

Abstract
Mitochondrial perturbations such as oxidative stress, increased fission/fusion dysfunction, and mitophagy are consistent features of Alzheimer’s disease (AD), yet the mechanisms that initiate these perturbations are unclear. One potential source for mitochondrial defects could be an imbalance in mitochondrial proteostasis. In this regard, studies indicate that a specialized mitochondrial unfolded protein response (mtUPR) is activated upon the aberrant accumulation of damaged or unfolded proteins in the mitochondrial matrix, resulting in the up-regulation of key genes involved in mitochondrial stabilization. To test whether mtUPR activation occurs in AD, we performed real-time quantitative PCR on postmortem frontal cortex samples from subjects classified as sporadic AD, familial AD linked to presenilin-1 mutations, or cognitively intact controls. Compared to controls, sporadic AD subjects exhibited a significant ~40-60% increase in expression levels of select genes activated by the mtUPR, including mitochondrial chaperones dnaja3, hspd1, and hspe1, mitochondrial proteases clpp and yme1l1, and txn2, a mitochondrial-specific oxidoreductase. Furthermore, levels of all six mtUPR genes were significantly up-regulated by ~70-90% in familial AD compared to controls, and these expression levels were significantly higher compared to sporadic AD. The increase in hspd1 (Hsp60) was validated by western blotting. These data support the concept that both sporadic and familial AD are characterized by mtUPR gene activation. Understanding the physiological consequences of this response may provide subcellular mechanistic clues to selective neuronal vulnerability or endogenous compensatory mechanisms during the progression of AD.
Keywords: Alzheimer, mitochondria, mitophagy, presenilin, unfolded protein response
INTRODUCTION
Mitochondrial perturbations are a consistent feature of sporadic Alzheimer’s disease (AD). Impairments in cellular respiration, mitochondrial oxidative stress, increased fission/fusion dysfunction, and mitophagy support the concept that pathogenic alterations in fundamental mitochondrial processes contribute to selective neuronal vulnerability during disease progression [1, 2]. The mechanisms that initiate these perturbations are unclear, yet one potential nexus for this spectrum of mitochondrial defects could be an imbalance in mitochondrial proteostasis [3, 4]. Interestingly, studies over the past decade indicate that a specialized unfolded protein response (UPR), similar to that described for the endoplasmic reticulum, is also present in mitochondria [5–7]. Specifically, this mitochondrial UPR (mtUPR) is activated upon the aberrant accumulation of misfolded or unfolded proteins in the mitochondrial matrix, which in turn triggers a mitochondria-to-nuclear signal that up-regulates several key genes involved in mitochondrial proteostasis, including those encoding the chaperones heat shock protein Hsp60 (chaperonin), Hsp10, and mitochondrial Hsp40/Tid1, the mitochondrial proteases ClpP and Yme1L1, the mitochondrial import protein Timm17, and mitochondrial enzymes such as thioredoxin-2 [5, 6]. Whether mtUPR activation is observed in sporadic AD is unknown; if so, then this would suggest that problems with maintaining mitochondrial proteostasis may be involved in disease pathogenesis. Moreover, familial AD-linked presenilin-1 (PS1) and amyloid-β (Aβ) precursor protein (APP) mutations have been linked to mitochondrial dysfunction [8–10], raising the possibility that mtUPR induction could be involved in genetic forms of AD, as well. To address these possibilities, we performed real-time quantitative PCR (qPCR) and western blotting studies on postmortem tissue samples from control, sporadic AD, and familial AD cases. Here, we show that multiple mtUPR markers are up-regulated in frontal cortex of subjects who died with sporadic or familial AD, suggesting that dysregulation of mitochondrial proteostasis is a common pathogenic process in these two AD subtypes.
MATERIALS AND METHODS
Subjects
Postmortem samples of frozen frontal cortex (Brodmann area 10) were harvested from subjects classified as cognitively intact controls (n = 9), sporadic AD (n = 8), or familial AD linked to PS1 (mutations = T115C, I143T, G209V, A260V, A431E; n = 8) (Table 1). All tissue samples were provided by Dr. Thomas Montine from the University of Washington Alzheimer’s Disease Research Center Brain Bank. The age at death for the familial AD group was significantly lower than the control and sporadic group (p < 0.01; Table 1), whereas both the sporadic and familial AD groups exhibited significantly lower Mini-Mental State Exam global cognitive scores (p < 0.0001) and higher Braak scores (p < 0.001) scores than the control group. There was no difference in the post mortem interval (Table 1).
