

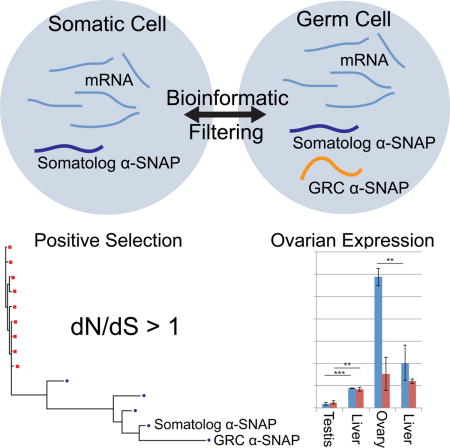
Summary
Developmentally programmed genome rearrangements are rare in vertebrates but have been reported in scattered lineages including the bandicoot, hagfish, lamprey, and zebra finch (Taeniopygia guttata) [1]. In the finch, a well-studied animal model for neuroendocrinology and vocal learning [2], one such programmed genome rearrangement involves a Germline-Restricted Chromosome, or GRC, which is found in germlines of both sexes but eliminated from mature sperm [3, 4]. Transmitted only through the oocyte, it displays uniparental female-driven inheritance, and early in embryonic development it is apparently eliminated from all somatic tissue in both sexes [3, 4]. The GRC comprises the longest finch chromosome at over 120 million basepairs [3] and previously, the only known GRC-derived sequence was repetitive and non-coding [5]. Because the zebra finch genome project was sourced from male muscle (somatic) tissue [6] the remaining genomic sequence and protein-coding content of the GRC remain unknown. Here we report the first protein-coding gene from the GRC: a member of the α-Soluble NSF Attachment Protein (α-SNAP) family hitherto missing from zebra finch gene annotations. In addition to the GRC-encoded α-SNAP, we find an additional paralogous α-SNAP residing in the somatic genome (a somatolog)—making zebra finch the first example in which α-SNAP is not a single-copy gene. We show divergent, sex-biased expression for the paralogs and also that positive selection is detectable across the bird α-SNAP lineage, including the GRC-encoded α-SNAP. This study presents the identification and evolutionary characterization of the first protein-coding GRC gene in any organism.
eTOC Blurb
Biederman et al. report the first gene on the zebra finch germ-line restricted chromosome (GRC): a gene encoding α-Soluble NSF Attachment Protein (α-SNAP). Positive selection and higher expression in ovaries suggest a novel biological role, and the discovery of a somatic paralog in this first known instance of the gene occurring in multiple copies.
Results and Discussion
To identify genes from the GRC we adopted a subtractive transcriptomic approach. We sequenced RNA from germline tissue of male and female adult birds, obtaining 10 million read pairs for each, and performed de novo assembly; we then performed computational elimination of sequences matching the published somatic (muscle) genome sequence [6], its raw (Sanger) read data, and a brain (somatic) transcriptome [7] (Figure 1A) to identify potential germline-limited sequences. During the filtering process we identified 936 proteins having strong (1e-20 or better) matches to either the Swiss-Prot database or Pfam-A (thus, strong candidates for bona fide new genes) that are nevertheless missing from the current finch gene annotation (version 3.2.4 [8]). These new genes help fill in several important gaps in finch biology. For example, we uncovered a member of the DNA Methyltransferase 1 (Dnmt1) family[9], an H1× linker histone [10], and the zeta subunit of the vesicle coat complex (COPI) [11], a member of the core eukaryotic orthologous family KOG3343.
Figure 1. Discovery of a paralogous α-SNAP gene pair.
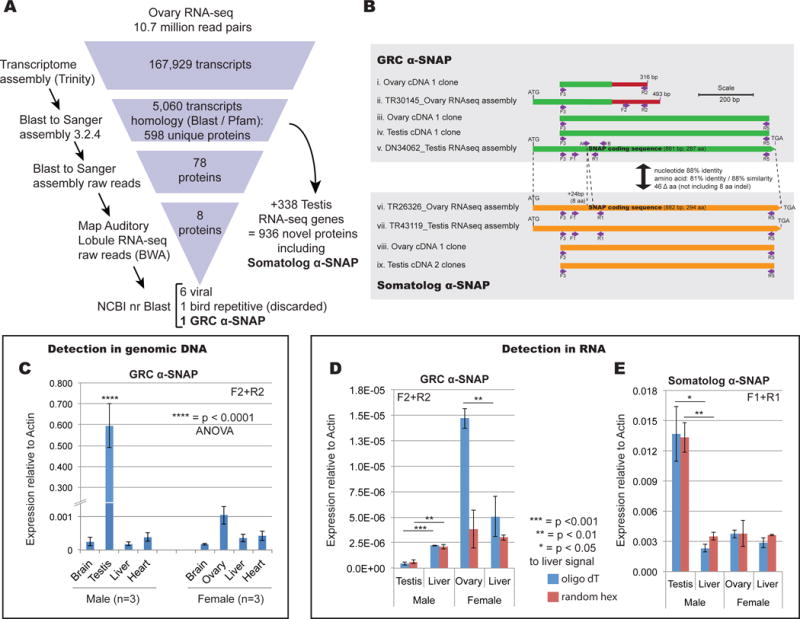
A. Subtractive transcriptomic analysis used in this study. B. Overview of sequence comparison between assembled GRC (green) and somatolog (orange) α-SNAP sequences along with confirmation by cloning. C. Genomic DNA qPCR analysis confirming GRC α-SNAP is only detected in testis or ovary (germline) tissue (Primers F2+R2, see panel B). Error bars represent standard error of the mean. Two-way ANOVA identified testis signal (**** = p < 0.0001) as highly statistically significant, with n = 3 individuals of each sex tested. D. RT-qPCR analysis of expression of GRC α-SNAP showing strong ovary expression. Statistical significance calculated with Students 2-tailed T-test. E. RT-qPCR analysis of somatolog α-SNAP showing strong testis expression. Statistical significance calculated with Students 2-tailed T-test. See also Figure S1.
