

SUMMARY
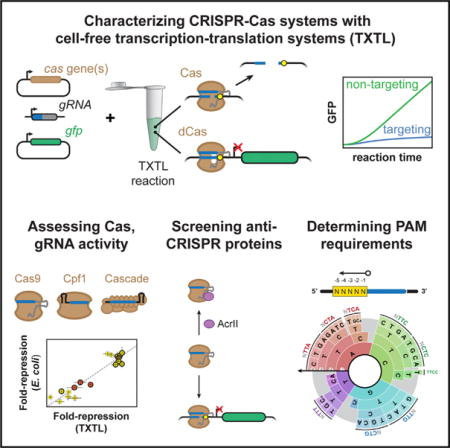
CRISPR-Cas systems offer versatile technologies for genome engineering, yet their implementation has been outpaced by ongoing discoveries of new Cas nucleases and anti-CRISPR proteins. Here, we present the use of E. coli cell-free transcription-translation (TXTL) systems to vastly improve the speed and scalability of CRISPR characterization and validation. TXTL can express active CRISPR machinery from added plasmids and linear DNA, and TXTL can output quantitative dynamics of DNA cleavage and gene repression—all without protein purification or live cells. We used TXTL to measure the dynamics of DNA cleavage and gene repression for single- and multi-effector CRISPR nucleases, predict gene repression strength in E. coli, determine the specificities of 24 diverse anti-CRISPR proteins, and develop a fast and scalable screen for protospacer-adjacent motifs that was successfully applied to five uncharacterized Cpf1 nucleases. These examples underscore how TXTL can facilitate the characterization and application of CRISPR technologies across their many uses.
Graphical abstract
In Brief Marshall et al. demonstrate that an E. coli cell-free transcription-translation (TXTL) system can be used to improve the speed and scalability of characterizing CRISPR nucleases and their accessory factors. The method will facilitate the discovery of uncharacterized CRISPR nucleases and anti-CRISPR proteins and aid the validation of designed gRNAs.
INTRODUCTION
CRISPR technologies have proven to be broadly useful genome-editing tools for biomolecular research, biotechnology, human health, and agriculture (Barrangou and Doudna, 2016). These technologies rely on RNA-guided nucleases derived from prokaryotic CRISPR-Cas immune systems (Mohanraju et al., 2016). The nuclease specifically cleaves DNA or RNA sequences complementary to the guide portion of the RNA and flanked by a protospacer-adjacent motif (PAM). For DNA targets, this ability has allowed programmable DNA damage or repair-mediated genome editing. Furthermore, by disrupting endonuclease activity, these nucleases can be readily converted into programmable nucleic acid-binding proteins and used for applications in gene activation (CRISPRa) or repression (CRISPRi), base editing, and real-time imaging (Chen et al., 2016; Dominguez et al., 2016; Komor et al., 2017; Nelles et al., 2016; Qi et al., 2013).
While the vast majority of CRISPR-based technologies have relied on the DNA-targeting Cas9 nuclease from Streptococcus pyogenes (SpyCas9) (Barrangou and Doudna, 2016; Jinek et al., 2012), nature boasts a diverse collection of CRISPR-Cas systems that are currently sub-divided into two classes, six types, and 33 subtypes (Koonin et al., 2017; Makarova et al., 2015; Shmakov et al., 2015). Interrogation of the emerging subtypes has revealed nucleases with widely varying properties that can be smaller, recognize different PAMs, degrade DNA, target RNA, exhibit reduced propensity for off-target effects, or be more amenable to multiplexing in comparison to SpyCas9 (Abudayyeh et al., 2016; Kim et al., 2017; Kleinstiver et al., 2016; Mulepati and Bailey, 2013; Zetsche et al., 2017). Separately, the discovery of anti-CRISPR proteins that inhibit Type I-E, I-F, II-A, and II-C CRISPR-Cas systems offers means to tightly control nuclease activity (Bondy-Denomy et al., 2015; Pawluk et al., 2016a, 2016b; Rauch et al., 2017), with the likely existence of similar inhibitors for the other subtypes. However, despite this diversity, the vast majority of these proteins have been slow to be adopted as CRISPR technologies.
One major bottleneck is the long and tedious process of characterizing these proteins’ basic properties and functions. To date, characterization has been performed with multiple methodologies based on in vitro biochemical assays or live cells. More recently, these assays have been modified for high-throughput analysis of PAM-binding requirements, off-target propensities, or large libraries of guide RNAs (gRNAs) through next-generation sequencing or imaging of arrayed nucleotides (Abudayyeh et al., 2016; Boyle et al., 2017; Jiang et al., 2013; Karvelis et al., 2015; Kleinstiver et al., 2016; Leenay et al., 2016; Pattanayak et al., 2013; Tsai et al., 2017). However, these assays consistently require days to weeks to perform due to the requirement for protein purification or for culturing and transforming live cells. Furthermore, these assays scale poorly when testing large sets of proteins or gRNAs. Given the growing abundance of known CRISPR nucleases, the ease in gRNA design, and the growing prevalence of factors that interface with CRISPR-Cas systems, there remains a pressing need to develop rapid and scalable characterization methodologies.
DESIGN
Here, we address this need using an Escherichia coli cell-free transcription-translation (TXTL) system (Garamella et al., 2016; Shin and Noireaux, 2012). The system is based on E. coli lysates prepared from exponentially growing cells to preserve the natural transcriptional, translational, and metabolic machinery. This machinery contrasts with traditional TXTL systems, which rely on RNA polymerases from bacteriophages such as T7 as well as purified ribosomes. DNA encoding genes of interest can be added to the lysates in different amounts, resulting in gene expression within minutes to hours. While plasmid DNA is commonly used, TXTL can also accommodate linear DNA as long as RecBCD is inhibited using GamS or linear DNA containing Chi sites (Marshall et al., 2017). The TXTL reactions can also be conducted in volumes as small as a few microliters, allowing TXTL reactions to be scalably used in microtiter plates. Note that although we generally prepared our own E. coli lysates, a system based on our lysate-production protocols is available commercially (STAR Methods).
To express CRISPR-Cas systems, DNA encoding the cas genes and gRNAs are added to the TXTL mix, resulting in the generation of active nucleoprotein complexes. To measure activity of the complexes in TXTL, we use a reporter construct expressing the fluorescent protein deGFP, a slightly modified version of eGFP with identical fluorescent properties (Shin and Noireaux, 2012). Targeting the construct results in changes in deGFP fluorescence levels that can be measured quantitatively in real time. While deGFP or other fluorescent proteins lend to rapid measurement using a fluorescence microplate reader, other standard colorimetric or luminescence readouts like violacein or firefly luciferase can be used. Here, we demonstrate the utility of TXTL across a diverse set of Cas nucleases and show how it can be used to predict gRNA activity in vivo, characterize anti-CRISPR proteins, and elucidate recognized PAMs. Based on these findings, we expect TXTL to provide a powerful characterization tool for the expanding universe of CRISPR-Cas systems and proteins, and we show the applicability of TXTL beyond biomanufacturing, diagnostics, and genetic circuit prototyping (Carlson et al., 2012; Dudley et al., 2015; Garamella et al., 2016; Gootenberg et al., 2017; Hockenberry and Jewett, 2012; Jewett et al., 2008; Kanter et al., 2007; Karzbrun et al., 2014; Pardee et al., 2016; Sun et al., 2014; Swartz, 2006; Takahashi et al., 2015a, 2015b; Tayar et al., 2015). See Methods S1, Protocol 1 for more information about how to conduct TXTL with CRISPR.
RESULTS
SpyCas9 and dSpyCas9 Exhibit Robust Activity in TXTL
We initially examined the activity of the SpyCas9 nuclease. To monitor the dynamics of DNA cleavage by SpyCas9, we designed single-guide RNAs (sgRNAs) that target within the promoter, 5′ UTR, and coding sequence of a deGFP reporter construct. We measured the dynamics of deGFP production following the addition of the SpyCas9 plasmid, linear DNA encoding the sgRNA, and the deGFP reporter plasmid (Figure 1A). We found that the three tested sgRNAs resulted in greatly reduced deGFP concentrations in comparison to a non-targeting sgRNA (Figure 1B). Measurable repression was observed beginning after less than 1 hr into the TXTL reaction, indicating that this time span was required to express and assemble the Cas9-sgRNA ribonucleoprotein (RNP) complex and for the complex to bind and cleave the target DNA (Figures S1A and S1B). The rate of deGFP production then dropped quickly to approximately zero, consistent with irreversible DNA cleavage as confirmed by PCR amplification of the target site (Figure S1C). Interestingly, the onset of deGFP repression and the rate at which deGFP production dropped varied depending on the sgRNA used (Figures S1A and S1B), suggesting that the dynamics by which different guides target Cas9 to DNA can vary. These results indicate that an active SpyCas9-sgRNA complex can be expressed directly in TXTL, providing a dynamic and quantitative readout of nuclease activity in a few hours.
Figure 1. S. pyogenes Cas9 Functions Efficiently in TXTL.
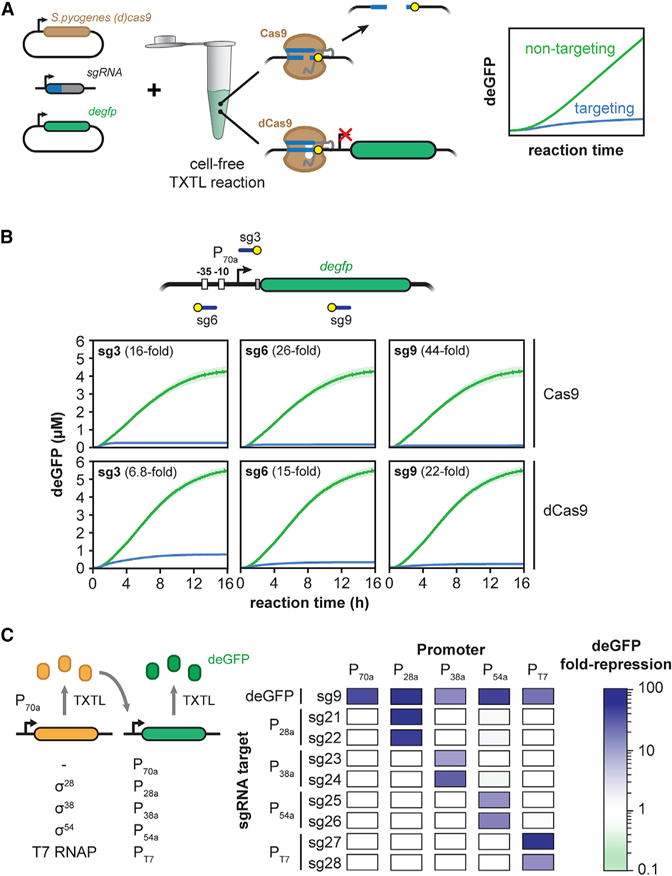
(A) Schematic of using TXTL to dynamically and quantitatively measure the activity of Cas9 and dCas9.
