

Abstract
Mammalian cells can respond to damage or stress by entering a state of arrested growth and altered function termed cellular senescence. Several lines of evidence suggest that the senescence response suppresses tumorigenesis. Cellular senescence is also thought to contribute to aging, but the mechanism is not well understood. We show that senescent human fibroblasts stimulate premalignant and malignant, but not normal, epithelial cells to proliferate in culture and form tumors in mice. In culture, the growth stimulation was evident when senescent cells comprised only 10% of the fibroblast population and was equally robust whether senescence was induced by replicative exhaustion, oncogenic RAS, p14ARF, or hydrogen peroxide. Moreover, it was due at least in part to soluble and insoluble factors secreted by senescent cells. In mice, senescent, much more than presenescent, fibroblasts caused premalignant and malignant epithelial cells to form tumors. Our findings suggest that, although cellular senescence suppresses tumorigenesis early in life, it may promote cancer in aged organisms, suggesting it is an example of evolutionary antagonistic pleiotropy.
Multicellular organisms have evolved mechanisms to prevent the unregulated growth and malignant transformation of proliferating cells. One such mechanism is cellular senescence, which arrests proliferation—essentially irreversibly—in response to potentially oncogenic events. Cellular senescence appears to be a major barrier that cells must overcome to progress to full-blown malignancy (1–3).
Cellular senescence was first described as a process that limits the proliferation of cultured human fibroblasts (replicative senescence). Proliferating cells progressively lose telomeric DNA, and short telomeres, which are potentially oncogenic, elicit a senescence response. In addition, DNA damage, oncogene expression, and supraphysiological mitogenic signals cause cellular senescence. Cellular senescence is controlled by tumor suppressor genes and seems to be a checkpoint that prevents the growth of cells at risk for neoplastic transformation (2, 3). In this regard, cellular senescence is similar to apoptosis. However, whereas apoptosis kills and eliminates damaged or potential cancer cells, cellular senescence stably arrests their growth.
Cellular senescence is also thought to contribute to aging (4–7), although how it does so is poorly understood. In addition to arresting growth, senescent cells show changes in function (2, 6, 7). Because senescent cells accumulate with age (8–10), they may contribute to age-related declines in tissue function. If so, cellular senescence may be an example of antagonistic pleiotropy. Aging phenotypes are thought to result from the declining force of natural selection with age. Consequently, traits selected to maintain early life fitness can have unselected deleterious effects late in life, a phenomenon termed antagonistic pleiotropy (11). The senescence-induced growth arrest may suppress the development of cancer in young organisms. The functional changes, by contrast, may be unselected consequences of the growth arrest and thus compromise tissue function as senescent cells accumulate.
Cellular senescence has been extensively studied in stromal fibroblasts from humans and mice. Upon senescence, such cells show striking changes in gene expression (7, 12), some of which relate to the growth arrest and senescent morphology. Other changes, however, relate to fibroblast function. Senescent fibroblasts secrete growth factors, cytokines, extracellular matrix, and degradative enzymes (2, 7, 13), all of which can alter tissue microenvironments and affect nearby epithelial cells. Interestingly, this secretory phenotype resembles that of fibroblasts adjacent to some carcinomas, although senescent and tumor-associated fibroblasts differ in growth potential, morphology, and other traits. Tumor-associated fibroblasts can stimulate epithelial tumorigenesis (14).
Because senescent cells can alter the tissue microenvironment, we and others proposed that senescent cells may contribute to the exponential rise in cancer that occurs with age (2, 4, 13, 15). It is now clear that nonmutational events, such as telomere dysfunction or epigenetic changes in gene regulation or the stromal milieu, are important for the development of late-life cancers (15). Changes in the stroma, which supports and maintains epithelial functions, may be particularly important in humans, where most age-related cancers arise from epithelial cells (15).
Here, we show that senescent human fibroblasts promote the proliferation and tumorigenesis of mutant epithelial cells. Our data suggest that cellular senescence is antagonistically pleiotropic, protecting from cancer early in life, but promoting carcinogenesis in aged organisms.
Materials and Methods
Cells.
WI-38 (American Type Culture Collection), 82-6 (J. Oshima, University of Washington, Seattle), and 184 (M. Stampfer and P. Yaswen, Lawrence Berkeley National Laboratory) fibroblasts senesce after ≈50, 40, and 25 doublings, respectively, and were cultured as described (8, 16). Presenescent and senescent cultures generally contained >70% and <10% proliferating cells and were <10% and >70% senescence-associated β-galactosidase positive, respectively (8, 16). HaCAT (A. Paller, Northwestern University, Evanston, IL), S1 (M. Bissell, Lawrence Berkeley National Laboratory), Ha(Pk) (L. Packer, University of California, Berkeley), and MDA231 (R. Lupu, Lawrence Berkeley National Laboratory) cells were cultured as described (17–20). Keratinocytes (Clonetics) were cultured as directed by the supplier and used 2–3 passages after receipt. 82-6 and WI-38 were immortalized with an hTERT-expressing retrovirus (21).