The subtractive genomic pipeline uncovered a single GRC gene, a member of the α-SNAP family (hereafter the ‘GRC α-SNAP’) (Figure 1A). Although our initial assembly captured a relatively short portion of the SNAP coding sequence that appeared to be an alternatively spliced isoform (Figure 1B(ii)); we were able to reconstruct the full α-SNAP coding sequence by de novo assembly of a 94 million-read finch testis RNA-seq dataset [12]. This assembled male-derived sequence matched the SNAP portion of the ovarian contig but encompasses a full SNAP coding sequence (Figure 1B compare (ii) to (v)). We confirmed both isoforms by cloning and sequencing (Figure 1B). Quantitative PCR from a tissue panel of genomic DNA detected this gene at a statistically significant level only in testis (Figure 1C and S1A). While the GRC α-SNAP was not detected in ovary DNA, we detected robust expression as RNA from this tissue (Figure 1D, S1C).
In the process of filtering the transcriptome data, we discovered a second α-SNAP gene. This one was filtered out from the raw Sanger reads in Figure 1A; thus, it is present in the somatic genome but is not present in the Sanger assembly [6]. Given we cannot use the term ‘gametolog’, which refers to an autosomal copy of a sex-linked gene [13], we coin ‘somatolog’ in reference to a somatic copy of a germline-limited gene. We suggest that the somatolog α-SNAP underwent an ancient duplication event (possibly at the genesis of the GRC itself) forming a germline-restricted copy, which has subsequently undergone significant evolutionary divergence.
Expression of this paralogous gene system is sex-biased. RT-qPCR of ovary and testis RNA revealed expression of the GRC gene is predominantly ovarian (Figure 1D), although gel analysis post-quantitation showed low-level detection also in testis (Figure S1C), as also confirmed by our assembly of the gene from testis RNAseq data and RT-PCR clone confirmation from both testis and ovary (Figure 1B). The somatolog α-SNAP is expressed in germlines and soma of both sexes, though most strongly in testis (Figure 1E).
We find that α-SNAP is in a particularly difficult-to-assemble genomic location, leading to annotation problems for this gene family. While β-SNAP genes have been deposited in Genbank for ten bird species, only two of these species had full-length α-SNAP genes available, and alignment with each other and the zebra finch somatolog highlights showed apparent discrepancies (Figure S2). Canary (Serinus canarius), accession XP_009098415.2, aligns with α-SNAP of other species but has 20 central amino acids that are completely divergent, while society finch (Lonchura striata domestica), accession XP_021401324, displays 30 altered amino acids at its carboxyl terminus (Figure S2). Ground tit (Pseudopodoces humilis), has an α-SNAP protein sequence (XP_005534295) that displays multiple problems: a 25 amino acid stretch is missing, as is the terminal 70 amino acids (Figure S2).
As we note above, no α-SNAP locus is present in zebra finch Sanger assembly [6]; however a recent (unannotated) PacBio haploid assembly [14] represents the locus as two allelic contigs, MUGN01000184.1 (386 kb) and MUGN01000615.1 (348 kb). Comparison of these with the α-SNAP scaffolds in both canary (unplaced scaffold NW_007931326.1, 84 kb) and society finch (unplaced scaffold NW_018657153.1, 496 kb) reveal that in all 3 species the exons are conserved, embedded within largely non-conserved repetitive micro- and minisatellite DNA. In both canary and society finch assembly gaps obscure portions of exonic α-SNAP sequence, but no gaps exist in the two zebra finch PacBio contigs. The lack of high-confidence full-length α-SNAP protein annotations from other avian species (including zebra finch) suggests that this region is problematic across birds, apparently both for assembly and for annotation.
To address this problem we performed de novo RNAseq assembly. Given that the zebra finch somatolog α-SNAP was robustly expressed within liver and testis (Figure 2AC), we identified high-quality, deeply sequenced liver RNAseq datasets from society finch (SRR5223631), canary (SRR2915372), and ground tit (SRR768235). (As in most birds, germline RNAseq datasets are not available for these species). After Trinity assembly we were able to retrieve a single full-length α-SNAP coding sequence for each bird that corrected the issues noted above (Figure S2). The canary, society finch, and ground tit α-SNAP genes were both identical, or within allelic variation, to the Genbank versions; however the problematic regions have been replaced with sequences that align confidently to other bird α-SNAPs (Figure S2). The transcriptomes of canary and ground tit also yielded full-length β-SNAP genes identical to those already deposited in Genbank, suggesting that our de novo assembly method is accurate (β-SNAP is a brain-enriched, though not brain-exclusive, gene [15]). We also assembled RNAseq datasets for great tit (Parus major) and golden-collared manakin (Manacus vitellinus) but these assemblies failed to yield full-length SNAP genes. Therefore, with the high-confidence zebra finch, canary, society finch, and ground tit α-SNAP gene sequences we performed evolutionary tree reconstruction and analysis.
We aligned 14 sequences derived from 9 bird species (society finch was omitted for reasons described below) and built both Bayesian (Figure 2A) and maximum likelihood (Figure S3) trees using chicken β-SNAP as outgroup. Both trees present nearly identical topologies and recover the α- and β-SNAP genes as separate highly supported clades (Figure 2A, S3). However, while β-SNAP genes are extremely well conserved among passerines, the α-SNAP genes are much more divergent, located on extended branches (Figure 2A, S3). Indeed the β-SNAP amino acid sequences of canary, ground tit, society finch, and zebra finch are 100% identical while manakin has only a single amino acid substitution (species-specific synonymous polymoprphisms are present in their mRNAs). This suggests that β-SNAP genes are under significant purifying selection across passerines. In contrast, α-SNAP genes are widely divergent in passerines, with 23 amino acid substitutions between the somatolog and canary α-SNAP and a somatolog-to-ground tit divergence of 44 amino acids. Remarkably the interparalog, intra-zebra finch divergence is greater than that between the somatolog and all other passerine α-SNAP genes (46 amino acids, ignoring the 8 amino acid deletion, Figure S1E). This results in an extremely long branch rivaling the one rooting the entire passerine clade (compare branches A and G in Figure 2A).
Figure 2. Avian dN/dS analysis of α-SNAP genes.