(B) Time series showing deGFP concentration for cell-free reactions expressing (d)Cas9 and a non-targeting sgRNA (green) or targeting sgRNAs (blue). Target locations include the sequence matching the guide (blue line) and the PAM (yellow circle). Error bars represent the SEM from at least six repeats.
(C) Alternative sigma factors σ28, σ38, and σ54 and the T7 polymerase can be expressed in TXTL from the P70a promoter and activate their cognate promoters P28a, P38a, P54a, and PT7, respectively. A matrix showing dSpyCas9-based repression of promoters dependent on σ28, σ38, σ54, and the T7 polymerase is shown. An sgRNA targeting each promoter or the gfp gene body was expressed along with each sigma factor or polymerase and a reporter gene driven by the sigma factor of its cognate promoter. Values represent the mean of at least three repeats.
We also interrogated the activity of the catalytically dead version of SpyCas9 (dSpyCas9) commonly used for programmable DNA binding and gene regulation (Gilbert et al., 2014; Qi et al., 2013). DNA binding by dSpyCas9 can block transcriptional initiation or elongation of RNA polymerase in bacteria, offering a means to link DNA binding with reporter expression in TXTL (Bikard et al., 2013; Qi et al., 2013). To assess the expression and regulatory activity of dSpyCas9, we measured the concentration of deGFP over time in TXTL reactions for the same three targeting sgRNAs and the non-targeting sgRNA. Similar to reactions expressing SpyCas9, reactions expressing dSpyCas9 exhibited consistent deGFP repression, with deGFP production dropping after less than an hour (Figures S1A and S1B). dSpyCas9 repressed expression less strongly and exhibited some deGFP production even at the end of the TXTL reaction (Figures 1B and S1A) due to lack of irreversible target cleavage (Figure S1B). We also observed different extents of deGFP fold repression across the three targeting sgRNAs, similar to repression strengths reported in bacteria (Bikard et al., 2013; Qi et al., 2013).
To begin assessing the scalability of TXTL reactions, we expanded from one promoter to four promoters, each targeted by two sgRNAs and driving expression of deGFP. Each promoter is dependent on a unique alternative sigma factor (σ28, σ38, and σ54) or T7 RNA polymerase, where each transcription factor is supplied on an added expression plasmid under the control of a σ70 promoter (Garamella et al., 2016; Shin and Noireaux, 2012) (Figure 1C). By including a degfp-targeting sgRNA, a non-targeting sgRNA, and a σ70 promoter, we tested a total of 50 promoter-sgRNA combinations. Each sgRNA was designed to target either strand across the recognition elements of its cognate promoter; the only exception was the P28a promoter, where both sgRNAs were designed to target the non-template strand due to the lack of an NGG PAM on the template strand. The TXTL reactions confirmed that measurable repression was only observed when an sgRNA was matched with its target (Figure 1C), with the strength of repression ranging between 7-fold and 105-fold. We also targeted binding sites for the NtrC operator sites within the σ54 promoter (Figure S1D), demonstrating that these sites were important for reporter expression.
Multiple Factors Impact the Measured Activity of dSpyCas9 in TXTL
After characterizing SpyCas9 and dSpyCas9 in TXTL, we sought to determine parameters that affect the measured activity. First, we found that dSpyCas9 was limiting based on more dSpyCas9 plasmid reducing the total amount of GFP produced in the TXTL reaction (Figure S2A). Surprisingly, adding more dSpyCas9 plasmid elevated deGFP production for the non-targeting sgRNA, underscoring the need to add the same total amount of DNA such as when comparing the activity of different targeting sgRNAs.
Second, we evaluated the impact of destabilizing deGFP to emulate reporter turnover or dilution in vivo. We appended an ssrA degron tag recognized by the ClpXP protease to the C terminus of deGFP (Figure S2B) (Schmidt et al., 2009). Appending the tag resulted in greater total dSpyCas9-based repression (Figure S2C) due to the inability of deGFP to accumulate in the TXTL reaction.
We next evaluated how CRISPR-based repression is influenced by linear versus plasmid DNA. Linear DNA can be generated without cloning and thus can be readily used in TXTL (Marshall et al., 2017; Sun et al., 2014). However, some Cas nucleases have been shown to require supercoiled DNA for binding (Westra et al., 2012), suggesting that targets encoded on relaxed, linear DNA may not allow for efficient repression. We measured the extent of dSpyCas9-based repression when targeting a plasmid or linear version of the deGFP reporter construct. We found that dSpyCas9 was capable of repressing gene expression when targeting the reporter gene on the linear template; however, negligible repression was observed when targeting the promoter region (Figure 2A). These results suggest that dSpyCas9 can block elongation, but not initiation, of the E. coli RNA polymerase on linear DNA. Regardless of the underlying mechanism, linear DNA should not be used when assessing the ability of dCas9 to block transcription initiation.
Figure 2. Multiple Factors Affect dSpyCas9-Based Repression of Reporter Gene Expression in TXTL.
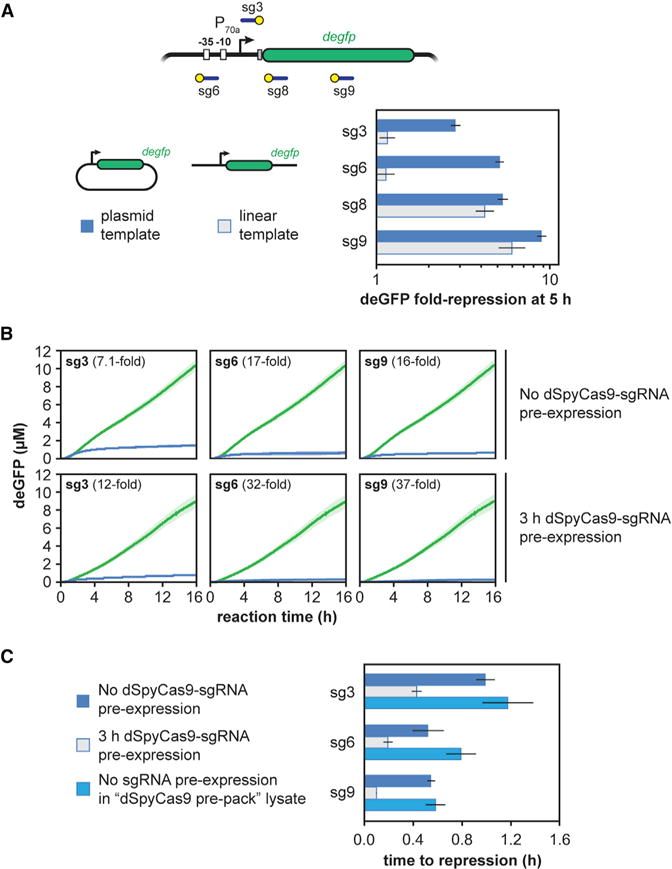
(A) Fold repression produced by a TXTL reaction when deGFP is expressed from either a targeted plasmid (dark) or linear (light) construct. Error bars represent the SEM from at least three repeats.
(B) Time series showing deGFP concentration in TXTL for cell-free reactions expressing dSpyCas9 and a targeting sgRNA. The reporter plasmid is added to the reaction either at the same time as dSpyCas9 and the sgRNA (top row) or after 3 hr (bottom row). Error bars represent the SEM from at least five repeats.
(C) Time to repression for the curves from (B), as well as for “dSpyCas9 pre-pack.” Error bars represent the SEM from at least five repeats.
When DNA expressing dSpyCas9, an sgRNA, and the fluorescent reporter were added together at the beginning of a TXTL experiment, we observed a transient period in which the reporter gene was expressed before the onset of dSpy-Cas9-based repression (Figure 1B). We hypothesized that this period of transient expression was due to the slow expression and assembly of the dSpyCas9-sgRNA RNP complex. To test this hypothesis, we varied our initial protocol to pre-express dSpyCas9 and the sgRNA before adding the reporter plasmid for 3 hr. We reasoned that this period of pre-expression would allow the RNP to be expressed and assemble and that this would shorten the time until we observed repression of the reporter gene. Consistent with our hypothesis, pre-expressing dSpyCas9 and the sgRNA reduced the time before the reporter gene was repressed (Figure 2B), with measurable repression occurring as fast as 6 min (Figures 2C and S2D). We also evaluated the effect of pre-expressing dSpyCas9 in cells prior to generating the lysate in a “dSpyCas9 pre-pack” mix (Figure S2E). Interestingly, reactions in this lysate exhibited similar times to repression as reactions with no dSpyCas9 or sgRNA pre-expression (Figure 2C), indicating that assembly of the RNP complex primarily contributes to the delayed onset of dSpyCas9-based repression. Taken together, these results suggest that dCas9 RNP assembly is relatively slow (on the order of 30 min), but DNA binding is fast (on the order of 5 min), in line with the timescales of Cas9:gRNA complex formation and DNA binding in vitro in the presence of non-specific RNA competitors (Mekler et al., 2016).
The Strength of dSpyCas9-Based Repression Strongly Correlates between TXTL and E. coli
Given the speed and scalability of employing TXTL to characterize CRISPR nucleases and gRNAs, an ensuing question is how well the quantified activity correlates to in vivo settings and recapitulates known phenomena (Chappell et al., 2013). To begin addressing this question, we compared dSpyCas9-based repression in TXTL and in E. coli by targeting 19 different locations within the deGFP reporter plasmid (Figure 3A). The particular locations were chosen to also evaluate the impact of strong PAMs (NGG) and weak PAMs (NAG) as well as targeting the template or non-template strand within the promoter or transcribed region. We chose these comparisons because Cas9 recognizes NAG PAMs more weakly than NGG PAMs (Boyle et al., 2017; Jiang et al., 2013) and exhibited impaired gene repression in bacteria when targeting the template strand within the transcribed region versus any other location (Bikard et al., 2013; Qi et al., 2013). The experiments were conducted by encoding the dSpyCas9, sgRNA, and deGFP reporter on separate, compatible plasmids for parallel testing in TXTL and in E. coli.