Table 1.
Clinical, demographic, and neuropathological characteristics by diagnostic category.
| Clinical Diagnosis | ||||||
|---|---|---|---|---|---|---|
| CTL (N=9) |
sAD (N=8) |
fAD (N=8) |
P-value | Pair-wise comparison | ||
| Age (years) at death: | Mean ± SD (Range) |
71.1 ± 5.9 (55–91) |
68.9 ± 4.2 (53–75) |
52.8 ± 7.1 (37–65) |
<0.01a | (CTL, sAD) > fAD |
|
| ||||||
| MMSE: | Mean ± SD (Range) |
27.1 ± 1.9* (25–30) |
11.6 ± 3.4* (4–17) |
12.1 ± 5.1** (0–16) |
<0.0001a | CTL > (sAD, fAD) |
|
| ||||||
| Post-mortem interval (hours): | Mean ± SD (Range) |
8.1 ± 3.3 (6.0–11.4) |
8.4 ± 2.8 (4.8–12.6) |
11.5 ± 4.5 (7.7–17.3) |
0.2a | – |
|
| ||||||
| Distribution of Braak scores: | 0 | 0 | 0 | 0 | <0.001a | (sAD, fAD) > CTL |
| I/II | 4 | 0 | 0 | |||
| III/IV | 5 | 2 | 1 | |||
| V/VI | 0 | 6 | 7 | |||
3 cases and
4 cases did not have scores available for this test
Kruskal-Wallis test, with Bonferroni correction for multiple comparisons.
qPCR
Total RNA was extracted (PureLink, Ambion) and RNA integrity and concentration was verified using Bioanalysis (Agilent). Samples were assayed on a real-time PCR cycler (ABI 7500, Applied Biosystems) in 96-well optical plates as described previously [11–13]. qPCR was performed using Taqman hydrolysis probe primer sets (Applied Biosystems) specific for amplification of the following transcripts: clpp (ClpP mitochondrial protease; probe set Hs00195655_m1), dnaja3 (mitochondrial hsp40, or tid1; Hs00170600_m1), hspa5 (GRP78, or BiP; Hs00607129_gH), hspa9 (mitochondrial mtHsp70, or mortalin; Hs00269818_m1), hspd1 (Hsp60; Hs01941522_u1), hspe1 (Hsp10; Hs01654720_g1), lonp1 (lon 1 mitochondrial peptidase; Hs00998404_m1), txn2 (thioredoxin 2; Hs00912509_g1), or yme1l1 (mitochondrial Yme1-like ATPase 1; Hs00204609_m1). We used primer sets specific for citrate synthase (clpp, dnaja3, hspa9, hspd1, hspe1, lonp1, txn2, and yme1l1; probe set Hs02574374_s1) or GAPDH (hspa5; Hs02758991_g1) as control housekeeping transcripts. The ddCT method was employed to determine relative levels of each amplicon [11–14]. Variance component analyses revealed relatively low levels of within-case variability, and the average value of the triplicate qPCR products from each case was used in subsequent analyses. Alterations in PCR product synthesis were analyzed by one-way ANOVA with Bonferroni correction for post-hoc comparison. The level of statistical significance was set at α =0.05 (two-sided).
Western Blotting
Frozen tissue was extracted and enriched as mitochondrial fractions (qProteome, Qiagen) for quantitative immunoblotting of Hsp60 protein. Mitochondrial protein levels were quantified by the BCA method (Pierce). Protein samples (25 μg) were solubilized in loading buffer (Pierce, Thermo) and separated by SDS-PAGE (10% gels, Lonza), transferred to Immobilon-P membranes (Millipore), blocked in Tris buffered saline (pH 7.4) containing 0.1% Tween-20 and 2% nonfat milk, and then incubated overnight at 4°C with rabbit polyclonal antiserum to Hsp60 (1:500; Chemicon). Blots were then incubated for one hour with horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antiserum (Bio-Rad; 1:5000) and reactivity was quantified using Kodak 1D image analysis software (Perkin-Elmer) [14].