Cocultures.
Presenescent (5 × 104) and senescent (1 × 105) fibroblasts were allowed to attach to 6-well culture dishes overnight and were incubated in serum-free medium for 1–3 days to generate lawns with similar cell numbers. Epithelial cells were incubated in growth factor-deficient medium for 2–3 days, plated (2 × 104/well) on fibroblast lawns, and maintained in growth factor-deficient medium for 8 days, unless noted otherwise. Growth factor-deficient medium contained KBM, 1.8 mM CaCl2, 5 μg/ml insulin, 0.5 μg/ml hydrocortisone for HaCAT and normal keratinocytes, DME/F12, 5 μg/ml insulin, 1.4 μM hydrocortisone, 5 μg/ml prolactin for SCp2 cells, DME/F12, 250 ng/ml insulin, 10 μg/ml transferrin, 2.6 ng/ml Na2SeO3, 1 ng/ml epidermal growth factor, 0.1 nM estradiol, 1.4 μM hydrocortisone, 5 μg/ml prolactin for S1 cells, and DME/F12, 5 μg/ml insulin for MDA231 cells. Cultures were fixed in 4% paraformaldehyde and were stained with 1% Rhodanile blue (Sigma) or 1 μg/ml 4,6-diamidino-2-phenylindole (DAPI).
Soluble and Matrix Factors.
Epithelial cells were seeded in upper chambers of Millicells-PCF (Millipore) containing presenescent or senescent lawns in the lower chambers, or onto matrices prepared by culturing fibroblasts in serum-free medium for 2–3 days, removing the cells with EDTA (Versene, GIBCO/BRL) at 37°C or 0.05% Nonidet P-40 at 23°C (with similar results), and washing with serum-free medium.
Quantification of Epithelial Cells.
Fluorescent images from five random fields/well were captured at 400× and analyzed by using a modification of SCIL_IMAGE 1.3 (Netherlands Organization for Applied Scientific Research, Institute of Applied Physics, University of Amsterdam). The program was customized to recognize differences in DAPI fluorescence between epithelial (smaller, more intense) and fibroblast (larger, less intense) nuclei and calculate epithelial fluorescence/field. We verified that the epithelial DAPI signal is proportional to cell number by expressing enhanced green fluorescence protein (EGFP) by using a retrovirus (LXSN) (22) and comparing epithelial DAPI and EGFP fluorescence in cocultures. EGFP was expressed in >90% of cells.
Senescence Induced by p14ARF, RAS-Ha(V12), and H2O2.
Fibroblasts infected with p14ARF (22) or RAS-Ha(V12) retroviruses (23) were selected in puromycin (22), incubated without puromycin for 2–3 (p14ARF) or 3–4 (RAS) days (22, 23), and replated for coculture as described above. Confluent presenescent fibroblasts were given 550 μM H2O2 in serum-containing medium for 2 h, left to recover overnight, then replated at subconfluence (24). After 5–7 d, cells developed a senescent phenotype and were replated for coculture.
Tumorigenesis Assays.
Nude (nu/nu) mice (5 weeks old) were injected (100 μl vol) s.c. into the dorsal flap with 1.5 × 106 HaCAT or Ha(Pk) cells, alone or with 1.5 × 106 fibroblasts, or into the nipple region with 1 × 106 SCp2 cells, alone or with 1 × 106 fibroblasts, or 2.5 × 105 MDA231 alone or with 1 × 106 fibroblasts. We measured three maximum diameters at x, y, and z axes to derive tumor volume (size). Tumors were fixed in buffered formalin or embedded in Tissue-Tek (Sakura) and snap-frozen, with or without prior fixation in 4% paraformaldehyde and passage through a sucrose gradient. Fixed frozen sections were blocked in 1% BSA and stained with pan-cytokeratin antibody (1:100; Dako) for 1 h and FITC secondary antibody for 45 min.
Results
Senescent Fibroblasts Stimulate Preneoplastic Epithelial Cell Growth.
We preincubated presenescent and replicatively senescent human fibroblasts in serum-free medium to arrest the presenescent cells and used equal numbers of nondividing presenescent and senescent cells to produce 50–80% confluent lawns, onto which epithelial cells were seeded. We used four epithelial cell lines. HaCAT human epidermal keratinocytes (17), S1 human mammary epithelial cells (18), and SCp2 mouse mammary epithelial cells (19) are immortal, harboring p53 mutations, but do not form tumors in immunocompromised mice (refs. 17, 18, and 25; see Fig. 4). Thus, they are preneoplastic, having acquired only some mutations that predispose to malignancy. In addition, we used MDA231, an aggressive human breast cancer cell line (20). We also used normal human keratinocytes from adult or neonatal donors. Like the fibroblasts, these normal strains have a finite replicative capacity and no known mutations that predispose to malignancy. We preincubated epithelial cells in growth factor-deficient medium before seeding them in this medium onto fibroblast lawns.