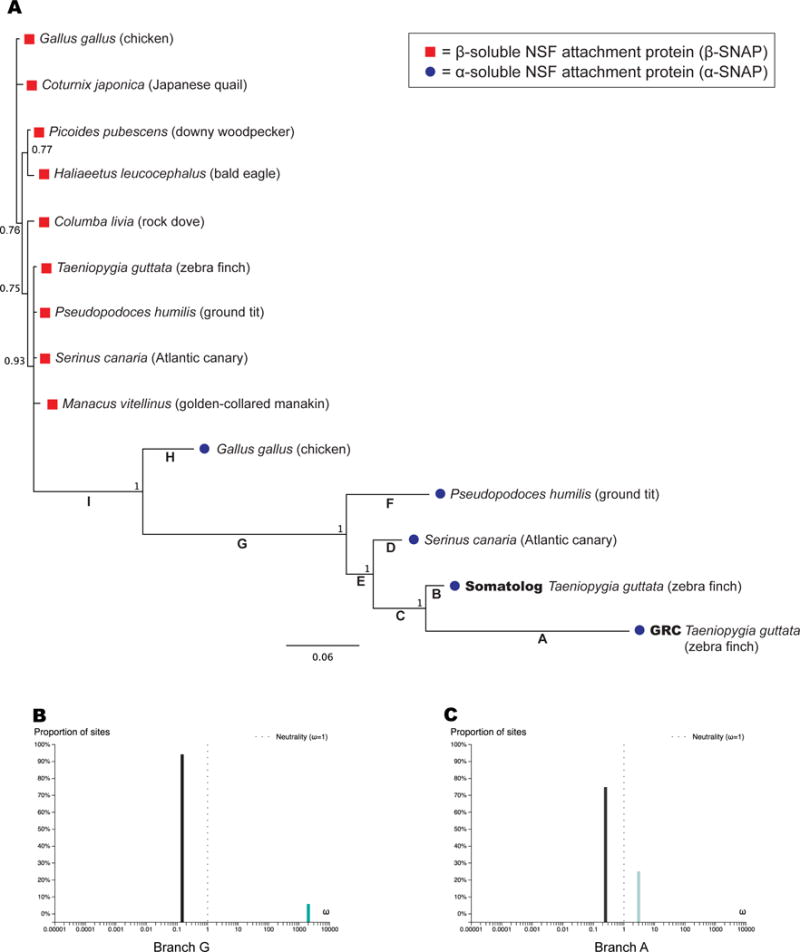
A. Bayesian tree of birds and dN/dS analysis. Red boxes represent β-SNAP; blue dots represent α-SNAP proteins. Branch numbers indicate posterior probabilities and scale bar represents substitutions per site. Branch letters A–I correspond to Table 1 for ω-ratio (dN/dS) estimation of selection pressure. B. Analysis of ω for branch G using aBSREL [24] showing two selective regimes. Positive selection on this branch was statistically significant (p-value 0.0045, Table 1). C. Analysis of ω for branch A using aBSREL [24] showing two selective regimes, with positive selection affecting 25% of sites but at a lower overall level than branch G. See also Figure S3.
Society finch produced an unanticipated complexity: its α-SNAP consistently and confidently groups with the zebra finch GRC α-SNAP rather than somatic genes (Figure 3). Society finch is the only passerine beside the zebra finch confirmed to have a GRC [16], but we derived this α-SNAP sequence from de novo assembled female liver (somatic) RNAseq data. Furthermore the gene is highly similar to a previous annotated version based on a blood-sourced (somatic) genome assembly (XP_021401324) (Figure S2). Due to the uncertainty surrounding this particular sequence, and the possibility that the unusual phylogenetic grouping is due to a long-branch attraction artifact [17, 18], we excluded society finch both from the trees in Figures 2A and S3, but we include it in Figure 3.
Figure 3. Multi-species Bayesian tree, confirming that both SNAP genes in finch are from the α-SNAP family.
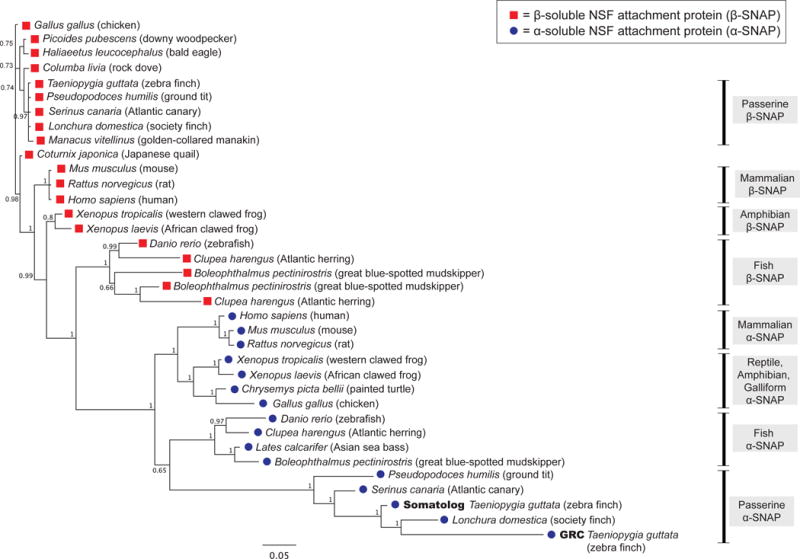
Red boxes, β-SNAP; blue dots, α-SNAP proteins. Branch numbers indicate posterior probabilities and scale bar represents substitutions per site. Related to Figure S2.
We analyzed the bird-only phylogenetic tree (Figure 2A) for evidence of positive selection by analyzing the ratio of nonsynonymous mutations (dN) relative to synonymous (dS) mutations. When the dN/dS ratio, ω, is equal to 1 it implies the sequence is evolving neutrally— suggesting a loss of function on a coding sequence. Purifying selection—the weeding out of deleterious mutations to retain function—is indicated by ω less than 1, while positive selection— the promotion of specific amino acid changes due to advantageous function—is indicated by ω greater than 1 [19]. Branch models estimate ω for a whole protein (averaged across all amino acid sites) while branch-site models allow ω to vary across the amino acid sites at a specific branch of a phylogenetic tree [20]. This is a more sensitive method because positive selection may only affect a few amino acids in a protein transiently during evolution, while most of the sites remain under purifying selection and mask the positive signal [19].