It has demonstrated by western blotting that mtHsp70/mortalin levels are unchanged in AD compared to control tissue samples [15], and here we observed that mtHSP70 mRNA levels were stable in our control and AD samples (Fig. 1), suggesting that mtHSP70 might serve as a loading control for the mitochondrial fractions. To validate this concept, we used a mouse monoclonal antibody to mtHsp70 (1:500, Abcam) to compare mtHsp70-immunoreactive signals in each 25 μg sample with coomassie staining of total protein in the same gels. mtHsp70 protein levels were unchanged and coomassie staining confirmed equal loading (data not shown). Hence, mtHsp70 was used as a loading control for the mitochondrial fractions. To this end, the membranes were stripped and re-probed with the mouse monoclonal mtHsp70 antibody (1:500) overnight followed by a one hour incubation with horseradish peroxidase-conjugated goat anti-mouse IgG (1:8,000; Pierce, IL) and reactivity was again quantified using Kodak 1D image analysis software (Perkin-Elmer, MA). Each sample was analyzed on three different western blots in independent experiments. Signals for Hsp60 were normalized to mtHsp70 for quantitative analysis [16, 17]. Alterations in normalized Hsp60 levels were analyzed by one-way ANOVA with Bonferroni correction for post-hoc comparison. The level of statistical significance was set at α = 0.05 (two-sided).
Fig. 1. Evidence for mtUPR gene activation in AD.
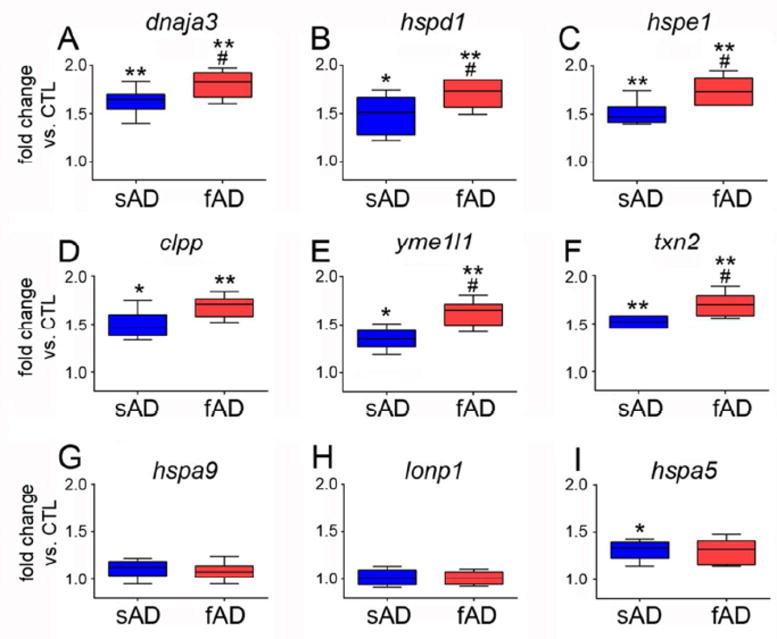
qPCR was performed on RNA extracted from frozen frontal cortex of sporadic AD (sAD) and familial AD (fAD) subjects and compared to control (CTL) subjects. Primer sets specific for mtUPR genes (A) dnaja3 (mitochondrial hsp40, or tid1), (B) hspd1 (hsp60), (C) hspe1 (hsp10), (D) clpp (ClpP mitochondrial protease), (E) yme1l1 (mitochondrial YME1-like ATPase 1), and (F) txn2 (thioredoxin 2) were compared among the groups. Primer sets specific for non-mtUPR genes (G) hspa9 (mitochondrial hsp70, or mortalin), (H) lonp1 (lon 1 mitochondrial peptidase), and (I) hspa5 (GRP78, or BiP) were also compared. Samples were run in triplicate and the ddCT method was employed to compare normalized PCR products. Box plots represent fold change in sAD and fAD compared to CTL. *p < 0.05 vs. CTL; ** p < 0.01 vs. CTL; # p < 0.05 vs. sAD.
