

Abstract
The large protein (L) of the human parainfluenza virus type 3 (HPIV3) is the functional RNA-dependent RNA polymerase, which possesses highly conserved residues QGDNQ located within motif C of domain III comprising the putative polymerase active site. We have characterized the role of the QGDNQ residues as well as the residues flanking this region in the polymerase activity of the L protein by site-directed mutagenesis and examining the polymerase activity of the wild-type and mutant L proteins by an in vivo minigenome replication assay and an in vitro mRNA transcription assay. All mutations in the QGDNQ residues abolished transcription while mutations in the flanking residues gave rise to variable polymerase activities. These observations support the contention that the QGDNQ sequence is absolutely required for the polymerase activity of the HPIV3 RNA-dependent RNA polymerase.
Keywords: Human parainfluenza virus type 3 (HPIV3), RNA-dependent RNA polymerase, Mutagenesis, In vitro transcription
THE large protein (L) of the human parainfluenza virus type 3 (HPIV3), an enveloped, nonsegmented, negative-stranded RNA virus with a genome of 15,462 nucleotides (4) comprises a protein of 2233 amino acids with a molecular mass of 255 kDa. The L protein represents a multifunctional enzyme and is the putative RNA-dependent RNA polymerase together with the phosphoprotein (P), a polypeptide of 603 amino acids that acts as a cofactor (4). Sequence alignment studies have predicted the existence of six highly conserved domains, I–VI, in all the L proteins of the negative-stranded RNA viruses (20,23,24,28). The central “polymerase” module comprising the motifs A, B, C, and D within domain III is probably the active site for RNA synthesis. The core residues GDN within the motif C of domain III are assumed to correspond to the GDD residues of positive-strand RNA viruses and M/V/IDD residues of retroviruses with the DN (DD) residues being required for cation binding, template specificity, or other catalytic processes (1). Tertiary structure comparison of the HIV-1 reverse transcriptase, a RNA-dependent DNA polymerase, with the L protein of the negative-stranded RNA viruses suggested that motifs A and C possess similar structures and are part of the catalytic site (16,20).
Extensive studies by mutations or deletions in the GDN residues of the polymerase subunit of several viruses, including HIV-1 reverse transcriptase (17,18), Bunyamwera virus L protein (15), vesicular stomatitis virus (29), influenza virus PB1 protein (2), rabies virus (25), bamboo mosaic virus (19), and rabbit hemorrhagic disease virus (30), have demonstrated the critical importance of these residues in sustaining the polymerase activity. Furthermore, within the GDN motif the only change tolerated was replacement of the asparagine (N) residue to the conserved aspartate (D) residue. This represents a conversion of the “DN” signature of mononegavirales to the “DD” signature of the reverse transcriptases and polymerases of positive-stranded RNA viruses.
In the present study, we have investigated the critical role of QGDNQ residues within the L protein of HPIV3 by performing site-directed mutagenesis to measure the polymerase activity of the mutant HPIV3 L proteins. In addition, we have also examined the role of the flanking residues on the functional integrity of the polymerase. We have employed the HPIV3 in vivo minigenome assay to measure polymerase activity by monitoring the luciferase reporter gene expression and an in vitro transcription assay that utilizes HPIV3 N-RNA as template to measure mRNA synthesis. We found that not only mutations of the GDN residues of HPIV3 L protein completely abolished polymerase activity, but the majority of the mutations in the flanking residues also resulted in nonfunctional polymerases.
MATERIALS AND METHODS
Cells and Viruses
HPIV3 (HA-1, NIH 47885) was propagated in CV-1 cells as described previously (7,8). Recombinant vaccinia virus (MVA) expressing T7 RNA polymerase was grown in HeLa cells.
Site-Directed Mutagenesis
Oligonucleotides harboring point mutations in the QGDNQ residues (Fig. 1) were employed in PCR using the Expand polymerase (Roche Biochemicals, Indianapolis, IN). The plasmid pGEM 4-LFLAG, with FLAG epitope (DYKDDDDK) fused to the 3′ terminus of the HPIV3 L gene was used as a template for the PCR. The amplified fragments containing the mutations were cloned into pGEM 4-LFLAG and dideoxy sequencing was done to ensure the presence of the mutations.