Figure 3. TXTL Can Be Used to Assess the Activity of sgRNAs.
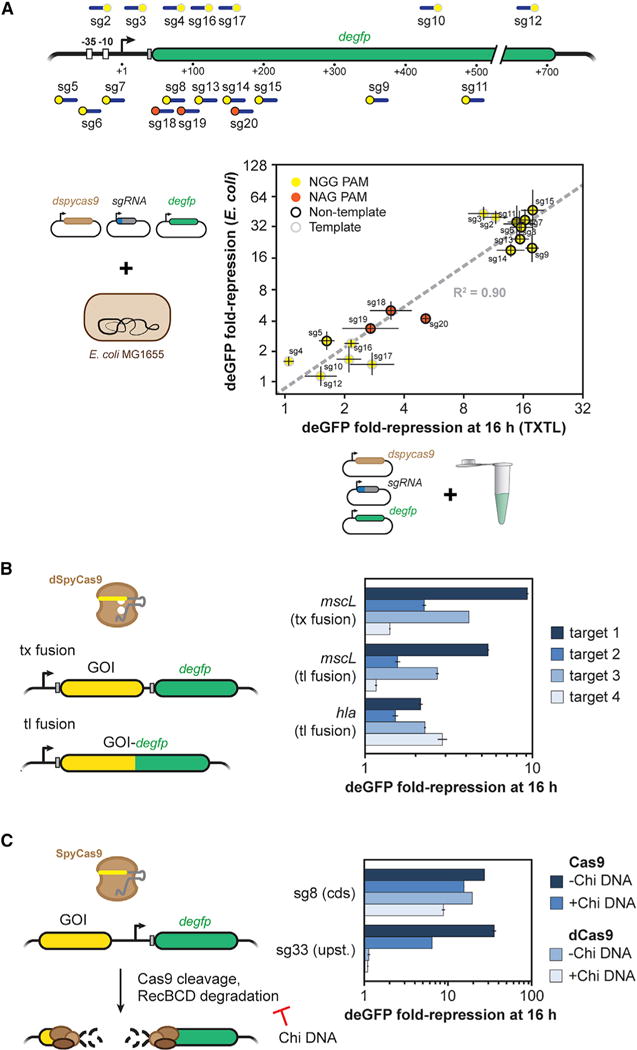
(A) A schematic of where each guide binds in the gfp promoter and gene body (top). The location of the target and PAM is indicated by a blue line and a yellow or orange dot, respectively. The fold repression of GFP production by dCas9-based repression for each sgRNA in vivo and in vitro (bottom). Points are colored by whether the guide is adjacent to an NGG (yellow) or NAG (orange) PAM, and whether the sgRNA targets the non-template strand (black ring) or template strand (gray ring). Error bars represent the SEM from at least three repeats.
(B) Assessing non-gfp targeting sgRNAs used by dCas9. The sequence or gene of interest is transcriptionally or translationally fused upstream of degfp. Fold repression was measured in TXTL for four targeting sgRNAs when degfp is fused to mscL or hla. Error bars represent the SEM from at least three repeats.
(C) Assessing non-gfp targeting sgRNAs used by Cas9. The sequence or gene of interest was inserted upstream of the promoter driving expression of deGFP. In the absence of a RecBCD inhibitor, cleavage by Cas9 leads to rapid degradation of the plasmid and loss of GFP expression. Fold repression was measured in TXTL when targeting in the gfp coding sequence (sg8) or upstream of the promoter (sg8) with Cas9 or dCas9, and in the presence or absence of the RecBCD inhibitor Chi site containing DNA. Error bars represent the SEM from at least three repeats.
These experiments revealed a strong correlation (R2 = 0.90) between the measurements in TXTL and in E. coli (Figure 3A). The correlation was based on a log-log plot rather than a linear-scale plot due to differences between continuous cellular processes and batch TXTL processes. Consistent with the strong correlation, we observed greatly reduced repression for targets flanked by NAG versus NGG PAMs. Furthermore, repression was consistently weaker when targeting the template strand of the transcribed region in comparison to targeting the non-template strand of the transcribed region or either strand within the vicinity of the promoter. This direct comparison therefore showed that TXTL can reasonably predict in vivo activity of CRISPR-Cas systems, at least for gene repression by dSpyCas9 in E. coli.
The Activity of Targeting Other Genes Can Be Measured with TXTL
To demonstrate that we can target virtually any sequence or gene, we devised schemes to indirectly link the activity of dCas9 or Cas9 to deGFP expression. In the case of dCas9, our scheme involved introducing the target sequence or gene as a transcriptional or translational fusion to the 5′ end of the degfp gene. Based on this setup, any sgRNA-mediated transcriptional silencing would downregulate the downstream expression of deGFP. As a proof-of-principle demonstration, we fused the Staphylococcus aureus alpha-hemolysin gene hla or the E. coli mechanosensory channel gene mscL to the N terminus of eGFP, or we introduced mscL upstream of deGFP as an operon-like DNA assembly with separate ribosome-binding sites. Two sets of four different sgRNAs were designed to target the non-template strand of hla or mscL. For each of the fusion constructs, we measured GFP fluorescence in TXTL reactions expressing dCas9 and a targeting or non-targeting sgRNA (Figure 3B). The TXTL reactions showed variable repression of deGFP in comparison to the non-targeting control. Our results also show the consistency of transcriptional repression between the MscL-eGFP transcriptional and translational fusions.
In the case of Cas9, our scheme involved targeting a sequence upstream of the degfp promoter on the reporter plasmid. Without an inhibitor of RecBCD, we hypothesized that if Cas9 cleaved the sequence, the cleaved plasmid would be rapidly degraded, leading to a loss of deGFP expression. To test this hypothesis, we designed two sgRNAs: one targeting upstream of the promoter and another targeting the coding sequence of degfp. Targeting upstream of the promoter greatly reduced GFP expression when using Cas9, but not dCas9, while targeting within the coding sequence of gfp reduced expression for both Cas9 and dCas9, thus demonstrating that cleavage was required for quenching (Figure 3C). Note that adding DNA containing Chi sites, a RecBCD inhibitor, reduced repression by Cas9 when targeting upstream of the promoter, which shows the mechanism of quenching was RecBCD degradation of the cleaved plasmid. These results illustrate how to employ TXTL with dCas9 or Cas9 to measure the activity of sgRNAs.
The Activity of Other CRISPR Nucleases Can Be Measured with TXTL
Our results showed that active SpyCas9 RNPs can be expressed in TXTL and suggested that other CRISPR-Cas systems would also function in TXTL. To test this possibility, we evaluated effector proteins from two other CRISPR-Cas systems: the single-effector nuclease Cpf1 (Cas12a) from the Type V-A system in F. tularensis subsp. Novicida U112 and the multi-subunit complex Cascade from the Type I-E system in E. coli (Figures 4A and 4B). These effector proteins were selected because Cpf1 exhibits desirable properties over Cas9, while Type I CRISPR-Cas systems are the most prevalent system type in nature but are more challenging to characterize because multiple proteins form the effector complex (Makarova et al., 2015; Zetsche et al., 2015). Previous studies have shown that both systems are capable of programmable gene repression in bacteria when using a catalytically dead version of the nuclease (dFnCpf1) or expressing Cascade in the absence of the Cas3 endonuclease, respectively (Leenay et al., 2016; Luo et al., 2015; Rath et al., 2015; Zetsche et al., 2015). We designed three gRNAs (a mature gRNA for Cpf1 and a repeat-spacer-repeat array for Cascade) targeting distinct sites within the P70a promoter, with each site flanked by an appropriate PAM.
Figure 4. Single Effector and Multi-protein Effector Cas Proteins Function Efficiently in TXTL.
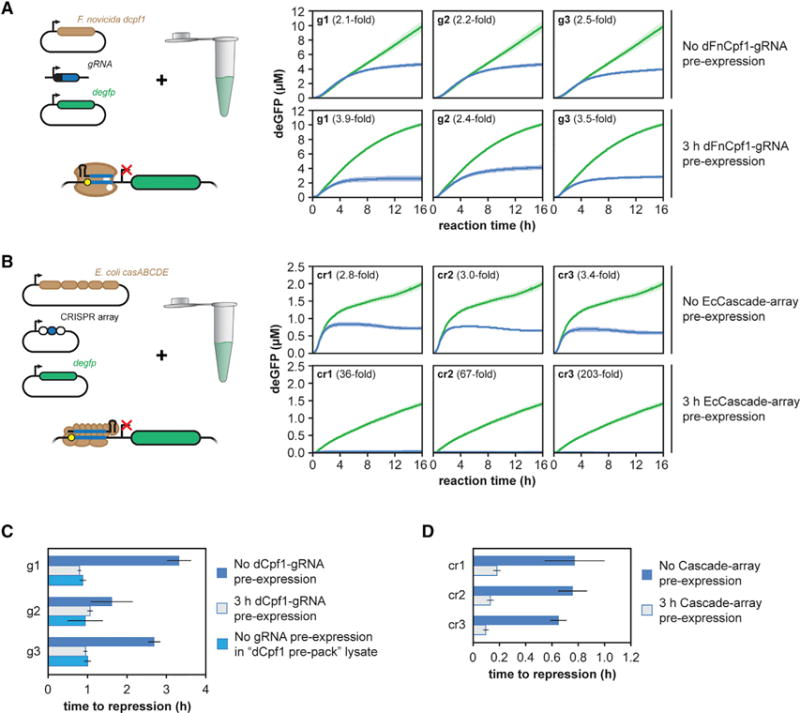
(A and B) Time series of reporter gene expression in TXTL for cell-free reactions expressing (A) a catalytically inactive version of the Type V-A Cpf1 nuclease from Francisella novicida or (B) the Type I-E Cascade complex from E. coli. The protein or set of proteins was expressed along with a non-targeting gRNA (green) or one of three gRNAs (blue) designed to target the promoter of the deGFP reporter construct. The reporter plasmid is added to the reaction either at the same time as the constructs expressing the Cas protein(s) and the gRNA (top row) or after 3 hr (bottom row). Error bars represent the SEM from six repeats.