Analyzing the tree in Figure 2A, we found all branches (A–I) were estimated to have ω between 0 and 1, suggesting purifying selection (Table 1, Branch Model). However, we observed significant variation in ω estimates along lineages, with branches A and B in particular being elevated (ω=0.548 and 0.827, Table 1). Of the nine branches tested, only four were statistically significantly under purifying selection (Table 1) suggesting that the remaining branches are potentially either under relaxed purifying selection or positive selection at some sites. For the GRC and somatolog α-SNAP, this may be attributed directly to their paralogy, since genome-wide studies of gene duplication report relaxed purifying selection on paralogs, at least initially [21, 22]. However, the relaxed selection pressure is usually evolutionarily brief, reverting to a strongly purifying regime for both paralogs [21]. The GRC-somatolog α-SNAP divergence appears to be ancient by two measures: large amino acid divergence resulting in long branch lengths already noted (Figure 2A) and by the synonymous (silent) mutations accruing between the copies, with pairwise dS = 0.26 by PAML. Most duplicate genes are lost (non-functionalized or turned into a pseudogene) by the time dS reaches a few percent [21], so the fact that both zebra finch genes produced by the α-SNAP duplication have been retained may indicate evolution of new function by the GRC copy.
Table 1.
Analysis of dN/dS ratios (ω) for branches of the tree in Figure 2A, with bolded values indicating positive selection (ω > 1).
| PAML Analysis | |||||||
|---|---|---|---|---|---|---|---|
| Branch Model | Branch-Site Model | ||||||
| Branch | ω Branch | ω2 (positive sites) | ω1 (purifying sites) | Fraction Sites under Positive Selection (ω>1) | Fraction Sites under Purifying Selection (ω<1) | LRT statistic (2 lnL)a | Significant Positively Selected Amino Acids (posterior probability) |
| A | 0.548 | 1.73 | 0.029 | 0.363 | 0.628 | 0.904 | 1 M (0.997) 14 N (0.997) 36 R (0.966) 107 R (0.999) 167 E (0.998) 266 W (0.998) |
| B | 0.827 | 70.60 | 0.043 | 0.021 | 0.971 | 0.854 | – |
| C | 0.173 | 3.27 | 0.041 | 0.057 | 0.933 | 1.154 | 130 E (0.988) |
| D | 0.048**** | 13.28 | 0.043 | 0.010 | 0.979 | 3.780 | 41 A (0.984) |
| E | 0.103 | 999.00 (dS =0) | 0.042 | 0.020 | 0.968 | 13.807** | – |
| F | 0.097**** | 1.21 | 0.039 | 0.098 | 0.890 | 0.035 | – |
| G | 0.128* | 4.53 | 0.042 | 0.060 | 0.928 | 0.707 | 1 M (0.974) 43 C (0.983) 171 R (0.980) 182 V (0953) |
| H | 0.066 | 1.00 | 0.044 | 0.000 | 0.988 | 0.000 | – |
| I | 0.053**** | 4.91 | 0.041 | 0.045 | 0.942 | 5.298 | 97 R (0.978) |
| aBSREL Analysis (Branch-Site Model) | |||||||
| Branch | – | ω2 (positive sites) | ω1 (purifying sites) | Fraction of Sites under Positive Selection | – | p-value | Significant after multiple hypothesis testing? |
| A | – | 3.03 | 0.249 | 0.250 | 0.1356 | no | |
| E | – | 15.2 | 0.00 | .024 | 0.0293 | no | |
| G | – | 2000 | 0.147 | 0.058 | – | 0.0045 | yes |
= p <0.05,
= p < 0.01,
= p <10−4, corrected for multiple hypothesis testing.
LRT, Likelihood Ratio Test statistic, used in χ2. Critical values are 3.84 (5%) and 6.63 (1%).
We hypothesized that if the long branch lengths reflect selection for novel function, the elevated branch ω values (Table 1) might reflect a mixture of positive and purifying selection acting at different sites in the protein. We therefore evaluated branch-site models, which detect different selection pressures at specific branches across sites in a protein [20, 23]. PAML analysis uncovered positive selection on branches A, B, C, D, E, F, G and I, all of which had some proportion of sites under ω2 > 1 (Table 1). The background purifying selection (ω1) was found to be extremely consistent across branches and to account for the majority of sites on all branches tested (Table 1). Specific positively selected amino acids were identified by an empirical Bayesian approach [23] at posterior probability 0.95 or better for branches A, C, D, G, and I (Table 1). Branch E stands out with 0.02 of sites at estimated ω2 of 999, which means no synonymous substitutions were observed (dS =0). While the dN/dS cannot be taken as a real value, likelihood ratios can still be accurately calculated for this branch, yielding a highly significant p < 0.01 for positive selection, and branch I was significant to p < 0.05 prior to multiple testing correction (Table 1). Branch A leading to the GRC α-SNAP yielded a relatively modest ω2 of 1.73, but it had by far the most sites under positive selection (0.363) possibly explaining why the branch ω was elevated (ω = 0.548).
To confirm these findings we ran the Adaptive Branch-Site Random Effects Likelihood (aBSREL) algorithm, which is similar to PAML but builds the tree and estimates the model complexity directly from the input sequence alignment [24]. Branch G showed statistically significant strong positive selection (ω2 = 2000 at 5.8% of sites, p = 0.0045, Table 1, Figure 2B) while E was also statistically significant (p = 0.03) before multiple hypothesis correction. (In all cases, we performed simple Bonferroni correction, which has been advocated in branch-sites analysis [25], but may be too stringent [26, 27]; correction of Bayesian posterior probabilities is not required [28, 29].) We conclude that the positive selection along the phylogeny in Figure 2A is of extremely variable strength and distribution among sites. Branches G and E exhibit strong selection at 2–6% of sites, while branch A (leading to the GRC α-SNAP) evidences a weaker positive selection across 25–36% of sites (Table 1, Figure 2B and 2C).
To evaluate the wider evolutionary context of α- and β-SNAP genes, we aligned 16 α-SNAP and 20 β-SNAP genes from birds, reptiles, mammals, and fish. Consistent with the bird tree (Figure 2A) we recover α- and β-SNAP genes as separate clades, and β-SNAPs have generally shorter branch lengths (Figure 3). Long β-SNAP branches occurs in fish, specifically Atlantic herring (Clupea harengus) and great blue-spotted mudskipper (Boleophthalmus pectinirostris), which also display β-SNAP paralogy, the only cases outside the zebra finch α-SNAP duplication described in this work (Figure 3) in which the SNAP genes are duplicated.
