

Abstract
Tau misprocessing to form aggregates and other toxic species has emerged as a major feature in our developing understanding of the etiology and pathogenesis of Alzheimer's Disease (AD). The significance of tau misprocessing in AD has been further emphasized by recent studies showing that tau can be secreted from neurons via exosomes and may itself be an important agent in the spreading of neurofibrillary lesions within the brain. Tau secretion occurs most readily under disease-associated conditions in cellular models, suggesting that cellular changes responsible for secretion, possibly including tau oligomerization, could play a key role in the propagation of neurofibrillary lesions in neurodegenerative disease. Here we show that overexpression of 4R0N human tau in neuroblastoma cells recruits mitochondrial and axonogenesis-associated proteins relevant to neurodegeneration into the exosomal secretion pathway via distinct mechanisms. The recruitment of mitochondrial proteins appears to be linked to autophagy disruption (exophagy) in multiple neurodegenerative conditions but but has few known direct links to AD and tau. By contrast, the involvement of synaptic plasticity and axonogenesis markers is highly specific to both tau and AD and may be relevant to the reactivation of developmental programs involving tau in AD and the recently demonstrated ability of secreted tau to establish tissue distribution gradients in CNS neuropil. We also found a highly significant correlation between genes that are significantly downregulated in multiple forms of AD and proteins that have been recruited to exosomes by tau, which we interpret as strong evidence for the central involvement of tau secretion in AD cytopathogenesis. Our results suggest that multiple cellular mechanisms may link tau secretion to both toxicity and and neurofibrillary lesion spreading in Alzheimer's disease and other tauopathies.
Keywords: tau, exosome, tau secretion, mitochondria, autophagy, tau lesion spreading, morphogen, gene downregulation, Alzheimer, endosome
Introduction
The central importance of the microtubule-associated protein tau in human neurodegenerative disease has made understanding its biological and pathological functions a major recent focus of biomedical research. This has resulted in an improved understanding of how the formation of hyperphosphorylated tau aggregates results in cytotoxicity [1-3] and has also expanded our view of tau biology and pathobiology beyond microtubule stabilization and aggregation (respectively) to include additional normal tau functions that may also play a role in disease. Such functions include interactions with the plasma membrane [4-8], signal transduction (via the N terminal projection domain of tau) [7] and diverse developmental functions including the cell cycle [8], tissue patterning [9] neuronal polarization and axon identity [9-11] and various aspects of axonogenesis [12-16]. All of these now appear to play some kind of role in the pathogenesis of diverse diseases that involve tau, including Alzheimer's Disease (AD) the various non AD tauopathies [17-21], Down's Syndrome [22] and even some forms of breast and prostate cancer [23]. Over the past few years it has also become generally accepted that tau can be secreted from neurons via multiple non-classic pathways [24-28] and that secretion may be an important biological function of tau protein, especially in disease [26, 29-35]. Surprisingly, the cellular mechanisms underlying tau secretion and uptake have as yet received little detailed attention, despite growing evidence that tau can be toxic when applied extracellularly [36-39], and the intense recent interest in a possible role for tau playing a “prion-like” role in the spreading of neurofibrillary lesions [40-51].
Tau lacks a signal sequence and is secreted unconventionally via exosomes [26] and/or microvesicle shedding mechanisms [24,27]. The importance of exosomal tau secretion is highlighted by our recent study suggesting that tau secretion via the exosome pathway may account for the elevated CSF tau levels typically seen in early AD [26, 34]. However, while it does appear that tau secretion occurs in conjunction with misprocessing events that are set in motion by its release from axonal MTs [27] it is still unclear what these events are, how they result in the accumulation of tau in exosomes and most importantly, how they are related to tauopathy cytopathogenesis. Our understanding of the role played by tau secretion in disease is particularly hampered by the paucity of direct experimental context at the cellular level, a circumstance which has also limited progress in important and exciting areas of study (e.g. tau lesion spreading mechanisms). In order to better understand how tau becomes membrane associated and shunted into unconventional secretory pathways, we have conducted a systems-based analysis of proteins recruited to exosomes in tau-overexpressing neuroblastoma cells designed to identify cellular events that can account for exosomal tau secretion in terms of known protein-protein interactions involving tau. In addition, we compared our control and tau-associated sets of exosomal proteins to online databases of genes whose expression is significantly affected by AD to assess the relevance of tau secretion via the exosome pathway to the cytopathogenesis of tau-associated neurodegeneration in AD. This approach is largely complementary to that taken in other studies in this Special Issue in that it does not address the subject of oligomer-associated tau toxicity directly, but rather aims to provide a cellular context against which tau oligomer toxicity and the possibility of “prionlike” lesion spreading can be evaluated.
We find that 4R0N tau overexpression in neuroblastoma cells recruits a characteristic set of proteins to the exosomal proteome. Tau-recruited proteins are associated with axon development and identity, synaptic structure and function and neurodegenerative disease. Many of these proteins are known tau interactors that are specifically associated with AD and (somewhat surprisingly) are themselves frequently found in exosomes. We also show that the proteins identified in exosomes from 4R0N tau expressing M1C cells are very strongly associated with the downregulation of gene expression in both familial and sporadic forms of AD. We propose that specific sets of recruited proteins associated with synaptic plasticity, autophagy abnormalities, axonal development, and pattern generation may be of particular significance to understanding the cellular mechanisms responsible for exosomal tau secretion and their relationship to the pathobiology of neurodegenerative disease.
Methods
Collection and purification of neuroblastoma and hippocampal homogenate exosomes
M1C neuroblastoma cells were cultured, induced to express 4R0N tau and the exosomes secreted during the induction period obtained using serial ultracentrifugation [52] and mass spectrometry as described [26] to generate a set of 662 proteins. The additional sucrose fractionation step for “viscous bodily fluids” for preparing exosomes was added to the basic protocol for the isolation of brain homogenate exosomal fractions. Membrane pellets isolated by ultracentifugation from frozen hippocampal samples were resuspended with 2 ml of isolation buffer and equilibrated in a linear sucrose gradient (2.0–0.25 M sucrose in 20 mM HEPES, pH 7.2) layered on top of the suspension of membranous vesicles, permitting the removal of the exosome fraction. Tryptic digests of peptides for mass spectrometry were performed using the In-Gel tryptic digest kit (Thermo Scientific). Samples of digested peptides from raw and exosomal fractions of CSF and hippocampal homogenates were desalted and analyzed by MALDI mass spectroscopy in the positive mode at the Chemical Instrumentation Center (Boston University) using standard reverse phase LC/MS (injection volume, 10 l) Electrospray ionization on a Waters Qtof API US instrument from 100 to 8,000 m/z. The optimized conditions were found as follows: capillary, 3,000 kV; cone, 35; source temperature, 120 °C; and desolvation temperature, 350 °C. The gradient was 100% A (water 0.1% formic Acid) to 100% B (acetonitrile 0.1% formic acid) at 0.5 ml/min in 30 min. The column (Inertsil ODS-4, GL Sciences) used was a 250 4.6 mm with a particle size of 5 m. Deconvolution was carried out by MaxEnt 1. Identification of proteins containing specific peptide sequences was accomplished by searching online data bases maintained on the ExPASy proteomics server using the peptide mass fingerprinting tool MASCOT (Matrix Science Ltd., London, UK). Only proteins with match ratios (based on Mascot data base searches) indicating a 95% or greater confidence in the identity of the specific protein listed were included. The MALDI and tandem spectra used for protein identification from tryptic fragments were searched against the NCBI protein.databases using the MASCOT search engine (http://www.matrixscience.com). Peptides parameters were set as monoisotopic, cutoff was one missed trypsin cleavage allowed, and Mass tolerance of 0.8-Da MS/MS. A suitable control (NBC) dataset was assembled from uninduced M1C supernatants, which were supplemented with an online neuroblastoma exosomal protein dataset (ExoCarta) [53] for the analyses presented, since these collectively represent a proteome of equivalent size (633 proteins) to that of the 4R0N-expressing M1C cells. Gene products with non-descriptive names (i.e. those described as ORFs and with prefixes such as FLJ, KIA, ENG etc) were removed from all datasets in this study to facilitate analyses with the String program, which has a 2000 protein limit.
Systems based analyses
Gene Ontology (GO) terms defining biological function and components and Kyoto Encyclopedia of Genes and Genomes (KEGG) categories of cellular pathways and function available as pulldowns (Enrichment) at the String site were used to compare and analyze neuroblastoma exosome and published AD gene expression datasets. The String 9.05 proteomic analysis program [54] was used for most GO term based analyses, supplemented by use of the GeneMania database for the analysis of physical interactions between proteins as described [55]. A collection of terms covering a variety of cellular and developmental functions typically involved in either normal or neurodegenerative disease associated tau function [29] were chosen for these analyses. We used KEGG pathway, cell component and cell process GO terms relevant to a) established tau functions and associations in the mature and developing nervous system (Alzheimer's Diasease, cytoskeletal/axonal and synaptic localization, synapse plasticity, axonogenesis and guidance) and b) vesicle trafficking pathways associated with exosome formation in our initial characterization of the tau-associated exosomal proteome in M1C cells. In order to assess the effect of the cellular background inherent in using a neuroblastoma model, we also used general cell cycle and “pathways of cancer” KEGG terms. We obtained internal connectivity information for the 4R0N tau and control proteomes and subsets of these that we generated – see below) using the network diagrams provided by String 9.05. In analyses involving the detailed differentiation of specific axon guidance pathways, the Reactome gene list analysis feature [56] was used. Significance values presented here (when not otherwise specified) were provided by the String and Reactome sites (Bonferroni correction). In some cases, statistical significance was determined using Fisher's Exact test, with the Chi Square test (Yates correction) being used when larger samples (500 proteins or more) were involved. String connectivity drawings were used to illustrate linkages between specific GO function distributions – this was done by superimposing the String protein functional connectivity diagrams for each term in Photoshop and altering the color of positives to permit direct comparisons. Overlap was indicated by using bicolored/multicolored protein icons as described in figure legends. String connectivity images were also used to delineate M1C subsets 2 and 3 as described below.
Segementation Analysis of 4R0N tau overexpression and NB control exosomal proteomes
String 9.05 and GeneMania were used to model the tau induced recruitment of proteins to exosomes based on known physical interactions between specific proteins and tau and to perform segmentation analysis of the M12C dataset based on the presence of physical protein-protein interactions involving tau (Set 1), the presence of functional protein-protein links excluding tau (Set 2) or the lack of any known interprotein interactions as defined by String connectivity maps as described below. Physical interaction information is available as a pulldown function at the GeneMania website (http://pages.genemania.org) and can be obtained from the String site (http://stringdb.org/newstring_cgi/show input page.pl) by using only the “Experimental data” channel (shown at the bottom of the connectivity diagram page) to generate linkage maps and lists. We used a threshold of 0.4 for both sites used to define a “significant” protein-protein interaction for the definition of Set 1. This was done using the “saturation” technique (Supplemental Figure 1) to define subsets of exosomal proteins that are tau-linked and which therefore might have been recruited to exosomes via direct and/or indirect physical interactions between overexpressed tau. Sets 2 and 3 were then defined by removing the 99 Set 1 proteins from the M1C protein list and generating the String connectivity map (for this purpose all information categories were used, not only the “Experimental Data” channel. Protein icons that were not connected to other proteins in the map at the 0.4 link strength (moderate) setting were then placed in Set 3 with the remaining linked proteins becoming Set 2.
Generation of target datasets
We generated datasets targeted at several proteins and cellular elements that were not adequately described by exisiting GO and KEGG terms for characterizing the relationship of these elements to tau. This was done via one of several approaches depending on the nature of the target. For APP, a well characterized candidate tau ligand protein, we used a set of high confidence physical interactors using the String interactor tool. This approach was also used with tau itself for examining the contribution of physical interactions between overexpressed tau and exosomal proteins versus the NB control exosomal proteome. A more elaborate approach was required to define the exosomal proteome itself, owing to the inadequacy of the available GO terms and the multiplicity of exosomal control datasets and even definitions of what constitutes an exosome [52-53]. In order to circumvent these difficulties, we determined the citation frequency in ExoCarta (mammalian protein datasets only, 12, 372 citations, 3082 proteins) for each of the proteins in our tau overexpression and NB control exosomal datasets. Citation frequencies determined in this way were within 5% of those of the “25 most frequently identified proteins in exosomes” list on the ExoCarta site [53]. This approach permitted us to distinguish between exosome “markers” (64 proteins with 20+ citations) and less frequently cited proteins, which vary among exosomal proteomes according to their source and (possibly) the mechods used to isolate and characterize them. We subdivided the ExoCarta set into groups with a single citation (1369 proteins), 2-4 citations (943 proteins) 5-19 (708 proteins) as well as the 64 proteins with 20+. The 373 ExoCarta proteins that were also present in the control (226) and tau overexpression (147) were analyzed on an individual basis; those with 5 or more cites are shown in Figure 1a. In order to control for the use of cell line-derived exosomes in the NB control set, which might be expected to show an artifactual enrichment of tumor and apoptosis-associated proteins over normal tissue, we used a salivary exosomal proteome of 714 proteins derived from the curated set on ExoCarta (456 proteins [53]) and 385 proteins from an additional published source [57] that was not listed with ExoCarta.
