

Abstract
The 39-kDa DNA polymerase β (polβ) is an essential enzyme in short-patch base excision repair pathway. A wild-type and a truncated forms of polβ proteins are expressed in primary colorectal and breast adenocarcinomas and in a primary culture of renal cell carcinoma. To test whether polβ has a contributory role in tumorigenicity of human tumor cell lines, we have undertaken a study to determine expression of polβ in colon, breast, and prostate tumor cell lines. Unlike primary colon tumor cells, three types of polβ mRNA have been identified in HCT 116, LoVo, and DLD1, colon tumor cell lines. A 111-bp-deleted polβ transcript was expressed in MCF7, a breast tumor cell line, but not in primary breast tumor cells. An expression of a smaller polβ transcript has been revealed in DU145, a prostate tumor cell line, whereas, a single base (T) deletion in mRNA at codon 191 was found in prostate cancer tissue. Interestingly, a wild-type polβ transcript was also expressed in all tumor cell lines similar to primary tumor cells. Furthermore, the cell extract of LoVo exhibited highest gap-filling synthesis function of polβ when the extract of DU145 showed lowest activity. MNNG, a DNA alkylating agent, enhanced the gap-filling synthesis activity in extracts of LoVo cell line. Furthermore, the cellular viability of LoVo and HCT116 cells is sensitive to MNNG when DU145 cells are resistant. These results demonstrate heterogeneity in polβ mRNA expression, which may be a risk factor related to tumorigenic activities of tumor cell lines.
Keywords: DNA polβ, Human tumor cell lines, mRNA, Protein, Gap-filling synthesis, DNA binding, MNNG
DNA polymerase β (polβ), one of the 16 eukaryotic polymerases, is recognized to be a major contributor in gap-filling synthesis of short-patch base excision repair (BER) pathway (28,29). It repairs 1–14 nucleotide gap in the damaged DNA strand. The long-patch BER pathway is largely dependent on polβ in cell extracts but can be reconstituted with polδ (15). The polp, having an intrinsic 5′-deoxyribose phosphate (dRP) lyase activity, can remove dRP from excised DNA strand at a preceding step of gap-filling synthesis (28). It may also function in DNA replication (25). DNA polβ is essential for embryogenesis and neurogenesis and has a role in meiosis, chromosomal stability, apoptosis, and in drug resistance mechanisms (1,12,18–20,24). The polβ protein consists of 335 amino acids with two distinct functional domains of an 8-kDa DNA binding domain and a 31-kDa catalytic domain. The catalytic domain is divided into fingers, palm, and thumb subdomains (28).
An 87-bp-deleted polβ mRNA is expressed in colorectal, breast, and lung cancers and in a primary culture of renal cell carcinoma (2,3,9,26,27). Furthermore, a T-deleted polβ mRNA was revealed in primary prostate adenocarcinomas, resulting in a frame shift (11,27). Altered mRNAs were also detected in bladder and gastric cancers (14,17). A wild-type (WT) polβ mRNA accompanied by the altered mRNA has been found in all tumors (2,3,9,11,14,17,26,27). Furthermore, both truncated and WT polβ proteins are expressed in colorectal and breast carcinomas and in a primary culture of renal cell carcinoma (2,4,9).
We hypothesized earlier that the truncated form of polβ might inhibit repair functions of the WT enzyme, which may have a role in carcinogenesis (26,27). The primary functions of polβ of 16.3 and HeLa cells (expressing the WT protein) were inhibited by nuclear extracts of 16.3ΔP and HeLapolβΔ cells, respectively, expressing the truncated polβ protein in terms of their gap-filling synthesis and DNA binding activities (4,5). These results indicate that the truncated polβ acts as a dominant negative mutant. More recently, evidence has been reported for an inhibitory role of a recombinant purified truncated polβ protein bound to XRCC1, a BER protein, on the functions of a purified WT polβ protein (6); it prevents polβ from forming DNA–protein complex and gap-filling synthesis. In this communication, we have chosen to study HCT116, LoVo, DLD1 (colon), MCF7 (breast), and DU145 (prostate), widely used human tumor cell lines. These cell lines express replication error phenotype (RER), an instability at microsatellites of the genome (7,16). A study has been initiated to evaluate for a possible contributory role of defective polβ in tumorigenicity of tumor cells, the expression and repair functions of polβ in HCT116, LoVo, DLD1, MCF7, and DU145 cell lines.
MATERIALS AND METHODS
Tumor Cell Lines
The tumor cell lines HCT116, LoVo, DLD1, MCF7, and DU145 were obtained from the American Type Culture Collection (Rockville, MD). The cell lines were grown following the instructions of the supplier.
PCR Amplification of polβ mRNA
Five micrograms of total RNA isolated from cells using RNAzol B reagent was reverse transcribed (2,27). For amplification, the first-strand cDNA was amplified using a pair of primers encompassing the entire coding sequence of human polβ (26). For reamplification, a second pair of primers flanking the codons for amino acids 149 to 297 was used (9). Isolation, purification, subcloning, and sequencing were done according to our routine protocols (2,3,9,26,27). The nucleotide sequences of the PCR products were reconfirmed.
Assay for Gap-Filling Synthesis
The gap-filling synthesis activity in nuclear extracts of all cell lines (50 μg protein) was determined using a 51-bp DNA template with a G:U mismatch at the 22 bp position (4,6). The 51-bp template was labeled at the 5′ end by [γ-32P]ATP (Amersham Pharmacia Biotech, Piscataway, NJ) and T4 polynucleotide kinase (Roche Molecular Biochemicals, Indianapolis, IN). The 5′-end-labeled oligonucleotide was annealed to a complementary strand with G opposite to U residue (6). The 51-bp product was separated by a 15% PAGE gel.