Figure 1.

Sequences of the oligonucleotides used for site-directed mutagenesis of the HPIV3 L gene. The exchanged nucleotides are shown in bold.
Coexpression and Immunoprecipitation of Proteins
HeLa cells in a 12-well plate were infected with MVA at a m.o.i of 3. One hour postinfection, the cells were transfected with the plamid DNA expressing pP and pLWT or pLMUT using Lipofectin (Life Technologies Inc., Rockville, MD) according to manufacturer’s instructions. Twelve hours posttransfection, the medium was replaced with 2 ml of methionine-free Dulbecco’s modified Eagle’s medium (DMEM), and incubation was continued at 37°C. At 14 h posttransfection the cells were labeled with 50 μCi of [35S]methionine (1175 Ci/mmol, NEN, Boston, MA) in 1 ml of methionine-free DMEM for 6 h. Cells were washed with PBS and lysed in 150 μl of luciferase lysis buffer. An aliquot of the lysate was used for SDS-PAGE and Western blot analysis to detect the P protein using polyclonal anti-RNP antibody. The rest of the lysate was immunoprecipitated with anti-FLAG antibody (Sigma, St. Louis, MO) conjugated to agarose beads following the manufacturer’s protocol. The immunoprecipitated proteins were analyzed by SDS-PAGE, stained with Coomassie blue, dried, and subjected to fluorography.
In Vivo Minigenome Replication and Luciferase Assay
In vivo minigenome replication assay was studied following the procedure of Hoffman and Banerjee (11). Briefly, HeLa cell monolayers in a 12-well plate, grown to 90% confluency, were infected with recombinant vaccinia virus MVA, which expresses T7 RNA polymerase, at a m.o.i of 3. After 1 h at 37°C, the HPIV3 minigenome plasmid pHPIV3-MG(−) 200 ng, and support plasmids pHPIV3-N (pN) 600 ng, pHPIV3-P (pP) 750 ng, and wild-type HPIV3-LWT (pLWT) or HPIV3 mutant L proteins (pLMUT) 100 ng were transfected using Lipofectin (Life Technologies Inc.) according to the manufacturer’s instructions. After 4 h, the transfection medium was replaced with 1 ml of DMEM/10% fetal calf serum. At 28 h post-transfection, the monolayers were lysed in 150 ja.l of lysis buffer, from which 1.5 ja.l of lysate (equivalent to 2.3 × 103 cells) was then used to determine luciferase activity (Luciferase Assay Kit; Roche Biochemicals, Indianapolis, IN) according to the manufacturer’s specifications using a Dynatech ML2250 luminometer.
In Vitro Transcription
HeLa cells in a six-well plate were transfected with pP alone or in presence of pLWT or pLMUT plasmids as described above except that six-well plate was used and all material amounts were increased proportionately. At 24 h posttransfection, the cell monolayers were washed with ice-cold PBS and harvested by scraping in PBS. The cells were pelleted by centrifugation at 800 × g for 10 min. Cytoplasmic extracts were prepared by lysing cells by three cycles of freezing and thawing in 40 μl of hypotonic buffer containing 10 mM Tris-HCl (pH 8.0), 10 mM NaCl, and 1 mM dithiothreitol (DTT). Nuclei and cell debris were removed by centrifugation for 5 min in an Eppendorf centrifuge at 40°C. The soluble cytoplasmic extract was estimated as 5 mg/ml.
N-RNA templates were prepared from HPIV3-infected CV-1 cells (108) following the procedure of Curran et al. (6) with some modifications. Briefly, the cells were harvested in PBS and resuspended in 5 ml of buffer containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 0.6% NP-40, 1% Triton X-100, and 1 mM DTT. The cells were lysed by vortexing, and nuclei and cell membranes were removed by centrifugation at 10,000 × g for 5 min. The cell extract was made 6 mM in EDTA and layered onto a 20–40% CsCl (w/w) gradient and centrifuged at 38,000 × g for 2 h at 12°C in an SW41 rotor. The visible N-RNA band was collected and repurified using the CsCl gradient. Finally, the N-RNA was sedimented through 40% glycerol in 50 mM HEPES-KOH (pH 8.0), 50 mM NaCl, 0.2% NP-40, 1 mM DTT onto a 100 μl cushion of 100% glycerol and stored in liquid nitrogen.