Figure 1. Tau overexpression results in significant changes in the exosomal proteome relative to exosome markers and neuroblastoma control exosomes.
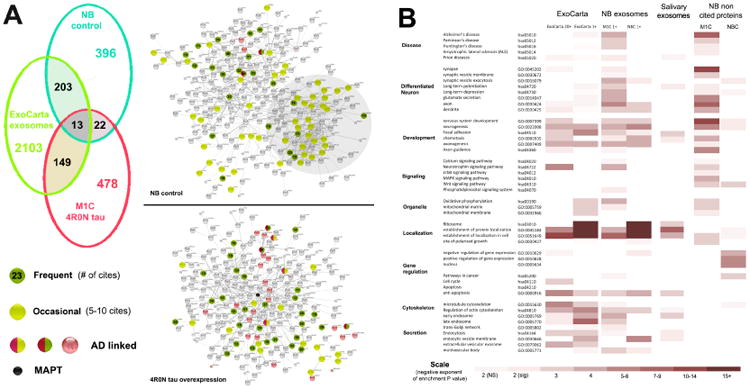
Panel A: Left top: Venn diagram showing the control neuroblastoma exosomal proteome (NB) with only a small degree of overlap with respect to specific proteins between the proteome obtained from exosomes released by M1C neuroblastoma cultures overexpressing 4R0N tau. Both sets of neuroblastoma exosome proteins overlapped with the large ExoCarta exosomal protein dataset of 2468 unique proteins identified once or more in a curated set of exosomal samples from a variety of mammalian sources. Right: Exosomal protein distributions within control and 4R0N tau datasets as seen in String connectivity diagrams. 415 control and 379 4R0N tau associated exosomal proteins with significant internal connectivity (0.2 or more, physical interactions only – see Methods) are shown. Proteins that are frequently seen in exosomes are shown as dark green icons with the number of ExoCarta cites. This group includes the “classical” exosome markers PDCD6IP (33 cites) TSG101 (19 cites) and the cytosolic enzymes ENO1 (37 cites) and GAPDH (46 cites), together with actins (ACTB 46 cites, ACTN1 15 cites), actin associated proteins (ACTR3 16 cites) and tubulins (TUBB, TUBB4, TUBA1A – 15-19 cites). Other proteins in this category were 14-3-3 proteins (5; 18-31 cites) and heat shock proteins (4; 21-51 cites). Proteins that are occasionally seen in exosomes include ribosomal subunit proteins include 28 ribosomal subunit proteins (6-10 cites) and 6 mitochondrial ATP synthases (5-10 cites) are shown in light green. Proteins in the in the KEGG set of Alzheimer's Disease (hsa30510) are also shown. Note that tau overexpression results in the displacement of certain classes of “occasional” exosomal proteins seen in the control set (e.g. ribosomal proteins) by tau-associated proteins with stronger links to AD. Panel B: Heat map representing the degree of enrichment seen for key GO component and Kegg pathway categories in the M1C and control sets. The 64 proteins that are most frequently cited in ExoCarta (leftmost column) and were also seen in neuroblastoma exosomes are very similar in the control and 4R0N tau datasets – they are combined here to illustrate the common pattern in enriched terms. Strong differences associated with tau overexpression begin to appear with the inclusion of 5-19 citation proteins (peripheral exosomal proteins). The NB exosomal GO term profile exhibits strong cell cycle and gene expression regulators that have not been identified in multiple ExoCarta exosome sets and which presumably represent elements specific to the cell line and the transformed state. Ribosome-associated proteins in the NB set are seen in a significant minority of exosome sets on ExoCarta and are associated with cellular localization terms; this presumably reflects strong microRNA and ribonucleoprotein representation in exosomes [68-69]. The absence of these proteins in the M1C proteome and their replacement by proteins associated directly (axonogenesis, axon, morphogenesis) and indirectly (mitochondrion, oxidative phosphorylation) with tau overexpression and secretion suggests active displacement by tau-associated proteins with 4R0N tau overexpression. Interestingly, structural proteins associated with axons and synapses – which are never seen in uninduced neuroblastoma cultures - were also recruited to exosomes by tau overexpression. Proteins not previously seen in exosomes show little enrichment in relevant GO terms in the NB control set, but are strongly enriched in AD, synaptic and development markers, presumably relecting their recruitment to exosomes via direct or indirect interactions with tau. Scale shows the negative exponent representing enrichment significance (p value) as determined using the enrichment tool in the String 9.05 proteomic analysis program.
Online target datasets
We used several online datasets to assess the relationship between tau associated (M1C) and neuroblasoma control (NBC) proteins in exosomes to a) changes in gene expression patterns seen during the early and middle stages of AD, b) changes seen in brain lipid rafts in the “triple transgenic” AD model mouse versus non TG control mice [58] and c) a set of AD/tauopathy associated proteins consisting of the union of the KEGG AD pathway dataset, the Alzheimer's Association list [59] and a compilation of toxicity-associated proteins derived from human tau overexpression in several fly model studies [60-63]. This last set is included in Supplemental file 2.
The extensively characterized dataset of up and downregulated genes from CA1 hippocampal homogenates in incipient, moderate and severe LOAD cases initially published by Blalock et. al. [64] and used in multiple subsequent studies [65-66]. We also made use of the excellent dataset of significantly up and downregulated mRNAs in posterior cingulate gyrus from early onset familal (PSEN1) and sporadic AD patients relative to age matched controls recently made available by Antonell et. al [67]. While all genes classified as “upregulated” or “downregulated” in these datasets were limited to those showing significant (p < 0.05) differences from control in the original studies [64, 67], we also examined more stringently defined subsets of the Blalock dataset in an attempt to correct for possible differences between CA1 and cingulate samples due to the somewhat earlier involvement of the hippocampus in the disease cascade of AD and thus increase the comparability of gene expression patterns. We therefore defined a “incipient-moderate only” subset of the Blalock dataset [59] that includes only those genes exhibiting significant increases/decreases relative to either incipient or moderate AD on the ground that changes in gene expression associated with large scale neuron death might become significant in hippocampal tissue by severe stages of AD and thus impair the usefulness of comparison made with the Antonell datasets, which are less likely to have this problem due to the source (i.e. posterior cingulate) of the material used. We also used more stringently defined subsets of the Blalock 04 dataset generated in other recent studies [65-66] to assess the robustness of our results [65]. Some of these results are shown in Supplemental figure 3.
We used the control and 3× AD model datasets published by Chadwick et. al. [58] to compare the effects of tauopathy on neuroblastoma exosomal and brain lipid raft proteomes. The 3× AD (900 proteins) and control mouse brain (1012 proteins) proteomes were used to construct heat map elements for comparison with tau overexpression, NB control and other datasets. Supplemental file 4 summarizes the heat map data generated by each of these datasets in comparison with the 4R0N tau and control NB datasets.
Tissue sections
Bright field images of lamprey CNS neurons that expressed full length (4R0N) and N terminal half (residues 1-255) human 4R0N tau are shown in Figures 4b. Tau expression was induced via transient transfection with microinjected plasmids as described elsewhere [27-28].
Figure 4. GO term based characterization of the M1C exosomal proteome vs tau associations and connectivity.
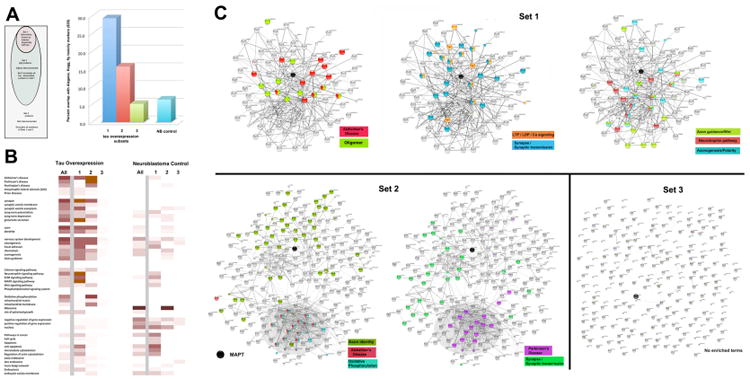
We used known physical interactions with tau (supplied by String 9.05 and GeneMania) and protein-protein interaction maps generated by String as criteria for sorting the 656 M1C and 633 control NB exosomal datasets into 3 classes. Panel A: Top - Schematic summarizing the criteria for segmentation. Bottom – Quantitative analysis of the relative significance (negative exponents of p values, Fisher's Exact Test) of enrichment of each set for members of the AD/fly tauopathy list of 835 proteins (Supplemental file 4) relative to the control NB set (light blue at right of graph). Both Set 1 and Set 2 were significantly enriched with proteins on this list. Panel B: Connectivity diagrams from String 9.05 for each subset of the 4R0N exosomal proteome. Proteins of Set 1 have known physical interactions (0.4 confidence or more, String) with tau and/or other proteins that interact with tau (up to 3 degrees of separation). Set 2 proteins are not connected with tau but exhibit 0.4+ level interconnections among themselves. Set 3 proteins were not interconnected either with each other or with tau at this level. GO and KEGG terms that were significantly enriched in terms relevant to neurodegenerative disease, axonal identity, axonal guidance during development and synaptic localization. Significant features of each group are color coded to highlight the strikingly selective enrichment of exosomal proteins with relevant GO and KEGG categories, An exception is made for proteins associated with tau oligomerization (Set 1 left) which did not have a GO term. Note that the mitochondrial/AD markers described in Figure 3 are completely segregated to Set 2 (shaded areas of connectivity diagram) and are not specific to AD. By contrast, markers of terminal neuronal differentiation (axon, synapse) are significantly enriched in both Sets 1 and 2. Specific markers for AD, axonal guidance, neurotrophins and synaptic plasticity are significantly enriched only in Set 1. Panel C: Heat map profiles for Sets 1-3 showing the significance of enrichment for these terms and others relative to the NB control set, which was segmented according to the same criteria.
Results
Initial characterization of tau overexpression and control exosomal datasets
The tau overexpression (M1C) and control (NB) exosomal datasets show broad functional similarities that reflect their shared exosomal and neuroblastoma origins. These included intrinsic and associated membrane proteins such as ion transporters and channels and phospholipid signaling pathway enzymes plus proteins associated with microtubule and actin cytoskeleton dynamics, endocytosis/exocytosis and neurite outgrowth, plus cytosolic proteins associated with cell cycle regulation and various housekeeping functions. These accounted for a significant minority of the proteins in both proteomes. However, only a relatively small proportion of the specific proteins identified (35, or 5-6% of each proteome) are common to both proteomes, consistent with our hypothesis that the overexpression of the 4R0N human tau isoform significantly alters the composition of cellular and secreted exosomes in neuroblastoma cells. A heat map summarizing the initial dataset analysis is given in Figure 1. Both control and tau overexpression exosomal proteomes include many of the proteins typically considered to be exosome markers (e.g. Alix, TSG101, Exosc9) [53] plus others typically found in endosomal populations (early endosomes, late endosomes, multivesicular bodies, autophagosomes) and in membranous organelles (Golgi apparatus, endoplasmic reticulum, mitochondria) that might contribute to exosome formation (Figure 1). Both the M1C and NB datasets contained over 10% of these markers as well as a large number of similar proteins, confirming the exosomal identity of the M1C dataset (Figure 2A). Because the exosome-mediated secretion pathway is not yet well defined in non-immune tissues, we used the citation frequency of each protein in the ExoCarta exosomal protein database (12,500 citations, 3087 unique proteins) as a proxy for its tendency to appear in exosomes, an approach used by the curators of the ExoCarta site [53]. By this measure, “classic” exosome markers” such as PDCP6IP (Alix), TSG101 and CD9 have citation frequencies ranging between 15 and 55. The ExoCarta dataset consists of 3000+ proteins with an average citation rate of 4.05. We found that 35% of control NB exosome proteins were cited at least once in the ExoCarta dataset. Tau overexpression reduced this significantly (p < 0.0001, chi square test) to 25%, but does not reduce the proportion of highly cited exosome markers seen (ExoCarta 25, HSPs, annexins, actins, tubulins). This suggests that tau overexpression and misprocessing may “recruit” non-exosomal proteins to exosomes, possibly displacing other proteins in the process.