Figure 4.

Tumor growth stimulated by fibroblasts. Nude mice were injected with epithelial cells alone (Control) or presenescent (Presn), senescent (Sen), or hTERT-immortalized (Telom) fibroblasts. At the indicated intervals (Days), tumor size was measured as described in Materials and Methods. The number of animals per group (n) is indicated. The last point on each line indicates when tumors were excised for histology. (A) HaCAT cells (1.5 × 106) alone or with 1.5 × 106 WI-38 fibroblasts. Shown are results from two experiments. The incidence of tumors >10 mm3 and average tumor size were greater in the senescent group (P < 0.05). (B) SCp2 cells (1 × 106) alone or with 1 × 106 WI-38 cells. Tumor incidence was greater in the senescent group (P < 0.05). (C) Ha(Pk) cells (1.5 × 106) alone or with 1.5 × 106 WI-38 cells. (D) MDA231 cells (2.5 × 105) alone or with 1 × 106 WI-38 cells. Tumor size was greater in the senescent group (P < 0.01).
We first evaluated preneoplastic and neoplastic epithelial cells cocultured with fibroblasts using Rhodanile blue, which preferentially stains epithelial colonies (Fig. 1A). Senescent fibroblasts, much more than presenescent fibroblasts, stimulated the growth of all four cell lines. To quantify this, we stained the cocultures with DAPI, a fluorescent DNA dye, and quantified the smaller, more intensely stained epithelial nuclei by image analysis (Fig. 1B). Alternatively, we expressed EGFP in the epithelial cells and measured EGFP fluorescence (see Fig. 3A). Compared with presenescent fibroblasts, senescent fibroblasts stimulated HaCAT, S1, and MDA231 cells 2- to 4-fold and SCp2 cells 3- to 7-fold (Fig. 1B). Preliminary data suggest that senescent fibroblasts also stimulate the growth of sarcoma cells (HT1080 fibrosarcoma, SOAS-2 osteosarcoma), but to a lesser extent (50–80% stimulation).
Figure 1.

Effects on preneoplastic and malignant epithelial cells. Epithelial cells were plated on WI-38 fibroblast lawns and cultured in growth factor-deficient medium for 8 days. (A) S1 (i and iv), SCp2 (ii and v), and HaCAT (iii and vi) cells, cultured on presenescent (i–iii) or senescent (iv–vi) lawns, stained with Rhodanile (240×). (B) Cocultures described in A, and coculture with MDA231 cells, stained with DAPI. Epithelial nuclei were quantified as described in Materials and Methods. The results (in arbitrary units) shown are from one of 3–5 experiments. Error bars = SEM of duplicate or triplicate wells. Gray bars, presenescent lawns; black bars, senescent lawns. (C) SCp2 cells cultured without fibroblasts (i) or with presenescent and senescent cultures at ratios 10:0 (ii), 9:1 (iii), 8:2 (iv), 5:5 (v), and 0:10 (vi), stained with Rhodanile (240×). (D) SCp2 cells cultured without fibroblasts (No Fb) or with ratios of fibroblasts described in C, quantified by DAPI fluorescence. Error bars = SEM from triplicate wells from one of two experiments.
Figure 3.

Characteristics of the growth stimulation. (A) EGFP-expressing HaCAT cells, seeded onto presenescent or senescent WI-38 lawns. EGFP fluorescence (in arbitrary units) was measured after 1–8 days, as indicated. Error bars = SEM from duplicate wells from one of two experiments. Gray bars, presenescent lawns; black bars, senescent lawns. (B) SCp2 cells, seeded onto WI-38 fibroblasts (Cells), plated in the upper chambers of Millicells containing fibroblasts in the lower chambers (Soluble), or plated onto matrices deposited by fibroblasts (Matrix), as described in Materials and Methods. Cell number was assessed after 8 days by DAPI fluorescence. Error bars = SEM from duplicate wells from one of two experiments. Gray bars, presenescent fibroblasts; black bars, senescent fibroblasts. (C) HaCAT cells, seeded onto lawns of control (Left) or p14ARF-expressing (Right) presenescent 82-6 fibroblasts, stained 8 days later with Rhodanile (250×). (D) HaCAT cells, quantified by EGFP fluorescence after coculture with control (−) or p14ARF-expressing (+) 82-6 fibroblasts (ARF); control (−) or p14ARF-expressing (+) hTERT-immortalized 82-6 cells (TERT-ARF); control (−) or RAS-Ha/V12-expressing (+) WI-38 cells (RAS); 82-6 cells untreated (−) or treated (+) with H2O2; or replicatively senescent WI-38 cells (R). Growth was assessed after 8 days, except for RAS panels, where growth was assessed after 5 days. Error bars = SEM from duplicate wells from one of two experiments. Gray bars, presenescent lawns; black bars, senescent lawns.