The in vitro transcription reaction was performed in a 50 μl total volume essentially as described previously by De et al. (9) and included 100 mM HEPES-KOH, 100 mM KCl, 5 mM MgCl2, 1 mM DTT, 1 mM each ATP, GTP, and CTP, 10 μM UTP, 20 μCi of [α-32P]UTP, 25 U of human placental RNase inhibitor, 5 μg of actinomycin D per ml, 2 μg of N-RNA, and, unless otherwise stated, 10 μg of cell extract containing L and P proteins.
RESULTS
Sequence Comparison of the L Proteins of Nonsegmented, Negative-Stranded RNA Viruses
Multiple sequence alignment of the L-proteins of the nonsegmented, negative-stranded RNA viruses reveals the presence of conserved residues QGDNQ within motif C of domain III. In the case of HPIV3 L protein these conserved residues occupy the amino acid positions 771–775 (Fig. 2A). In addition, the alignment exhibits a variable degree of conservation of the flanking residues. Thus, for HPIV3 L protein, methionine and valine occupy the upstream positions 769 and 770, respectively, while alanine and isoleucine are present downstream, at positions 776 and 777, respectively.
Figure 2.

(A) Sequence alignment of the conserved QGDNQ motif and the flanking residues of the L protein of negative stranded RNA viruses. Numbers at the left indicate the position in the respective L protein. The conserved QGDNQ residues are underlined. PIV3, human parainfluenza virus type 3 (10); SV, Sendai virus (27); MV, measles virus (3); CDV, canine distemper virus (28); SV5, simian virus 5 (24); MuV, mumps virus (21); NDV, Newcastle disease virus (31); VSV, vesicular stomatitis virus (26); and RV, rabies virus (5). (B) Sequence of the HPIV3 L protein showing the conserved QGDNQ motif and the flanking residues. The corresponding mutants are shown by arrows. Numbers above the sequence indicate the position of each residue on the L protein.
In order to investigate the role of the QGDNQ and the flanking residues on the polymerase activity of the HPIV3 L protein, point mutations were introduced to alter these residues to alanine as well as alter the flanking sequences by a conserved or a non-conserved residue (Fig. 2B). One of the alterations N→D resembled the GDD sequence of the active polymerase site of a positive-strand RNA virus (13,14). The expression of the mutant FLAG-tagged L proteins was monitored by coexpression with the nontagged P protein in HeLa cells followed by metabolic labeling and immunoprecipitation with anti-FLAG antibody. All the mutant L proteins were expressed in amounts similar to the wild-type and formed normal L–P complexes. These results clearly demonstrated that none of the mutations had any effect on its stability, expression, and interaction with the P protein (Fig. 3). Thus, the mutant L proteins were further analyzed for polymerase activity both in vivo and in vitro.
Figure 3.