Figure 2. Tau overexpression enriches exosomes with APP related proteins and proteins associated with synaptic transmission, nervous system development and AD.
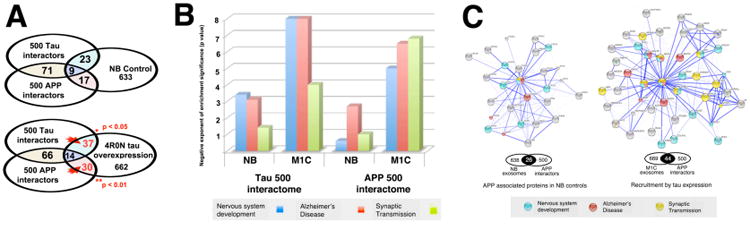
Panel A: Comparison of the NB and M1C proteomes with sets of the 500 strongest tau and APP interactors showed significant increases in both sets with tau overexpression (Fisher's Exact Test). Panel B: NB and M1C exosome proteins intersecting the tau 500 and APP 500 interactomes are enriched for developmental, AD and synaptic GO terms. While both NB and M1C proteomes show significant enrichment for each term, the effect of tau overexpression has a very strong effect on both APP and tau interacting proteins, especially those associated with nervous system development, (GO 0007399), synaptic transmission (GO 0007268) and Alzheimer's Disease (hsa 05010). The degree of enrichment (vertical axis) is given as the negative exponent of the P value provided (no correction) for each GO term. Panel C: Connectivity analysis illustrates tau-driven enrichment of APP interactors. The increased number of APP and synapse associated proteins in the tau overexpression proteome have significantly (p < 0.001, Fisher's Exact Test) greater functional and text database connectivity than controls, indicates that tau misprocessing may have significant functional effects on APP biology. These might involve APP postranslational processing by BACEs or presenilins during endocytosis induced by tau localization to endosomes, via a) the direct or indirect sequestation of APP secretases and/or b) synergistic effects on the oligomerization of A beta or other aggregation prone proteins.
Both M1C and NB proteomes include neuroblastoma-specific categories (i.e. cancer associated and early neurodifferentiation proteins) as well as lipid raft, trans-Golgi network (TGN) vesicle and exosome associated proteins. Proteins associated with cancer and the cell cycle and some proteins that are peripherally associated with exosomes (e.g. ribosomal proteins, ribonucleoproteins) were enriched only in the NB dataset, presumably because they were displaced in part by recruited tau interactors. Tau overexpression induced major changes in the identity and the degree of enrichment in several pathway and component categories. By GO term, the most enriched category of proteins was with the KEGG “Alzheimer” term, which was only marginally significant in controls and highly enriched in the tau associated proteome (Figure 2B), even when highly relevant overlapping proteins (including APP, APLP2) were removed to make the sets unique (not shown). In order to characterize the effects of tau overexpression on exosome protein components in terms of current literature on tau interactors and neurodegenerative disease (NDD) marker terms, we compared both the M1C and NB datasets with a “tau associated” dataset consisting of the 500 proteins most strongly associated with tau (Figure 2C). This “tau 500” dataset is highly enriched in proteins associated with AD (KEGG hsa05010), axonal development (hsa04360, axonogenesis) and identity (GO: 0030424). We found that both of these terms are also strongly enriched in the M1C relative to the NB exosomal proteome (Figure 2B), consistent with our hypothesis that tau secretion plays a role in both AD and in developmental tau functions associated with axonogenesis. The strong AD “signal” in M1C exosomes was confirmed by the observation that M1C proteome was strongly enriched in the AlzGene dataset of 616 AD-associated proteins [59] and in a set of 106 tau-interacting proteins identified in various recent publications in the fly tauopathy model as well [60-63] (Supplemental File 2).
Additional GO categories enriched by tau overexpression in exosomes comprehensively reflected the influence of normal and disease-associated tau characteristics, localization and function (Figure 2B). Among the most enriched tau overexpression correlates were localization/cell component terms reflecting tau cellular distribution, including axonal (GO: 0030424) relative to somatodendritic (GO: 0030425). Post synaptic localization (GO: 0045202, GO: 0045202) and synaptic transmission (GO 0007268) were particularly enriched, especially in characteristics specifically associated with glutamatergic synapses [70-71], which presumably reflects the role played by tau in synaptogenesis and (increasingly) in synaptic plasticity associated with learning and memory (GO 0007611). Signal transduction pathway elements associated with MAP kinases (REACT_12058, 9.20E-03) and neurotrophins (i.e. EGF (REACT_9417, 5.50E-03) and NGF (REACT_1106, 7.30E-03)) were also enriched somewhat by tau overexpression over the control set, which was itself strongly enriched for these terms, as might be expected for neuroblastoma cell lines. It is worth noting that this tau-associated enrichment in synaptic and axonal localization terms occurred in the absence of effective neurite polarization and synapse formation, which did not occur under the culture conditions used [26].
Exosomal APP associated proteins are enriched by tau overexpression
Much of the AD-associated signal seen with tau overexpression in the exosomal proteome was due to enrichment with proteins associated with the generation of A-beta from APP. This appeared to suggest that tau misprocessing can occur “upstream” of correlated events involving APP and A beta, contrary to the general consensus that A-beta acts upstream of tau in AD pathogenesis. Many of the tau exosome proteins associated with A-beta related mechanisms in AD (APP, APLP2, APH1A, APBA1 APBA2 AVRP2, PSEN1, ARRB2) were also members of synapse-associated and early endosome associated GO categories, consistent with known synapse and endosome-associated aspects of APP misprocessing. Segmentation of tau vs APP- associated proteins with respect to the ExoCarta list of proteins that have been identified in exosomes (described further below) showed that both APP and tau have strong physical interactions with proteins that are very frequently (20 identifications or more) seen in exosomes (e.g. heat shock and 14-3-3 proteins). These fail to account for the strong tau-APP link in neuroblastoma exosomes, since many of these are also present in the control NB set; the two sets show no significant difference in the number of tau interacting proteins present. However, proteins that have significant physical interactions with both APP and tau are significantly overrepresented with tau overexpression vs NB controls (p < 0.05, Fisher's Exact Test) in more “peripheral” exosome-associated proteins with between 1-19 citations in ExoCarta, suggesting that APP-tau interactions are a plausible candidate for facilitating tau diversion to exosomes.
Axon guidance pathways involving morphogen secretion are selectively overrepresented in tau-associated exosomes
Screening of the M1C and NB datasets for developmentally relevant signal transduction revealed an enrichment for a variety of developmentally relevant signal transduction pathways, including those associated with the development and the generation of morphogen gradients as well as synaptic plasticity, and Alzheimer's Disease. While comparisons between control and tau overexpression exosomes showed significantly elevated representation of proteins involved in a variety of developmental functions, those associated with axonal guidance via pathways involving diffusible morphogens (robo/slit, semaphorin, ephrin, netrin) were most consistently enriched (Figure 3A). The axon guidance signal was verified using an additive probe dataset (Appendix 1) targeting semaphorin, ephrin netrin and slit pathways (Figure 3A), which yielded a highly significant enrichment vs the control (NB) dataset (Fisher's Exact test p < 001). The selective enrichment for axon-associated morphogen pathways suggested a possible link between tau secretion and its functions in normal axonogenesis, which is highly consistent with the gradient-like distribution patterns of secreted tau protein within the CNS neuropil in the lamprey tauopathy model shown in Figure 3B. The dependence of the slope of tau tissue gradients on the presence of the MTBR suggests that MTBR-dependent interactions of tau with extracellular matrix elements (e.g. heparan sulfate proteoglycan) may mediate gradient formation in a manner akin to that seen with other developmental morphogen gradients [28, 69, 72].
Figure 3. Tau overexpression causes exosomes to be enriched in axonogenesis, axon guidance and macroautophagy markers.
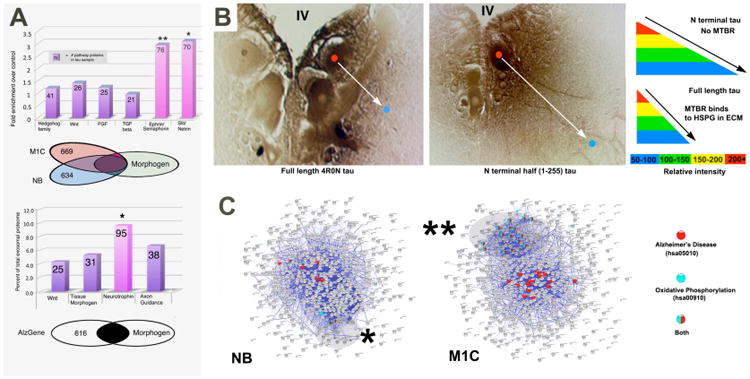
Panel A: The tissue distribution of secreted tau suggest a link between tau secretion and developmental morphogen pathways.Tau overexpression and control proteomes were subdivided into subsets associated with tissue morphogenesis (hedgehog, notch, Hox, n=1467), growth factor pathways (EGF, NGF, FGF, PDGF, TGFb, n= 1675) and axonogenesis (ephrin, semaphorin, netrin slit, n = 1481) and tau toxicity (n= 1035). Axonal guidance pathway proteins, but not the others, were significantly enriched. This differs from the relationship between tau associated and AD/tau toxicity datasets, which overlapped significantly more with with neurotrophin pathway proteins than with other morphogen pathways. The Venn diagram shows the degree of specific protein overlap between developmental proteomes (see Appendix) and the M1C and NB exosomal proteomes. The degree of enrichment relative to controls is highly significant for both morphogen associated and axonal development pathways (asterisk, p ≪ 0.001, chi square test). Predicted sets of tau interactors were generated by using canonical members for each pathway (via STRING) and then determining the proportion of them present in the control and tau overexpression proteomes. Surprisingly, tau recruitment appears to be specific to signaling pathways associated with both secreted morphogens and axonal development. The degree of enrichment in each of these pathways was significantly greater (* p < 0.03, ** p < 0.01, Fisher's Exact Test) than tissue patterning pathways such as hh, Notch, and even the Wnt pathway, which has established links to both exosome-mediated secretion and AD. Panel B: Tissue distribution of full length (left) and N terminal fragments lacking the microtubule binding domain (right) exhibit immunolabel gradients typical of classic morphogens such as wingless, hedgehog, semaphorins and netrins, which generate short (top right) or long (bottom right) range gradients depending on their interactions with ECN elements such as HSPGs. Such similarities suggest that that tau may be secreted by common or similar secretion mechanisms (characterized further in Le et. al 12). Panel C: Connectivity diagrams of the entire NB and M1C exosomal proteomes. Proteins with exclusively mitochondrial functions (GO: oxidative phosphorylation (blue) – highlighted) form a functional group associated with AD (red). We interpret this as a marker for exophagy – the diversion of auptophagosome contents to the exosome pathway.
Mitochondrial protein diversion to tau-associated exosomes is associated with AD and other NDDs
We used GO and Kegg pathway terms specific for intrinsic elements (mitochondrial membrane, mitochondrial matrix) and function (oxidative phosphorylation) exclusive to mitochondria to further characterize the enrichment of tau-associated exosomes in mitochondrial proteins noted in our initial analysis (Figure 1B). Superimposition of String connectivity maps with AD and oxidative phosphorylation pathway proteins (i.e. mitochondrial inner membrane) showed colocalization of nearly half of the AD markers with 90% of the mitochondrial inner membrane markers (Figure 3C). This pattern was highly non-random and indicates a close linkage between tau secretion and the disruption of organelle turnover mechanisms via autophagy, which has a long established association with pre-tangle tau pathology in AD [73]. These proteins were also associated with KEGG terms for Huntington's Disease (HD hsa05016) and Parkinson's disease (hsa05012), but not prion diseases (hsa05020). String connectivity diagrams illustrating these relationships are shown in Figures 3C and 4C.