DNA Binding Activity
Gel mobility shift assay was used to evaluate the DNA binding affinity of polβ protein in nuclear extracts (15 μg protein) of tumor cell lines (4,6). The 32P-labeled 51-bp double-stranded oligonucleotide served as a substrate.
Treatment of Cells
Cells (2 × 106) were plated in 100-mm dishes and allowed to grow for 18 h. A stock solution of MNNG in DMSO diluted serially with medium was made prior to adding it to the cells. For the gap-filling synthesis study and the DNA binding study, cells were exposed to 15 μM of MNNG in serum-free medium for 60 min.
Effect of MNNG on Survival of Tumor Cells
To further investigate whether tumor cells are hypersensitive to chemicals, the degree of survival of cells treated with MNNG was also determined. Survival was measured by colony-forming assay (4,5). Two hundred cells were seeded in a 60-mm tissue culture dish and incubated overnight. A freshly made stock solution of MNNG (Aldrich, >99% pure) in DMSO was serially diluted with DMEM. MNNG (1–50 μM) was added to cells treated with MNNG for 60 min, followed by washing with PBS buffer to remove the chemical. Cells were allowed to grow for 5 days in fresh medium. Finally, cells were fixed in 70% methanol, stained with Giemsa, and scored for colonies of a minimum of 50 cells.
Expression of polβ Protein
Expression of the polβ enzyme was determined in extracts containing 50 μg protein by Western blot analysis (4,6) using a purified monoclonal anti-polβ antibody (Neo Markers, Inc., Fremont, CA).
DNA polβ Activity
DNA polβ activity in cell extracts of 50 μg protein was determined by a gel activity assay as described previously (4,5). The extracts were separated on a 12% SDS-PAGE containing 150 μg/ml of activated salmon sperm gapped DNA. The gapped DNA was prepared following Spanos and Hubscher (22). After electrophoresis, SDS was removed by washing the gel, proteins were renatured using 50 mM Tris-HCl buffer, pH 7.5, 1 mM EDTA, 5 mM β-mercaptoethanol (βME) for 2 h. The renatured gel was incubated for 18 h in 50 mM Tris-HCl, pH 7.5, 7 mM MgCl2, 3 mM βME, 12 mM each of TTP, dGTP, dATP, and 1 μCi/ml of [α-32P]dCTP. The gel was thoroughly washed with 5% TCA and 10% sodium pyrophosphate for 5 h and fixed in 40% methanol and 10% acetic acid for 1 h. The intensities of the bands on the dried gel were monitored by a PhosphorImager (Amersham Pharmacia Biotech, Piscataway, NJ).
RESULTS AND DISCUSSION
Expression of polβ mRNA in Tumor Cell Lines
To determine expression of polβ mRNA in colon tumor cells (LoVo, DLD1, and HCT116), we analyzed mRNA by RT-PCR. Two mRNA species (∼1 kb and ∼800 bp) were identified in these cells based on DNA size markers (Fig. 1). These products, isolated, and purified from the gel, were subcloned into the TA cloning system (27). To investigate a potential alteration in the sequences of two mRNA expressed in colon tumor cells, we sequenced the sub-cloned plasmids. The sequence of the 1-kb product matched that of the WT polβ mRNA (26). Three types of alterations in the polβ coding sequence were detected in these inserts: deletions of 87 and 140 bp (18 colonies), an insertion of 105 bp and deletion of 87 bp in 12 colonies, and a point mutation GGA→AGA was detected in 6 colonies (Fig. 2). All the altered sequences of the PCR products were confirmed by direct sequencing.
Figure 1.
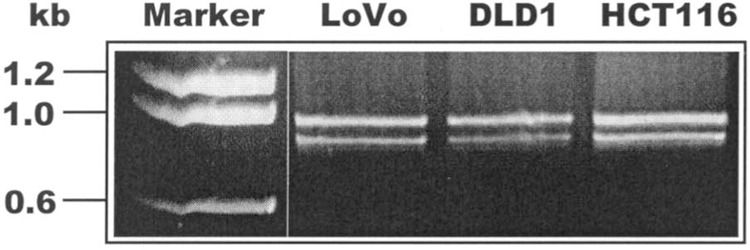
Expression of polβ mRNA in colon tumor cell lines. The PCR-amplified polβ cDNA from LoVo, DLD1, and HCT116 cells was separated by 1% agarose gel electrophoresis. Two polβ mRNA segments, of ∼1 kb and ∼800 bp, were identified in these cells. The molecular weight markers in kilobases are shown on the left side of the figure.
Figure 2.