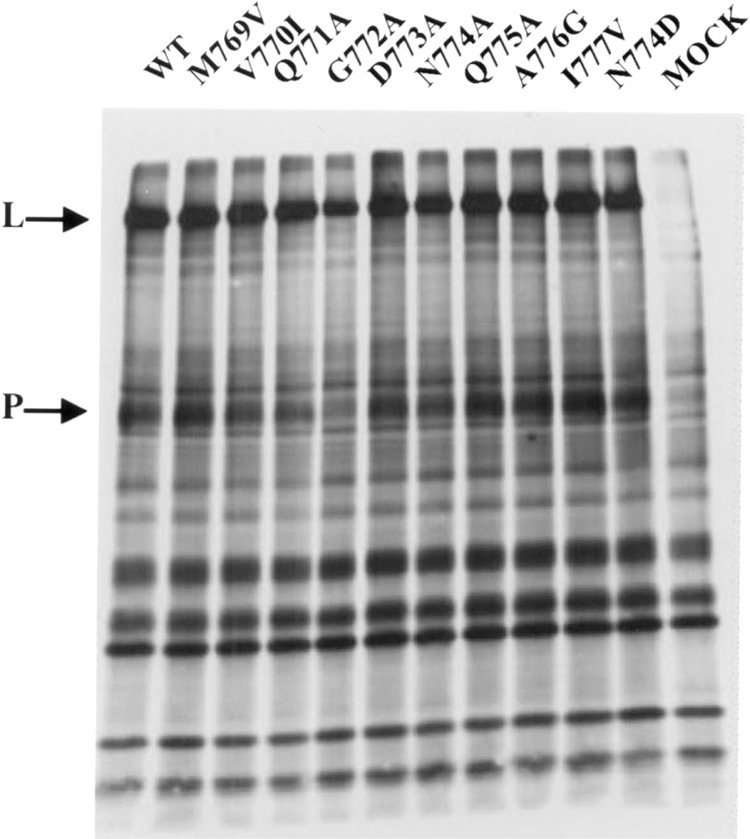
Analysis of L–P complexes by immunoprecipitation using FLAG-tagged assay. The wild-type and mutant L proteins were coexpressed with P protein in a recombinant vaccinia virus expression system. Cells were metabolically labeled with [35S]methionine and the labeled proteins were immunoprecipitated with anti-FLAG antibody conjugated to agarose beads and analyzed by SDS-PAGE followed by fluorography. Arrows indicate the positions of the L and P proteins.
Mutant Polymerases Exhibit Variable Activities
To investigate the polymerase activity of different mutants, HPIV3 minigenome assay measuring the expression of the luciferase gene flanked by the HPIV3 promoter and trailer elements (11) were utilized. HeLa cells infected with a recombinant vaccinia virus, expressing T7 RNA polymerase, were transfected with the minigenome plasmid pHPIV-3 MG(−), and support plasmids pN, pP, and pLWT or pLMUT, under the control of T7 promoter. Following transfection, the resulting encapsidated negative sense minigenome RNA is transcribed by the support plasmids to generate the sense luciferase mRNA, which subsequently translated to luciferase protein. Thus, measurement of luciferase activity directly indicates the replication efficiency of the minigenome in vivo. The assay was performed in duplicate and results from three individual experiments were calculated as percent activity relative to wild-type. The luciferase gene expression was amplified approximately 200-fold over background levels (lacking L plasmid) in the presence of all three support plasmids and is represented as 100% expression (Fig. 4). It is quite evident from Figure 4 that mutations within the QGDNQ resulted in almost total abrogation of the polymerase activity. Thus, the mutants Q771A, G772A, D773A, N774A, and Q775A failed to transcribe the luciferase mRNA and hence no luciferase expression. In addition, one mutant, N774D, wherein asparagine was replaced with aspartate so as to resemble the conserved GDD sequence of a positive-strand RNA virus polymerase, virtually exhibited no polymerase activity. Interestingly, this observation is similar to the one reported for the rabies virus L protein where a similar replacement of asparagine to aspartate resulted in an inactive polymerase (25). However, in contrast to rabies and our observation, replacement of the similar asparagine to aspartate in VSV resulted in 27% polymerase activity as measured in an in vitro transcription assay (29). Thus, our results clearly demonstrate that the QGDNQ motif of HPIV3 L protein is absolutely indispensable to maintain its polymerase activity.
Figure 4.