The placement of fish and bird α-SNAP as sister clades is surprising (Figure 3). We do not have high confidence in this arrangement, as the branch support is lower. Instead we attribute this grouping to the extremely long branch-length of bird α-SNAP genes creating long-branch attraction [17] and causing them to root basal to the mammal-reptile-chicken clade (Figure 3). This has been reported to be a risk of Bayesian reconstruction specifically in cases of rapidly evolving lineages with rate heterogeneity among sites [18]. However, a maximum likelihood (RAxML) tree built from the same data displayed the same topology (not shown). Finally, we note the placement of society finch α-SNAP with the zebra finch GRC as a sister clade, an arrangement discussed above and which is extremely well supported, and may also be due to long-branch attraction.
In this work we have identified the first gene from the germline-restricted chromosome in zebra finch, and the first case of α-SNAP paralogy in any organism. We confirmed this by searching the avian α-SNAP genes deposited in Genbank, representing 25 bird species, and noting that any duplicates we found were redundant copies of the same α-SNAP gene. To uncover potentially missed SNAP genes we performed a tblastn search of the RefSeq passerine genomes and only uncovered single-copy α- and β- SNAP genes, consistent with the literature [15, 30].
The GRC-to-somatalog α-SNAP amino acid divergence (81% identity, 88% similarity) is comparable in scale to the divergence between zebra finch α- (somatolog) and β- SNAPs (72% identity, 90% similarity). This is reflected also by the branch leading to the GRC α-SNAP being nearly as long as the branches separating α and β SNAP clades (Figure 2). Therefore we cannot exclude the possibility that the GRC-encoded gene is a pioneering member of a new SNAP family [31]. Demonstrating this will require isolating more GRC SNAP genes from other birds, and to date germline genomic data are sorely lacking.
Since the duplicated gene in zebra finch, the GRC α-SNAP, is present on a germline-limited sequence, the paralogy causes an effective doubling of the α-SNAP copy number in the germline only. Perhaps in response to this, the two paralogous genes have diverged to a high degree under positive selection. We also demonstrate that the two genes have sex-dimorphic expression in germline, with the GRC α-SNAP more highly expressed in ovary than testis. These data suggest the finch germline-restricted chromosome is likely playing an important biological role, in agreement with other studies showing that germline-restricted sequences are often involved in sex-determination or germline function [1, 32], and we predict that more GRC-encoded genes are awaiting discovery. Finally, we note that if the gene duplication event leading to the zebra finch α-SNAP paralogy was the genesis of the GRC itself, our data imply that the GRC is relatively old and may be present in more bird lineages than originally expected.
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, John Bracht (jbracht@american.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Adult zebra finches (Taeniopygia guttata) were obtained from a commercial breeder and housed in groups (15–25 per cage) in same-sex aviaries. The colony room was maintained at 20C, 70% humidity and a 14:10 L:D cycle. Food, water and grit were available ad libitum. All animal husbandry was approved by the American University Animal Care and Use Committee.
RNA and DNA used in this study were extracted from germline and somatic tissue of five male and five female young adult birds.
METHOD DETAILS
RNA extraction & sequencing
Subjects were rapidly decapitated and tissue was removed and flash frozen on dry ice. Samples were then weighed and stored at −80 degrees until further processing. For RNA extraction, tissues were homogenized in 500 uL 100 mM Phosphate Buffer pH 7.4, and RNA was extracted from 100 uL of resultant homogenate using the RNeasy Mini Kit (Qiagen) according to manufacturer instructions. The purity and concentration of each RNA sample was analyzed on a NanoDrop ND-100 spectrophotometer. Only extracts that exceeded a 260/280 ratio of 1.9 were used. Contaminating genomic DNA was eliminated by treatment with Turbo DNAse (ThermoFisher Cat #AM2238) and 15–20 g of RNA was submitted to Eurofins Genomics (Huntsville AL). Total DNA was purified from homogenates using the Nucleospin Tissue kit (Macherey-Nagel, Düren, Germany) according to manufacturer’s instructions.
Sequencing
Paired-end, expression-normalized, and strand-specific Illumina sequencing was performed by Eurofins Genomics (Huntsville AL). Read lengths were 300 basepair (bp) from a MiSeq, and the total number of read pairs obtained was 10,704,971 for Ovary and 9,703,220 for Testis.
Error Correction and Assembly
After sequencing the paired reads were stitched together with PEAR[33] to generate high-quality merged raw reads with a mean length of 300bp. Read error correction was performed using Reptile[34], followed by assembly on AU’s Zorro High Performance Computing Cluster using Trinity[35] run in default mode and specifying the –SS_lib_type parameter for strand-specific libraries. Following assembly the longest open reading frames were identified using TransDecoder.LongOrfs.
Assembly of publically available RNA-seq datasets
Zebra finch testis: Datasets SRR2299402, SRR2299403, and SRR2299404 were downloaded from NCBI’s Sequence Read Archive database (https://www.ncbi.nlm.nih.gov/sra). The fastq files were combined into a single file and Trinity was run using default settings and the ‘–trimmomatic’ flag.
For society finch data set SRR5223631 was downloaded (177 million reads) and assembled with the ‘—trimmomatic’ and ‘–SS_lib_type FR’ flags.
For canary, data set SRR2915372 was downloaded (123 million reads) and assembled with the ‘—trimmomatic’ flag.
For ground tit, data set SRR768235 was downloaded (28 million reads), and assembled with the ‘—trimmomatic’ flag.
dN/dS analysis
PAML Branch model
Codeml (PAML v.4.9)[36, 37] was used to estimate ω using the branch model setting (runmode = 0, seqtype = 1, model =2, NSsites = 0). Branches to be estimated were specified in the newick tree file (for the Bayesian tree), one at a time. Each branch was estimated twice: once with a neutral model (above settings plus fix_omega = 1 and omega=1) and using a purifying selection model (fix_omega =0, omega = 1). The P-values were determined using the likelihood ratio test (LRT) statistic 2Δl [38] compared against χ2 with critical values of 3.84, 5% significance level, and 6.63, for 1% significance [31]. Correction for multiple hypothesis testing was performed.