RESULTS AND DISCUSSION
Compared to controls, sporadic AD subjects exhibited a significant ~40-60% increase in frontal cortex expression levels of select genes activated by the mtUPR, including mitochondrial chaperones dnaja3, hspd1, and hspe1, mitochondrial proteases clpp and yme1l1, and txn2, a mitochondrial-specific oxidoreductase that also plays a role in maintenance of the mitochondrial inner membrane potential [5, 7] (Fig 1). Furthermore, frontal cortex levels of all six mtUPR genes were significantly up-regulated by ~70-80% in familial AD compared to controls, and in most instances these expression levels were significantly higher compared to sporadic AD (Fig. 1). In this regard, the hspd1 gene product Hsp60 was also differentially expressed in mitochondrial protein fractions prepared from sporadic and familial AD cases. Hsp60 protein levels were significantly elevated by ~50% in sporadic AD compared to control, whereas Hsp60 levels were up-regulated an additional ~35% in familial AD (Fig. 2). By contrast, two mitochondrial genes not up-regulated by the mtUPR, hspa9 (mtHsp70/mortalin) and lon1p1 (the Lon1 mitochondrial protease) [5, 7] were unchanged across the groups (Fig. 1). The endoplasmic reticulum stress-mediated UPR gene hspa5 (BiP) was up-regulated ~35% in both sporadic and familial AD (Fig. 1), consistent with previous findings in sporadic cases [18]. Previous gene expression studies have implicated several pathways in AD pathogenesis, including those regulating synaptic function, inflammation, and endosomal/lysosomal activity [19–23]. Here, our data suggest, for the first time, that sporadic and familial AD are also characterized by mtUPR gene activation.
Fig. 2. Hsp60 levels are up-regulated in AD.

(A) Representative western blot (lower panel) shows a relative increase in Hsp60-immunoreactive signals in sporadic AD (sAD) and familial (fAD) cases compared to controls (CTL). The blot was stripped and re-probed for mtHsp70 as a loading control (see Materials and Methods). Note that mtHsp70 signals on the same blot (upper panel) remain constant across the cases. (B) Quantitative analysis of Hsp60-immunoreactive signals normalized to mtHsp70 reveals a step-wise increase in Hsp60 protein levels in sAD and fAD, respectively, compared to CTL. Hsp60 levels in fAD were significantly higher compared to sAD. *p < 0.05 vs. CTL; ** p < 0.01 vs. CTL; # p < 0.05 vs. sAD.
The mtUPR is a conserved pathway that is critical for maintaining mitochondrial protein homeostasis and has also been implicated in lifespan extension [3]. For instance, experimental disturbances in mitochondrial protein balance induce mtUPR genes encoding not only molecular chaperones and proteases but also genes involved in ROS scavenging machinery, mitochondrial fission, and ubiquinone biosynthesis [5, 24]. The current observations provide evidence that this physiologically important cellular response may be chronically activated in AD, perhaps as a compensatory neuroprotective response to a sustained accumulation of unfolded, misfolded and damaged mitochondrial proteins. Although the physiological consequence of chronic mtUPR activation remains unclear, it may be similar to the endoplasmic reticulum UPR, where sustained activation shifts a normally protective pathway to a deleterious one [12]. In this regard, a sustained mtUPR may be related to increased mitochondrial fission and mitophagy events observed in AD [25–27]. This might provide a cellular mechanistic link between mitochondrial proteostatic stress and lysosomal dysregulation prevalent during AD pathogenesis [28–30]. Notably, the greater amplification of mtUPR in PS1-linked AD is reminiscent of increased lysosomal pathology in familial compared to sporadic AD cases [29]. Ultimately, understanding whether this response occurs during the earliest stages of sporadic disease and whether this increased steady-state response is protective or pathogenic may provide critical sub-cellular mechanistic clues to selective neuronal vulnerability during the progression of AD.