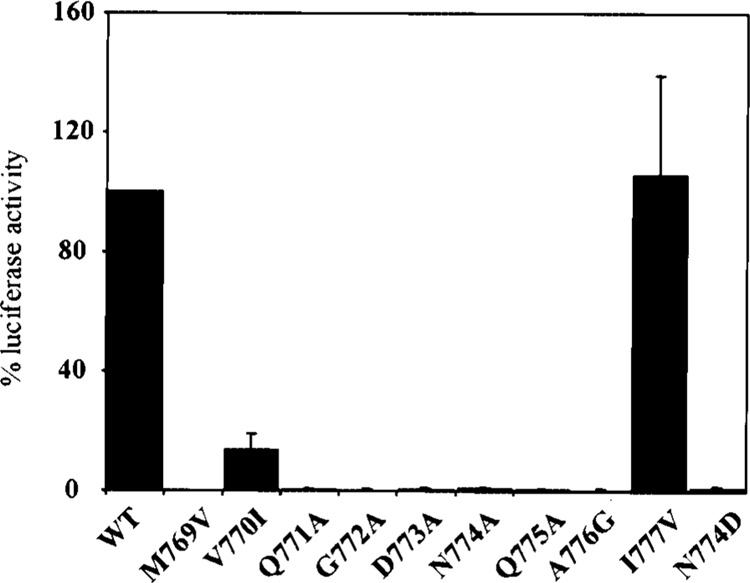
Acitvity of mutant L proteins in minigenome replication in vivo. The wild-type and mutant L proteins were coexpressed with N, P proteins in the minigenome replication system along with pHPIV3-MG(−) plasmid as described. At 24 h postinfection, cell lysates were prepared and luciferase activity was measured and expressed as mean of three independent experiments.
To gain insight into the role of the flanking residues on the polymerase activity of the L protein four additional mutants were designed. The upstream residues, methionine and valine, of HPIV3 L protein were replaced by valine and isoleucine, respectively, to obtain mutants M769V and V770I. Similarly, alanine and isoleucine residues present downstream of the HPIV3 L protein were replaced by glycine and valine, respectively, to generate the mutants A776G and I777V. The measurement of the polymerase activity of these flanking residue mutants revealed that two mutants, M769V and A776G, were totally inactive while V770I retained 13% activity. Surprisingly, the polymerase activity of the mutant I777V was similar to or greater than wild-type HPIV3 L protein (Fig. 4). In the case of VSV and rabies virus similar variations in polymerase activities have been reported as a result of mutations in the flanking residues (25,29).
mRNA Synthesis Is Defective in Mutant Polymerases In Vitro
The role of L and P proteins in mRNA synthesis as well as the functions of the various domains of the two proteins can also be studied by an in vitro reconstitution system that utilizes purified HPIV3 N-RNA depleted of endogenous RNA polymerase complex, and recombinant L and P proteins. The in vitro transcription system described by De et al. (9) was employed to study the effect of mutations in the L protein on the synthesis of viral mRNA. HeLa cells were infected with recombinant vaccinia virus and transfected with either the LWT or LMUT in the presence of P protein. At 24 h postinfection, cytoplasmic extract was prepared and used directly in an in vitro transcription reaction containing purified HPIV3 N-RNA template in the presence of [α-32P]UTP as described in Materials and Methods. The RNA products were analyzed in a 5% urea-acrylamide gel followed by autoradiography. As shown in Figure 5, the N-RNA template alone (MOCK) had no RNA-synthesizing activity while addition of an extract containing coexpressed LWT and P protein (WT) resulted in an efficient transcription of the genomic RNA. Cytoplasmic extracts of transfected LMUT and P proteins were then analyzed for the ability to synthesize mRNA in vitro. As shown in Figure 5, four mutants, G772A, D773A, N774A, and N774D, were totally inactive in mRNA synthesis. In addition, two mutants where the flanking residues were mutated, M769V and A776G, were unable to support mRNA synthesis. The in vitro activity of two mutants, Q771A and Q775A, was close to 5% compared with the wild-type. Interestingly, the in vitro activities of two mutants, V770I and I777V, were 10% and 105%, respectively. These values correlated with their in vivo luciferase activity values, thus confirming the fact that these mutations exerted their effect on both replication and transcription. Western blot analysis of the cytoplasmic extracts containing coexpressed L and P proteins using anti-HPIV3 antibody confirmed a uniform level of expression of both L and P proteins similar to that shown in Figure 3 (data not shown).
Figure 5.