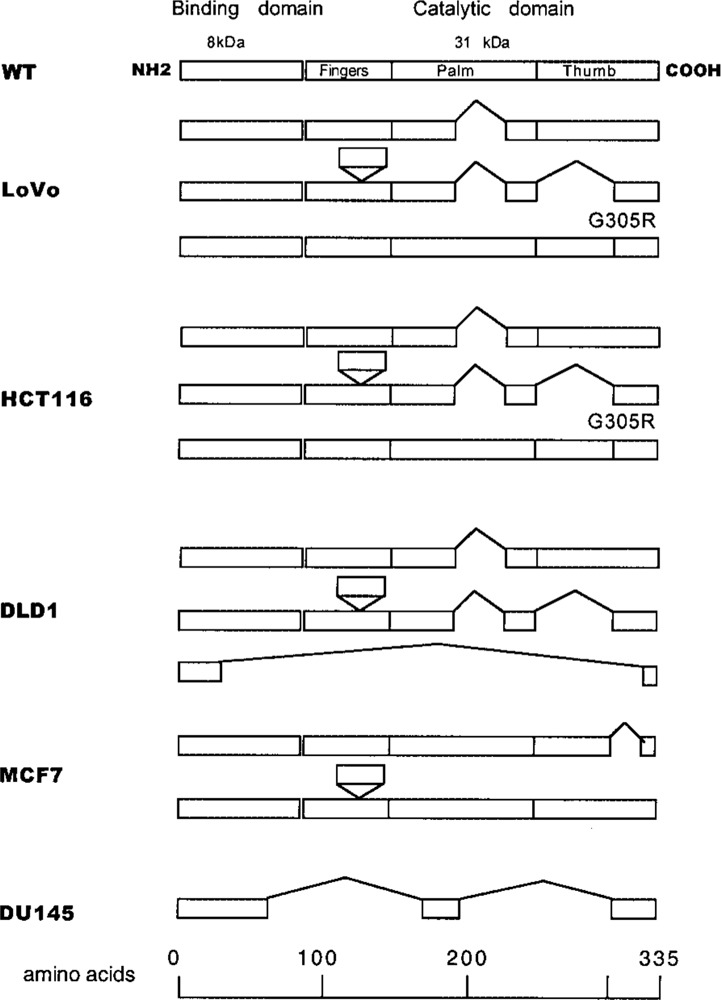
Schematic illustrations of expression of polβ mRNA (in primary structure) in tumor cell lines. The upward “V” indicates deletions and the downward “envelope” shows insertion in mRNA of tumor cell lines. The putative truncations in the polβ protein are localized in the catalytic domain of the enzyme.
An insertion of 105 bp in the 5′ region of the polβ mRNA has been reported in cell lines and primary lung tumors (3,9,21). To further determine a potential expression of this unique insertion in tumor cell lines, we amplified this specific region of RNA (10). As shown in schematic form in Figure 2, all tumor cell lines that we examined, except DU145, expressed a 105-bp insertion in polβ mRNA. This sequence presumably would reside between exons 6 and 7 (10). In addition, a 58-bp deletion in the cDNA sequence, which would correspond to exon 2 of the polβ gene, was identified only in the DLD1 colon tumor cell line. Due to this deletion, an in-frame shift may occur in the cDNA sequence of DLD1 cells. A presumptive polβ protein of 26 amino acids may be expressed in these cells. A segment of mRNA with a 58-bp deletion was expressed in colorectal tumors, normal mucosa, normal renal cells, renal cell carcinoma cells, and in cells from a patient with Werner syndrome (9,21,27). It is interesting to note that all tumor cells expressing variant forms of polβ mRNA also expressed the WT mRNA. A segment of mRNA with an 87-bp deletion (29-amino acid truncation) (Fig. 2) occurs in LoVo, HCT116, and DLD1 cells, resulting in a putative open reading frame of polβ protein consisting of 306 amino acids. Deletions of 87 and 140 bp (truncation of 76 amino acids) and insertion of 105 bp (addition of 35 amino acids) in an mRNA of colon tumor cells may encode a protein of 294 amino acids instead of 335 amino acids. These deletions are localized in the palm and thumb regions of the catalytic domain of the polβ protein (Fig. 2). Unlike these colon tumor cell lines, primary colorectal tumors predominantly express mRNA with an 87-bp deletion of polβ cDNA (26,27). Occurrences of deletions of 87 and 140 bp in the 3′ region of polβ cDNA in colon tumor cell lines are similar to those found in primary lung tumors (3). Furthermore, a point mutation (missense mutation) in the 3′ region of polβ (GGA→AGA at codon 305, conversion of glycine to arginine) was detected in LoVo and HCT116 cell lines. The number of amino acids of the resultant protein is expected to remain unchanged. The location of these alterations in mRNA sequence is summarized in Figure 2.
The sequences of polβ mRNA of MCF7 and DU145 cell lines were found to be different than sequences from colon tumor cell lines. A deletion of 111 bp (from 1008 to 1118 nt) of the polβ cDNA sequence encoding 37 amino acids occurs in breast tumor MCF7 cells (Fig. 2) with a putative open reading frame of 298 amino acids. Due to this deletion, exon 14 in its entirety is presumed to be deleted from the 3′ region of the polβ gene, residing in the thumb region of catalytic domain of the polβ protein. Interestingly, the primary breast tumor cells express polβ mRNA with an 87-bp deletion and a 36-kDa protein (2,27). In DU145 prostate tumor cells, deletions of 650 bp (at 304 to 662 nt and 735 to 1026 nt) of the polβ mRNA encoding a 217-amino acid truncation were identified, causing a frame shift with a predicted polβ protein of 118 amino acids (Fig. 2). This alteration may cause deletions of exons 4 to 9 and exons 11 to 13 of the polβ gene and would encompass a part of the binding domain and most of the catalytic domain of the polβ protein. In contrast to cells of the prostrate tumor cell line, a single base T deletion at codon 181 of the polβ has been identified in primary prostate adenocarcinoma, resulting in a putative smaller polypeptide of 198 amino acids (27). As shown in Figure 2, all the alterations in the polβ transcripts identified in five tumor cell lines reside in the catalytic domain of the polβ enzyme. An insertion of 105 bp also was detected in LoVo, HCT116, DLD1, and MCF7 cells (Fig. 2).