The growth stimulation was not caused by epithelial cells attaching better to senescent fibroblasts. One day after seeding, DAPI fluorescence showed that epithelial cells attached equally well to presenescent and senescent lawns (not shown). This differential growth stimulation was seen with three human fibroblast strains: WI-38 (fetal lung, Fig. 1), 82-6 (adult skin, Fig. 3), and 184 (adult breast, not shown).
Cells expressing a senescence marker (8) accumulate with age (8–10) but remain relatively rare, even in old tissues. To determine whether senescent fibroblasts stimulate preneoplastic epithelial cells even when abundant presenescent fibroblasts are present, we cultured SCp2 cells alone or on lawns containing varying fractions of senescent fibroblasts and stained with Rhodanile (Fig. 1C) or DAPI (Fig. 1D) 8 days later. SCp2 proliferation was minimal in the absence of fibroblasts (Fig. 1 Ci, and D, No Fb). Presenescent fibroblasts stimulated growth 5- to 8-fold (Fig. 1 Cii, and D, 10:0), which could be caused by presenescent fibroblasts per se, or by the 10–30% senescent cells always present in presenescent cultures. Whatever the case, increasing proportions of senescent fibroblasts progressively stimulated additional SCp2 growth, even when senescent cultures were only 10% of the fibroblast population (Fig. 1 C and D). This experiment also suggests that presenescent fibroblasts do not inhibit the growth of preneoplastic epithelial cells but, rather, that senescent cells facilitate their growth.
Senescent Fibroblasts Do Not Stimulate Normal Epithelial Cells.
In striking contrast to preneoplastic and neoplastic epithelial cells, genetically normal keratinocytes grew equally well on presenescent and senescent fibroblasts. After 8 days in coculture, there was no statistical difference between growth on presenescent and senescent lawns. This was true for neonatal and adult human keratinocytes (Fig. 2) and human mammary epithelial cells (not shown). Thus, although senescent fibroblasts stimulated preneoplastic and malignant epithelial cells, they did not differentially stimulate normal epithelial cells.
Figure 2.
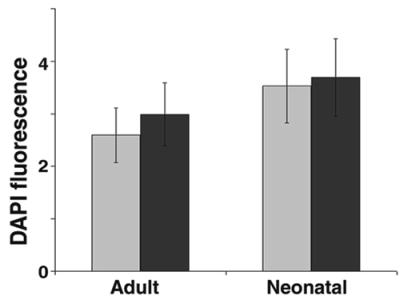
Effects on normal epithelial cells. Normal adult (Adult) or neonatal (Neonatal) human keratinocytes were seeded onto WI-38 lawns, cultured for 8 days, and quantified by DAPI fluorescence. Error bars = SEM from duplicate wells from one of two experiments. Gray bars, presenescent lawns; black bars, senescent lawns.
Kinetics.
To follow the kinetics of the growth stimulation, we seeded EGFP-expressing HaCAT cells onto presenescent or senescent fibroblast lawns and monitored EGFP fluorescence with time (Fig. 3A). Growth on senescent fibroblasts surpassed that on presenescent fibroblasts within 4 days (Fig. 3A), and continued to do so until the epithelial cells reached confluence (after 9–10 days; not shown). Unless noted otherwise, subsequent experiments were terminated within 8 days.
Contribution of Secreted Factors.
Senescent fibroblasts might stimulate epithelial cell growth by direct cell–cell interaction or by secreting diffusible factors or an insoluble extracellular matrix.
To test these possibilities, we cultured cells in two-chamber dishes. These separated fibroblast lawns from the epithelial cells by a porous membrane (0.4 μm pore size), preventing direct contact but permitting exchange of soluble diffusible factors. Soluble factors secreted by senescent fibroblasts were 2- to 3-fold more potent in stimulating HaCAT (not shown) and SCp2 (Fig. 3B) growth than those secreted by presenescent fibroblasts. Overall, however, senescent fibroblast-derived soluble factors were 10-fold less potent than direct cell contact (Fig. 3B, soluble vs. cells, black bars), even though epithelial cells attached equally well to the membrane and fibroblasts (not shown).