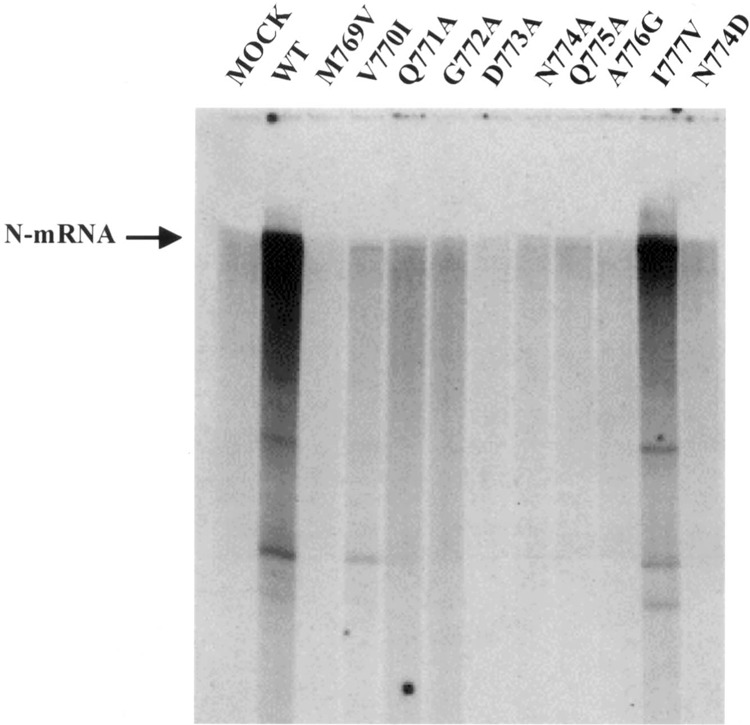
In vitro transcription of mutant L proteins. The wild-type and mutant L proteins indicated were coexpressed with P protein in a recombinant vaccinia virus expression system. Cell extracts (total protein 6 μg) containing coexpressed L and P proteins were used in transcription reactions containing N-RNA template in the presence of [α-32P]UTP, and the mRNA products were analyzed on a 5% polyacrylamide-urea gel.
DISCUSSION
The pentapeptide QGDNQ constitutes the longest continuous stretch of invariant amino acids that are present in domain III of the RNA-dependent RNA polymerases of the negative-stranded RNA viruses. Sequence alignment has shown that the GDN motif is a variant of the GDD motif of the polymerase of positive-stranded RNA viruses, and also closely resembles the SDD/MDD motif of influenza virus and human immunodeficiency virus (2,17,18). In the present study we assessed the role of these conserved residues in the HPIV3 L protein by site-directed mutagenesis and measuring the polymerase activity of the mutant proteins in vivo and in vitro. Our results have demonstrated that mutations in any of the critical residues give rise to inactive polymerases, incompetent in replication and transcription, thus reinforcing the fact that these residues are indispensable for polymerase function. Mutational analysis of polio virus (13,14), Bunyamwera virus (15), and Qβ bacteriophage (12) and rabies virus (25) polymerases resulted in similar loss of activity. On the other hand, replacement of asparagine (N) to aspartate (D) within the GDN motif of VSV polymerase retained 27% transcriptional activity compared with the wild-type polymerase.
We also investigated the effect of mutations of the flanking residues on the polymerase activity. Among the two mutants that harbored mutations preceding the QGDNQ motif, only one mutant, V770I, exhibited 13% activity while the other mutant was inactive. A similar scenario was observed with poliovirus (12,13) and influenza virus (2), where it was shown that the upstream residues could be variable. In contrast, rabies virus was shown to tolerate only one change in the flanking residues. Mutation of the downstream isoleucine to valine, in HPIV3 L protein, to yield the mutant I777V resulted in a fully functional polymerase. In the case of VSV (29), influenza virus (2), and rabies virus (25), several conservative changes downstream of the GDN/GDD motif of the polymerase also resulted in variable polymerases activities.