Gap-Filling Synthesis Activity of polβ in Tumor Cell Lines
It is conceivable that deletions and insertion in polβ mRNA of tumor cells may cause adverse effect on the functions of the enzyme. Thus, next we evaluated the gap-filling synthesis activities of polβ protein in these cell lines. Examining colon tumor cell lines, a comparatively higher activity in gap-filling synthesis activity was exhibited in LoVo cell extract (Fig. 3A, lane 3) than in extracts of DLD1 cells (lane 4) and HCT116 cells (lane 5) represented by the bar diagram. Comparing with all other tumor cell extracts, the gap-filling synthesis activity was identified at a lowest level in extract of DU145 (lane 6 and bar diagram). This activity appeared to be more in MCF7 extract (lane 7) than DU145 (lane 6). However, the activity appears to be significantly less in extract of MCF7 than in three colon tumor cell lines. Activities in extracts of HeLa and fresh normal mucosa used as controls are shown in lanes 1 and 2, respectively. Thus, results presented in this figure demonstrate a higher level of gap-filling synthesis activity is associated with LoVo cells when compared with other tumor cells that we examined. In contrast, the level of functional activity of polβ is lowest in prostate tumor cells, DU145. A markedly lowest level of activity in DU145 cells may be due to a large deletion (almost entirely) mapped to the finger, palm, and thumb regions of the catalytic domain of the polβ enzyme in comparison to other cells. It is highly likely that the enzyme would be significantly deficient in filling one base gap of the substrate, primary function of the polβ protein.
Figure 3.
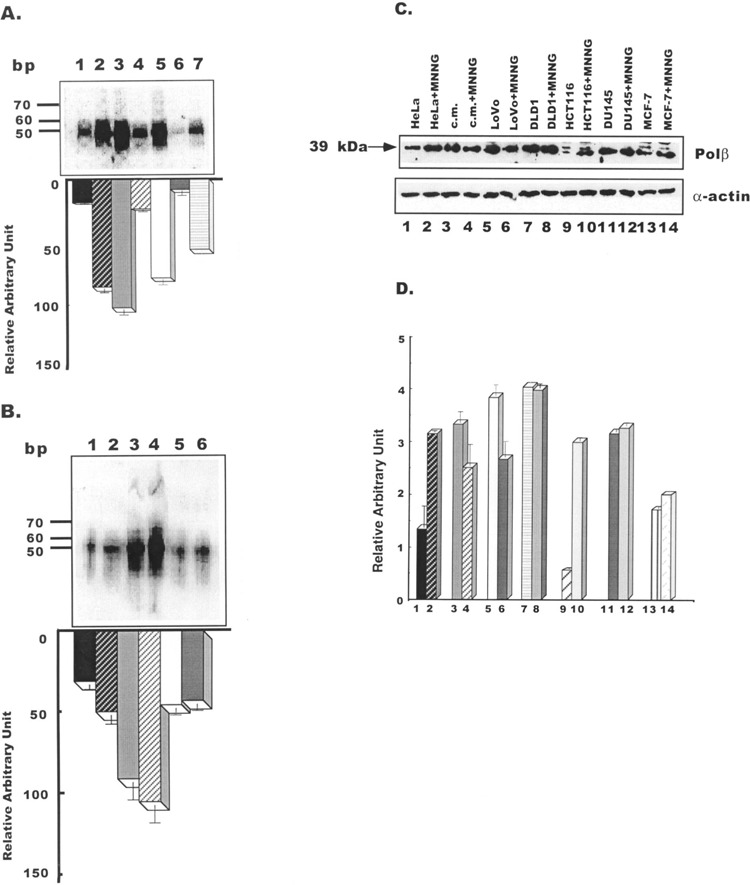
(A) Gap-filling synthesis activities in nuclear extracts of tumor cell lines. The 51-bp repaired products revealed in HeLa (lane 1), normal colon mucosa (lane 2), LoVo (lane 3), DLD1 (lane 4), HCT116 (lane 5), DU145 (lane 6), and MCF7 (lane 7) cells are shown. The relative ± SE activities in these cell lines are represented by a bar diagram. The molecular markers are in base pairs. (B) Effect of MNNG on the gap-filling synthesis function. Lane 1: gap-filling synthesis activity in HeLa cells; lane 2: gap-filling synthesis activity in HeLa cells treated with MNNG; lane 3: gap-filling synthesis activity in LoVo cells; lane 4: gap-filling synthesis activity in LoVo cells treated with MNNG; lane 5: gap-filling synthesis activity in DU145 cells; lane 6: gap-filling synthesis activity in DU145 cells treated with MNNG. The relative gap-filling synthesis activities ± SE in these cell lines are represented by a bar diagram. (C) Western blot analysis of polβ in HeLa, normal mucosa tissue, and tumor cell lines. Fifty micrograms of protein from cell extract was used for the assay. Proteins were separated by 12.5% SDS/PAGE. A comparison is given of each group’s response in the absence and presence of MNNG. Lanes 1 and 2: HeLa cell extracts; lanes 3 and 4: colon mucosa extracts; lanes 5 and 6: LoVo cell extracts; lanes 7 and 8: DLD1 cell extracts; lanes 9 and 10: HCT116 cell extracts; lanes 11 and 12: DU145 cell extracts; lanes 13 and 14: MCF7 cell extracts. Lower panel shows equal loading of protein in each lane by actin expression. (D) The relative effect of MNNG on polβ expression ± SE in cell lines is represented by a bar diagram.
