 Ulf von Euler Lecture. The L-arginine: nitric oxide pathway. Acta Physiol. Scand. 1992;145:201–227. doi: 10.1111/j.1748-1716.1992.tb09359.x. [DOI] [PubMed] [Google Scholar]
- Nagamani S.C., Erez A., Lee B. Argininosuccinate lyase deficiency. Genet. Med. 2012;14:501–507. doi: 10.1038/gim.2011.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogilvy S., Elefanty A.G., Visvader J., Bath M.L., Harris A.W., Adams J.M. Transcriptional regulation of vav, a gene expressed throughout the hematopoietic compartment. Blood. 1998;91:419–430. [PubMed] [Google Scholar]
- Oshima T., Jordan P., Grisham M.B., Alexander J.S., Jennings M., Sasaki M., Manas K. TNF-alpha induced endothelial MAdCAM-1 expression is regulated by exogenous, not endogenous nitric oxide. BMC Gastroenterol. 2001;1:5. doi: 10.1186/1471-230X-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegg A.E. Functions of Polyamines in Mammals. J. Biol. Chem. 2016;291:14904–14912. doi: 10.1074/jbc.R116.731661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premkumar M.H., Sule G., Nagamani S.C., Chakkalakal S., Nordin A., Jain M., Ruan M.Z., Bertin T., Dawson B., Zhang J. Argininosuccinate lyase in enterocytes protects from development of necrotizing enterocolitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2014;307:G347–G354. doi: 10.1152/ajpgi.00403.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath M., Müller I., Kropf P., Closs E.I., Munder M. Metabolism via Arginase or Nitric Oxide Synthase: Two Competing Arginine Pathways in Macrophages. Front. Immunol. 2014;5:532. doi: 10.3389/fimmu.2014.00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg I., Cherayil B.J., Isselbacher K.J., Pillai S. Mac-2-binding glycoproteins. Putative ligands for a cytosolic beta-galactoside lectin. J. Biol. Chem. 1991;266:18731–18736. [PubMed] [Google Scholar]
- Sahu B.D., Kumar J.M., Sistla R. Fisetin, a dietary flavonoid, ameliorates experimental colitis in mice: Relevance of NF-κB signaling. J. Nutr. Biochem. 2016;28:171–182. doi: 10.1016/j.jnutbio.2015.10.004. [DOI] [PubMed] [Google Scholar]
- Sambuy Y., De Angelis I., Ranaldi G., Scarino M.L., Stammati A., Zucco F. The Caco-2 cell line as a model of the intestinal barrier: influence of cell and culture-related factors on Caco-2 cell functional characteristics. Cell Biol. Toxicol. 2005;21:1–26. doi: 10.1007/s10565-005-0085-6. [DOI] [PubMed] [Google Scholar]
- Seyfried T.N., Flores R., Poff A.M., D’Agostino D.P., Mukherjee P. Metabolic therapy: a new paradigm for managing malignant brain cancer. Cancer Lett. 2015;356(2 Pt A):289–300. doi: 10.1016/j.canlet.2014.07.015. [DOI] [PubMed] [Google Scholar]
- Shah V., Lyford G., Gores G., Farrugia G. Nitric oxide in gastrointestinal health and disease. Gastroenterology. 2004;126:903–913. doi: 10.1053/j.gastro.2003.11.046. [DOI] [PubMed] [Google Scholar]
- Soliman K.F.A., Mazzio E.A. In vitro attenuation of nitric oxide production in C6 astrocyte cell culture by various dietary compounds. Proc. Soc. Exp. Biol. Med. 1998;218:390–397. doi: 10.3181/00379727-218-44309. [DOI] [PubMed] [Google Scholar]
- Soufli I., Toumi R., Rafa H., Touil-Boukoffa C. Overview of cytokines and nitric oxide involvement in immuno-pathogenesis of inflammatory bowel diseases. World J. Gastrointest. Pharmacol. Ther. 2016;7:353–360. doi: 10.4292/wjgpt.v7.i3.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suschek C.V., Schnorr O., Kolb-Bachofen V. The role of iNOS in chronic inflammatory processes in vivo: is it damage-promoting, protective, or active at all? Curr. Mol. Med. 2004;4:763–775. doi: 10.2174/1566524043359908. [DOI] [PubMed] [Google Scholar]
- Thaker A.I., Shaker A., Rao M.S., Ciorba M.A. Modeling colitis-associated cancer with azoxymethane (AOM) and dextran sulfate sodium (DSS) J. Vis. Exp. 2012;67:4100. doi: 10.3791/4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Lugt B., Khan A.A., Hackney J.A., Agrawal S., Lesch J., Zhou M., Lee W.P., Park S., Xu M., DeVoss J. Transcriptional programming of dendritic cells for enhanced MHC class II antigen presentation. Nat. Immunol. 2014;15:161–167. doi: 10.1038/ni.2795. [DOI] [PubMed] [Google Scholar]
- Vareille M., Rannou F., Thélier N., Glasser A.L., de Sablet T., Martin C., Gobert A.P. Heme oxygenase-1 is a critical regulator of nitric oxide production in enterohemorrhagic Escherichia coli-infected human enterocytes. J. Immunol. 2008;180:5720–5726. doi: 10.4049/jimmunol.180.8.5720. [DOI] [PubMed] [Google Scholar]
- Vatassery G.T., SantaCruz K.S., DeMaster E.G., Quach H.T., Smith W.E. Oxidative stress and inhibition of oxidative phosphorylation induced by peroxynitrite and nitrite in rat brain subcellular fractions. Neurochem. Int. 2004;45:963–970. doi: 10.1016/j.neuint.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Whittem C.G., Williams A.D., Williams C.S. Murine colitis modeling using dextran sulfate sodium (DSS) J. Vis. Exp. 2010;35:1652. doi: 10.3791/1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijnands K.A.P., Vink H., Briedé J.J., van Faassen E.E., Lamers W.H., Buurman W.A., Poeze M. Citrulline a more suitable substrate than arginine to restore NO production and the microcirculation during endotoxemia. PLoS ONE. 2012;7:e37439. doi: 10.1371/journal.pone.0037439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witthöft T., Eckmann L., Kim J.M., Kagnoff M.F. Enteroinvasive bacteria directly activate expression of iNOS and NO production in human colon epithelial cells. Am. J. Physiol. 1998;275:G564–G571. doi: 10.1152/ajpgi.1998.275.3.G564. [DOI] [PubMed] [Google Scholar]
- Zigmond E., Bernshtein B., Friedlander G., Walker C.R., Yona S., Kim K.-W., Brenner O., Krauthgamer R., Varol C., Müller W., Jung S. Macrophage-restricted interleukin-10 receptor deficiency, but not IL-10 deficiency, causes severe spontaneous colitis. Immunity. 2014;40:720–733. doi: 10.1016/j.immuni.2014.03.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.