In the present study, using a mutagenesis approach we have analyzed the critical role of the invariant QGDNQ motif on the functional activity of the HPIV3 polymerase. Currently, the three-dimensional structure based on crystallographic data is unavailable for RNA-dependent RNA polymerases. In the absence of the tertiary structural data on the nonsegmented, negative-stranded RNA-dependent RNA polymerases, results from our study together with the earlier observations clearly ascertain the importance of the QGDNQ residues and thus provide invaluable insight in elucidating the functions of other domains of this multifunctional protein.
ACKNOWLEDGMENT
This work was supported in part by a grant from N.I.H, AI3207 (A.K.B).
REFERENCES
- 1. Beese L.; Steitz T. A. Structural basis for the 3′-5′ exonuclease activity of Escherichia coli DNA polymerase I: A two metal ion mechanism. EMBO J. 10:25–33; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Biswas S. K.; Nayak D. P. Mutational analysis of the conserved motifs of influenza A virus polymerase basic protein 1. J. Virol. 68:1819–1826; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Blumberg B. M.; Crowley J. C.; Silverman J. I.; Menonna J.; Cook S. D.; Dowling P. C. Measles virus L protein evidences elements of ancestral RNA polymerase. Virology 164:487–497; 1988. [DOI] [PubMed] [Google Scholar]
- 4. Collins P. L.; Chanock R. M.; McIntosh K. Parainfluenza viruses. In: Fields B. M.; Knipe D. M.; Howley P. M., eds. Fields virology, 3rd ed. Philadelphia: Lippincott-Raven Publishers; 1996:1205–1241. [Google Scholar]
- 5. Conzelmann K. K.; Cox J. H.; Schneier L. G.; Thiel H. J. Molecular cloning and complete nucleotide sequence of the attenuated rabies virus SAD B19. Virology 175:485–499; 1990. [DOI] [PubMed] [Google Scholar]
- 6. Curran J.; Pelet T.; Kolakofsky D. An acidic activation-like domain of the Sendai virus P protein is required for RNA synthesis and encapsidation. Virology 202:875–884; 1994. [DOI] [PubMed] [Google Scholar]
- 7. De B. P.; Lesoon A.; Banerjee A. K. Human parainfluenza virus type 3 transcription in vitro: Role of cellular actin in mRNA synthesis. J. Virol. 65:3268–3275; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. De B. P.; Burdsall; A. L.; Banerjee A. K. Role of cellular actin in human parainfluenza virus type 3 genome transcription. J. Biol. Chem. 268:5703–5710; 1993. [PubMed] [Google Scholar]
- 9. De B. P.; Hoffman M. A.; Choudhary S.; Huntley C. C.; Banerjee A. K. Role of the NH2-and COOH-terminal domains of the P protein of human parainfluenza virus type 3 in transcription and replication. J. Virol. 74:5886–5895; 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Galinski M. S.; Mink M. A.; Pons M. W. Molecular cloning and sequence analysis of the human parainfluenza 3 virus gene encoding the L protein. Virology 165:499–510; 1988. [DOI] [PubMed] [Google Scholar]
- 11. Hoffman M. A.; Banerjee A. K. Precise mapping of the replication and transcription promoters of human parainfluenza virus type 3. Virology 269: 201–211; 2000. [DOI] [PubMed] [Google Scholar]
- 12. Inocuchi Y.; Hirashima A. Interference with viral replication by defective RNA replicase. J. Virol. 61:3946–3949; 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jablonski S. A.; Luo M.; Morrow C. D. Enzymatic activity of polio virus RNA polymerase mutants with single amino acid changes in the conserved YGDD amino acid motif. J. Virol. 65:4565–4572; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jablonski S. A.; Morrow C. D. Enzymatic activity of polio virus RNA polymerase with mutations at the tyrosine residue of the conserved YGDD motif: Isolation and characterization of polio viruses containing RNA polymerases with FGDD and MGDD sequences. J. Virol. 67:373–381; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jin H.; Elliott R. M. Mutagenesis of the L protein encoded by Bunyamwera virus and production of mono-specific antibodies. J. Gen. Virol. 67:1334–1339; 1992. [DOI] [PubMed] [Google Scholar]