Effect of MNNG on Gap-Filling Synthesis Activity of polβ
MNNG is a known DNA alkylating agent. Being a potent electrophilic species, it readily reacts with nucleophilic sites of DNA and forms adduct. Cells expressing a WT and a 29-amino acid truncated polβ protein are hypersensitive to MNNG (4,5). To determine whether MNNG has adverse effect on polβ in tumor cells, functions of the polβ enzyme were determined by gap-filling synthesis and DNA binding in colon and prostate tumor cells in the presence of MNNG. For this experiment we chose LoVo cells expressing highest gap-filling synthesis activity and DU145 cells exhibiting lowest activity (Fig. 3A). Quantitation by ImageQuant program (Molecular Dynamics) shows a low level of enhancement in gap-filling synthesis activity by MNNG identified in LoVo cell extract (Fig. 3B, lane 4 and bar diagram) in comparison to the activity of untreated cells (lane 3). It also appears from results that MNNG does not alter the gap-filling synthesis activity of DU145 cell extract (lane 6), when MNNG induces the functional activity of HeLa cells at a low level (lane 2). MNNG induces repair synthesis catalyzed by defective polβ in HeLapolβΔ, a stable cell line expressing both WT and 29-amino acid truncated polβ protein (5). Furthermore, repair synthesis by polβ has been induced by this agent in human fibroblast cells (13). The results from this experiment indicate that MNNG does not affect catalytic function of the polβ enzyme expressed in DU145 or LoVo cells.
Expression of polβ Protein in Tumor Cell Lines
Figure 3C shows the levels of expression of polβ in tumor cell lines in the absence or presence of MNNG. Levels of expression of polβ are similar in all cell lines. A relatively larger nonspecific band was found in HCT116 and MCF7 cell extracts (lanes 9 and 13). Among five tumor cell lines, the expression level appeared to be significantly induced by MNNG only in HCT116 cells (Fig. 3C and D, lane 10 and bar diagram). There was no appreciable change in polβ expression in DLD1, DU145, or MCF7 cells exposed to MNNG (Fig. 3C, lanes 8, 12, and 14). On the other hand, the expression level was upregulated by MNNG in HeLa cells used as a control (indicated in lane 2 and bar diagram). As it does in HeLa cells, MNNG also induces polβ expression in Chinese hamster ovarian cells (23). Interestingly, polβ is downregulated in LoVo cells exposed to MNNG (Fig. 3C and D, lane 6 and bar diagram). Equal protein loading in each lane was determined by expression of α-actin.
DNA Binding Activity of the polβ Protein in Tumor Cell Lines
To assess whether the defective polβ in tumor cell lines can form a protein–DNA complex with a gapped 51-bp substrate, DNA binding activity of polβ was examined by gel mobility shift assay. Two bands were visible on the gel (Fig. 4). Results showed that the level of the polβ–DNA complex is uniform in nuclear extracts of all cell lines, suggesting that the DNA binding activity of polβ remains unaltered. These results were expected, as the NH2-terminal of the 8-kDa binding domain of the polβ enzyme is unaffected in the colon, breast, and prostate (75% of the domain) tumor cell lines (Fig. 2). Consequently, the defective polβ retains its affinity for the damaged cognate DNA substrate. The slow-moving band represents the polβ–DNA complex. The more quickly moving band is nonspecific. This postulation is based on our previous observation in 16.3 cells expressing the wild-type polβ mRNA and protein (4). Using the same DNA substrate used in this study, two DNA– protein bands were visible. The polβ–DNA complex was blocked by cold consensus oligonucleotides and supershifted by anti-polβ antibody. Two similar bands also have been reported in HCT116, DLD1, and HCT115 cell lines (8).
Figure 4.
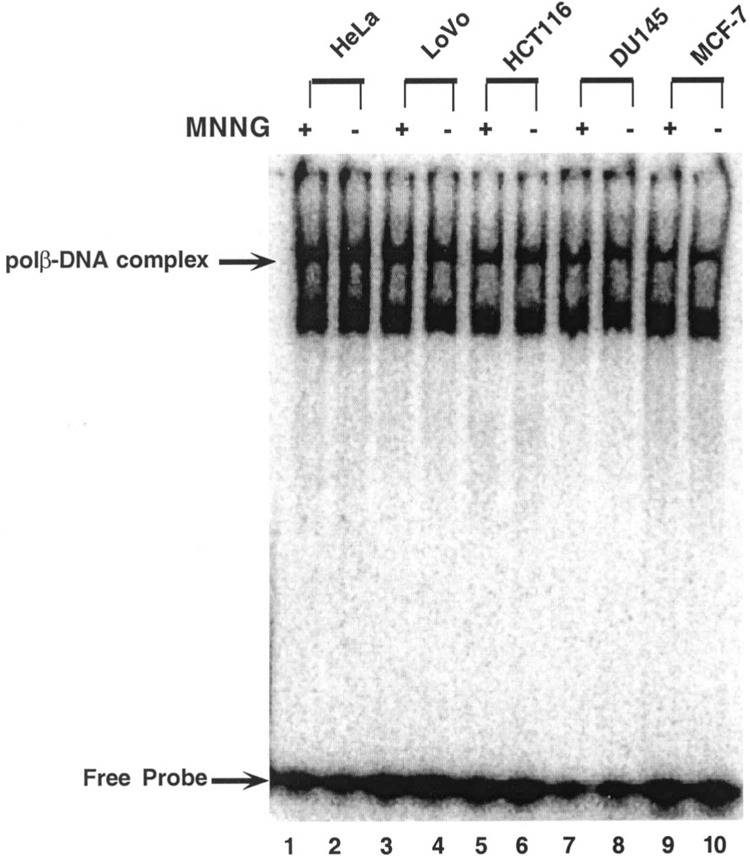
DNA binding activity of polβ protein in the presence (+) or absence (−) of MNNG in different tumor cell lines.