(C) Time to repression for the curves from (A), as well as for “dFnCpf1 pre-pack.” Error bars represent the SEM from at least five repeats.
(D) Time to repression for the curves from (B). Error bars represent the SEM from at least four repeats.
We first found that dFnCpf1 was capable of repressing gene expression with each of the three gRNAs that targeted the reporter gene promoter (Figures 4A and S3A). Although dFnCpf1 eventually blocked deGFP expression to a similar extent as dCas9, the measured repression by dFnCpf1 was less than dSpyCas9 due to the longer delay in the onset of repression (Figures 4C and S3A). Consistent with this, pre-expressing dFnCpf1 and its gRNA for 3 hr reduced the delay in measurable repression by approximately 2 hr. Interestingly, a “dFnCpf1 pre-pack” TXTL lysate yielded a similar reduction in the time to repression. These results indicate that, in contrast to dCas9, the expression of dFnCpf1 is a major contributor to the delay in repression.
We also found that EcCascade could elicit gene repression in TXTL despite the need to coordinately express five proteins. The total reduction of GFP produced by the reaction was modest because of the delay in the onset of strong repression (Figures 4B, 4D, and S3B). However, pre-expressing EcCascade and the CRISPR RNA strongly reduced the total amount of GFP produced due to strong repression shortly after the addition of the reporter plasmid (Figures 4B, 4D, and S3B), indicating that the complex rapidly binds DNA and efficiently blocks RNA polymerase recruitment. Unexpectedly, deGFP production was lower when expressing EcCascade versus any of the other effector proteins (Figure 4B). This decrease in deGFP production was even more prominent when EcCascade was pre-expressed. Adding pure recombinant eGFP to a TXTL reaction expressing EcCascade resulted in only marginally lower fluorescence than a TXTL reaction expressing SpyCas9 or a no-nuclease control (Figure S3C). Therefore, the non-specific, inhibitory effect of EcCascade can be traced to deGFP expression rather than its stability or fluorescence properties. In sum, we showed that TXTL can be extended to the characterization of CRISPR-Cas systems requiring both single-effector and multi-protein effector complexes.
TXTL Can Quantify the Inhibitory Activity of Anti-CRISPR Proteins
Recently, the discovery of anti-CRISPR proteins that bind and inhibit Cascade and Cas3 from Type I-E and Type I-F CRISPR-Cas systems and Cas9 from Type II-A and II-C systems raised the potential of using these proteins to tightly regulate genome editing and gene regulation (Bondy-Denomy et al., 2015; Pawluk et al., 2016a, 2016b; Rauch et al., 2017). We tested if TXTL could be used to rapidly assess the inhibitory activity of potential anti-CRISPR proteins. We initially focused on AcrIIA2 and AcrIIA4, two anti-CRISPR proteins recently reported to inhibit the activity of SpyCas9 in vitro and in human cells (Rauch et al., 2017). Both proteins counteracted gene repression by dSpyCas9 (Figures 5A and S2E), although AcrIIA4 was a more potent inhibitor than AcrIIA2 in line with prior measurements in E. coli (Rauch et al., 2017).
Figure 5. TXTL Can Be Used to Rapidly Characterize Anti-CRISPR Proteins.
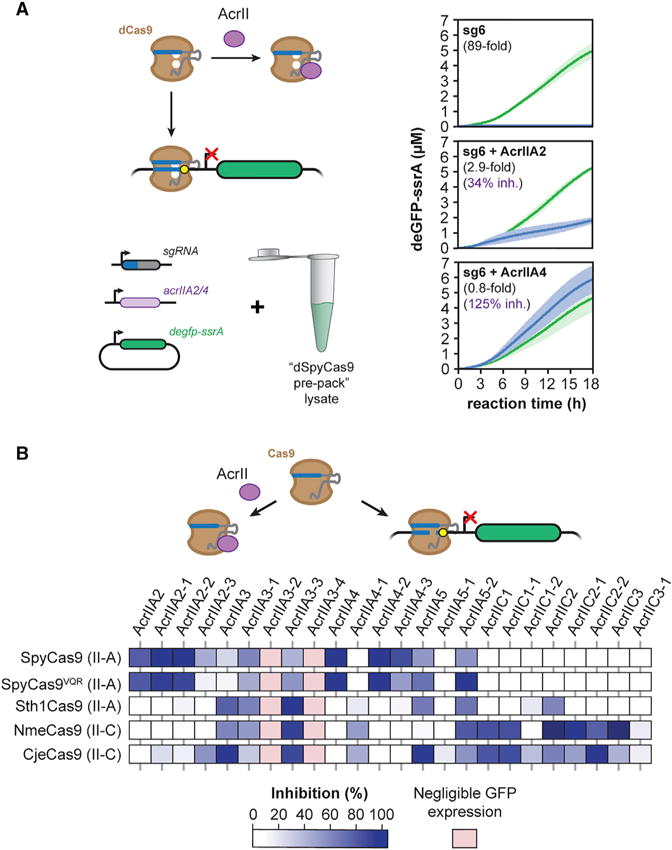
(A) Time series of deGFP-ssrA expression in TXTL for cell-free reactions also expressing dSpyCas9, an sgRNA, and one of two anti-CRISPR proteins, AcrIIA2 and AcrIIA4, shown to inhibit SpyCas9 activity. Each reaction was performed with a targeting sgRNA (blue) or a non-targeting sgRNA (green). Error bars represent the SEM from at least three repeats.
(B) A matrix showing the percentage inhibition for 24 different anti-CRISPR proteins on five different Cas9. Samples with no appreciable GFP expression in the presence of the anti-CRISPR protein are designated with light red. Values represent the mean of at least three technical replicates.
We next tested a panel of 24 anti-CRISPR proteins, including seven previously reported proteins from defined classes (e.g., AcrIIA2 and AcrIIC1) (Hynes et al., 2017; Pawluk et al., 2016a), as well as up to four homologs of each protein. The panel of anti-CRISPR proteins was tested against five different Cas9 nucleases: the Type II-A SpyCas9, the VQR variant of SpyCas9 that recognizes an altered NGA PAM (SpyCas9VQR) (Kleinstiver et al., 2015), the Type II-A CRISPR1 Cas9 from Streptococcus thermophilus (Sth1Cas9), the Type II-C Cas9 from Neisseria meningitidis (NmeCas9), and the Type II-C Cas9 from Campylobacter jejuni (CjeCas9). In total, we tested 120 different combinations (Figure 5B).
Across the set of tested anti-CRISPR proteins and Cas9 nucleases, we identified widely ranging specificities and activities. Many of the anti-CRISPR proteins inhibited a single nuclease or subtype of nucleases, such as AcrIIA2 and two of its homologs inhibiting only SpyCas9 and SpyCas9VQR or AcrIIC1 and one of its homologs inhibiting both of the Type II-C Cas9 nucleases, NmeCas9 and CjeCas9, in line with a recent study of AcrIIC1 (Harrington et al., 2017). Finally, some anti-CRISPR proteins appeared to differentiate between SpyCas9 and SpyCas9VQR: two (AcrIIA2-3 and AcrIIA4-3) preferred SpyCas9 while one (AcrIIA5-2) preferred SpyCas9VQR.
Surprisingly, the TXTL-based measurements suggested that other anti-CRISPR proteins can inhibit beyond these boundaries. For instance, AcrIIA3 and AcrIIC2 inhibited the activity of the II-A Sth1Cas9 and both II-C Cas9 nucleases. AcrIIC2 and AcrIIC3 showed much stronger inhibition of NmeCas9 than CjeCas9, paralleling recent in vitro cleavage experiments with NmeCas9 and CjeCas9 (Harrington et al., 2017). Most strikingly, three of the tested homologs (AcrIIA3-1, AcrIIA3-3, and AcrIIA5-2) appeared to inhibit all tested Cas9 nucleases. While highly intriguing, these anti-CRISPR proteins strongly reduced deGFP expression even with the non-targeting sgRNA (Figure S4). As a result, these anti-CRISPR proteins may have limited expression of Cas9 and the targeting sgRNA, thereby confounding any definitive conclusions about inhibitory activity. Similarly, two of the tested anti-CRISPR proteins (AcrIIA3-2 and AcrIIA3-4) completely inhibited GFP expression even with the non-targeting sgRNA, preventing us from discerning any inhibitory activity. Overall, these experiments demonstrate that TXTL can be used to rapidly and scalably assess the inhibitory activity of diverse anti-CRISPR proteins, and they revealed anti-CRISPR proteins with widely ranging specificities.
TXTL Offers a Rapid Means of Elucidating CRISPR PAMs
One of the major barriers to the functional characterization of new Cas nucleases is determining the recognized PAM sequences. While numerous experimental methods have been developed for PAM determination, they consistently rely on in vitro assays that require protein purification or on cell-based assays that require culturing and transforming of live cells (Karvelis et al., 2017; Leenay and Beisel, 2017). We therefore asked if TXTL could also be used as the basis of PAM determination assays. Paralleling prior in vitro and in vivo DNA cleavage assays (Jiang et al., 2013; Zetsche et al., 2015), our devised assay relies on introducing a library of potential PAM sequences flanking a site targeted by an expressed gRNA and Cas nuclease (Figure 6A). After incubating the pooled PAM library and DNA encoding the Cas nuclease and gRNA, the pool of uncleaved target sequences was PCR-amplified and subjected to next-generation sequencing. By comparing the relative frequency of individual sequences within the library before and after cleavage, we could quantify the depletion of each library member and therefore how well the nuclease recognized each sequence as a PAM. Critically, the assay was completed in 10–20 hr from when the DNA constructs were in hand to when PCR products were submitted for sequencing. See Methods S1, Protocol 2 for more information about how to conduct the TXTL-based PAM determination assay.
Figure 6. TXTL Can Be Used to Determine CRISPR PAMs.