Segmentation analysis of the M1C and NBC exosomal proteomes
The finding that proteins in the 4R0N tau overexpression set had significanly stronger direct physical interactions with tau than did those in the NB control set (0.840 vs 0.654, p < 0.01, Students t test) suggested that both direct and indirect physical interactions with overexpressed tau in the exosome pathway might account for the observed differences seen in the tau+ and tau- exosomal proteomes. In order to assess comprehensively the ability of physical interactions between overexpressed 4R0N tau to account for the diversion of non-exosomal proteins into the M1C exosomal proteome, we performed segmentation analyses on the M1C exosomal proteome based on the intrinsic tendency of each protein to a) associate with exosomes, b) interact physically with tau and c) its degree of interconnectedness based on String connectivity diagrams at 0.4 confidence. The analysis of exosome association was carried out as described (Methods) and as illustrated in Supplemental Figure 1. Segmentation based on tau association and interconnection and was done by sorting the 656 M1C proteins into 3 groups based on whether each protein a) has been shown to interact physically with tau and b) Set 1 includes 99 proteins that physically interact with tau directly or indirectly (at up to 3 degrees of separation) as determined by Gene Mania and String physical interaction database searches (Methods). To define Sets 2 and 3, we subtracted all Set 1 proteins from the total M1C exosomal proteome and generated a String 9.05 connectivity (confidence links) diagram at 0.4 (moderate) stringency from the list of the remaining 563 proteins. Set 2 was then defined as the interconnected protein set (282 proteins), whereas Set 3 includes the remaining non-interconnected proteins. Since all of these proteins were in fact identified in the dataset of exosomal proteins seen with tau overexpression, this segmentation effectively divided the M1C exosomal proteome into groups representing a) known tau-associated functions b) cellular functions hitherto not thought to involve tau that interact with misprocessed tau resulting in exosomal diversion and secretion and c) proteins whose diversion to exosomes with tau overexpression is entirely novel or fortuitous. Connectivity and GO term-based analysis of these sets is shown in Figure 4.
As expected for proteins with established physical links of some kind to tau, the GO term analysis of Set 1 proteins yielded a strong and specific signal for AD (hsa 05010) and for normal established tau functions in the development and function of the CNS. These included MT cytoskeleton (GO:0015630,1.39E-04), synapse markers and in particular synaptic plasticity. Strong development-associated signals include neurotrophin (e.g. NGF, EGF, FGF – hsa4722), axon guidance (including morphogen pathways semaphorin, robo/slit and netrin) and Wnt (hsa04310) pathways. The neurotrophin and synaptic plasticity signals were also present in the NB Set 1 control, suggesting a role in neuroblastoma cell lines, such as responsiveness to NGF and initial stages of neuronal differentiation. the The AD signal was quite specific and appears to be primarily represented by APP and associated proteins (APLP2, APBA1, PSEN1, BACE1). Chaperone proteins with known disease-associated tau interactions (STUB1, HSPA5, HSPA8) and disease-associated proteins known or thought to co-oligomerize with tau (SNCA, YWHAZ) were strongly represented, as were caspases (CASP7, CASP8) and disease-associated tau kinases (GSK3b, FYN), which also play roles in endocytosis-associated oligomerization [74-79] are also present in Set 1 (Figure 4b, top left panel).
The 282 proteins in Set 2 were identified as having no direct (i.e. within 3 degrees of separation) physical interactions with tau, but were part of the network of interacting proteins (see Figure 4b). also showed markers for synapses (SNAP25, STX1A, SYT1), but not synaptic plasticity. Despite the absence of tau links, this set had a significant AD signal (2.12E-03) via the KEGG AD term. This was primarily due to the presence of mitochondrial markers (mitochondrial ATP synthases, cytochrome oxidases and NADH dehydrogenases among others) that are associated significantly with Parkinson's Disease and Huntington's Disease as well as AD (Figure 4). Almost all of the “exophagy” (mitochondrial/NDD) markers were localized to Set 2, together with the strongest oligomerization signal present in any of the M1C exosomal subsets. Set 2 also contained a marginal axonal guidance signal involving the ephrin and Wnt pathways. By contrast, the 281 unconnected proteins in Set 3 showed no enrichment at all for pathways associated with tau or AD, indicating that these proteins have no known common involvement in cellular pathways that is directly discoverable by the analysis of existing databases.
Tau-associated exosomal proteins are downregulated in AD
We used online microarray datasets of genes up and downregulated in LOAD [64], FAD and early onset sporadic AD (EOSP) [67] to assess the relevance of tau-induced exosomal protein content changes to gene expression changes seen in tauopathy (Figure 5). We found a remarkable and unexpectedly strong positive correlation between the tau-recruited proteins in exosomes and reduced gene expression in AD. Proteins with reduced expression in AD brain were 4× more likely to be present in tau overexpression associated exosomes, a relationship that was highly significant (Figure 5a). The downregulated proteins included both the specific AD signal, synaptic plasticity and differentiated neuron (axon, dendrite, synapse) markers that we saw in Set 1 and the mitochondrial/general NDD markers of Set 2, suggesting strong direct and indirect connections (respectively) between these changes and AD pathogenesis. These correlations were largely independent of variations in age of onset and etiology between LOAD and FAD, although certain terms enriched in NB cells with tau overexpression such as axon guidance (hsa04360) and mitochondrial matrix (GO 0005759) were downregulated selectively in FAD/EOS and LOAD, respectively. The correlation between AD-associated downregulation and diversion to NB exosomes by tau overexpression was unaffected by the exclusion of severe AD data from the Blalock et. al. LOAD dataset, and thus seems quite robust. Overall, this analysis indicates a strong link between tau misprocessing and related changes resulting in exosomal tau secretion in AD [26] and the core mechanisms reponsible for AD cytopathogenesis. There was also a much weaker but still significant correlation between cancer pathway and gene expression regulation signals in the control NB dataset and those showing increased expression in AD (Figure 5a). However, these terms were not enriched in exosome sets from non-transformed sources (e.g. salivary exosomes) or in the enrichment profile of the ExoCarta exosomal proteome taken as a whole (Figure 5b), suggesting that their enrichment in NB control exosomes may be due to source-specific factors and may not reflect diseases relevance.
Figure 5. Tau associated proteins in M1C cell exosomes are strongly correlated with downregulated proteins in AD.
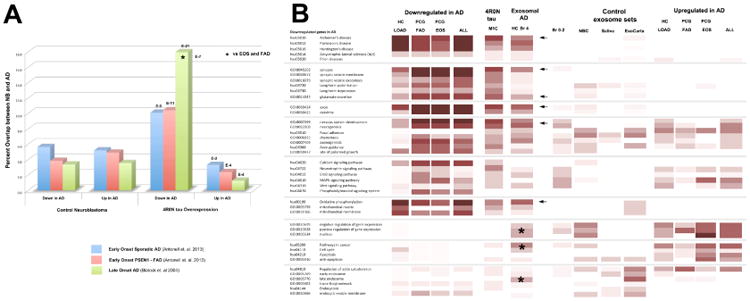
Panel A: Comparison of the exosomal proteomes with online databases of AD-influenced genes from late onset sporadic, early onset sporadic and early onset familial AD patients (Blalock 04, Antonell 13) show a highly significant increase in the proportion of proteins whose genes are downregulated in AD. Panel B: Heat map illustrating the results summarized in A. The most prominent feature in the common GO profile of downregulated genes in AD is the dominance of key markers of the differentiated neuron (axon, dendrites and synapses), genes involved in neuron development and mitochondrial genes. These features are shared by the M1C exosomal proteome to a remarkable extent. They also appear on the GO profile of exosomal proteins seen in hippocampal homogenates from 3 Braak Stage 4 cases (arrows) but not in control samples from low Braak stage (0-2) patients used in a previous analysis [26], strongly suggesting that tau secretion via the exosome pathway is highly relevant to specific aspects of AD cytopathogenesis. Interestingly, some of the other GO terms that showed increased enrichment with disease progression in AD exosomes (asterisks) were also enriched in the profile of upregulated genes in AD as well as in NB and other control exosome sets. The meaning of this is currently unclear.
Comparison of tau induced changes in exosomes with changes in lipid raft content in the 3×AD mouse model
In order to better understand the nature and disease relevance of tau-induced changes in the exosomal proteome, we compared the results of Chadwick et. al. [58], who used a bioinformatic analysis of the effects of the familial AD and tau mutations in the “triple transgenic” mouse model of AD, to our results. We compared these datasets with 4R0N tau overexpression and NB control with String 9.05 analysis as described above, with the results shown in Figure 6a (rightmost heat maps). Our rationale for doing this is based on the following considerations: a) perisynaptic lipid rafts are a known source of endosomes bearing synapse, synaptic plasticity and signal transduction/axon guidance signals, and thus a likely source of these proteins in exosomes [70] and c) the FAD and tauopathy mutations used in the 3×AD mouse should greatly increase the supply of both vesicular, abnormally phosphorylated tau and A beta. Furthermore, the presence of a control dataset in the Chadwick et. al. study should make it possible to test the implication of our segmentation analysis that the mitochondrial and synaptic plasticity/axon guidance markers that we found in 4R0N tau exosomes came from synapse-derived endosomes and mitochondria, respectively. One of the major results reported by Chadwick et. al. was the significant enrichment of synaptic plasticity, neurodevelopment and axonal markers relative to control mice. General synapse and axonal guidance markers were also significantly enriched in 3×AD rafts but were reduced relative to controls. This is consistent with a synaptic endosomal source for these proteins in exosomes with tau overexpression, and raises the possibility that axon guidance markers (in particular) in such endosomes might be diverted to exosomes by tauopathy mutant tau and increased A beta levels, thereby recapitulating their functions in developmental tissue polarization [69]. The list of control raft proteins provided in the Chadwick et. al. study contained none of the characteristic mitochondrial inner membrane markers (i.e. COX, NDUF and ATP5 family proteins) that we saw associated with the AD signal (KEGG “oxidative phosphorylation”) in exosomes with tau overexpression. Other mitochondrial markers (especially mitochondrial matrix – GO 0005759) were present in controls, but reduced in the 3×AD mice, leaving no clear implications as to the source of mitochondrial markers in NB exosomes. Overall, the comparison between 3×AD rafts and the effects of tau overexpression in neuroblastoma cultures is consistent with a direct recruitment of tau-interacting proteins from raft domains (including synaptic endosomes) to exosomes via tau overexpression as proposed below (Figure 6b).
Figure 6.
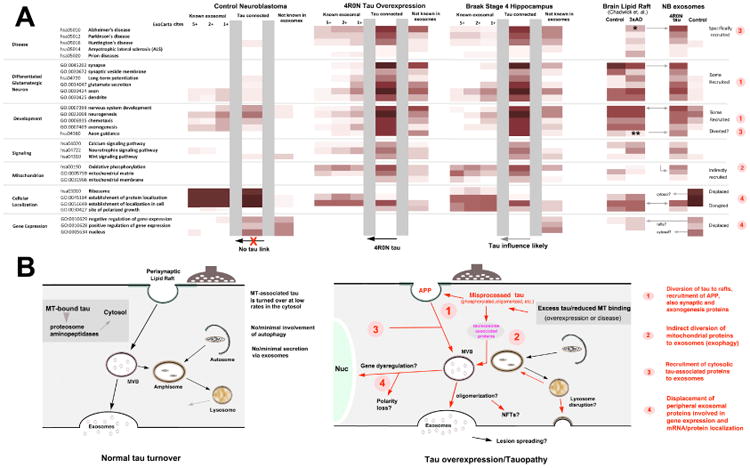
Model of tau-mediated effects on the exosomal proteome based on known protein-protein interactions between tau and exosome marker proteins. Panel A: Heat maps of neuroblastoma control and tau overexpression exosome proteins as a function of citation frequency on the ExoCarta list of 2458 characterized proteins identified in mammalian exosomes. GO term classifications for proteins with the highest Exocarta score are shown at left for the control (leftmost set) and 4R0N tau (center) proteomes, with the effect of adding proteins with progressively fewer ExoCarta citations (5+, 2+ 1+) are shown in adjacent columns to the right. GO term profiles for the subsets of proteins that have interactions with tau (i.e. NBC and M1C Set 1 proteins) and non-exosomal proteins are shown for NB control and tau overexpression proteomes, Exosomal, interconnected and non-exosomal sets are separated by gray vertical bars. Note that exosomal, tau-connected and non-exosomal sets exhibit a continuum of common features in the overall exosomal proteome associate with tau overexpression, but not in the neuroblastoma controls, suggesting that the observed effects of tau overexpression on exosome protein content can be largely accounted for by known protein-protein interactions. The similar GO profiles seen in an exosomal protein set from hippocampal homogenates of AD patients (Braak stage 4) suggest that exosomal recruitment due to tau misprocessing may have significant disease relevance. Right: Comparison of GO term profiles of neuroblastoma exosomes and 3×AD mouse brain lipid raft proteins are consistent with an endosomal origin for many previously non-ExoCarta cited (non exosomal) proteins recruited by tau overexpression. The presence of the 3×AD mutations modulate GO term signals characteristic of tau overexpression in ways that could reflect a merging of membrane raft and exosomal proteomes (asterisks, arrows) or a unidirectional transfer of proteins either into (axon guidance) or out of (ribosomal proteins) exosomes. Panel B: Schematic summarizing the observed and hypothesized cellular effects of tau misprocessing. Tau becomes under conditions that preclude its normal association with axonal MTs (e.g. AD, non AD tauopathies and overexpression), which saturate cellular MTs with tau and force the accumulation of non-MT associated tau at alternative tau binding cellular loci Non MT-associated tau becomes abnormally phosphorylated and associated with membranous cellular elements, eventually giving rise to both cytotoxicity and abnormal secretion via the exosome pathway. Numbers refer to: 1 association of non-MT bound tau with actin and actin-associated proteins in the subcortical cytoskeleton associated with perisynaptic lipid rafts, permitting interactions with APP, co-oligomerization candidates, morphogen pathway elements and synaptic markers, 2: diversion of mitochondrial AD markers away from lysosomes toward exosomes (exophagy) plus related effects on lysosomal acidification and transport resulting in extracellular release of incompletely proteolyzed contents, 3: abnormal interactions with cytosolic elements, including MT (DCX, MAP1A) and intermediate filament (NEFL, NEFH, GFAP) associated proteins, some with their own links to exosomes and 4: possible disruption of the “normal” exosomal proteome by the influx of tau-associated and exophagy related proteins, resulting in altered exosomal functions (protein localization, polarity) and/or changes induced by the displaced proteins themselves (possibly altered gene expression). Tau oligomerization might be a cause (e.g. driving interacting proteins to exosomes) or a consequence of these interactions with the exosome pathway, resulting in NFT formation and the spread of fibrillar tau lesions.