PAML Branch-sites model
Branch-sites ω were estimated by adding NSsites = 2 to the Codeml control file and estimating one branch at a time. The P-values were calculated as for the branch model by LRT statistic.
PAML Pairwise
A pairwise alignment of GRC and somatolog coding sequences was provided to Codeml with runmode=−2 and CodonFreq=2.
aBSREL
For aBSREL [24] the 14-sequence bird alignment that was used for tree building was input into the online interface (www.datamonkey.org). The relevant foreground branches were selected as indicated in Table 1.
Phylogenetic Trees
All sequences for comparison were obtained from NCBI or assembled de novo and curated for length, with short (incomplete) sequences discarded. Alignment was performed using the MAFFT algorithm [39] implemented within the Geneious software package (www.biomatters.com). The tree was generated with Mr. Bayes [40] in Geneious, using the Rate Matrix= equalin and Rate Variation= invgamma settings. The maximum likelihood (ML) tree was built using RAxML version 8.2.11 [41] with the GAMMA JTT protein model and 200 bootstrap replicates. The outgroup was chicken β-SNAP. Acccession numbers used are given below.
Accession numbers for α-SNAP mRNA are: human- NM_003827, mouse- NM_025898, rat- NM_080585, chicken- XM_015272486, painted turtle- XM_005310524, western clawed frog- NM_001011280, African clawed frog- NM_001092405, Atlantic herring- XM_012832758, Asian sea bass- XM_018665555, zebrafish- NM_199766, and great-blue spotted mudskipper-XM_020928551.
Accession numbers for α-SNAP proteins are: human- NP_003818.2, mouse-NP_080174.1, rat- NP_542152.1, chicken- XP_015127972.1, painted turtle- XP_005310581.1, western clawed frog- NP_001011280.1, African clawed frog- NP_001085874.1, Atlantic herring- XP_012688212.1, Asian sea bass- XP_018521071.1, zebrafish- NP_956060.1, and great-blue spotted mudskipper- XP_020784210.1.
Accession numbers for β-SNAP mRNA are: human- NM_001283018, mouse-NM_019632, rat- NM_001191966, chicken- NM_001199430, zebra finch- XM_002199762, ground tit- XM_005525483, canary- XM_009093739, rock dove- XM_005513170, downy woodpecker- XM_009902073, eagle- XM_010574905, Japanese quail- XM_015856683, golden-collared manakin- XM_018077509, western clawed frog- NM_001079098, zebrafish-NM_001080702, Atlantic herring- XM_012826735 and XM_012838056, African clawed frog-XM_018265067, and great-blue spotted mudskipper- XM_020921395 and XM_020933727.
Accession numbers for β-SNAP proteins are: human- NP_001269947.1, mouse-NP_062606, rat- NP_001178895.1, chicken- NP_001186359.1, zebra finch- XP_002199798.1, ground tit- XP_005525540.1, canary- XP_009091987.1, rock dove- XP_005513227.1, downy woodpecker- XP_009900375.1, eagle- XP_010573207.1, Japanese quail- XP_015712169.1, golden-collared manakin- XP_017932998.1, western clawed frog- NP_001072566.1, zebrafish-NP_001074171.2, Atlantic herring- XP_012682189.1 and XP_012693510.1, African clawed frog- XP_018120556.1, and great-blue spotted mudskipper- XP_020777054.1 and XP_020789386.1.
Subtractive genomics for zebra finch
Phase 1
The Sanger finch genome (GCA_000151805.1 Taeniopygia_guttata-3.2.4) and the mitochondrial sequence (MT) were downloaded from NCBI and combined into a single Basic Local Alignment Search Tool (blast) nucleotide database [42]. The Trinity ovary and testis assemblies were used as queries for local blastn against this combined genome+MT database, with default settings in order to provide maximal confidence in the remaining sequences’ uniqueness. A custom python script was used to segregate the non-matching sequences (i.e., those with no blastn matches). Open reading frames were identified using TransDecoder.LongestOrfs,(supplied with Trinity software package) and we used custom python scripts to remove redundant protein isoforms by selecting for the longest protein-coding sequence from each gene. Potential protein-coding homologs were identified by 1) blastp against the uniprot-swissprot database (evalue 1e-20) or 2) Hmmer3.1b2 [43] search against the Pfam-A database (Pfam 28.0) (also requiring evalue 1e-20).
Phase 2
The 936 proteins identified in Phase 1 were more stringently filtered by blast against the raw Sanger data, downloaded from the NCBI Trace Database (ftp://ftp-private.ncbi.nlm.nih.gov/pub/TraceDB/taeniopygia_guttata/). Default tblastn settings were used but we checked to confirm that evalues were highly significant and represented true matches. For example, we filtered out 520 Ovary hits (out of an inital set of 598, see Figure 1) giving 78 potential GRC genes. Of the 520 blast hits against Sanger raw reads, 517 (99.4%) occurred with an e-value of 2e-6 or better. Similarly our blast against Sanger raw reads filtered out 614 from a Testis set of 705 (keeping 91 genes) and of those blast hits 605 (98.5%) were of evalue 1.25e-6 or better.
This dataset was further filtered by mapping raw reads from a very large Auditory Lobule (brain) dataset generated by the Balakrishnan lab [7] (SRA archive SRS576610, SRS576611, and SRS576612), totaling approximately 70 million reads, onto the germline gene coding sequences with BWA[44] (bwamem, default settings). We eliminated any candidates with matching reads from the AL read mapping. Remarkably, this eliminated all but 8 of the 78 ovary transcripts: six were viral in nature (suggesting an unrecognized and apparently asymptomatic infection); of the remaining two, one was clearly repetitive in sequence and not considered further. The remaining gene was the novel SNAP protein (TR30145) confirmed to be GRC derived based on qPCR off genomic DNA and described further here. The testis dataset did not yield any GRC genes but yielded a contig encoding the somatolog α-SNAP, which was also assembled independently in the ovary transcriptome.
qPCR and PCR
Unless otherwise noted, all qPCR reactions (PowerSYBR, ThermoFisher Cat # 4367659) were run as a 2-stage cycle with 95°C for 10 min initial melt, then 40 cycles of 95°C for 30 sec, 60°C for 1 min and measurement of DNA concentration. Primers F1 + R1 cannot be used for qPCR off genomic DNA owing to a 689bp intron situated between them, necessitating the construction of primers A + B used instead. To gain specificity with the A+B primer set required a customized 2-step cycle of 95°C for 30 sec, followed by 64°C for 10 sec (still run for 40 total cycles).