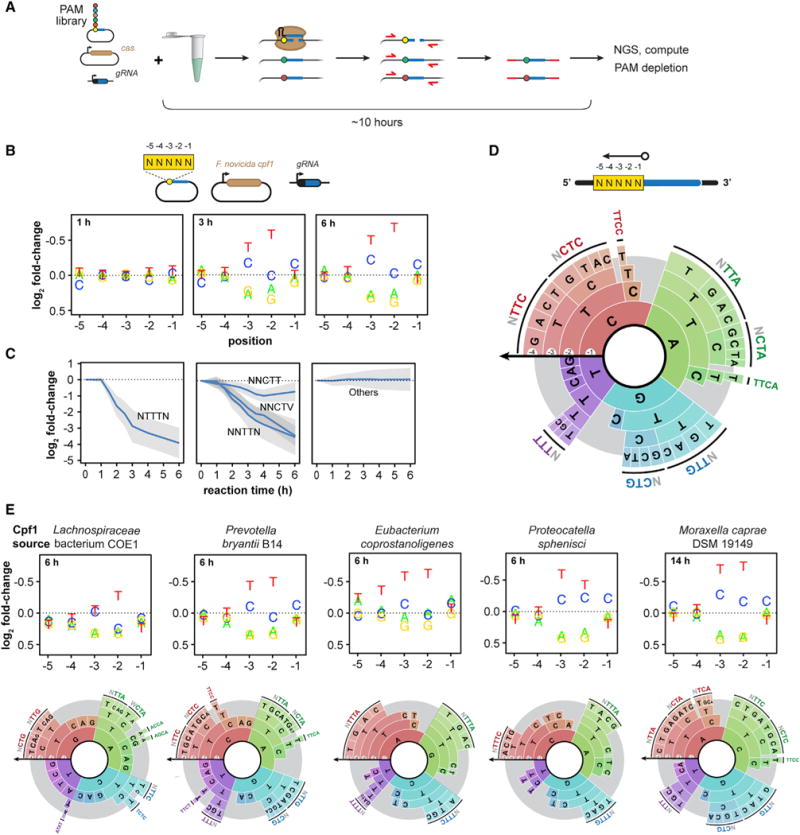
(A) Schematic of a TXTL-based cleavage assay to determine the PAM sequences recognized by Cas nucleases.
(B) Plots showing the fold change in the representation of a nucleotide at each variable position in the PAM library as a result of FnCpf1 activity in comparison to the original PAM library. Note that the y axis is inverted to highlight nucleotides that are depleted.
(C) Time series showing the depletion of selected motifs by FnCpf1 matching the consensus sequence in the sequencing libraries is shown. Error bars show the SD of the fold change.
(D) A PAM wheel showing the determined PAM sequences recognized by FnCpf1. PAM sequences are read proceeding from the outside to the inside of the circle, and the arc length directly correlates with the extent of PAM depletion. The −5 position was not shown for clarity.
(E) Plots showing the fold change in the representation of a nucleotide at each variable position in the PAM library in comparison to the original PAM library (top) and PAM wheels showing the determined PAM sequences (bottom) for five uncharacterized Cpf1 nucleases. A sixth Cpf1 nuclease (MbCpf1) previously characterized in Zetsche et al. (2015) is also reported in Figure S5.
As a proof-of-principle demonstration, we assessed the PAM requirements of the well-characterized FnCpf1 nuclease. Previous assays revealed that FnCpf1 prefers a TTN motif on the 5′ end of the target for efficient cleavage using the gRNA-centric orientation, although CTN can also be recognized (Fonfara et al., 2016; Leenay et al., 2016; Zetsche et al., 2015). In vitro PAM assays are known to yield less specific PAM sequences for higher nuclease concentrations (Karvelis et al., 2015), so we assessed the determined PAMs in our assay for reaction times ranging from 1 to 6 hr following the addition of the FnCpf1 and gRNA expression constructs (Figure S5A). Figure 6 depicts the identified PAM sequences based on the depletion of individual nucleotides at each position in a 5-nt library (Figure 6B) or specific motifs (Figure 6C), as well as a PAM wheel capturing the relative depletion of each sequence across the library (Figure 6D) (Leenay et al., 2016). The assay recapitulated the canonical 5′ TTN PAM while revealing NTTTN as the most active motif, with ATTTA as the most depleted by ~3-fold more than the next most depleted sequences (Data S1). We further found that NNCTN supported efficient cleavage in line with previous reports (Fonfara et al., 2016; Zetsche et al., 2015) and a T at the −1 position was detrimental to cleavage, indicating a consensus YTV PAM. NTCN also supported cleavage if there was a T at the −4 position and a C or an A at the −1 position. Importantly, the PAM distribution did not become less specific at later cleavage times unlike prior in vitro PAM assays (Karvelis et al., 2015).
To further demonstrate the applicability of the TXTL-based PAM assay, we performed the assay on five phylogenetically diverse and previously uncharacterized Cpf1 nucleases as well as one Cpf1 that had undergone limited characterization (Figure S5B). Initial analyses revealed that the nucleases exhibited varying levels of activity on the TTTC PAM when targeting the GFP plasmid (Figure S5C). Interestingly, the Cpf1 from Moraxella bovoculi 237 (MbCpf1) exhibited greatly enhanced activity at a reaction temperature of 37°C than the standard 29°C (Figure S5C). The resulting PAM assays revealed two general consensus PAMs following that of FnCpf1 (YTV) or AsCpf1 (TTTV) (Figures 6E and S4D) (Zetsche et al., 2015). Interestingly, the PAM preferences did not correlate with the extent of homology (Figures 6E and S4B). Furthermore, some Cpf1 nucleases exhibited PAM preferences within these consensus sequences, such as the Cpf1 from Lachnospiraceae bacterium COE1 (Lb6Cpf1) accommodating ACCA and AGCA PAMs (Figure 6E). We proceeded to test four of these Cpf1 nucleases (McCpf1, EcCpf1, Pb2Cpf1, and Lb6Cpf1) for genome-editing activity using an established recombination system in rice callus (Begemann et al., 2017). All four exhibited detectable indel formation at a targeted site downstream of a TTTC PAM site based on T7EI assays and Sanger sequencing of the edited loci from antibiotic-resistant rice callus (Figures S5E and S5F). Taken together, these results indicate that a TXTL-based assay can elucidate PAM requirements for CRISPR nucleases, opening the door to the characterization of PAM requirements across both natural and engineered CRISPR nucleases.
DISCUSSION
We have demonstrated that E. coli cell-free TXTL can be used for the rapid and scalable characterization of CRISPR nucleases, gRNAs, and anti-CRISPR proteins. Unlike in vitro biochemical assays or cell-based assays, TXTL does not require any protein purification or cell culturing and transformation. TXTL also allows exquisite control over the reaction conditions and the amount of the DNA templates, and it can provide a dynamic and quantitative readout of nuclease activity within a few hours. With recent advances in DNA synthesis as well as liquid handling systems, the approaches here could be readily scaled to hundreds of reaction conditions or constructs.
CRISPR-Cas systems are remarkably diverse, with significant sequence, structural, and functional diversity (Koonin et al., 2017). This diversity exists even within a single subtype; for instance, Cas9 proteins from the well-characterized Type II-A subtype can exhibit less than 10% sequence homology at the amino acid level and show a range of recognized PAM lengths and sequences (Fonfara et al., 2014). However, only a few representative nucleases have been characterized for the other subtypes. This is particularly striking for Type I and III CRISPR-Cas systems, the most prevalent types in prokaryotes. These systems rely on multiple proteins to form the effector complex, requiring the purification or expression of multiple proteins in defined stoichiometries that has complicated their widespread characterization. TXTL is ideally suited to characterize the multi-subunit effector complexes from these systems because linear, chemically synthesized DNA encoding each subunit can be combined in a single TXTL reaction. The expressed complex can then be characterized in a variety of ways, such as determining its assembly kinetics and PAM requirements. Evaluating numerous systems from one subtype could help reveal how CRISPR-Cas systems evolved and the structural basis of PAM recognition through mapping sequence-function relationships.
While our results demonstrate the promise of TXTL for characterizing CRISPR-Cas systems, there are multiple opportunities to further expand the utility of TXTL. For example, TXTL may be explored to investigate spacer acquisition across the diversity of Cas acquisition proteins found in nature. While it remains to be seen if acquisition can be recapitulated in TXTL, PCR-based or next-generation sequencing-based approaches could be readily used to assess the frequency and composition of integrated spacers (Fineran et al., 2014; Levy et al., 2015). We also found that dCas9-based repression correlated between TXTL and E. coli and devised different means of assessing the activity of any sgRNA, opening the possibility of using TXTL to validate gRNA designs. While more work is needed to determine which factors primarily determine gRNA activity in different cellular settings (e.g., chromatin state or sequence biases in DNA repair), factors that impact gRNA stability, the assembly of RNP complexes, or efficiency of DNA binding and cleavage would be identified in TXTL. Finally, measuring the time to repression with or without pre-expression of the CRISPR machinery opens the potential of using TXTL to rapidly measure the kinetics of RNP complex assembly and function under in vivo-like conditions. Through the demonstrations reported here and through further extensions, TXTL has the potential to make a widespread impact on the characterization of CRISPR-Cas systems and their transition into a new generation of CRISPR technologies.