To determine the contribution of secreted matrices, we allowed fibroblasts in serum-free medium to deposit extracellular matrix onto culture dishes for 2–3 days. We then removed the cells by calcium chelation or mild detergent. Immunocytochemistry showed that fibroblast-depleted dishes contained abundant fibronectin (not shown), suggesting that at least this matrix component survived cell removal. We plated SCp2 cells onto the matrices and quantified cell number by DAPI fluorescence. Matrix produced by senescent fibroblasts was 3- to 4-fold more stimulatory than matrix produced by presenescent fibroblasts (Fig. 3B). This difference was not caused by differences in epithelial cell attachment (not shown).
Together, these results indicate that about 10% of the growth stimulation caused by senescent fibroblasts was caused by secreted soluble factors, whereas 40% was caused by secreted extracellular matrix. These are minimal estimates because cells may experience higher levels of soluble factors in direct coculture, and/or matrix components may be lost or inactivated during cell removal. Thus, at least 50% of the growth stimulation was attributable to the secretory phenotype of senescent fibroblasts.
Growth Stimulation Is Independent of the Senescence Inducer.
Overexpression of certain oncogenes or tumor suppressor genes or DNA damage can induce a phenotype that closely resembles replicative senescence (2, 12). We therefore asked to what extent the ability to stimulate preneoplastic epithelial cells depended on replicative exhaustion.
First, we induced senescence by overexpressing p14ARF, a tumor suppressor that mediates a senescence response (22). We seeded HaCAT cells onto lawns of presenescent or p14ARF-arrested fibroblasts and assessed HaCAT number by EGFP fluorescence. p14ARF-arrested fibroblasts stimulated HaCAT cell growth 2- to 3-fold relative to presenescent fibroblasts (Fig. 3 C and D).
We next tested 82-6 fibroblasts that we immortalized with hTERT (21), the catalytic subunit of human telomerase (26, 27). The immortal cells had completed >80 doublings, or twice the replicative lifespan of unmodified control cells. HaCAT cells grew equally well on immortal and presenescent control fibroblasts [Fig. 3D (−, ARF) vs. (−, TERT-ARF)]. Thus, cell division per se was not responsible for the stimulation caused by replicatively senescent fibroblasts. In response to p14ARF, the immortal fibroblasts arrested growth with a senescent morphology (not shown), whereupon they stimulated HaCAT growth 2- to 3-fold [Fig. 3D (+, ARF) vs. (+, TERT-ARF)].
We also induced senescence by expressing oncogenic RAS [RAS-Ha(V12)] (23) and seeded HaCAT cells onto presenescent or RAS-arrested fibroblasts. RAS-arrested fibroblasts stimulated HaCAT growth 1.5-fold compared with presenescent fibroblasts (Fig. 3D, RAS). This stimulation may be an underestimate because we analyzed the cocultures after only 5 days, owing to poor survival of RAS-expressing cells after >5 days in growth factor-deficient medium.
Finally, we treated fibroblasts with a sublethal dose (550 μM) of H2O2, allowed them to develop a senescent phenotype (24), and seeded HaCAT cells onto untreated or H2O2-treated fibroblasts. HaCAT cells proliferated 2- to 3-fold more readily on the H2O2-arrested fibroblasts (Fig. 3D, H2O2).
Thus, fibroblasts induced to senesce by several means stimulated the proliferation of preneoplastic epithelial cells. This stimulation was 50–70% of that caused by replicatively senescent fibroblasts (Fig. 3D, R), indicating that, at least qualitatively, the growth stimulation was independent of the senescence inducer.
Senescent Fibroblasts Stimulate Tumorigenesis.
To test the idea that senescent cells create a microenvironment that promotes the growth of potentially or frankly neoplastic cells in vivo, we injected epithelial cells, alone or with fibroblasts, into immunocompromised (nu/nu) mice (Fig. 4).
HaCAT cells alone did not form tumors after 40 (Fig. 4Ai) or 180 (not shown) days, as reported (17). When injected with an equal number of presenescent fibroblasts, three of 20 animals developed small tumors (<20 mm3); one regressed and two were excised for histology (Fig. 4Aii). Most of the animals (17 of 20), however, were tumor-free after >100 days. By contrast, when HaCAT were injected with senescent fibroblasts, seven of 15 animals developed tumors that averaged twice the size of those formed by presenescent fibroblasts (P < 0.01) (Fig. 4Aiii). Most of the tumors began to regress before excision, suggesting they were not malignant. This was not the case with SCp2 cells. SCp2 cells also did not form tumors (Fig. 4Bi), as reported (25). Moreover, when injected with presenescent fibroblasts, they did not form tumors after 120 (Fig. 4Bii) or 160 (not shown) days. However, when injected with senescent fibroblasts, they formed very large (400–2,000 mm3) tumors in four of six animals (Fig. 4Biii) with a latency of >80 days. These tumors would have killed the animals if not excised. Thus, senescent fibroblasts stimulated hyperproliferation and neoplastic progression, respectively, of HaCAT and SCp2 cells in vivo.
