

Abstract
αNAC is a transcriptional coactivator known to interact with the N-terminal activation domain of the c-Jun transcription factor. In this article, we describe the identification of the c-Jun interaction domain within the αNAC protein. Deletion analysis of αNAC indicated that the c-Jun binding site was located in the middle part of the protein, between residues 89 and 129. The deletion of the C-terminal end of αNAC, including the c-Jun interacting domain, induced a nuclear translocation of the mutated coactivator. Despite its presence in the nucleus, this deletion mutant did not retain the capacity to coactivate an AP-1 response. These results demonstrate that the interaction between αNAC and c-Jun was necessary for the potentiation of the AP-1 transcriptional activity. These data are consistent with a mechanism by which αNAC acts as a coactivator for c-Jun-dependent transcription by interacting with the c-Jun N-terminal activation domain.
Keywords: αNAC, c-Jun, Coactivation, Transcriptional activation
THE Fos and Jun proteins, members of the AP-1 family of transcription factors, regulate a wide variety of cellular processes including cell proliferation, differentiation, apoptosis, and oncogenesis (6,9,20). Fos and Jun proteins function as dimeric transcription factors that bind AP-1 regulatory elements in the promoter and/or enhancer regions of numerous genes (8). Jun family members (c-Jun, JunB, JunD) can homodimerize, as well as form heterodimers among themselves or with partners of the Fos or ATF families (14,17). Fos proteins, on the other hand, are obligate heterodimers. The dimeric complexes bind DNA on AP-1 sites with high affinity and CRE elements with low affinity.
Both Fos and Jun proteins interact with coactivators to potentiate transcription. The proteins CBP (CREB binding protein) (3), JAB-1 (Jun-activation domain binding protein 1) (7), SRC-1 (steroid receptor coactivator-1) (18), TRBP/ASC-2 (activating signal cointegrator-2) (19), and αNAC (nascent poly-peptide associated complex and coactivator alpha) (21) were characterized as coactivators of AP-1-mediated transcription. Several coactivators characterized to date share functional properties. But some of them have unique functions such as histone ace-tyltransferase for CBP, p300 (4), and SRC-1, kinase for hTAFII250 (10), or specific DNA binding for dTAFII150 (24). αNAC also appears to exhibit specific DNA binding activity (27), although the physiological relevance of this property remains to be explored.
The αNAC gene was first identified as a modulator of translation (25) and purified as an heterodimer with βNAC/BTF3b, previously identified as a transcriptional factor in yeast (16) and higher eukaryotes (28). α and β NAC subunits were, moreover, shown to enter the nucleus in yeast (12), and αNAC enters the nucleus in eukaryotic cells (26). We characterized the αNAC subunit as a transcriptional coactivator of the chimeric Gal4-VP16 activator and of c-Jun homo-dimers in vivo (21,26). αNAC provides a protein bridge between these transcription factors and the basal transcriptional machinery by contacting the general transcription factor TBP (TATA binding protein) (26). The current model of the αNAC coactivator function is to promote an interaction between the transcription factors bound to DNA and the basal transcriptional machinery, therefore stabilizing the transcription factors on DNA and resulting in an enhanced transcription rate. To exert its coactivation function, αNAC enters the nucleus (12), and the sub-cellular localization of the protein appears regulated (26). We have previously identified the 1-89 c-Jun N-terminal domain as the region interacting with αNAC (21). We were then interested in characterizing the domain of αNAC that interacts with c-Jun.
In this report, we identified the c-Jun-interacting domain of αNAC within the mid-part of the molecule, more precisely between residues 89 and 129. Deletion of the C-terminal part of the molecule, including the c-Jun-interacting domain, induces the nuclear translocation of αNAC. This nuclear patterning presumably occurs by passive diffusion due to the small size of the mutant molecule. Despite its nuclear patterning, this deletion mutant molecule does not retain the coactivating ability of αNAC, showing that the interaction between αNAC and c-Jun is required for the potentiation of the c-Jun transcriptional response.
MATERIALS AND METHODS
Plasmids and Constructs (Subcloning Details and Vector Maps Available on Request)
Full-length αNAC (WT) and mutants cDNAs (see Fig. 1) were subcloned in-frame at their C-termini with the Intein-Chitin Binding Domain (ICBD) of the pTYB2 expression vector (NEB, Mississauga, ON) to give pTYB2-NAC plasmids. The Flag epitope was inserted into the pSI mammalian expression vector (Promega, Madison, WI) to give the pSI-Flag plasmid. The cDNAs encoding wild-type or m4 mutated αNAC were inserted in-frame into pSI-Flag to yield the pSI-NAC-Flag expression vectors. The c-Jun cDNA was cloned into the pCI mammalian expression vector (Promega) to yield pCI-c-Jun. The expression vector for the constitutively active form of ILK (pcDNA3.1/V5-His-ILK S343D) was kindly provided by Dr. S. Dedhar (23).