Limitations
While TXTL can greatly accelerate the characterization of CRISPR-Cas systems, TXTL does possess limitations that restrict its use. For instance, gene expression in TXTL is limited to 25°C–42°C, with an optimal temperature of 29°C (Shin and Noireaux, 2010). While this range allowed us to enhance the activity of the MbCpf1 (Figure S5C), it could limit the characterization of CRISPR-Cas systems native to thermophilic prokaryotes. TXTL reactions also lose activity after ~16 hr, potentially limiting the use of GFP as a readout for poorly expressed nucleases. Additionally, there may be some variability in reaction rates between different batches of TXTL. However, this limitation is overcome by including internal standards for each batch (e.g., non-targeting sgRNAs) when measuring CRISPR-based activity. Next, TXTL is missing factors common to eukaryotic cells, such as long and diverse DNA sequences that impact the dynamics of target search, chromatin and other nucleoid proteins that impact the structure and availability of DNA, and proteins responsible for repair involved in eukaryotic genome editing. Finally, factors for CRISPR RNA processing besides RNase III would need to be identified (e.g., tracrRNAs), as E. coli would be unlikely to naturally express these factors.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial Strains | ||
| BL21 Rosetta 2 | Novagen | 71405 |
| MG1655 | Table S1 | Table S1 |
| 5-alpha Electrocompetent Cells (High Efficiency) | NEB | C2987I |
| Chemicals, Peptides, and Recombinant Proteins | ||
| reGFP | Cell Biolabs | STA-201 |
| Taq DNA Polymerase | NEB | M0273 |
| Q5 DNA Hot Start High-Fidelity DNA Polymerase | NEB | M0493 |
| Potassium Glutamate | Sigma | G1149 |
| Magnesium Glutamate | Sigma | 49605 |
| Maltodextrin | Sigma | 419672 |
| PEG8000 | Sigma | P2139 |
| Critical Commercial Assays | ||
| myTXTL | Arbor Biosciences | 507024 |
| ZymoPURE Plasmid Midi Prep Kit | Zymo Research | D4200 |
| NEBuilder HiFi DNA Assembly Cloning Kit | NEB | E5520S |
| Q5 Site-Directed Mutagenesis Kit | NEB | E0554S |
| Deposited Data | ||
| Cpf1 NGS data | Table S1 | Table S1 |
| Oligonucleotides | ||
| Primers | Table S1 | Table S1 |
| Nextera Indexing primers | Illumina | https://support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/experiment-design/illumina-adapter-sequences-1000000002694-03.pdf |
| Recombinant DNA | ||
| Plasmids and gBlocks | Table S1 | Table S1 |
| Software and Algorithms | ||
| PAM counting script (Python) | This paper | https://bitbucket.org/csmaxwell/crispr-txtl-pam-counting-script |
| Other | ||
| Ampure XP beads | Beckman Coulter | A63880 |
| Detailed protocols (TXTL reaction and PAM assay) | Methods S1 | Methods S1 |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests should be directed to the Lead Contact, Vincent Noireaux (noireaux@umn.edu).
METHOD DETAILS
Preparation of TXTL lysates and reactions
The E. coli cell-free TXTL lysate was prepared from BL21 Rosetta 2 from Novagen as described previously (Caschera and Noireaux, 2014). Currently, the TXTL lysate is also commercially available through the company Arbor Biosciences under the product name myTXTL. The dSpyCas9-loaded and dFnCpf1 TXTL lysates followed the same procedure, only the E. coli cells harbored a plasmid constitutively expressing dSpyCas9 or dFnCpf1 from a J23108 promoter. TXTL reactions were composed of 33% volume crude extract and the remaining 67% volume with the following components: energy mix, amino acid mix, cofactors, ions, and DNA. A typical TXTL reaction contains 50 mM HEPES pH 8, 1.5 mM ATP and GTP, 0.9 mM CTP and UTP, 0.2 mg/mL tRNA, 0.26 mM coenzyme A, 0.33 mM NAD, 0.75 mM cAMP, 0.068 mM folinic acid, 1 mM spermidine, 30 mM 3-PGA, 1.5% PEG8000, 30 mM malto-dextrin, 3 mM of each of the 20 amino acids, 90 mM K-glutamate, and 4 mM Mg-glutamate. TXTL reactions were conducted in volumes of 5 μl at 29-30°C. When expressing from linear DNA template, 2 μM of annealed oligos containing six copies of the χ-site sequence (Chi6; for details see Marshall et al., 2017) was added to the reaction. The linear DNA construct for expressing deGFP used to produce the data for Figure 2A was generated by PCR and included flanks over one kilobase away from the closest target site, mitigating potential effects from the end of the dsDNA molecule. To measure the inhibitory activity of AcrIIA2 and AcrIIA4 against SpyCas9 in TXTL, each protein was encoded on a linear expression construct that was expressed for two hours in the “dSpyCas9 pre-pack” lysate (Figures 5A and S2E). Then, DNA encoding the deGFP reporter plasmid and linear DNA encoding a targeting or non-targeting sgRNA was added and deGFP fluorescence was measured over time. To test the panel of anti-CRISPR proteins against the five Cas9 nucleases, we pre-expressed for three hours the catalytically active Cas9 from a plasmid, a targeting or non-targeting sgRNA and the anti-CRISPR protein from linear DNA. Then we added plasmid DNA encoding the GFP reporter. We measured the endpoint GFP expression after an additional 18 hours for each combination of Cas9 and anti-CRISPR protein (Figure 5B)
Assembly of expression constructs
Plasmids were constructed using standard techniques. The sequence of each gBlock and plasmid used in this experiment is available in the supporting information (Table S1). The plasmid expressing catalytically active SpyCas9 was generated by amplifying the transcriptional unit expressing SpyCas9 from pCas9 but excluding the CRISPR array from using primers CSMpr1132/1157 and cloning it into the backbone pCSM117. pCas9 was a gift from Luciano Maraffini (Addgene plasmid #42876). gBlocks were ordered from IDT and amplified with CSMpr1105/1106 before being PCR purified. All constructed plasmids were verified by Sanger sequencing of the inserted sequences.
Fluorescence time-course measurements in TXTL
Fluorescence kinetics measurements were performed principally using the reporter plasmid P70a-deGFP expressing a truncated version of eGFP (deGFP, 25.4 kDa, 1 mg/mL = 39.38 μM) (Shin and Noireaux, 2012). Fluorescence was measured on a Biotek H1m plate reader using a 96-well V-bottom plate (Corning Costar 3357) and Ex 485 nm, Em 528 nm. Time-course measurements were run for at least 16 hours at 29-30°C, with an interval of 3 minutes between reads.
Amplification of DNA targets in TXTL
TXTL reactions with the SpyCas9 or dSpyCas9 plasmid, sgRNA DNA template, and the P70a-deGFP expression plasmid were incubated at 29°C for three hours, then diluted 200X in water. 1 μl of this dilution was then used as the DNA template in a 50 μl PCR reaction with 55°C annealing temperature, 45 s extension time, and 25 cycles. The PCR reaction was conducted using Taq polymerase and primers RM01s and RM05as to produce a 1074-bp amplicon. PCR products were visualized on a 0.8% agarose gel.
Fluorescence measurements in E. coli
E. coli K-12 MG1655 cells with plasmids expressing dSpyCas9 and deGFP was transformed by electroporation with plasmids encoding the sgRNAs targeting sites (g1-19 targeting) on degfp, as well as a non-targeting control (g-nt). To measure each strain, three colonies from a freshly streaked plate was inoculated into 2 mL of LB with 34 μg/ml chloramphenicol (Cm), 50 μg/ml ampicillin (Amp), and 50 μg/ml kanamycin (Kan). The strains were cultured for 16-hours at 37°C shaking at 250 rpm. The cultures were then diluted 1:10,000 in LB with Cm, Amp, and Kan to a final volume of 100 μL within a black 96-well assay plate. Each culture was diluted into two wells as technical replicates. Cultures were incubated at 37°C with single orbital shaking at 425 rpm on a 3 mm diameter circle within a BioTek Synergy H1 for 20 hours at which cultures were in stationary phase. Cultures were resuspended with a multichannel pipette and loaded back into the BioTek Synergy H1. Single-point fluorescence at 485/528 nm excitation/emission as well as OD600 were measured from each well.
TXTL-based PAM assay
The pET vector expressing FnCpf1 was a kind gift of Benson Hill Biosystems. A PAM library with five randomized nucleotides flanking the spacer sequence was prepared as described previously (Leenay et al., 2016). The crRNA expressing the FnCpf1 crRNA targeting the PAM library was expressed from the gBlock CSM-GB099. A TXTL reaction was assembled as described above except the reaction contained 0.5 mM IPTG and the following DNA components: 0.2 nM P70a-T7RNAP, 2 nM pET-FnCpf1, 2 μM of Chi6 annealed oligos, 0.5 nM of the 5N PAM library, 2 nM of linear DNA expressing the crRNA. The reaction was split into 5 μL reactions as above and incubated at 29°C. Samples were then collected at the specified time by cutting away the cap mat sealing individual reactions and immediately freezing the reaction at −20°C until subsequent use. The adapters were attached to the PAM library by PCR amplification of the TXTL reaction using NEB’s Q5 Hot Start High-Fidelity DNA Polymerase (NEB, M0493) with RL133 and RL134. The PCR reaction was then purified using Ampure XP (Beckman Coulter, A63880) beads before attaching unique Nextera indices to each sample by PCR amplification. After a final PCR purification using Ampure XP beads, each sample was normalized to 10 nM in a total volume of 20 uL and submitted for next-generation sequencing with 150 single-end reads on an Illumina MiSeq machine. For the other six Cpf1s, the assay was conducted in the same manner, where the cpf1 genes were expressed either in a T7 expression plasmid (Lb6Cpf1, Pb2Cpf1, EcCpf1, and PsCpf1) or from linear DNA (McCpf1 and MbCpf1), while a processed crRNA derived from the organism’s native CRISPR array was expressed from linear DNA. See Methods S1, Protocol 2 for more information about conducting the TXTL-based PAM determination assay.
Plant Transformation Construct Cloning
Cpf1 coding sequences were optimized for monocot codon usage preferences and synthesized by GenScript. Cloning of Cpf1 constructs and guide RNA constructs was performed as described previously (Begemann et al., 2017). Briefly, each Cpf1 open reading frame was expressed from an enhanced Cauliflower Mosaic Virus 35S promoter (2X35S promoter); guide RNAs were expressed from the rice U6 promoter (OsU6 promoter). Rice (Oryza sativa cv. Kitaake) callus was transformed biolistically as described previously (Begemann et al., 2017). Briefly, gold microcarriers (0.6 μm in diameter) were coated with plasmids encoding the Cpf1 gene of interest and a guide RNA targeting the rice CAO1 gene, along with a hygromycin resistance cassette containing a hygromycin resistance gene expressed from the maize ubiquitin promoter (ZmUbi promoter). Bombarded rice callus was placed on selection medium containing hygromycin (50mg/L) for 3 weeks, and resistant callus pieces were subcultured.