To determine effects on malignant epithelial cells, we used Ha(Pk), a weakly tumorigenic HaCAT derivative (unpublished data), and MDA231, an aggressive human breast cancer cell line (20). Ha(Pk) cells alone did not form tumors after 40 days (Fig. 4Ci) but did form tumors in four of five animals after 90 days (not shown). Presenescent fibroblasts accelerated tumorigenesis; within 25 days, three of five mice developed small (30–50 mm3) tumors that grew slowly over the next 2 weeks (Fig. 4Cii). Senescent fibroblasts accelerated tumorigenesis further; four of five mice developed tumors, two of which appeared in <10 days and grew rapidly (>100 mm3) (Fig. 4Ciii). More striking results were obtained with MDA231 cells. Small numbers (2 × 105) of MDA231 cells produced tumors that reached 300–400 mm3 in two of five mice in 45 days (Fig. 4Di). hTERT-immortalized fibroblasts slightly accelerated tumorigenesis, causing tumors that reached 100–500 mm3 in four of five mice (Fig. 4Dii). Senescent fibroblasts greatly accelerated tumorigenesis. Four of five mice developed tumors that reached 300–1,600 mm3 (Fig. 4Diii). Thus, at least for HaCAT, Ha(Pk), and MDA231, presenescent or hTERT-immortalized fibroblasts did not inhibit tumorigenesis. More important, senescent fibroblasts strongly stimulated or facilitated tumorigenesis.
Characteristics of Tumors.
We used EGFP-expressing HaCAT cells to confirm that tumors originated from the injected epithelial cells (Fig. 5 A and B). HaCAT tumors typically were firm nodules. In the presenescent group, tumors were often keratinized with sharply demarcated margins (Fig. 5C). By contrast, the senescent group frequently showed poorly defined margins with little keratinization (Fig. 5D). Tumors formed by Ha(Pk) cells were high-grade, dysplastic epidermoid cysts, with 5- to 10-fold more mitoses in the senescent group (not shown).
Figure 5.

Characterization of tumors induced by fibroblasts. (A) Fluorescence image of a frozen section of a tumor formed by EGFP-expressing HaCAT cells (80×). Tumors formed in the presence of presenescent or senescent fibroblasts were similarly fluorescent. (B) Section shown in A, counterstained with DAPI (80×). (C) HaCAT tumor formed in the presence of presenescent WI-38 cells (fixed section, stained by hematoxylin and eosin, 160×). Arrow shows a well demarcated tumor margin. (D) HaCAT tumor formed in the presence of senescent WI-38 cells (fixed section, stained by hematoxylin and eosin) (160×). Arrow shows a poorly demarcated tumor margin. (E) Fluorescence image of a frozen section of an SCp2 tumor formed by senescent WI-38 cells, stained for cytokeratin (160×). Solid arrow shows positive staining in the host mammary duct; open arrow shows lack of staining in surrounding tumor cells. (F) Frozen section adjacent to that shown in E, stained by hematoxylin and eosin (160×). Solid arrow, host ductal epithelial cells; open arrow, surrounding anaplastic tumor cells. (G) Paraffin-embedded section of an MDA231 tumor formed in the presence of presenescent WI-38 cells, stained with hematoxylin and eosin (40×). (H) Paraffin-embedded section of an MDA231 tumor formed in the presence of senescent WI-38 cells, stained with hematoxylin and eosin (40×).
SCp2 tumors, which formed only in the presence of senescent fibroblasts (Fig. 4B), were highly anaplastic, with frequent mitoses and features of malignant sarcoma (Fig. 5F). A striking feature was the loss of cytokeratin (Fig. 5 E and F), in contrast to abundant cytokeratin in the starting SCp2 cells (19, 25) and host mammary epithelium (Fig. 5 E and F). Senescent fibroblasts may induce an epithelial to mesenchymal transition in these cells or selectively stimulate variants that lost epithelial characteristics.
MDA231 cells formed poorly differentiated, aggressive tumors with characteristics of invasive ductal carcinoma, as reported (20). Aside from size, there were no discernible differences among tumors formed by presenescent and senescent fibroblasts (Fig. 5 G and H). Thus, senescent fibroblasts accelerated the growth, but did not influence the phenotype, of these tumors.
Discussion
Several lines of evidence indicate that cellular senescence suppresses tumorigenesis in vivo (1–3). First, many tumors contain cells that have partially or completely overcome senescence. Second, several oncogenes act at least partly by disabling the senescence checkpoint. Third, the senescence response requires p53 and pRB, the two most commonly lost tumor suppressors in malignant tumors. Finally, germ-line inactivation of the p53 or pRB pathways results in senescence-defective cells and cancer-prone organisms. Despite species differences in whether and how cells respond to specific senescence-inducing stimuli, cellular senescence very likely protects mammals from cancer, at least early in life.