Discussion
A distinctive feature of neurodegenerative tauopathies is the strong inverse correlation between tau toxicity and its localization to axonal microtubules. Tau overexpression provides an unusually good model for disease-associated tau misprocessing because it saturates cellular MTs with tau and causes excess tau to accumulate in the cytosol, thereby driving abnormal post-translational processing (hyperphosphorylation, proteolytic cleavage etc.), tau secretion and tau toxicity thereby facilitating the characterization of disease-associated cellular mechanisms [27]. We have used this approach to characterize the unconventional secretion of tau via exosomes in neuroblastoma cell cultures and the relationship of exosome-mediated tau secretion to cellular dysfunction in AD. We find that the overexpression of a readily secreted tau isoform (4R0N) [25] and its localization to exosomes [26] is accompanied by the displacement of specific groups of proteins that are normally common in exosomes (e.g. ribosomal components) by known tau interactors associated with non-MT tau functions that could mediate specific disease-associated events (e.g. Abeta toxicity, neuropil thread formation, autophagy disruption) associated with tau misprocessing in tauopathy. These changes in the exosomal proteome induced by 4R0N tau overexpression greatly resemble those seen in exosomes from hippocampal homogenates with the progression of AD and are consistent with the effects of FAD/tauopathy mutations in the 3×AD on the contents of brain lipid raft preparations [58]. Segmentation based on known physical associations with tau, APP and other proteins in the exosomal proteome suggests that distinct pathways account for the presence of synaptic, axon outgrowth and AD associated proteins (synapses-associated endosomes) versus mitochondrial proteins (exophagy). Perhaps most striking of all is the correlation between tau-induced alterations to the exosomal proteome and the progression of AD - we find that 80% of the proteins that tau recruits to exosomes in neuroblastomas are also significantly downregulated in AD and that the GO term profiles of 4R0N tau exosomes is very similar to that. The strength of this relationship (p values range between 10-12 – 10-25 –see Figure 5a) clearly suggest the existence of strong links between exosomal tau secretion and tauopathy cytopathogenesis. The cellular changes that accompany tau overexpression and NDD-linked tau misprocessing and which might be responsible for tau secretion are summarized in Figure 6 and are discussed further below.
The relevance of tau-associated protein recruitment in neuroblastoma exosomes to human neurodegenerative disease
In this study, we have assumed that neuroblastoma-derived exosomes are the result of the same endocytotic pathways responsible for exosome formation in neurons in situ and can be used as a model for the cellular events responsible for the presence of tau in CSF exosomes [26] and for tau lesion spreading as demonstrated in a variety of tauopathy models [40-49]. While it has been noted that active neurite outgrowth may cause the release of exosomes containing differentiation and lysosome-associated proteins from growth cones [70], this cannot plausibly account for the large increases in enrichment for AD-specific, axonal synaptic and mitochondrial markers seen with tau overexpression on exosome contents relative to those from control neuroblastoma cultures and in exosomes from Braak Stage 4 hippocampal homogenates (Figure 5b, arrows). The high degree of similarity between the tau-induced effects of tau overexpression in neuroblastomal exosomes and the exosomal contents in AD brain, together with the very high congruence between the GO term profiles of both and those of genes downregulated in AD provides a very strong argument for the importance of both membrane-associated tau misprocessing and the disruption of endocytic pathways in the cytopathogenesis of both sporadic and familial AD and very likely of non-AD tauopathies as well.
Physical interactions between non-cytoskeletal tau and exosome markers favor but cannot account for the diversion of tau to exosomes
A somewhat surprising finding in this study was the high frequency at which proteins known to play important roles in tau pathobiology have been identified in the mammalian exosomal proteomes curated by ExoCarta. We found 38 neuroblastomal exosome proteins with strong links to tau that are in the top 10% of Exocarta cites, including 4 heat shock proteins (21-51 cites), 4 “14-3-3” proteins (19-32 cites), 6 “classical” exosome markers (flotillins, TSG101, alix – 9-33 cites), and 6 proteins in the actin-rich subcortical cytoskeleton (15-46 cites). Set 1 (i.e. known tau-interacting) proteins accounted for 67% and 50% of the top 10% ExoCarta proteins in the tau-induced and control sets respectively. The difference between control and tau induced sets was not significant; this may have been due either to the small sample size or to the inability of tau misprocessing to alter the disposition of strongly exosome-associated proteins. Such results suggest that exosomal links to tau via these proteins could influence its relocalization to the exosome pathway (or vice versa), but their strong representation in the NB control dataset, which showed no significant enrichment for AD terms, suggests the involvement of additional factors. Tau overexpression also had no apparent effects on a number of less frequently cited protein classes (e.g tubulins, proteosome subunits, integrins). By contrast, other less frequently cited proteins varied significantly with the presence of physical links to tau, even in the absence of direct interactions with tau. In particular, ribosomal proteins (2-14 cites) were strongly represented in the NB control (43 proteins) but not in the 4R0N tau set (1 protein), while mitochondrial proteins (4-13 cites) showed the reverse pattern (1 in the control set, 11 in the 4R0N tau set), suggesting that tau overexpression might displace some “peripheral” exosomal proteins with others via indirect means.
Increases in APP and synapse-associated proteins with tau overexpression
One major AD-relevant class of proteins that had increased representation in exosomes with tau overexpression was the proteins associated with APP, which had 5 ExoCarta cites, In particular, APP misprocessing to A beta has been linked to endocytosis and co-localizes with tau in endosomes [80] APP in lipid rafts also appears to interact with fyn and GSK3b resulting in disease-associated alterations to raft composition [79] that might favor its inclusion in exosomes [81] and might also involve tau [82-84]. Thus it is easy to see how interactions between non-MT associated tau, kinases known to phosphorylate tau and separate it from MTs (GSK3b, MARK1, CDK5, possibly fyn) and HSPs with established links to tau misprocessing (STUB1, HSPA8?) might bring tau and other tau-associated proteins to exosomes. The presence of fyn and non-exosomal synaptic proteins known to bind tau and favor tau oligomer formation (e.g. SNCA) [85-86] may also favor diversion of endosomes to the exosomal pathway [74], consistent with recent suggestions that tau oligomerization might favor interneuronal propagation of “prionlike” misprocessed tau species [40-49]. Even proteins (e.g. tubulins, neurofilament subunits) that are typically viewed as exclusively cytosolic may participate in tau localization to vesicles and redistribution to dendrites in conjunction with tau secretion [27]. Other proteins with strong intrinsic exosomal links that might account for tau-associated dysregulation include CDC42 (20 cites) which plays a central role in signal transduction pathways associated with axonogenesis, axon guidance and the establishment of neuronal polarity [87], all of which are key developmental functions of tau that appear to be involved in one way or another in tauopathy pathogenesis. The hypothesis that such interactions between tau and tau-associated proteins with strong (high citation) and/or “peripheral” (low citation) links to exosomes can account for the recruitment of proteins that have not hitherto been observed in exosomes is consistent with our observations in this study and is summarized in Figure 6a.
Exosomal tau secretion and the involvement of axonal guidance pathways in tauopathy - is tau a morphogen?
Diverse members of morphogen-associated signaling pathways were enriched in M1C exosomal proteins that were recruited using tau/exosome links in the String interactome [72, 87-90]. These include early endosome markers (FLOT1, EEA1), cell surface receptors (e.g. FZD10, EPHA4, EPHA7) [87, 91-95], and signal transduction elements (e.g. GSK3b, CDC42, PAKs, NRP2) that has been explicitly linked to axonal growth and guidance associated morphogens (see Figures 2B, 4A and 5) [69, 89, 92-96]. This raises the possibility that secreted tau may have morphogen-like features that might mediate some of the various roles tau plays in axonal morphogenesis and in the spreading of neurofibrillary lesions. We observed curated members of Wnt and other pathways linked to axonal guidance (e.g. slit, netrin, semaphorin, ephrin) were significantly enriched together with APP associated proteins in Set 1, together with other axonogenesis associated proteins (NF proteins, MAP1A). While axonal development is an established developmental function of tau (and is thus expected to be represented in the tau interactome), the enrichment in morphogens seen with exosomal tau secretion is novel, and suggests a possible developmental mechanism linking tau secretion in neurodegeneration with the recapitulation of developmental tau functions [29]. A morphogen-like function for tau secretion might mediate aspects of normal axonogenesis and neuronal polarization and also induce analogous abnormal events (growth of axonlike neuropil threads from dendrites, polarity loss) in tauopathy [97-98] and suggests a possible link between AD and risk factors such as head trauma via trauma associated proximal axonal injury and polarity loss [99-103] and the toxic consequences of somatodendritic tau accumulation and AD risk [105-106]. This hypothesis has been outlined in more detail elsewhere [106-107].
Tau and exophagy
The common disease signal for AD, PD and HD in Set 2 proteins was tightly linked to mitochondrial proteins as well as glutamatergic synaptic transmission, suggesting an important role for excitotoxicity and associated Ca++ dysregulation mechanisms that affect mitochondria in a variety of NDDs [108-109]. In particular, the exosomal enrichment of mitochondrial inner membrane proteins (cytochrome oxidases, ATP synthases, NADH dehydrogenases) in Set 2 proteins is strongly suggestive of an NDD-associated link between tau secretion and abnormalities in the autophagy pathway. Mitochondria are uniquely targeted by autophagy and play a central role in autophagy disruption in tau-associated NDDs [110], where associated tau misprocessing can cause increased colocalization of tau with autophagosomes and/or autophagolysosomes [111], with important implications for tau secretion and its role in tauopathy cytopathogenesis [112]. Lysosomal tau accumulation is a major cytopathological feature in inclusion body myositis [113] and in chloroquine induced myopathy [114]. Exosomal enrichment in autophagosomal markers has been attributed to a hybrid protein turnover mechanism (“exophagy”) in yeast [115], suggesting a similar origin for exosomal mitochondrial proteins in this study. Disease induced interactions between misprocessed tau in endosomes and autosomes (or mitochondria undergoing autophagy) might reverse the usual flow from MVBs to autophagosomes (pathway 2 in figure 6b) causing tau release by exophagy. Such a diversion would be consistent with early stage disruption of the endosome-lysosome system and autophagy pathways characteristically seen in AD [73, 110], nonAD tauopathies [113] and with tau overexpression [111] and could thus account for the colocalization of NDD signal with mitochondrial markers. The involvement of Ca++ dysregulation and excitotoxicity-related mechanisms suggested by exophagy are consistent with the glutamatergic synaptic signal seen in Set 2 proteins as well as Set 1 proteins [116].