QUANTIFICATION AND STATISTICAL ANALYSIS
Fluorescence time-course measurements in TXTL
Fluorescence intensity measurements were quantified using a linear standard calibration curve, spanning over three orders of magnitude, produced with pure recombinant eGFP (Cell Biolabs, STA-201, San Diego, CA). GFP production rates were calculated by two-point numerical differentiation and smoothed with a five-point quadratic polynomial. All fold-repression values for plasmid reporter constructs represent the ratio of deGFP concentrations after 16 hours of reaction for the non-targeting over the targeting sgRNA. For the linear reporter construct (Figure 2A), we measured the fold-repression of the deGFP produced by the reaction after five hours because linear templates incubated with Chi site-containing DNA degrade after that time in our TXTL system (Marshall et al., 2017). The time to repression was calculated based on the earliest time in which the rates of deGFP production diverge for the targeting sgRNA and the non-targeting sgRNA. For the experiments measuring the inhibitory activity of the anti-CRISPR proteins, inhibition was calculated from endpoint expression values after 18 hours of expression, using the following formula:
See Methods S1, Protocol 1 for more information about assessing the activity of CRISPR nucleases using TXTL.
Fluorescence measurements in E. coli
Endpoint fluorescence values were background subtracted using the fluorescence from cells lacking deGFP and then normalized by the endpoint, background-subtracted OD600 value. Fold-repression was then calculated as the normalized fluorescence of the non-targeting sgRNA strain divided by the normalized fluorescence of the targeting sgRNA strain.
TXTL-based PAM assay
The PAM wheel was generated as described previously (Leenay et al., 2016). Next-generation sequencing data is accessible through NCBI Biosample under BioProject: PRJNA415429. Links to the accession #’s are located in Table S1. See Methods S1, Protocol 2 for more information about conducting the TXTL-based PAM determination assay.
Sequence Alignments and Phylogenetic Trees
A multiple-sequence alignment of Cpf1 nuclease amino acid sequences was performed using MUSCLE with default parameters. A phylogenetic tree of Cpf1 protein sequences was built from this alignment using FigTree version 1.4.3. Midpoint tree rooting and cladogram transformation of branches were used for final tree visualization.
DATA AND SOFTWARE AVAILABILITY
Next-generation sequencing data for the PAM assay is accessible through NCBI Biosample under BioProject: PRJNA415429. Links to the accession #’s are located in Table S1. Krona plots used to generate the PAM wheels are found in Data S1.
Supplementary Material
Supplemental Data S1. Related to Figure 6. Krona plots used to generate the PAM wheels. The plots capture the output of the TXTL-based PAM determination assays conducted on FnCpf1, Lb6Cpf1, Pb2Cpf1, EcCpf1, PsCpf1, McCpf1, and MbCpf1.
Supplemental Protocol S1. Related to Figure 1. Protocol for measuring the activity of CRISPR nucleases using TXTL.
Supplemental Protocol S2. Related to Figure 6. Protocol for performing the TXTLbased PAM determination assay.
Supplemental Table S1. Related to Figures 1 - 6 and S1 - S5. Plasmid, gBlocks, bacterial cell stocks, and primers used in this study.
Highlights.
Active expression of multiple CRISPR nucleases and gRNAs in an E. coli TXTL system
Repression activity of dCas9 strongly correlated between TXTL and E. coli
A TXTL-based PAM assay allowed characterization of five Cpf1 nucleases
Determined specificities of 24 anti-CRISPR proteins against five Cas9 nucleases
Acknowledgments
We thank Ryan Leenay for help generating the FnCpf1 PAM wheel, Jennie Fagan for cloning the plasmid expressing SpyCas9, and Luciano Maraffini for providing the pCas9 plasmid (Addgene plasmid #42876). Preliminary experiments were performed during the 2016 CSHL Synthetic Biology summer course. This material is based upon work supported by the Defense Advanced Research Projects Agency (contract HR0011-16-C-01-34, V.N. and C.L.B.), the Office of Naval Research (award N00014-13-1-0074, V.N.), the NIH (grant 1R35GM119561-01 to C.L.B.), and the National Science Foundation (grant CBET-1403135 to C.L.B.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes five figures, one table, one data file, and one methods file and can be found with this article online at https://doi.org/10.1016/j.molcel.2017.12.007.
AUTHOR CONTRIBUTIONS
V.N., C.L.B., R.M., and M.L.L. conceived of the original project idea. V.N., C.L.B., R.M., C.S.M., and M.L.L. designed the TXTL characterization experiments. R.M. and C.S.M. performed the TXTL characterization experiments. S.P.C. performed the comparisons between TXTL and E. coli. S.P.C. and R.M. designed and performed the anti-CRISPR experiments. C.S.M. and T.J. designed and performed the PAM determination assays. M.B.B., B.N.G., E.J., A.S., and Y.H. identified the novel Cpf1 nucleases and designed and performed the genome-editing experiments in rice callus. V.N., C.L.B., R.M., and C.S.M. wrote the paper.
DECLARATION OF INTERESTS
C.L.B. is a co-founder and scientific advisory board member of Locus Biosciences and submitted provisional patent applications on CRISPR technologies. M.B.B., B.N.G., E.J., A.S., and Y.H. are current employees of Benson Hill Biosystems, which has proprietary CRISPR technologies on which patent applications have been filed. Noireaux laboratory receives research funds from Arbor Biosciences, a distributor of myTXTL cell-free protein expression kit.
References
- Abudayyeh OO, Gootenberg JS, Konermann S, Joung J, Slaymaker IM, Cox DB, Shmakov S, Makarova KS, Semenova E, Minakhin L, et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016;353:aaf5573. doi: 10.1126/science.aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R, Doudna JA. Applications of CRISPR technologies in research and beyond. Nat Biotechnol. 2016;34:933–941. doi: 10.1038/nbt.3659. [DOI] [PubMed] [Google Scholar]
- Begemann MB, Gray BN, January E, Gordon GC, He Y, Liu H, Wu X, Brutnell TP, Mockler TC, Oufattole M. Precise insertion and guided editing of higher plant genomes using Cpf1 CRISPR nucleases. Sci Rep. 2017;7:11606. doi: 10.1038/s41598-017-11760-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikard D, Jiang W, Samai P, Hochschild A, Zhang F, Marraffini LA. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 2013;41:7429–7437. doi: 10.1093/nar/gkt520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondy-Denomy J, Garcia B, Strum S, Du M, Rollins MF, Hidalgo-Reyes Y, Wiedenheft B, Maxwell KL, Davidson AR. Multiple mechanisms for CRISPR-Cas inhibition by anti-CRISPR proteins. Nature. 2015;526:136–139. doi: 10.1038/nature15254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle EA, Andreasson JOL, Chircus LM, Sternberg SH, Wu MJ, Guegler CK, Doudna JA, Greenleaf WJ. High-throughput biochemical profiling reveals sequence determinants of dCas9 off-target binding and unbinding. Proc Natl Acad Sci USA. 2017;114:5461–5466. doi: 10.1073/pnas.1700557114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson ED, Gan R, Hodgman CE, Jewett MC. Cell-free protein synthesis: applications come of age. Biotechnol Adv. 2012;30:1185–1194. doi: 10.1016/j.biotechadv.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caschera F, Noireaux V. Synthesis of 2.3 mg/ml of protein with an all Escherichia coli cell-free transcription-translation system. Biochimie. 2014;99:162–168. doi: 10.1016/j.biochi.2013.11.025. [DOI] [PubMed] [Google Scholar]
- Chappell J, Jensen K, Freemont PS. Validation of an entirely in vitro approach for rapid prototyping of DNA regulatory elements for synthetic biology. Nucleic Acids Res. 2013;41:3471–3481. doi: 10.1093/nar/gkt052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Guan J, Huang B. Imaging specific genomic DNA in living cells. Annu Rev Biophys. 2016;45:1–23. doi: 10.1146/annurev-biophys-062215-010830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez AA, Lim WA, Qi LS. Beyond editing: repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat Rev Mol Cell Biol. 2016;17:5–15. doi: 10.1038/nrm.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley QM, Karim AS, Jewett MC. Cell-free metabolic engineering: biomanufacturing beyond the cell. Biotechnol J. 2015;10:69–82. doi: 10.1002/biot.201400330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fineran PC, Gerritzen MJ, Suárez-Diez M, Künne T, Boekhorst J, van Hijum SA, Staals RH, Brouns SJ. Degeneratetarget sites mediate rapid primed CRISPR adaptation. Proc Natl Acad Sci USA. 2014;111:E1629–E1638. doi: 10.1073/pnas.1400071111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonfara I, Le Rhun A, Chylinski K, Makarova KS, Lécrivain AL, Bzdrenga J, Koonin EV, Charpentier E. Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR-Cas systems. Nucleic Acids Res. 2014;42:2577–2590. doi: 10.1093/nar/gkt1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonfara I, Richter H, Bratovic M, Le Rhun A, Charpentier E. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature. 2016;532:517–521. doi: 10.1038/nature17945. [DOI] [PubMed] [Google Scholar]
- Garamella J, Marshall R, Rustad M, Noireaux V. The all E. coli TX-TL toolbox 2.0: a platform for cell-free synthetic biology. ACS Synth Biol. 2016;5:344–355. doi: 10.1021/acssynbio.5b00296. [DOI] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. Genome-scale CRISPR-mediated control of gene repression and activation. Cell. 2014;159:647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gootenberg JS, Abudayyeh OO, Lee JW, Essletzbichler P, Dy AJ, Joung J, Verdine V, Donghia N, Daringer NM, Freije CA, et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science. 2017;356:438–442. doi: 10.1126/science.aam9321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington LB, Doxzen KW, Ma E, Liu JJ, Knott GJ, Edraki A, Garcia B, Amrani N, Chen JS, Cofsky JC, et al. A broad-spectrum inhibitor of CRISPR-Cas9. Cell. 2017;170:1224–1233.e15. doi: 10.1016/j.cell.2017.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockenberry AJ, Jewett MC. Synthetic in vitro circuits. Curr Opin Chem Biol. 2012;16:253–259. doi: 10.1016/j.cbpa.2012.05.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes AP, Rousseau GM, Lemay ML, Horvath P, Romero DA, Fremaux C, Moineau S. An anti-CRISPR from a virulent streptococcal phage inhibits Streptococcus pyogenes Cas9. Nat Microbiol. 2017;2:1374–1380. doi: 10.1038/s41564-017-0004-7. [DOI] [PubMed] [Google Scholar]