We found that senescent human fibroblasts stimulated hyperproliferation and progression of preneoplastic epithelial cells and accelerated tumorigenesis by neoplastic epithelial cells. These results may seem at odds with the tumor suppression function of cellular senescence. They are, however, consistent with the evolutionary theory of antagonistic pleiotropy (11), which predicts that some genes, selected to enhance the fitness of young organisms, can have unselected deleterious effects in aged organisms. Our findings suggest that cellular senescence, despite protecting from cancer in young adults, may promote cancer progression in aged organisms. We speculate that the growth arrest was selected to ensure that damaged, mutant, or inappropriately stimulated cells—cells at risk for neoplastic transformation—do not proliferate. By contrast, the functional changes may be unselected consequences of the growth arrest, having little impact on young organisms where senescent cells are rare (8–10). However, as damage, telomere attrition, or errors cause senescent cells to accumulate with age, their influence, particularly their secretory phenotype, may become significant and deleterious.
Senescent fibroblasts had little impact on the growth of normal epithelial cells, although they can disrupt tissue architecture and function (28). However, they clearly stimulated preneoplastic and neoplastic cell growth, largely because of the secretion of both soluble and insoluble factors. Our results suggest that senescent cells produce multiple factors, which act together, to stimulate epithelial cells with oncogenic mutations.
Somatic mutations increase with age (29, 30), and some are potentially oncogenic. For example, loss of heterozygosity and mutations in p53 and RAS-Ha accumulate in normal adult tissue (31–33). We suggest that, with age, there is an increasing probability that senescent cells and cells with oncogenic mutations occur in close proximity. Senescent cells, then, may create a microenvironment that facilitates the growth and progression of the mutant cells. Although tumors in older organisms tend to be more indolent, neoplastic cells were more likely to form tumors in older animals (34).
The senescent microenvironment may synergize with multiple factors to contribute to late-life cancers (15). In addition to mutations, these include telomere dysfunction, hormonal and immune status, and angiogenic potential of the tissue. It is still not clear why most late-life cancers are epithelial. Sarcomas may be less prone to stimulation by senescent fibroblasts, but other factors (e.g., a greater need for mutations or a young hormonal or angiogenic milieu) may explain the relative paucity of sarcomas.
In summary, our results suggest a link between cancer and aging and a plausible mechanism by which genetic (oncogenic mutations) and epigenetic (accumulation of senescent cells) events synergize to generate the exponential rise in cancer that occurs with aging.
Acknowledgments
We thank R. Weinberg for the hTERT cDNA, S. Lowe for the RAS-Ha(V12) vector, D. Yip for preliminary experiments, W. Chou for help with computer analyses, the S. Fisher laboratory for help with tumor analyses, V. Band and R. Lupu for advice on tumor studies, L. Frappart for pathological evaluation of the tumors, and S. Hayward, G. Cunha, and T. Tlsty for discussions. This work was supported by Department of Defense Fellowship DAMD17-98-1-8063 (to A.K.) and National Institutes of Health Grants AG09909 (to J.C.) and CA82548 (to P.-Y.D.).
Abbreviations
- DAPI
4,6-diamidino-2-phenylindole
- EGFP
enhanced green fluorescence protein
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Yeager T R, DeVries S, Jarrard D F, Kao C, Nakada S Y, Moon T D, Bruskewitz R, Stadler W M, Meisner L F, Gilchrist K W, et al. Genes Dev. 1998;12:163–174. doi: 10.1101/gad.12.2.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campisi J. In Vivo. 2000;14:183–188. [PubMed] [Google Scholar]
- 3.Reddel R R. Carcinogenesis. 2000;21:477–484. doi: 10.1093/carcin/21.3.477. [DOI] [PubMed] [Google Scholar]
- 4.Campisi J. J Am Geriatr Soc. 1997;45:482–488. doi: 10.1111/j.1532-5415.1997.tb05175.x. [DOI] [PubMed] [Google Scholar]
- 5.Smith J R, Smith O M. Science. 1996;273:63–67. [Google Scholar]
- 6.Campisi J. Cell. 1996;84:497–500. doi: 10.1016/s0092-8674(00)81023-5. [DOI] [PubMed] [Google Scholar]