Are the downregulation and diversion of tau-linked proteins to exosomes both part of a dysfunctional neuroprotective mechanism?
Our finding that the proteins diverted to exosomes with tau overexpression are typically downregulated in AD suggests that exosomal tau secretion is highly relevant to tauopathy pathogenesis, but offers no direct clue as to its nature. It has been persuasively argued that exosomes may be used by at least some cell types as a route for rapid disposal of proteins that are no longer needed for dynamic functions and which might become toxic if retained within the cell [70, 117]. This would be particularly applicable to postmitotic cells (such as neurons) that lack the option of diluting the effects of such proteins via mitosis and most especially to neurons specialized to perform functions (such as episodic synaptic plasticity) requiring highly dynamic changes in gene expression. If so, it is tempting to suppose that early tau mediated cytotoxic changes in AD that affect synaptic functions in glutamatergic neurons might induce both the downregulation and diversion to exosomes of plasticity-associated genes, especially those which mediate A beta-induced excitotoxicity [70, 118-119]. Small amounts of post-synaptically localized tau appear to play important roles in both synaptic plasticity and early disease-linked neurotoxic events [70, 104-105, 120-121], and may even play essential roles in mediating A beta synaptotoxicity [105,122]. The downregulation of such genes as part of a cytoprotective program could easily produce progressive dysregulation of proteins with multiple regulatory functions (e.g. GSK3b, fyn, CDK5, CDC42, PAK kinases) leading to other AD/tauopathy linked changes (cell cycle re-entry) [123] that could result in axonal transport failure, relocation of tau to dendrites and an amplification of tau mediated toxicity [106-107].
Where does tau oligomerization fit in exosome-mediated tau secretion?
Many of the tau-interacting proteins that we found in M1C exosomes have known (SNCA, GSK3b, APP/A beta) or suspected (YWHAZ, STUB1) roles in AD-relevant tau oligomerization, while other such proteins (fyn, MARK1, CDC42, ADAM22), could plausibly do so as well. For example, we found a significant enrichment of YWHAZ-interacting proteins [124] with 4R0N tau overexpression relative to NB controls (30% vs 4% of a total of 29, FET p = 0.01). Moreover, many proteins that might mediate tau oligomerization are themselves strongly linked to exosomes (e.g. 14-3-3 proteins and HSPs). Thus it seems likely that tau oligomerization might be a significant factor in the recruitment of such proteins to exosomes, especially since oligomerized tau has been identified in CSF exosomes in AD [26] and that oligomerization of membrane-associated proteins is a key event in multiple steps of the exosomal secretion pathway [74, 76, 79, 89] There is also reason to believe that tau oligomerization might play a subtle role in a “neuroprotective” exosomal secretion pathway, tau secretion in identified neurons in situ induced by anti-aggregation compounds (NNI3, NNI3b) has been linked to neuroprotection in the cells secreting tau [125-126]. NNI3 and similar compounds might conceivably favor secretion of some oligomers but not others depending on their relative level of oligomerization [74]. However, it is not known whether this treatment affected gene expression or whether the secreted tau was either oligomeric or exosomal; these remain attractive subjects for future study. Another point of uncertainty here involves the role of N terminal tau toxicity – N terminal tau fragments have been shown to mediate important aspects of A beta toxicity, especially in regard to glutamatergic synapses, but are unlikely candidates for oligomerization, since they lack the MTBR domain [127-130]. This makes it difficult (or at least premature) to assign a universal role to tau oligomerization in the cellular mechanism of tau mediated toxicity.
Conclusions
This study is part of an ongoing attempt to integrate exosomal tau secretion into what little is currently known about tau secretion at the cellular level with the goal of generating a cohesive view of cellular mechanisms in neurodegenerative tauopathy. We have used protein:protein interaction data from online databases and GO term profiling to characterize the changes in the exosomal protein population seen with 4R0N tau overexpression in neuroblastoma cells and from a database of exosomal fractions from low Braak stage (0-2) non AD and Braak Stage 4 AD patients [26]. We found that the normally non-exosomal proteins recruited to exosomes by tau are linked to multiple aspects of the pathogenesis of AD and some other NDDs, suggesting that tau secretion plays a central role in multiple neurofibrillary degenerative diseases. The involvement of morphogen pathways in tauopathy-associated exosomal tau secretion is consistent with tau oligomers and toxic proteolytic tau fragments diverting TGN vesicle trafficking mechanisms toward exosomes, resulting in the amplification of tau secretion in neurofibrillary disease. Such mechanisms might mediate tau lesion spreading by either oligomer-mediated or receptor-mediated tau toxicity pathways discussed in the recent tauopathy literature. Our results indicate a novel link between the dysregulation of gene expression and tau-associated exosomal secretion that may reflect neuroprotective changes in response to tauopathy or A beta-induced synaptic damage. Further investigation of these possibilities using direct experimental approaches should increase our understanding of the causes and consequences of exosomal tau secretion in the pathogenesis of neurofibrillary disease.
Supplementary Material
Abbreviations
- AD
Alzheimer's Disease
- PD
Parkinson's Disease
- NDD
neurodegenerative disease
- NFT
neurofibrillary tangle
- A beta
beta amyloid peptide
- MT
microtubule
- ECF
extracellular fluid
- MAP
microtubule associated protein
- PrP
prion protein
- YWHAZ
14-3-3-zeta
- CNS
central nervous system
- HD
Huntington's Disease
- APP
amyloid precursor protein
- 4R0N
human tau isoform with 4 MTBRs and 0 N terminal inserts
- GO
gene ontology
- CSF
cerebrospinal fluid
- MTBR
microtubule binding repeat
- TGN
trans Golgi network
- SNCA
alpha synuclein
References
- 1.Stoothoff WH, Johnson GV. Tau phosphorylation: physiological and pathological consequences. Biochim Biophys Acta. 2005;1739:280–297. doi: 10.1016/j.bbadis.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 2.Sergeant N, Bretteville A, Hamdane M, Hamdane M, Caillet-Boudin ML, Grognet P, Bombois S, Blum D, Delacourte A, Pasquier F, Vanmechelen E, Schraen-Maschke S, Buée L. Biochemistry of Tau in Alzheimer's disease and related neurological disorders. Expert Rev Proteomics. 2008;5:207–224. doi: 10.1586/14789450.5.2.207. [DOI] [PubMed] [Google Scholar]
- 3.Avila J. Tau phosphorylation and aggregation in Alzheimer's disease pathology. FEBS Lett. 2006;580:2922–2927. doi: 10.1016/j.febslet.2006.02.067. [DOI] [PubMed] [Google Scholar]
- 4.Brandt R, Leger J, Lee G. Interaction of tau with the neural plasma membrane mediated by tau's amino-terminal projection domain. J Cell Biol. 1995;131:1327–1340. doi: 10.1083/jcb.131.5.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee G, Newman ST, Gard DL, Band H, Panchamoorthy G. Tau interacts with src-family non-receptor tyrosine kinases. J Cell Sci. 1998;111:3167–3177. doi: 10.1242/jcs.111.21.3167. [DOI] [PubMed] [Google Scholar]
- 6.Maas T, Eidenmuller J, Brandt R. Interaction of tau with the neural membrane cortex is regulated by phosphorylation sites that are modified in paired helical filaments. J Biol Chem. 2000;275:15733–15740. doi: 10.1074/jbc.M000389200. [DOI] [PubMed] [Google Scholar]
- 7.Lee G, Thangavel R, Sharma VM, Litersky JM, Bhaskar K, Fang SM, Do LH, Andreadis A, Van Hoesen G, Ksiezak-Reding H. Phosphorylation of tau by fyn: implications for Alzheimer's disease. J Neurosci. 2004;24:2304–2312. doi: 10.1523/JNEUROSCI.4162-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andorfer C, Acker CM, Kress Y, Hof PR, Duff K, Davies P. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci. 2005;25:5446–5454. doi: 10.1523/JNEUROSCI.4637-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tian AG, Deng WM. Par-1 and Tau regulate the anterior-posterior gradient of microtubules in Drosophila oocytes. Dev Biol. 2009;327:458–464. doi: 10.1016/j.ydbio.2008.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caceres A, Kosik KS. Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature. 1990;343:461–463. doi: 10.1038/343461a0. [DOI] [PubMed] [Google Scholar]
- 11.Dawson HN, Cantillana V, Jansen M, Wang H, Vitek MP, Wilcock DM, Lynch JR, Laskowitz DT. Loss of tau elicits axonal degeneration in a mouse model of Alzheimer's disease. Neuroscience. 2010;169:516–531. doi: 10.1016/j.neuroscience.2010.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DiTella M, Feiguin F, Morfini G, Caceres A. Microfilament associated growth cone component depends upon tau for its intracellular localization. Cell Motil Cytoskeleton. 1994;29:117–130. doi: 10.1002/cm.970290204. [DOI] [PubMed] [Google Scholar]
- 13.Biernat J, Wu YZ, Timm T, Zheng-Fischhofer Q, Mandelkow E, Meijer L, Mandelkow EM. Protein kinase MARK/PAR-1 is required for neurite outgrowth and establishment of neuronal polarity. Mol Biol Cell. 2002;13:4013–4028. doi: 10.1091/mbc.02-03-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zmuda JF, Rivas RJ. Actin disruption alters the localization of tau in the growth cones of cerebellar granule neurons. J Cell Sci. 2000;113:2797–2809. doi: 10.1242/jcs.113.15.2797. [DOI] [PubMed] [Google Scholar]
- 15.Klein C, Kramer EM, Cardine AM, Schraven B, Brandt R, Trotter J. Process outgrowth of oligodendrocytes is promoted by interaction of fyn kinase with the cytoskeletal protein tau. J Neurosci. 2002;22:698–707. doi: 10.1523/JNEUROSCI.22-03-00698.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Belkadi A, LoPresti P. Truncated Tau with the Fyn-binding domain and without the microtubule-binding domain hinders the myelinating capacity of an oligodendrocyte cell line. J Neurochem. 2008;107:351–360. doi: 10.1111/j.1471-4159.2008.05600.x. [DOI] [PubMed] [Google Scholar]
- 17.Spillantini MG, Bird TD, Ghetti B. Frontotemporal dementia and parkinsonism linked to chromosome 17: a new group of tauopathies. Brain Pathol. 1998;8:387–402. doi: 10.1111/j.1750-3639.1998.tb00162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 19.Sergeant N, Wattez A, Delacourte A. Neurofibrillary degeneration in progressive supranuclear palsy and corticobasal degeneration: tau pathologies with exclusively “exon 10” isoforms. J Neurochem. 1999;72:1243–1249. doi: 10.1046/j.1471-4159.1999.0721243.x. [DOI] [PubMed] [Google Scholar]
- 20.Binder LI, Guillozet-Bongaarts AL, Garcia-Sierra F, Berry RW. Tau, tangles, and Alzheimer's disease. Biochim Biophys Acta. 2005;1739:216–223. doi: 10.1016/j.bbadis.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 21.Goedert M, Jakes R. Mutations causing neurodegenerative tauopathies. Biochim Biophys Acta. 2005;1739:240–250. doi: 10.1016/j.bbadis.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 22.Wegiel J, Gong CX, Hwang YW. The role of DYRK1A in neurodegenerative diseases. FEBS J. 2011;278:236–245. doi: 10.1111/j.1742-4658.2010.07955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Souter S, Lee G. Microtubule-associated protein tau in human prostate cancer cells: Isoforms, phosphorylation, and interactions. J Cell Biochem. 2009;108:555–564. doi: 10.1002/jcb.22287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim W, Lee S, Jung C, Ahmed A, Lee G, Hall GF. Interneuronal transfer of human tau between lamprey central neurons in situ. J Alzheimers Dis. 2010;19:647–664. doi: 10.3233/JAD-2010-1273. [DOI] [PubMed] [Google Scholar]
- 25.Kim W, Lee S, Hall GF. Secretion of human tau fragments resembling CSF-tau is modulated by the presence of the exon 2 insert. FEBS Lett. 2010;584:3085–3088. doi: 10.1016/j.febslet.2010.05.042. [DOI] [PubMed] [Google Scholar]