- Jewett MC, Calhoun KA, Voloshin A, Wuu JJ, Swartz JR. An integrated cell-free metabolic platform for protein production and synthetic biology. Mol Syst Biol. 2008;4:220. doi: 10.1038/msb.2008.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol. 2013;31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanter G, Yang J, Voloshin A, Levy S, Swartz JR, Levy R. Cell-free production of scFv fusion proteins: an efficient approach for personalized lymphoma vaccines. Blood. 2007;109:3393–3399. doi: 10.1182/blood-2006-07-030593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karvelis T, Gasiunas G, Young J, Bigelyte G, Silanskas A, Cigan M, Siksnys V. Rapid characterization of CRISPR-Cas9 protospacer adjacent motif sequence elements. Genome Biol. 2015;16:253. doi: 10.1186/s13059-015-0818-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karvelis T, Gasiunas G, Siksnys V. Methods for decoding Cas9 protospacer adjacent motif (PAM) sequences: a brief overview. Methods. 2017;121–122:3–8. doi: 10.1016/j.ymeth.2017.03.006. [DOI] [PubMed] [Google Scholar]
- Karzbrun E, Tayar AM, Noireaux V, Bar-Ziv RH. Synthetic biology. Programmable on-chip DNA compartments as artificial cells. Science. 2014;345:829–832. doi: 10.1126/science.1255550. [DOI] [PubMed] [Google Scholar]
- Kim E, Koo T, Park SW, Kim D, Kim K, Cho HY, Song DW, Lee KJ, Jung MH, Kim S, et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat Commun. 2017;8:14500. doi: 10.1038/ncomms14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT, Zheng Z, Gonzales AP, Li Z, Peterson RT, Yeh JR, et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015;523:481–485. doi: 10.1038/nature14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstiver BP, Tsai SQ, Prew MS, Nguyen NT, Welch MM, Lopez JM, McCaw ZR, Aryee MJ, Joung JK. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat Biotechnol. 2016;34:869–874. doi: 10.1038/nbt.3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komor AC, Badran AH, Liu DR. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. 2017;168:20–36. doi: 10.1016/j.cell.2016.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Makarova KS, Zhang F. Diversity, classification and evolution of CRISPR-Cas systems. Curr Opin Microbiol. 2017;37:67–78. doi: 10.1016/j.mib.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leenay RT, Beisel CL. Deciphering, communicating, and engineering the CRISPR PAM. J Mol Biol. 2017;429:177–191. doi: 10.1016/j.jmb.2016.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leenay RT, Maksimchuk KR, Slotkowski RA, Agrawal RN, Gomaa AA, Briner AE, Barrangou R, Beisel CL. Identifying and visualizing functional PAM diversity across CRISPR-Cas systems. Mol Cell. 2016;62:137–147. doi: 10.1016/j.molcel.2016.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy A, Goren MG, Yosef I, Auster O, Manor M, Amitai G, Edgar R, Qimron U, Sorek R. CRISPR adaptation biases explain preference for acquisition of foreign DNA. Nature. 2015;520:505–510. doi: 10.1038/nature14302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo ML, Mullis AS, Leenay RT, Beisel CL. Repurposing endogenous type I CRISPR-Cas systems for programmable gene repression. Nucleic Acids Res. 2015;43:674–681. doi: 10.1093/nar/gku971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJ, Charpentier E, Haft DH, et al. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol. 2015;13:722–736. doi: 10.1038/nrmicro3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall R, Maxwell CS, Collins SP, Beisel CL, Noireaux V. Short DNA containing χ sites enhances DNA stability and gene expression in E. coli cell-free transcription-translation systems. Biotechnol Bioeng. 2017;114:2137–2141. doi: 10.1002/bit.26333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekler V, Minakhin L, Semenova E, Kuznedelov K, Severinov K. Kinetics of the CRISPR-Cas9 effector complex assembly and the role of 3′-terminal segment of guide RNA. Nucleic Acids Res. 2016;44:2837–2845. doi: 10.1093/nar/gkw138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanraju P, Makarova KS, Zetsche B, Zhang F, Koonin EV, van der Oost J. Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science. 2016;353:aad5147. doi: 10.1126/science.aad5147. [DOI] [PubMed] [Google Scholar]
- Mulepati S, Bailey S. In vitro reconstitution of an Escherichia coli RNA-guided immune system reveals unidirectional, ATP-dependent degradation of DNA target. J Biol Chem. 2013;288:22184–22192. doi: 10.1074/jbc.M113.472233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelles DA, Fang MY, O’Connell MR, Xu JL, Markmiller SJ, Doudna JA, Yeo GW. Programmable RNA tracking in live cells with CRISPR/Cas9. Cell. 2016;165:488–496. doi: 10.1016/j.cell.2016.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardee K, Slomovic S, Nguyen PQ, Lee JW, Donghia N, Burrill D, Ferrante T, McSorley FR, Furuta Y, Vernet A, et al. Portable, on-demand biomolecular manufacturing. Cell. 2016;167:248–259.e12. doi: 10.1016/j.cell.2016.09.013. [DOI] [PubMed] [Google Scholar]
- Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol. 2013;31:839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawluk A, Amrani N, Zhang Y, Garcia B, Hidalgo-Reyes Y, Lee J, Edraki A, Shah M, Sontheimer EJ, Maxwell KL, Davidson AR. Naturally occurring off-switches for CRISPR-Cas9. Cell. 2016a;167:1829–1838.e9. doi: 10.1016/j.cell.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawluk A, Staals RH, Taylor C, Watson BN, Saha S, Fineran PC, Maxwell KL, Davidson AR. Inactivation of CRISPR-Cas systems by anti-CRISPR proteins in diverse bacterial species. Nat Microbiol. 2016b;1:16085. doi: 10.1038/nmicrobiol.2016.85. [DOI] [PubMed] [Google Scholar]
- Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath D, Amlinger L, Hoekzema M, Devulapally PR, Lundgren M. Efficient programmable gene silencing by Cascade. Nucleic Acids Res. 2015;43:237–246. doi: 10.1093/nar/gku1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch BJ, Silvis MR, Hultquist JF, Waters CS, McGregor MJ, Krogan NJ, Bondy-Denomy J. Inhibition of CRISPR-Cas9 with bacteriophage proteins. Cell. 2017;168:150–158.e10. doi: 10.1016/j.cell.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt R, Bukau B, Mogk A. Principles of general and regulatory proteolysis by AAA+ proteases in Escherichia coli. Res Microbiol. 2009;160:629–636. doi: 10.1016/j.resmic.2009.08.018. [DOI] [PubMed] [Google Scholar]
- Shin J, Noireaux V. Efficient cell-free expression with the endogenous E. Coli RNA polymerase and sigma factor 70. J Biol Eng. 2010;4:8. doi: 10.1186/1754-1611-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, Noireaux V. An E. coli cell-free expression toolbox: application to synthetic gene circuits and artificial cells. ACS Synth Biol. 2012;1:29–41. doi: 10.1021/sb200016s. [DOI] [PubMed] [Google Scholar]
- Shmakov S, Abudayyeh OO, Makarova KS, Wolf YI, Gootenberg JS, Semenova E, Minakhin L, Joung J, Konermann S, Severinov K, et al. Discovery and functional characterization of diverse class 2 CRISPR-Cas systems. Mol Cell. 2015;60:385–397. doi: 10.1016/j.molcel.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun ZZ, Yeung E, Hayes CA, Noireaux V, Murray RM. Linear DNA for rapid prototyping of synthetic biological circuits in an Escherichia coli based TX-TL cell-free system. ACS Synth Biol. 2014;3:387–397. doi: 10.1021/sb400131a. [DOI] [PubMed] [Google Scholar]
- Swartz J. Developing cell-free biology for industrial applications. J Ind Microbiol Biotechnol. 2006;33:476–485. doi: 10.1007/s10295-006-0127-y. [DOI] [PubMed] [Google Scholar]
- Takahashi MK, Chappell J, Hayes CA, Sun ZZ, Kim J, Singhal V, Spring KJ, Al-Khabouri S, Fall CP, Noireaux V, et al. Rapidly characterizing the fast dynamics of RNA genetic circuitry with cell-free transcription-translation (TX-TL) systems. ACS Synth Biol. 2015a;4:503–515. doi: 10.1021/sb400206c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi MK, Hayes CA, Chappell J, Sun ZZ, Murray RM, Noireaux V, Lucks JB. Characterizing and prototyping genetic networks with cell-free transcription-translation reactions. Methods. 2015b;86:60–72. doi: 10.1016/j.ymeth.2015.05.020. [DOI] [PubMed] [Google Scholar]
- Tayar AM, Karzbrun E, Noireaux V, Bar-Ziv RH. Propagating gene expression fronts in a one-dimensional coupled system of artificial cells. Nat Phys. 2015;11:1037–1041. [Google Scholar]
- Tsai SQ, Nguyen NT, Malagon-Lopez J, Topkar VV, Aryee MJ, Joung JK. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat Methods. 2017;14:607–614. doi: 10.1038/nmeth.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra ER, van Erp PB, Künne T, Wong SP, Staals RH, Seegers CL, Bollen S, Jore MM, Semenova E, Severinov K, et al. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Mol Cell. 2012;46:595–605. doi: 10.1016/j.molcel.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche B, Heidenreich M, Mohanraju P, Fedorova I, Kneppers J, DeGennaro EM, Winblad N, Choudhury SR, Abudayyeh OO, Gootenberg JS, et al. Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat Biotechnol. 2017;35:31–34. doi: 10.1038/nbt.3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data S1. Related to Figure 6. Krona plots used to generate the PAM wheels. The plots capture the output of the TXTL-based PAM determination assays conducted on FnCpf1, Lb6Cpf1, Pb2Cpf1, EcCpf1, PsCpf1, McCpf1, and MbCpf1.
Supplemental Protocol S1. Related to Figure 1. Protocol for measuring the activity of CRISPR nucleases using TXTL.
Supplemental Protocol S2. Related to Figure 6. Protocol for performing the TXTLbased PAM determination assay.
Supplemental Table S1. Related to Figures 1 - 6 and S1 - S5. Plasmid, gBlocks, bacterial cell stocks, and primers used in this study.