- 7.Campisi J, Dimri G P, Hara E. In: Handbook of the Biology of Aging. Schneider E, Rowe J, editors. New York: Academic; 1996. pp. 121–149. [Google Scholar]
- 8.Dimri G P, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano E E, Linskens M, Rubelj I, Pereira-Smith O, et al. Proc Natl Acad Sci USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pendergrass W R, Lane M A, Bodkin N L, Hansen B C, Ingram D K, Roth G S, Yi L, Bin H, Wolf N S. J Cell Physiol. 1999;180:123–130. doi: 10.1002/(SICI)1097-4652(199907)180:1<123::AID-JCP14>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 10.Mishima K, Handa J T, Aotaki-Keen A, Lutty G A, Morse L S, Hjelmeland L M. Ophthamol Visual Sci. 1999;40:1590–1593. [PubMed] [Google Scholar]
- 11.Rose M R. The Evolutionary Biology of Aging. New York: Oxford Univ. Press; 1991. [Google Scholar]
- 12.Shelton D N, Chang E, Whittier P S, Choi D, Funk W D. Curr Biol. 1999;9:939–945. doi: 10.1016/s0960-9822(99)80420-5. [DOI] [PubMed] [Google Scholar]
- 13.Rinehart C A, Torti V R. Mol Carcinog. 1997;18:187–192. [PubMed] [Google Scholar]
- 14.Olumi A F, Grossfeld G D, Hayward S W, Carroll P R, Tlsty T D, Cunha G R. Cancer Res. 1999;59:5002–5011. doi: 10.1186/bcr138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DePinho R. Nature (London) 2000;408:248–254. doi: 10.1038/35041694. [DOI] [PubMed] [Google Scholar]
- 16.Seshadri T, Campisi J. Science. 1990;247:205–209. doi: 10.1126/science.2104680. [DOI] [PubMed] [Google Scholar]
- 17.Skobe M, Fusenig N E. Proc Natl Acad Sci USA. 1998;95:1050–1055. doi: 10.1073/pnas.95.3.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Briand P, Petersen O W, van Deurs B. In Vitro Cell Dev Biol. 1987;23:181–188. doi: 10.1007/BF02623578. [DOI] [PubMed] [Google Scholar]
- 19.Desprez P Y, Roskelley C, Campisi J, Bissell M J. Mol Cell Differ. 1993;1:99–110. [Google Scholar]
- 20.Cailleau R, Olive M, Cruciger Q V J. In Vitro. 1978;14:911–915. doi: 10.1007/BF02616120. [DOI] [PubMed] [Google Scholar]
- 21.Kim S H, Kaminker P, Campisi J. Nat Genet. 1999;23:405–412. doi: 10.1038/70508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dimri G P, Itahana K, Acosta M, Campisi J. Mol Cell Biol. 2000;20:273–285. doi: 10.1128/mcb.20.1.273-285.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Serrano M, Lee A W, McCurrach M E, Beach D, Lowe S W. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 24.Chen Q M, Bartholomew J C, Campisi J, Acosta M, Reagan J D, Ames B N. Biochem J. 1998;332:43–50. doi: 10.1042/bj3320043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Desprez P-Y, Lin C Q, Thomasset N, Sympson C J, Bissell M J, Campisi J. Mol Cell Biol. 1998;18:4577–4588. doi: 10.1128/mcb.18.8.4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bodnar A G, Ouellette M, Frokis M, Holt S E, Chiu C-P, Morin G B, Harley C B, Shay J W, Lichtsteiner S, Wright W E. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 27.Meyerson M, Counter C M, Eaton E L, Ellisen L W, Steiner P, Caddle S D, Ziaugra L, Beijersbergen R L, Davidoff M, Liu Q, et al. Cell. 1997;90:785–795. doi: 10.1016/s0092-8674(00)80538-3. [DOI] [PubMed] [Google Scholar]
- 28.Funk W D, Wang C K, Shelton D N, Harley C B, Pagon G D, Hoeffler W K. Exp Cell Res. 2000;258:270–278. doi: 10.1006/excr.2000.4945. [DOI] [PubMed] [Google Scholar]
- 29.Dollé M E, Giese H, Hopkins C L, Martus H J, Hausdorff J M, Vijg J. Nat Genet. 1997;17:431–434. doi: 10.1038/ng1297-431. [DOI] [PubMed] [Google Scholar]
- 30.Dolle M E, Snyder W K, Gossen J A, Lohman P H, Vijg J. Proc Natl Acad Sci USA. 2000;97:8403–8408. doi: 10.1073/pnas.97.15.8403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deng G, Lu Y, Zlotnikov G, Thor A D, Smith H S. Science. 1996;274:2057–2059. doi: 10.1126/science.274.5295.2057. [DOI] [PubMed] [Google Scholar]
- 32.Jonason A S, Kunala S, Price G T, Restifo R J, Spinelli H M, Persing J A, Leffell D J, Tarone R E, Brash D E. Proc Natl Acad Sci USA. 1996;93:14025–14029. doi: 10.1073/pnas.93.24.14025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cha R S, Thilly W G, Zarbl H. Proc Natl Acad Sci USA. 1994;91:3749–3753. doi: 10.1073/pnas.91.9.3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCullough K D, Coleman W B, Smith G J, Grisham J W. Cancer Res. 1997;57:1807–1813. [PubMed] [Google Scholar]