- 16. Kohlstaedt L. A.; Wang J.; Friedman J. M.; Rice P. A.; Steitz T. A. Crystal structure at 3.5 Å resolution of HIV-1 reverse transcriptase complex with an inhibitor. Science 256:1783–1790; 1992. [DOI] [PubMed] [Google Scholar]
- 17. Larder B. A.; Purifoy D. J. M.; Powell K. L.; Darby G. Site-specific mutagenesis of AIDS virus reverse transcriptase. Nature 327:716–717; 1987. [DOI] [PubMed] [Google Scholar]
- 18. Larder B. A.; Kemp S. D.; Purifoy J. M. Infectious potential of human immunodeficiency virus type 1 reverse transcriptase mutants with altered inhibitor sensitivity. Proc. Natl. Acad. Sci. USA 86:4803–4807; 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li Y.; Cheng Y.; Huang Y.; Tsai C.; Hsu Y.; Meng M. Identification and characterization of the Escherichia coli-expressed RNA-dependent RNA polymerase of bamboo mosaic virus. J. Virol. 72:10093–10099; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Müller R.; Poch O.; Delarue M.; Bishop D. H. L.; Bouloy M. Rift valley fever virus L segment: Correction of the sequence and possible functional role of newly identified regions conserved in RNA-dependent polymerases. J. Gen. Virol. 75:1345–1352; 1994. [DOI] [PubMed] [Google Scholar]
- 21. Okazaki K.; Tanabayashi K.; Takeuchi K.; Hishiyama M.; Okazaki K.; Yamada A. Molecular cloning and sequence analysis of the mumps virus gene encoding the L protein and the trailer sequence. Virology 188:926–930; 1992. [DOI] [PubMed] [Google Scholar]
- 22. Poch O.; Sauvaget I.; Delarue M.; Tordo N. Identification of four conserved motifs among RNA-dependent polymerase encoding elements. EMBO J. 8:3867–3874; 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Poch O.; Blumberg B. M.; Bouguerelet L.; Tordo N. Sequence comparison of five polymerases (L proteins) of unsegmented negative-strand RNA viruses; theoretical assignment of functional domains. J. Gen. Virol. 71:1153–1162; 1990. [DOI] [PubMed] [Google Scholar]
- 24. Parks G. D.; Ward C. D.; Lamb R. A. Molecular cloning of the NP and L genes of simian virus 5: Identification of highly conserved domains in paramyxovirus NP and L proteins. Virus Res. 22:259–279; 1992. [DOI] [PubMed] [Google Scholar]
- 25. Schnell M. J.; Conzelmann K. K. Polymerase activity of in vitro mutated rabies virus L protein. Virology 214:522–530; 1995. [DOI] [PubMed] [Google Scholar]
- 26. Schubert M.; Harmison G. G.; Meier E. Primary structure of the vesicular stomatitis virus polymerase (L) gene: Evidence for a high frequency of mutations. J. Virol. 51:505–514; 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shioda T.; Iwasaki K.; Shibuta H. Determination of the complete nucleotide sequence of the Sendai virus genome RNA and the predicted amino acid sequences of the F, HN and L proteins. Nucleic Acids Res. 14:1545–1563; 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sidhu M. S.; Menonna J. P.; Cook S. D.; Dowling P. C.; Udem S. A. Canine distemper virus L gene: Sequence and comparison with related viruses. Virology 193:50–65; 1993. [DOI] [PubMed] [Google Scholar]
- 29. Sleat D. E.; Banerjee A. K. Transcriptional activity and mutational analysis of recombinant vesicular stomatitis virus RNA polymerase. J. Virol. 67:1334–1339; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vazquez A. L.; Alonso J. M. M.; Parra F. Mutation analysis of the GDD sequence motif of a calcivirus RNA-dependent RNA polymerase. J. Virol. 74:3888–3891; 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yussoff K.; Millar N. S.; Chambers P.; Emmerson P. T. Nucleotide sequence analysis of the L gene of Newcastle disease virus: Homologies with Sendai and vesicular stomatitis viruses. Nucleic Acids Res. 15:3961–3976; 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]