Acknowledgments
The authors would like to thank Dr. Thomas Montine and Aimee Schantz at the University of Washington Alzheimer’s Disease Research Center (P50 AG05136) for tissue samples and case demographics. This work was supported by NIH AG14449, AG42146, the Saint Mary’s Foundation, Mercy Health Saint Mary’s Hospital, Grand Rapids, MI, the Barrow Neurological Institute Barrow and Beyond Foundation, and the Arizona Alzheimer’s Disease Center, Phoenix, AZ.
Biography

Scott E. Counts
Footnotes
AUTHOR CONTRIBUTIONS
SEC and EJM conceived of the study and participated in case selection. JSB and SEC participated in study design and performed the experiments including statistical analysis. JSB, SEC and EJM helped to draft the manuscript. All authors read and approved the final manuscript.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
References
- 1.Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, et al. Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci. 2001;21:3017–23. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu X, Perry G, Moreira PI, Aliev G, Cash AD, Hirai K, et al. Mitochondrial abnormalities and oxidative imbalance in Alzheimer disease. J Alzheimers Dis. 2006;9:147–53. doi: 10.3233/jad-2006-9207. [DOI] [PubMed] [Google Scholar]
- 3.Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–7. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burte F, Carelli V, Chinnery PF, Yu-Wai-Man P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol. 2015;11:11–24. doi: 10.1038/nrneurol.2014.228. [DOI] [PubMed] [Google Scholar]
- 5.Aldridge JE, Horibe T, Hoogenraad NJ. Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS One. 2007;2:e874. doi: 10.1371/journal.pone.0000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haynes CM, Fiorese CJ, Lin YF. Evaluating and responding to mitochondrial dysfunction: the mitochondrial unfolded-protein response and beyond. Trends Cell Biol. 2013;23:311–8. doi: 10.1016/j.tcb.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002;21:4411–9. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science. 2004;304:448–52. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 9.Schon EA, Area-Gomez E. Is Alzheimer’s disease a disorder of mitochondria-associated membranes? J Alzheimers Dis. 2010;20 (Suppl. 2):S281–92. doi: 10.3233/JAD-2010-100495. [DOI] [PubMed] [Google Scholar]
- 10.Schuessel K, Frey C, Jourdan C, Keil U, Weber CC, Muller-Spahn F, et al. Aging sensitizes toward ROS formation and lipid peroxidation in PS1M146L transgenic mice. Free Radic Biol Med. 2006;40:850–62. doi: 10.1016/j.freeradbiomed.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 11.Alldred MJ, Che S, Ginsberg SD. Terminal continuation (TC) RNA amplification without second strand synthesis. J Neurosci Methods. 2009;177:381–5. doi: 10.1016/j.jneumeth.2008.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Counts SE, He B, Che S, Ikonomovic MD, DeKosky ST, Ginsberg SD, et al. Alpha7 nicotinic receptor up-regulation in cholinergic basal forebrain neurons in Alzheimer disease. Arch Neurol. 2007;64:1771–6. doi: 10.1001/archneur.64.12.1771. [DOI] [PubMed] [Google Scholar]
- 13.Ginsberg SD. Transcriptional profiling of small samples in the central nervous system. Methods Mol Biol. 2008;439:147–58. doi: 10.1007/978-1-59745-188-8_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weinberg RB, Mufson EJ, Counts SE. Evidence for a neuroprotective microRNA pathway in amnestic mild cognitive impairment. Front Neurosci. 2015;9:430. doi: 10.3389/fnins.2015.00430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Osorio C, Sullivan PM, He DN, Mace BE, Ervin JF, Strittmatter WJ, et al. Mortalin is regulated by APOE in hippocampus of AD patients and by human APOE in TR mice. Neurobiol Aging. 2007;28:1853–62. doi: 10.1016/j.neurobiolaging.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 16.Counts SE, Nadeem M, Lad SP, Wuu J, Mufson EJ. Differential expression of synaptic proteins in the frontal and temporal cortex of elderly subjects with mild cognitive impairment. J Neuropathol Exp Neurol. 2006;65:592–601. doi: 10.1097/00005072-200606000-00007. [DOI] [PubMed] [Google Scholar]