Effect of MNNG on DNA Binding
Next we determined DNA binding activities of polβ expressed in tumor cell lines are potentially modulated by MNNG. The polβ of LoVo, HCT116, DU145, and MCF7 cells forms a protein–DNA complex and binds to a 51-bp substrate at a comparable level (Fig. 4, lanes 4, 6, 8, and 10) in all cell lines. In the presence of MNNG, the levels of formation of protein–DNA complexes mediated by the polβ enzyme of these tumor cells remained unaltered (lanes 3, 5, 7, and 9). Two protein–DNA bands were visible in all nuclear extracts in the absence and presence of MNNG (Fig. 4). Thus, these results suggest DNA binding activities of polβ of tumor cell lines, especially LoVo, DU145, HCT116, or MCF7 cells, are resistant to MNNG.
The polβ Enzyme Activity of Tumor Cell Lines and Effect of MNNG on the Enzymatic Activity
The polβ activity of tumor cells was examined by activity gel assay. This assay determines not only the enzymatic activity but also indicates whether the protein is in active form. For this experiment, we also chose LoVo cells expressing highest gap-filling synthesis activity and DU145 cells exhibiting lowest activities. Represented in Figure 5, the polβ activity in extract of LoVo cells (lane 3) revealed approximately 60% higher activity than that expressed in HeLa (lane 1), indicated by the bar diagram. Using ImageQuant Program, it appears that enzyme activity in DU145 extract is approximately six times less than in LoVo cells (lane 5). The polβ activity in cell extract of this cell line is comparable with HeLa cells. These results suggest that the polβ enzyme is active in both colon and prostate tumor cells with differential level of activities. Examining a potential effect of MNNG on polβ activity, results shown in lanes 2, 4, and 6 indicate that MNNG does not affect polβ activity in HeLa, LoVo, or DU145 cell extract.
Figure 5.
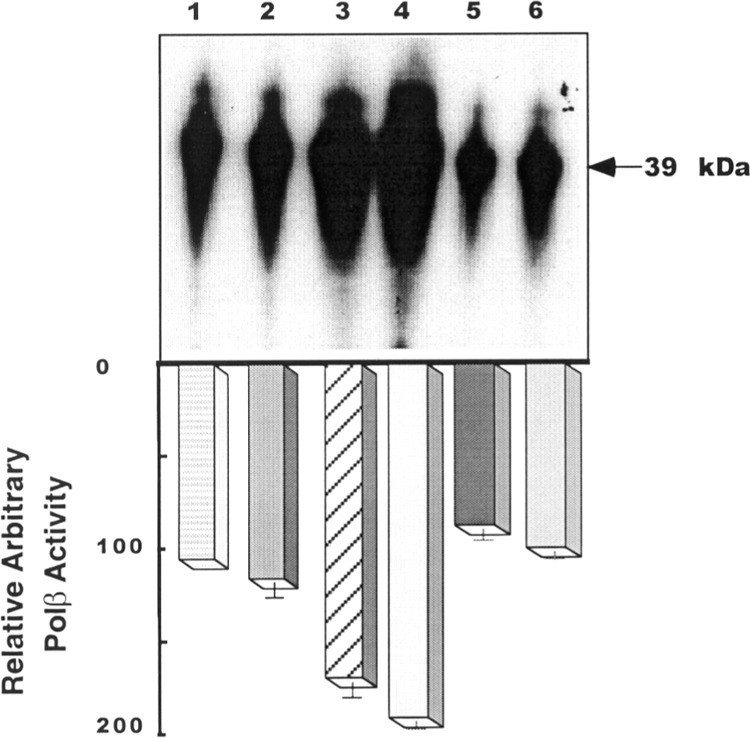
The polβ activities in HeLa (lane 1), LoVo (lane 3), and DU145 (lane 5) cells are shown. Lanes 2, 4, and 6 represent polβ activities in HeLa, LoVo, and DU145 cells treated with MNNG, respectively. The size of the polβ protein is 39 kDa. The bar diagram shows the relative polβ activity ± SE.
Sensitivity of Cells to MNNG
Sensitivity of tumor cells to MNNG was determined by survival. Survival of HeLa, LoVo, HCT116, and DU145 cells (Fig. 6) was progressively decreased with the increasing concentrations of MNNG. When concentration of MNNG was increased from 5 to 10 μM, the survival of colon tumor cells, LoVo and HCT116, was decreased from 50% to 30%. MNNG at 20 μM concentration was 100% lethal to LoVo cells. At the same concentration, 10% of HCT116 cells survived and formed colonies. However, 50 μM of MNNG was needed for 100% lethal effect. Results also indicate that, among three tumor cells used in this experiment, DU145 cells were most insensitive to MNNG. For example, at 20 μM MNNG, ∼65% of DU145 cells survived whereas only 0–10% LoVo and HCT116 cells survived.
Figure 6.