- 26.Saman S, Kim W, Raya M, Visnick Y, Miro S, Saman S, Jackson B, McKee AC, Alvarez VE, Lee NC, Hall GF. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem. 2012;287:3842–3849. doi: 10.1074/jbc.M111.277061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee S, Kim W, Li Z, Hall GF. Accumulation of vesicle-associated human tau in distal dendrites drives degeneration and tau secretion in an in situ cellular tauopathy model. Int J Alzheimers Dis. 2012;2012:172837. doi: 10.1155/2012/172837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Le MN, Kim W, Lee S, McKee AC, Hall GF. Multiple mechanisms of extracellular tau spreading in a non-transgenic tauopathy model. Am J Neurodegener Dis. 2012;1:316–333. [PMC free article] [PubMed] [Google Scholar]
- 29.Hall GF. The biology and pathobiology of tau protein. In: Kavallaris M, editor. Chapter 15 in “The Cytoskeleton and Human Disease”. Springer; 2012. pp. 285–313. [Google Scholar]
- 30.Simon D, Garcia-Garcia E, Royo F, Falcon-Perez JM, Avila J. Proteostasis of tau. Tau overexpression results in its secretion via membrane vesicles. FEBS Lett. 2012;586:47–54. doi: 10.1016/j.febslet.2011.11.022. [DOI] [PubMed] [Google Scholar]
- 31.Yamada K, Cirrito JR, Stewart FR, Jiang H, Finn MB, Holmes BB, Binder LI, Mandelkow EM, Diamond MI, Lee VM, Holtzman DM. In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci. 2011;31:13110–13117. doi: 10.1523/JNEUROSCI.2569-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barten DM, Cadelina GW, Hoque N, DeCarr LB, Guss VL, Yang L, Sankaranarayanan S, Wes PD, Flynn ME, Meredith JE, Ahlijanian MK, Albright CF. Tau transgenic mice as models for cerebrospinal fluid tau biomarkers. J Alzheimers Dis. 2011;24(2):127–141. doi: 10.3233/JAD-2011-110161. [DOI] [PubMed] [Google Scholar]
- 33.Plouffe V, Mohamed NV, Rivest-McGraw J, Bertrand J, Lauzon M, Leclerc N. Hyperphosphorylation and cleavage at D421 enhance tau secretion. PLoS One. 2012;7:e36873. doi: 10.1371/journal.pone.0036873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hall GF, Saman S. Secretion or Death? What is the Significance of elevated CSF-tau in early AD? Comm & Integ Biol. 2012;5(6):1–4. doi: 10.4161/cib.21437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hall GF. What is the common link between protein aggregation and interneuronal lesion propagation in neurodegenerative disease? In: Chang R, editor. Neurodegenerative Diseases - Processes, Prevention, Protection and Monitoring. 2011. pp. 1–17. InTech. [Google Scholar]
- 36.Gomez-Ramos A, Diaz-Hernandez M, Cuadros R, Hernandez F, Avila J. Extracellular tau is toxic to neuronal cells. FEBS Lett. 2006;580:4842–4850. doi: 10.1016/j.febslet.2006.07.078. [DOI] [PubMed] [Google Scholar]
- 37.Gomez-Ramos A, Diaz-Hernandez M, Rubio A, Diaz-Hernandez JI, Miras-Portugal MT, Avila J. Characteristics and consequences of muscarinic receptor activation by tau protein. Eur Neuropsychopharmacol. 2009;19:708–717. doi: 10.1016/j.euroneuro.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 38.Gomez-Ramos A, Diaz-Hernandez M, Rubio A, Miras-Portugal MT, Avila J. Extracellular tau promotes intracellular calcium increase through M1 and M3 muscarinic receptors in neuronal cells. Mol Cell Neurosci. 2008;37:673–681. doi: 10.1016/j.mcn.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 39.Díaz-Hernández M, Gómez-Ramos A, Rubio A, Gómez-Villafuertes R, Naranjo J, Miras-Portugal MS, Avila J. Tissuenonspecific Alkaline Phosphatase Promotes the Neurotoxicity Effect of Extracellular Tau. J Biol Chem. 2010;285:32539–32548. doi: 10.1074/jbc.M110.145003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Novak P, Prcina M, Kontsekova E. Tauons and prions: infamous cousins? J Alzheimers Dis. 2011;26:413–430. doi: 10.3233/JAD-2011-110194. [DOI] [PubMed] [Google Scholar]
- 41.Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, Jucker M, Goedert M, Tolnay M. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–913. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Soto C, Estrada L. Protein misfolding and neurodegeneration. Arch Neurol. 2008;65:184–189. doi: 10.1001/archneurol.2007.56. [DOI] [PubMed] [Google Scholar]
- 43.de Calignon A, Polydoro M, Suarez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, Spires-Jones TL, Hyman BT. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron. 2012;73:685–697. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu JW, Herman M, Liu L, Simoes S, Acker CM, Figueroa H, Steinberg JI, Marigttai M, Kayed R, Zurzolo C, Di Paolo G, Duff KE. Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J Biol Chem. 2013;288:1856–1870. doi: 10.1074/jbc.M112.394528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VM. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer's-like tauopathy. J Neurosci. 2013;33:1024–1037. doi: 10.1523/JNEUROSCI.2642-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284:12845–12852. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo JL, Lee VM. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem. 2011;286:15317–15331. doi: 10.1074/jbc.M110.209296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K. Trans-synaptic spread of tau pathology in vivo. PLoS One. 2012;7:e31302. doi: 10.1371/journal.pone.0031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI. Trans-cellular propagation of tau aggregation by fibrillar species. J Biol Chem. 2012;287:19440–19451. doi: 10.1074/jbc.M112.346072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vingtdeux V, Sergeant N, Buée L. Potential contribution of exosomes to the prion-like propagation of lesions in Alzheimer's disease. Front Physiol. 2012;3:229. doi: 10.3389/fphys.2012.00229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hall GF, Patuto BA. Is tau now ready for admission to the Prion club? Prion. 2012;6:223–233. doi: 10.4161/pri.19912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and bio- logical fluids. Curr Protoc Cell Biol. 2006 doi: 10.1002/0471143030.cb0322s30. Chapter 3, Unit 3.22. [DOI] [PubMed] [Google Scholar]
- 53.Mathivanan S, Fahner CJ, Reid GE, Simpson RJ. ExoCarta 2012: database of exosomal proteins, RNA and lipids. Nucleic Acids Res. 2012;40(Database issue):D1241–1244. doi: 10.1093/nar/gkr828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M, Bork P, von Mering C. Nucleic Acids Res. 2009;37(Database issue):D412–416. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.http://pages.genemania.org
- 56.http://www.reactome.org
- 57.Palanisamy V, Sharma S, Deshpande A, Zhou H, Gimzewski J, Wong DT. Nanostructural and transcriptomic analyses of human saliva derived exosomes. PLoS One. 2010;5:e8577. doi: 10.1371/journal.pone.0008577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chadwick W, Brenneman R, Martin B, Maudsley S. Complex and multidi- mensional lipid raft alterations in a murine model of Alzheimer's disease. Int J Alzheimers Dis. 2010;2010:604792. doi: 10.4061/2010/604792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.http://www.alzgene.org/
- 60.Ambegaokar SS, Jackson GR. Functional genomic screen and network analysis reveal novel modifiers of tauopathy dissociated from tau phosphorylation. Hum Mol Genet. 2011;20:4947–4977. doi: 10.1093/hmg/ddr432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blard O, Feuillette S, Bou J, Chaumette B, Frebourg T, Campion D, Lecourtois M. Cytoskeleton proteins are modulators of mutant tau-induced neurodegeneration in Drosophila. Hum Mol Genet. 2007;16:555–566. doi: 10.1093/hmg/ddm011. [DOI] [PubMed] [Google Scholar]
- 62.Fulga TA, Elson-Schwab I, Khurana V, Steinhilb ML, Spires TL, Hyman BT, Feany MB. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat Cell Biol. 2007;9:139–148. doi: 10.1038/ncb1528. [DOI] [PubMed] [Google Scholar]
- 63.Shulman JM, Feany MB. Genetic modifiers of tauopathy in Drosophila. Genetics. 2003;165:1233–1242. doi: 10.1093/genetics/165.3.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW. Incipient Alzheimer's disease Microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci U S A. 2004;101:2173–2178. doi: 10.1073/pnas.0308512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Blalock EM, Buechel HM, Popovic J, Geddes JW, Landfield PW. Microarray analyses of laser-captured hippocampus reveal distinct gray and white matter signatures associated with incipient Alzheimer's disease. J Chem Neuroanat. 2011;42:118e126. doi: 10.1016/j.jchemneu.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gomez Ravetti M, Rosso OA, Berretta R, Moscato P. Uncovering molecular biomarkers that correlate cognitive decline with the changes of hippocampus' gene expression profiles in Alzheimer's disease. PLoS One. 2010;5:e10153. doi: 10.1371/journal.pone.0010153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Antonell A, Lladó A, Altirriba J, Botta-Orfila T, Balasa M, Fernández M, Ferrer I, Sánchez-Valle R, Molinuevo JL. A preliminary study of the whole-genome expression profile of sporadic and monogenic early-onset Alzheimer's disease. Neurobiol Aging. 2013;34:1772–1778. doi: 10.1016/j.neurobiolaging.2012.12.026. [DOI] [PubMed] [Google Scholar]
- 68.Michael A, Bajracharya SD, Yuen PST, Zhou H, Star RA, Illei GG, Alevizos I. Exosomes from human saliva as a source of microRNA biomarkers. Oral Diseases. 2010;16:34–38. doi: 10.1111/j.1601-0825.2009.01604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lakkaraju A, Boutan E. Itinerant exosomes: emerging roles in cell and tissue polarity. Trends in Cell Biology. 2008;18(5) doi: 10.1016/j.tcb.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lachenal G, Pernet-Gallay K, Chivet M, Hemming FJ, Belly A, Bodon G, Blot B, Haase G, Goldberg Y, Sadoul R. Release of exosomes from differentiated neurons and its regulation by synaptic glutamatergic activity. Mol Cell Neurosci. 2011;46:409–418. doi: 10.1016/j.mcn.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 71.Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rogers KW, Schier AF. Morphogen gradients: from generation to interpretation. Annu Rev Cell Dev Biol. 2011;27:377–407. doi: 10.1146/annurev-cellbio-092910-154148. [DOI] [PubMed] [Google Scholar]
- 73.Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–122. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 74.Fang Y, Wu N, Gan X, Yan W, Morrell JC, Gould SJ. Higher-order oligomerization targets plasma membrane proteins and HIV gag to exosomes. PLoS Biol. 2007;5:e158. doi: 10.1371/journal.pbio.0050158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tournaviti S, Hannemann S, Terjung S, Kitzing TM, Stegmayer C, Ritzerfeld J, Walther P, Grosse R, Nickel W, Fackler OT. SH4-domain-induced plasma membrane dynamization promotes bleb-associated cell motility. J Cell Sci. 2007;120:3820–3829. doi: 10.1242/jcs.011130. [DOI] [PubMed] [Google Scholar]
- 76.Lindwasser OW, Resh MD. Multimerization of human immunodeficiency virus type 1 Gag promotes its localization to barges, raft-like membrane microdomains. J Virol. 2001;75:7913–7924. doi: 10.1128/JVI.75.17.7913-7924.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sverdlov M, Shajahan AN, Minshall RD. Tyrosine phosphorylation-dependence of caveolae-mediated endocytosis. J Cell Mol Med. 2007;11:1239–1250. doi: 10.1111/j.1582-4934.2007.00127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kikuchi A, Yamamoto H, Kisheda S. Multiplicity of the interactions of Wnt proteins and their receptors. Cell Signal. 2007;19:659–671. doi: 10.1016/j.cellsig.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 79.Williamson R, Usardi A, Hanger DP, Anderton BH. Membrane-bound beta-amyloid oligomers are recruited into lipid rafts by a fyn-dependent mechanism. FASEB J. 2008;22:1552–1559. doi: 10.1096/fj.07-9766com. [DOI] [PubMed] [Google Scholar]