All qPCR signal was measured relative to actin by Ct: we calculated average and standard deviation of 2 for all cases. Statistical significance was measured by Student’s 2-tailed T-test or 2-way ANOVA.
Normal (nonquantitative) PCR was carried out using AccuStart II polymerase (QuantaBio, Beverly, MA) and used according to manufacturer’s instructions, with annealing at 58°C, extension for 1 minute, and 35 cycles. Template was cDNA constructed as described below.
Reverse Transcription
SuperScript III First Strand Synthesis System (ThermoFisher Cat #18080051) was used in accordance with manufacturer’s instructions, with 4 g of total RNA that had been DNAse-treated with Turbo DNAse (ThermoFisher, Cat #AM2238) and phenol extracted. cDNAs were diluted 10× prior to use. Minus-RT controls were always tested in parallel to ensure no contaminating genomic DNA was present in the samples.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical tests for Figure 1 are described in the legend of that figure and include Student’s T-test and ANOVA. For Figure 1C and S1A, three birds (n = 3) were tested per sex, and the SNAP/actin ratio was measured by qPCR for each tissue in triplicate, yielding nine overall measurements per tissue. The graph shows the average and standard error of the mean for these nine measurements. Two-way ANOVA was performed with the XLSTAT (www.xlstat.com) Excel add-on software package. For Figure 1D, and 1E the graphs represent the average and error bars represent standard deviation of triplicate measurements, with statistical significance obtained by Student’s 2-tailed T-test.
For Figure S1 analysis was identical to Figure 1 except that for each of n=3 birds per sex, the tissue was measured six times, yielding 18 overall measurements per tissue. The graph represents average and standard error of the mean for these 18 measurements. All other analysis as in Figure 1.
For Figure 2 the statistical significance was obtained by PAML and by aBSREL; however PAML required running twice per branch (once for null and alternative), obtaining likelihood ratios, and testing these ratios by chi-square as described in Method Details.
DATA AND SOFTWARE AVAILABILITY
The zebra finch ovary RNA-seq reads have been deposited in SRA under accession # XXXXXX and the assembled data in TSA under accession # YYYYYYY.
The 936 high-confidence genes identified in this study have been deposited in Genbank under accession # ZZZZZZZ.
The α-SNAP genes from zebra finch GRC α-SNAP, somatolog, canary, society finch, and ground tit have been deposited in Genbank under accession #GGGGGGGG, #SSSSSSSSS, #CCCCCCC, #FFFFFFF, and #GGGGGGG, respectively.
Supplementary Material
Highlights.
- Discovery of the first germ-line restricted gene, α-Soluble NSF Attachment Protein.
- Discovery of a somatic paralog (somatolog) of the α-SNAP.
- Positive selection and long branch-length across α-SNAPs suggest novel function.
- The α-SNAP pair exhibits sex-dimorphic expression, with GRC greater in ovaries.
Acknowledgments
The authors wish to acknowledge Naden Krogan for insightful comments on the manuscript and Evgeny Bisk for Zorro computing cluster system administration. We are grateful to the Balakrishnan laboratory for the generation of two key transcriptomic resources used in this study: the testis (SRR2299402-4) and auditory lobule (SRS576610,11,12) RNAseq datasets. The American University High Performance Computing System was funded in part by a grant from the National Science Foundation, BCS-1039497. This work was supported by NIH grants 1K22CA184297 to J.R.B, NIH NS 047267 to C.J.S, and by an AU Faculty Research Support grant to C.J.S and J.R.B.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
M.M.N performed RNA isolation, sequencing, error correction, assembly, and initial validation qPCR. M.K.B performed tissue panel genomic DNA qPCR and RT-qPCR. K.C.A. performed codeml analysis on phylogenetic trees and made Figure 2A. A.L.P and C.J.S. performed tissue isolation and initial RNA extractions. J.R.B. and C.J.S. conceived the study and C.J.S provided biological material for sequencing. J.R.B oversaw the study, performed assembly of public transcriptomic data and phylogenetic analysis, made most figures, and wrote the paper.
Declaration of Interests
The authors declare no competing interests.
References
- 1.Wang J, Davis RE. Programmed DNA elimination in multicellular organisms. Curr Opin Genet Dev. 2014;27:26–34. doi: 10.1016/j.gde.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gurney ME, Konishi M. Hormone-induced sexual differentiation of brain and behavior in zebra finches. Science. 1980;208:1380–1383. doi: 10.1126/science.208.4450.1380. [DOI] [PubMed] [Google Scholar]
- 3.Pigozzi MI, Solari AJ. Germ cell restriction and regular transmission of an accessory chromosome that mimics a sex body in the zebra finch, Taeniopygia guttata. Chromosome research: an international journal on the molecular, supramolecular and evolutionary aspects of chromosome biology. 1998;6:105–113. doi: 10.1023/a:1009234912307. [DOI] [PubMed] [Google Scholar]
- 4.Pigozzi MI, Solari AJ. The germ-line-restricted chromosome in the zebra finch: recombination in females and elimination in males. Chromosoma. 2005;114:403–409. doi: 10.1007/s00412-005-0025-5. [DOI] [PubMed] [Google Scholar]
- 5.Itoh Y, Kampf K, Pigozzi MI, Arnold AP. Molecular cloning and characterization of the germline-restricted chromosome sequence in the zebra finch. Chromosoma. 2009;118:527–536. doi: 10.1007/s00412-009-0216-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Warren WC, Clayton DF, Ellegren H, Arnold AP, Hillier LW, Kunstner A, Searle S, White S, Vilella AJ, Fairley S, et al. The genome of a songbird. Nature. 2010;464:757–762. doi: 10.1038/nature08819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balakrishnan CN, Lin YC, London SE, Clayton DF. RNA-seq transcriptome analysis of male and female zebra finch cell lines. Genomics. 2012;100:363–369. doi: 10.1016/j.ygeno.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.The Zebra Finch Genome. Washington University Genome Sequencing Center; 2016. https://www.ncbi.nlm.nih.gov/genome?term=taeniopygia%20guttata. [Google Scholar]