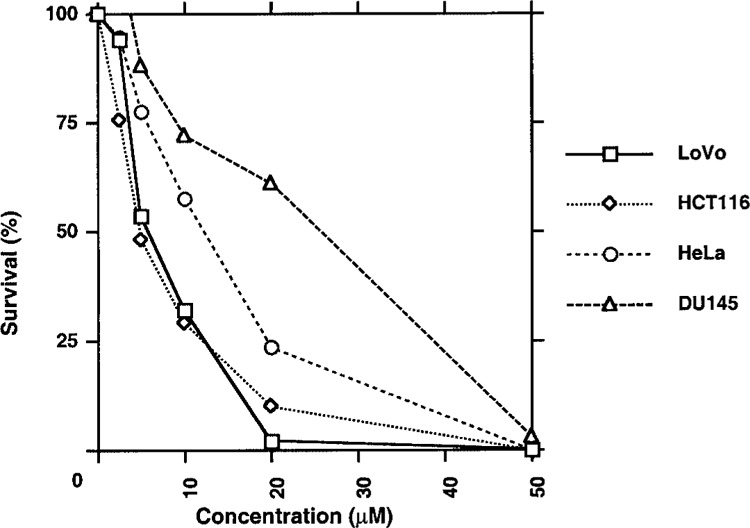
Survival assay of LoVo, HCT116, HeLa, and DU145 cells in response to MNNG. Cells were treated with 0–50 μM of MNNG for 1 h. The chemical was removed by washing with PBS. The cells were allowed to grow until they formed colonies. Surviving cells were counted by colony formation.
In summary, results presented in this article provide evidence for expression of aberrant transcripts of polβ in human LoVo, HCT116, DLD1, MCF7, and DU145 tumor cell lines. All cell lines that we examined except MCF7 also express defective mismatch repair genes (7). For example, hMSH2 and hMLH1 are defective in LoVo, HCT116, and DU145 cells. The hPMSH2 protein is also altered in DU145 cell line. Mutations in GTBP have been reported in DLD1 cells. At this time, expression of mismatch repair proteins in MCF7 cells remains unknown. Wild-type p53 mRNA is expressed in HCT116 cells when C→T conversion in p53 mRNA has been reported in DLD1 cells (30). Due to a large deletion in the polβ mRNA of DU145 cells, the gap-filling synthesis activity is markedly diminished in DU145 cells compared with LoVo, HCT116, DLD1, MCF7, or normal colon mucosa. A reduced level of catalytic function such as gap-filling synthesis of the polβ enzyme is also evident in MCF7 cells.
The polβ of tumor cell lines can efficiently bind to gapped DNA. As shown in Figure 2, the 8-kDa binding domain remains intact in all tumor cell lines except DU145. However, due to deletions in the catalytic domain of the enzyme and impaired gap-filling synthesis function, the polβ expressed in DU145 cells is unable to repair gapped DNA strand. The DNA binding activity of polβ appears to be unchanged in DU145 cells, although approximately 25% of this region has been deleted; perhaps the remaining 75% of the 8-kDa domain is sufficient to bind to DNA template. Thus, these results presented suggest that the defective polβ expressed in tumor cell lines that we tested may have a contributory role in tumorigenicity of these tumor cell lines.
ACKNOWLEDGMENTS
This study was supported by NIH grant 1R01CA83768 to S.B. We thank Christine Kassuba for editing the manuscript.
REFERENCES
- 1. Ali-Osman F.; Berger M. S.; Rairkar A.; Stein D. E. Enhanced repair of a cisplatin-damaged reporter chloramphenicol-O-acetyltransferase gene and altered activities of DNA polymerases α and β, and DNA ligase in cells of a human malignant glioma following in vivo cisplatin therapy. J. Cell. Biochem. 54:11–19; 1994. [DOI] [PubMed] [Google Scholar]
- 2. Bhattacharyya N.; Chen H.-C.; Grundfest-Broniatowski S.; Banerjee S. Alteration of hMSH2 and DNA polymerase β genes in breast carcinomas and fibroadenomas. Biochem. Biophys. Res. Commun. 259:429–435; 1999. [DOI] [PubMed] [Google Scholar]
- 3. Bhattacharyya N.; Chen H.-C.; Comhair S.; Erzurum S. C.; Banerjee S. Variant forms of DNA polymerase β in primary lung carcinomas. DNA Cell Biol. 18: 549–554; 1999. [DOI] [PubMed] [Google Scholar]
- 4. Bhattacharyya N.; Banerjee S. A variant DNA polymerase β acts as a dominant-negative mutant. Proc. Natl. Acad. Sci. USA 94:10324–10329; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bhattacharyya N.; Banerjee T.; Patel U.; Banerjee S. Impaired repair activity of a truncated DNA polymerase β protein. Life Sci. 69:271–280; 2001. [DOI] [PubMed] [Google Scholar]
- 6. Bhattacharyya N.; Banerjee S. A novel role of XRCC1 in the functions of a DNA polymerase β variant. Biochemistry 40:9005–9013; 2001. [DOI] [PubMed] [Google Scholar]
- 7. Boyer J. C.; Umar A.; Risinger J. I.; Lipford J. R.; Kane M.; Yin S.; Barrett J. C.; Kolodner R. D.; Kunkel T. A. Microsatellite instability, mismatch repair deficiency, and genetic defects in human cancer cell lines. Cancer Res. 55:6063–6070; 1995. [PubMed] [Google Scholar]
- 8. Branch P.; Hampson R.; Karran P. DNA mismatch binding defects, DNA damge tolerance, and mutator phenotypes in human colorectal carcinoma cell lines. Cancer Res. 55:2304–2309; 1995. [PubMed] [Google Scholar]
- 9. Chen H.-C.; Bhattacharyya N.; Wang L.; Recupero A. J.; Klein E. A.; Harter M. L.; Banerjee S. Defective DNA repair genes in a primary culture of human renal cell carcinoma. J. Cancer Res. Clin. Oncol. 126: 185–190; 2000. [DOI] [PubMed] [Google Scholar]