- 80.Fein JA, Sokolow S, Miller CA, Vinters HV, Yang F, Cole GM, Gylys KH. Co-localization of amyloid beta and tau pathology in Alzheimer's disease synaptosomes. Am J Pathol. 2008;172:1683–1692. doi: 10.2353/ajpath.2008.070829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rajendran L, Honsho M, Zahn TR, Keller P, Geiger KD, Verkade P, Simons K. Alzheimer's disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci USA. 2006;103:11242–11247. doi: 10.1073/pnas.0603838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rank KB, Pauley AM, Bhattacharya K, Wang Z, Evans DB, Fleck TJ, Johnston JA, Sharma SK. Direct interaction of soluble human recombinant tau protein with Abeta 1-42 results in tau aggregation and hyperphosphorylation by tau protein kinase II. FEBS Lett. 2002;514:263–268. doi: 10.1016/s0014-5793(02)02376-1. [DOI] [PubMed] [Google Scholar]
- 83.Guo JT, Arai T, Miklossy J, McGeer PL. Abeta and tau form soluble complexes that may promote self aggregation of both into the insoluble forms observed in Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:1953–1958. doi: 10.1073/pnas.0509386103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pérez M, Cuadros R, Benítez MJ, Jiménez JS. Interaction of Alzheimer's disease amyloid beta peptide fragment 25-35 with tau protein, and with a tau peptide containing the microtubule binding domain. J Alzheimers Dis. 6:461–467. doi: 10.3233/jad-2004-6501. [DOI] [PubMed] [Google Scholar]
- 85.Giasson BI, Forman M, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ, Lee VM. Initiation and synergistic fibrillization of tau and alpha synuclein. Science. 2003;300:636–640. doi: 10.1126/science.1082324. [DOI] [PubMed] [Google Scholar]
- 86.Waxman EA, Giasson BI. Induction of intracellular tau aggregation is promoted by α-synuclein seeds and provides novel insights into the hyperphosphorylation of tau. J Neurosci. 2011;31:7604–7618. doi: 10.1523/JNEUROSCI.0297-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Arimura N, Kaibuchi K. Neuronal polarity: from extracellular signals to intracellular mechanisms. Nat Rev Neurosci. 2007;8:194–205. doi: 10.1038/nrn2056. [DOI] [PubMed] [Google Scholar]
- 88.Robinson R. An axonal growth pathway requires an Alzheimer's protein. PLoS Biol. 2013;11:e1001559. doi: 10.1371/journal.pbio.1001559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Taelman VF, Dobrowolski R, Plouhinec JL, Fuentealba LC, Vorwald PP, Gumper I, Sabatini DD, De Robertis EM. Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell. 2010;143:1136–1148. doi: 10.1016/j.cell.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Boonen RA, van Tijn P, Zivkovic D. Wnt signaling in Alzheimer's disease: up or down, that is the question. Ageing Res Rev. 2009;8:71–82. doi: 10.1016/j.arr.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 91.Uchida Y, Ohshima T, Sasaki Y, Suzuki H, Yanai S, Yamashita N, Nakamura F, Takei K, Ihara Y, Mikoshiba K, Kolattukudy P, Honnorat J, Goshima Y. Semaphorin3A signalling is mediated via sequential Cdk5 and GSK3beta phosphorylation of CRMP2: implication of common phosphorylating mechanism underlying axon guidance and Alzheimer's disease. Genes Cells. 2005;10:165–179. doi: 10.1111/j.1365-2443.2005.00827.x. [DOI] [PubMed] [Google Scholar]
- 92.Tamagnone L, Artigiani S, Chen H, He Z, Ming GI, Song H, Chedotal A, Winberg ML, Goodman CS, Poo M, Tessier-Lavigne M, Comoglio PM. Plexins are a large family of receptors for transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell. 1999;99:71–80. doi: 10.1016/s0092-8674(00)80063-x. [DOI] [PubMed] [Google Scholar]
- 93.Rashid T, Upton AL, Blentic A, Ciossek T, Knöll B, Thompson ID, Drescher U. Opposing gradients of ephrin-As and EphA7 in the superior colliculus are essential for topographic mapping in the mammalian visual system. Neuron. 2005;47:57–69. doi: 10.1016/j.neuron.2005.05.030. [DOI] [PubMed] [Google Scholar]
- 94.Kao TJ, Law C, Kania A. Eph and ephrin signaling: lessons learned from spinal motor neurons. Semin Cell Dev Biol. 2012;23:83–91. doi: 10.1016/j.semcdb.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 95.Chilton JK. Molecular mechanisms of axon guidance. Dev Biol. 2006;292:13–24. doi: 10.1016/j.ydbio.2005.12.048. [DOI] [PubMed] [Google Scholar]
- 96.Mudher A, Shepherd D, Newman TA, Mildren P, Jukes JP, Squire A, Mears A, Drummond JA, Berg S, MacKay D, Asuni AA, Bhat R, Lovestone S. GSK-3beta inhibition reverses axonal transport defects and behavioural phenotypes in Drosophila. Mol Psychiatry. 2004;9:522–530. doi: 10.1038/sj.mp.4001483. [DOI] [PubMed] [Google Scholar]
- 97.Ihara Y. Massive somatodendritic sprouting of cortical neurons in Alzheimer's disease. Brain Res. 1988;459:138–144. doi: 10.1016/0006-8993(88)90293-4. [DOI] [PubMed] [Google Scholar]
- 98.Perry G, Kawai M, Tabaton M, Onorato M, Mulvihill P, Richey P, Morandi A, Connolly JA, Gambetti P. Neuropil threads of Alzheimer's disease show a marked alteration of the normal cytoskeleton. J Neurosci. 1991;11:1748–1755. doi: 10.1523/JNEUROSCI.11-06-01748.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hall GF, Poulos A, Cohen MJ. Sprouts emerging from the dendrites of axotomized lamprey central neurons have axonlike ultrastructure. J Neurosci. 1989;9:588–599. doi: 10.1523/JNEUROSCI.09-02-00588.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hall GF, Yao J, Selzer M, Kosik KS. Cytoskeletal correlates to cell polarity loss following axotomy of lamprey central neurons. J Neurocytol. 1997;26:733–753. doi: 10.1023/a:1018562331003. [DOI] [PubMed] [Google Scholar]
- 101.Rose PK, MacDermid V, Joshi M, Neuber-Hess M. Emergence of axons from distal dendrites of adult mammalian neurons following a permanent axotomy. Eur J Neurosci. 2001;13:1166–1176. doi: 10.1046/j.0953-816x.2001.1490.x. [DOI] [PubMed] [Google Scholar]
- 102.Singleton RH, Zhu J, Stone JR, Povlishock JT. Traumatically induced axotomy adjacent to the soma does not result in acute neuronal death. J Neurosci. 2002;22:791–802. doi: 10.1523/JNEUROSCI.22-03-00791.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fenrich KK, Skelton N, MacDermid VE, Meehan CF, Armstrong S, Neuber-Hess MS, Rose PK. Axonal regeneration and development of de novo axons from distal dendrites of adult feline commissural interneurons after a proximal axotomy. J Comp Neurol. 2007;502:1079–1097. doi: 10.1002/cne.21362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 105.Zempel H, Thies E, Mandelkow E, Mandelkow EM. A beta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010;30:11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bhatia N, Hall GF. Untangling the role of tau in AD - A unifying hypothesis. Translational Neuroscience. 2013;4(2) doi:10.2478. [Google Scholar]
- 107.Gendreau K, Hall GF. The Tao of Tangles-Tau pathobiology beyond neurofibrillary degeneration. Frontiers in Neurology. 2013 in press. [Google Scholar]
- 108.Müller WE, Eckert A, Kurz C, Eckert GP, Leuner K. Mitochondrial dysfunction: common final pathway in brain aging and Alzheimer's disease- therapeutic aspects. Mol Neurobiol. 41:159–171. doi: 10.1007/s12035-010-8141-5. [DOI] [PubMed] [Google Scholar]
- 109.Swerdlow RH, Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer's disease. Med Hypotheses. 2004;63:8–20. doi: 10.1016/j.mehy.2003.12.045. [DOI] [PubMed] [Google Scholar]
- 110.Boland BA, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J Neurosci. 2008;28:6926–6937. doi: 10.1523/JNEUROSCI.0800-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hamano T, Gendron TF, Causevic E, Yen SH, Lin WL, Isidoro C, Deture M, Ko LW. Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur J Neurosci. 2008;27:1119–1130. doi: 10.1111/j.1460-9568.2008.06084.x. [DOI] [PubMed] [Google Scholar]
- 112.Funk KE, Kuret J. Lysosomal fusion dysfunction as a unifying hypothesis for Alzheimer's disease pathology. Int J Alzheimers Dis. 2012;2012:752894. doi: 10.1155/2012/752894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Askanas V, Engel WK. Inclusion-body myositis: a myodegenerative conformational disorder associated with Abeta, protein misfolding, and proteasome inhibition. Neurology. 2006;66:S39–48. doi: 10.1212/01.wnl.0000192128.13875.1e. [DOI] [PubMed] [Google Scholar]
- 114.Murakami N, Oyama F, Gu Y, McLennan IS, Nonaka I, Ihara Y. Accumulation of tau in autophagic vacuoles in chloroquine myopathy. J Neuropathol Exp Neurol. 1998;57:664–673. doi: 10.1097/00005072-199807000-00003. [DOI] [PubMed] [Google Scholar]
- 115.Abrahamsen H, Stenmark H. Protein secretion: unconventional exit by exophagy. Curr Biol. 2010;20:R415–418. doi: 10.1016/j.cub.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 116.Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11:1107–1117. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- 117.Arantes RM, Andrews NW. A role for synaptotagmin VII-regulated exocytosis of lysosomes in neurite outgrowth from primary sympathetic neurons. J Neurosci. 2006;26:4630–4637. doi: 10.1523/JNEUROSCI.0009-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mondragon-Rodriguez S, Trillaud-Doppia E, Dudilot A, Bourgeois C, Lauzon M, Leclerc N, Boehm J. Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-D-aspartate receptor-dependent tau phosphorylation. J Biol Chem. 2012;287:32040–32053. doi: 10.1074/jbc.M112.401240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dobrowolski R, Vick P, Ploper D, Gumper I, Snitkin H, Sabatini DD, de Robertis EM. Presenilin deficiency or lysosomal inhibition enhance Wnt signaling through relocalization of GSK3 to the late endosomal compartment. Cell Rep. 2012;2:1316–1328. doi: 10.1016/j.celrep.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tackenberg C, Brandt R. Divergent pathways mediate spine alterations and cell death induced by amyloid-beta, wild-type tau, and R406W tau. J Neurosci. 2009;29:14439–50. doi: 10.1523/JNEUROSCI.3590-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Boekhoorn K, Terwel D, Biemans B, Borghgraef P, Wiegert O, Ramakers GJ, de Vos K, Krugers H, Tomiyama T, Mori H, Joels M, van Leuven F, Lucassen PJ. Improved long-term potentiation and memory in young tau-P301L transgenic mice before onset of hyperphosphorylation and tauopathy. J Neurosci. 2006;26:3514–3523. doi: 10.1523/JNEUROSCI.5425-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 123.Yang Y, Mufson EJ, Herrup K. Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer's disease. J Neurosci. 2003;23:2557–2563. doi: 10.1523/JNEUROSCI.23-07-02557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Heverin M, Brennan G, Koehler C, Achim Treumann A, Henshall D. Proteomic analysis of 14-3-3 zeta binding proteins in the mouse hippocampus. Int J Physiol Pathophysiol Pharmacol. 2012;4(2):74–83. 2012. [PMC free article] [PubMed] [Google Scholar]
- 125.Hall GF, Lee S, Yao J. Neurofibrillary degeneration can be arrested in an in vivo cellular model of human tauopathy by application of a compound which inhibits tau filament formation in vitro. J Mol Neurosci. 2002;19:253–60. doi: 10.1385/jmn:19:3:251. [DOI] [PubMed] [Google Scholar]
- 126.Honson NS, Jensen JR, Abraha A, Hall GF, Kuret J. Small-molecule mediated neuroprotection in an in situ model of tauopathy. Neurotoxicity Res. 2009;15:274–83. doi: 10.1007/s12640-009-9028-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.King ME, Kan H, Baas PW, Erisir A, Glabe C, Bloom GS. Tau-dependent microtubule disassembly initiated by prefibrillar beta-amyloid. J Cell Biol. 2006;175:541–546. doi: 10.1083/jcb.200605187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Amadoro G, Serafino AL, Barbato C, Ciotti MT, Sacco A, Calissano P, Canu N. Role of N-terminal tau domain integrity on the survival of cerebellar granule neurons. Cell Death Differ. 2004;11:217–230. doi: 10.1038/sj.cdd.4401314. [DOI] [PubMed] [Google Scholar]
- 129.Park SY, Ferreira A. The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates beta-amyloid-induced neurodegeneration. J Neurosci. 2005;25:5365–5375. doi: 10.1523/JNEUROSCI.1125-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Amadoro G, Ciotti MT, Costanzi M, Cestari V, Calissano P, Canu N. NMDA receptor mediates tau-induced neurotoxicity by calpain and ERK/MAPK activation. Proc Natl Acad Sci U S A. 2006;103:2892–2897. doi: 10.1073/pnas.0511065103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.