- 9.Cheng XD, Blumenthal RM. Mammalian DNA methyltransferases: A structural perspective. Structure. 2008;16:341–350. doi: 10.1016/j.str.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hergeth SP, Schneider R. The H1 linker histones: multifunctional proteins beyond the nucleosomal core particle. EMBO Rep. 2015;16:1439–1453. doi: 10.15252/embr.201540749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Futatsumori M, Kasai K, Takatsu H, Shin HW, Nakayama K. Identification and characterization of novel isoforms of COP I subunits. J Biochem. 2000;128:793–801. doi: 10.1093/oxfordjournals.jbchem.a022817. [DOI] [PubMed] [Google Scholar]
- 12.Singhal S, Leffler EM, Sannareddy K, Turner I, Venn O, Hooper DM, Strand AI, Li Q, Raney B, Balakrishnan CN, et al. Stable recombination hotspots in birds. Science. 2015;350:928–932. doi: 10.1126/science.aad0843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garcia-Moreno J, Mindell DP. Rooting a phylogeny with homologous genes on opposite sex chromosomes (gametologs): a case study using avian CHD. Molecular biology and evolution. 2000;17:1826–1832. doi: 10.1093/oxfordjournals.molbev.a026283. [DOI] [PubMed] [Google Scholar]
- 14.Korlach J, Gedman G, Kingan SB, Chin CS, Howard JT, Audet JN, Cantin L, Jarvis ED. De novo PacBio long-read and phased avian genome assemblies correct and add to reference genes generated with intermediate and short reads. GigaScience. 2017;6:1–16. doi: 10.1093/gigascience/gix085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whiteheart SW, Griff IC, Brunner M, Clary DO, Mayer T, Buhrow SA, Rothman JE. SNAP family of NSF attachment proteins includes a brain-specific isoform. Nature. 1993;362:353–355. doi: 10.1038/362353a0. [DOI] [PubMed] [Google Scholar]
- 16.del Priore L, Pigozzi MI. Histone modifications related to chromosome silencing and elimination during male meiosis in Bengalese finch. Chromosoma. 2014;123:293–302. doi: 10.1007/s00412-014-0451-3. [DOI] [PubMed] [Google Scholar]
- 17.Bergsten J. A review of long-branch attraction. Cladistics. 2005;21:163–193. doi: 10.1111/j.1096-0031.2005.00059.x. [DOI] [PubMed] [Google Scholar]
- 18.Kolaczkowski B, Thornton JW. Long-branch attraction bias and inconsistency in Bayesian phylogenetics. PloS one. 2009;4:e7891. doi: 10.1371/journal.pone.0007891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang Z. Inference of selection from multiple species alignments. Curr Opin Genet Dev. 2002;12:688–694. doi: 10.1016/s0959-437x(02)00348-9. [DOI] [PubMed] [Google Scholar]
- 20.Zhang JZ, Nielsen R, Yang ZH. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Molecular biology and evolution. 2005;22:2472–2479. doi: 10.1093/molbev/msi237. [DOI] [PubMed] [Google Scholar]
- 21.Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290:1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- 22.Kondrashov FA, Rogozin IB, Wolf YI, Koonin EV. Selection in the evolution of gene duplications. Genome biology. 2002;3:RESEARCH0008. doi: 10.1186/gb-2002-3-2-research0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang ZH, Wong WSW, Nielsen R. Bayes empirical Bayes inference of amino acid sites under positive selection. Molecular biology and evolution. 2005;22:1107–1118. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]
- 24.Smith MD, Wertheim JO, Weaver S, Murrell B, Scheffler K, Pond SLK. Less Is More: An Adaptive Branch-Site Random Effects Model for Efficient Detection of Episodic Diversifying Selection. Molecular biology and evolution. 2015;32:1342–1353. doi: 10.1093/molbev/msv022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anisimova M, Yang Z. Multiple hypothesis testing to detect lineages under positive selection that affects only a few sites. Molecular biology and evolution. 2007;24:1219–1228. doi: 10.1093/molbev/msm042. [DOI] [PubMed] [Google Scholar]
- 26.Perneger TV. What’s wrong with Bonferroni adjustments. BMJ. 1998;316:1236–1238. doi: 10.1136/bmj.316.7139.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Noble WS. How does multiple testing correction work? Nature biotechnology. 2009;27:1135–1137. doi: 10.1038/nbt1209-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gelman A, Hill J, Yajima M. Why We (Usually) Don’t Have to Worry About Multiple Comparisons. J Res Educ Eff. 2012;5:189–211. [Google Scholar]
- 29.Westfall PH, Johnson WO, Utts JM. A Bayesian perspective on the Bonferroni adjustment. Biometrika. 1997;84:419–427. [Google Scholar]
- 30.Stenbeck G. Soluble NSF-attachment proteins. Int J Biochem Cell B. 1998;30:573–577. doi: 10.1016/s1357-2725(97)00064-2. [DOI] [PubMed] [Google Scholar]
- 31.Clary DO, Griff IC, Rothman JE. SNAPs, a family of NSF attachment proteins involved in intracellular membrane fusion in animals and yeast. Cell. 1990;61:709–721. doi: 10.1016/0092-8674(90)90482-t. [DOI] [PubMed] [Google Scholar]
- 32.Kloc M, Zagrodzinska B. Chromatin elimination - an oddity or a common mechanism in differentiation and development? Differentiation. 2001;68:84–91. doi: 10.1046/j.1432-0436.2001.680202.x. [DOI] [PubMed] [Google Scholar]
- 33.Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics. 2014;30:614–620. doi: 10.1093/bioinformatics/btt593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang X, Dorman KS, Aluru S. Reptile: representative tiling for short read error correction. Bioinformatics. 2010;26:2526–2533. doi: 10.1093/bioinformatics/btq468. [DOI] [PubMed] [Google Scholar]
- 35.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature biotechnology. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- 37.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Molecular biology and evolution. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 38.Yang ZH. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Molecular biology and evolution. 1998;15:568–573. doi: 10.1093/oxfordjournals.molbev.a025957. [DOI] [PubMed] [Google Scholar]
- 39.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- 41.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 43.Eddy SR. Accelerated Profile HMM Searches. PLoS Comput Biol. 2011;7:e1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.