- 10. Chyan Y. J.; Strauss P. R.; Wood T. G.; Wilson S. H. Identification of novel mRNA isoforms for human DNA polymerase β. DNA Cell Biol. 15:653–659; 1996. [DOI] [PubMed] [Google Scholar]
- 11. Dobashi Y.; Shuin T.; Tsuruga H.; Uemura H.; Torigoe S.; Kubota Y. DNA polymerase P gene mutation in human prostate cancer. Cancer Res. 54:2827–2829; 1994. [PubMed] [Google Scholar]
- 12. Gu H.; Marth J. D.; Orban P. C.; Mossmann H.; Rajewsky K. Deletion of a DNA polymerase β gene segment in T cells using cell type-specific gene targeting. Science 265:103–106; 1994. [DOI] [PubMed] [Google Scholar]
- 13. Hammond R. A.; McClung J. K.; Miller M. R. Effect of DNA polymerase inhibitors on DNA repair in intact and permeable human fibroblasts: Evidence that DNA polymerases δ and β are involved in DNA repair synthesis induced by N-methyl-N′-nitro-N-nitrosoguanidine. Biochemistry 29:286–291; 1990. [DOI] [PubMed] [Google Scholar]
- 14. Iwanaga A.; Ouchida M.; Miyazaki K.; Hori K.; Mukai T. Functional mutation of DNA polymerase P found in human gastric cancer—inability of the base excision repair in vitro. Mutat. Res. 435:121–128; 1999. [DOI] [PubMed] [Google Scholar]
- 15. Klungland A.; Lindahl T. Second pathway for completion of human DNA base excision-repair: Reconstitution with purified proteins and requirement for DNase IV (FEN1). EMBO J. 16:3341–3348; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Levinson G.; Gutman G. A. Slipped-strand mispairing: A major mechanism for DNA sequence evolution. Mol. Biol. Evol. 4:203–221; 1987. [DOI] [PubMed] [Google Scholar]
- 17. Matsuzaki J.; Dobashi Y.; Miyamoto H.; Ikeda I.; Fujinami K.; Shuin T.; Kubota Y. DNA polymerase β gene mutations in human bladder cancer. Mol. Carcinogen. 15:38–43; 1996. [DOI] [PubMed] [Google Scholar]
- 18. Miscia S.; Di Baldassarre A.; Alba Rana R.; Di Pietro R.; Cataldi A. Engagement of DNA polymerases during apoptosis. Cell Prolif. 30:325–340; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ochs K.; Sobol R. W.; Wilson S. H.; Kaina B. Cells deficient in DNA polymerase β are hypersensitive to alkylating agent-induced apoptosis and chromosomal breakage. Cancer Res. 59:1544–1551; 1999. [PubMed] [Google Scholar]
- 20. Plug A. W.; Clairmont C. A.; Sapi E.; Ashley T.; Sweasy J. B. Evidence for a role for DNA polymerase β in mammalian meiosis. Proc. Natl. Acad. Sci. USA 94:1327–1331; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sadakane Y.; Maeda K.; Kuroda Y.; Hori K. Identification of mutations in DNA polymerase beta mRNAs from patients with Werner syndrome. Biochem. Biophys. Res. Commun. 200:219–225; 1994. [DOI] [PubMed] [Google Scholar]
- 22. Spanos A.; Hubscher U. Recovery of functional proteins in sodium dodecyl sulfate gels. Methods Enzymol. 91:263–277; 1983. [DOI] [PubMed] [Google Scholar]
- 23. Srivastava D. K.; Rawson T. Y.; Showalter S. D.; Wilson S. H. Phorbol ester abrogates up-regulation of DNA polymerase β by DNA-alkylating agents in Chinese hamster ovary cells. J. Biol. Chem. 270:16402–16408; 1995. [DOI] [PubMed] [Google Scholar]
- 24. Sugo N.; Aratani Y.; Nagashima Y.; Kubota Y.; Koyama H. Neonatal lethality with abnormal neurogenesis in mice deficient in DNA polymerase β. EMBO J. 19:1397–1404; 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sweasy J. B.; Loeb L. A. Detection and characterization of mammalian DNA polymerase β mutants by functional complementation in Escherichia coli . Proc. Natl. Acad. Sci. USA 90:4626–4630; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang L.; Patel U.; Ghosh L.; Banerjee S. DNA polymerase β mutations in human colorectal cancer. Cancer Res. 52:4824–4827; 1992. [PubMed] [Google Scholar]
- 27. Wang L.; Banerjee S. Mutations in DNA polymerase β occur in breast, prostate and colorectal tumors. Int. J. Oncol. 6:459–463; 1995. [PubMed] [Google Scholar]
- 28. Wilson S. H. Mammalian base excision repair and DNA polymerase beta. Mutat. Res. 407:203–215; 1998. [DOI] [PubMed] [Google Scholar]
- 29. Wood R. D.; Mitchell M.; Sgouros J.; Lindahl T. Human DNA repair genes. Science 291:1284–1289; 2001. [DOI] [PubMed] [Google Scholar]
- 30. Yoshikawa H.; Nagashima M.; Khan M. A.; McMenamin M. G.; Hagiwara K.; Harris C. C. Mutational analysis of p73 and p53 in human cancer cell lines. Oncogene 18:3415–3421; 1999. [DOI] [PubMed] [Google Scholar]