- 17.Counts SE, Nadeem M, Wuu J, Ginsberg SD, Saragovi HU, Mufson EJ. Reduction of cortical TrkA but not p75(NTR) protein in early-stage Alzheimer’s disease. Ann Neurol. 2004;56:520–31. doi: 10.1002/ana.20233. [DOI] [PubMed] [Google Scholar]
- 18.Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Scheper W. Activation of the unfolded protein response is an early event in Alzheimer’s and Parkinson’s disease. Neurodegener Dis. 2012;10:212–5. doi: 10.1159/000334536. [DOI] [PubMed] [Google Scholar]
- 19.Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW. Incipient Alzheimer’s disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci USA. 2004;101:2173–8. doi: 10.1073/pnas.0308512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Colangelo V, Schurr J, Ball MJ, Pelaez RP, Bazan NG, Lukiw WJ. Gene expression profiling of 12633 genes in Alzheimer hippocampal CA1: transcription and neurotrophic factor down-regulation and up-regulation of apoptotic and pro-inflammatory signaling. J Neurosci Res. 2002;70:462–73. doi: 10.1002/jnr.10351. [DOI] [PubMed] [Google Scholar]
- 21.Counts SE, Alldred MJ, Che S, Ginsberg SD, Mufson EJ. Synaptic gene dysregulation within hippocampal CA1 pyramidal neurons in mild cognitive impairment. Neuropharmacology. 2013;79C:172–9. doi: 10.1016/j.neuropharm.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mufson EJ, Counts SE, Che S, Ginsberg SD. Neuronal gene expression profiling: uncovering the molecular biology of neurodegenerative disease. Prog Brain Res. 2006;158:197–222. doi: 10.1016/S0079-6123(06)58010-0. [DOI] [PubMed] [Google Scholar]
- 23.Ginsberg SD, Alldred MJ, Counts SE, Cataldo AM, Neve RL, Jiang Y, et al. Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer’s disease progression. Biol Psychiatry. 2010;68:885–93. doi: 10.1016/j.biopsych.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pellegrino MW, Nargund AM, Haynes CM. Signaling the mitochondrial unfolded protein response. Biochim Biophys Acta. 2013;1833:410–6. doi: 10.1016/j.bbamcr.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moreira PI, Siedlak SL, Wang X, Santos MS, Oliveira CR, Tabaton M, et al. Increased autophagic degradation of mitochondria in Alzheimer disease. Autophagy. 2007;3:614–5. doi: 10.4161/auto.4872. [DOI] [PubMed] [Google Scholar]
- 26.Santos RX, Correia SC, Wang X, Perry G, Smith MA, Moreira PI, et al. A synergistic dysfunction of mitochondrial fission/fusion dynamics and mitophagy in Alzheimer’s disease. J Alzheimers Dis. 2010;20 (Suppl. 2):S401–12. doi: 10.3233/JAD-2010-100666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cardoso SM, Pereira CF, Moreira PI, Arduino DM, Esteves AR, Oliveira CR. Mitochondrial control of autophagic lysosomal pathway in Alzheimer’s disease. Exp Neurol. 2010;223:294–8. doi: 10.1016/j.expneurol.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 28.Cataldo AM, Barnett JL, Berman SA, Li J, Quarless S, Bursztajn S, et al. Gene expression and cellular content of cathepsin D in Alzheimer’s disease brain: evidence for early up-regulation of the endosomal- lysosomal system. Neuron. 1995;14:671–80. doi: 10.1016/0896-6273(95)90324-0. [DOI] [PubMed] [Google Scholar]
- 29.Nixon RA, Cataldo AM. Lysosomal system pathways: genes to neurodegeneration in Alzheimer’s disease. J Alzheimers Dis. 2006;9:277–89. doi: 10.3233/jad-2006-9s331. [DOI] [PubMed] [Google Scholar]
- 30.Mufson EJ, Counts SE, Ginsberg SD. Gene expression profiles of cholinergic nucleus basalis neurons in Alzheimer’s disease. Neurochem Res. 2002;27:1035–48. doi: 10.1023/a:1020952704398. [DOI] [PubMed] [Google Scholar]