Figure 1.
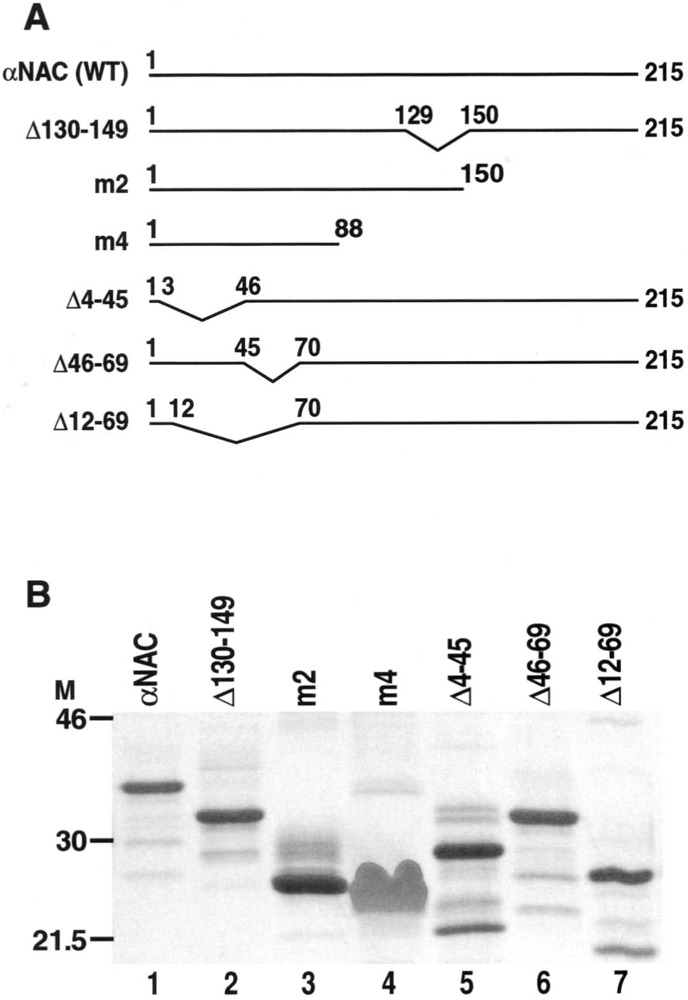
αNAC mutants. (A) Schematic representation of αNAC wild-type (WT) and deletion mutant proteins. (B) The WT, C-terminal, and N-terminal deletion mutants of αNAC were cloned into pTYB2 expression vector and the proteins produced and purified following the manufacturer’s procedure (NEB). An aliquot of each purified protein was run onto a 12% SDS-PAGE and revealed by Gel code blue staining. Lane 1: αNAC (WT), lane 2: Δ130–149, lane 3: m2 (Δ150–215), lane 4: m4 (Δ89–215), lane 5: Δ4–45, lane 6: Δ46–69, lane 7: Δ12–69.
Cell Culture and Transfection
COS-7 African green monkey kidney cells were maintained in low-glucose DMEM supplemented with 10% fetal bovine serum at 37°C in 5% CO2. All transient transfections were performed using 5 μl per μg of DNA of the GenePorter transfection reagent, according to the manufacturer’s procedure (Gene Therapy System, San Diego, CA).
C2C12 cells, a pluripotent mesenchymal cell line, were grown in high-glucose DMEM supplemented with 10% fetal bovine serum at 37°C in 5% CO2. Transient transfections were performed with Fugene 6 (3 μl/μg DNA) (Roche, Laval, QC).
Protein Production
The pTYB2 and pTYB2-NAC constructs were transformed into E. coli ER2566 cells. The ICBD from pTYB2, αNAC WT, and mutant proteins were produced and purified following the manufacturer’s procedure (NEB). Briefly, after IPTG induction, the bacterial cell extract was passed through a chitin column. Following extensive washes, the αNAC moiety was cleaved from the ICBD moiety in the presence of DTT and eluted from the column. The eluted proteins were concentrated using Centricon 10 columns (Millipore, Bedford, MA) and protein concentration was measured by Bradford assay (Bio-Rad, Hercules, CA). The purified proteins were run onto a 12% SDS-polyacrylamide gel and stained with the Gel code blue staining reagent (Pierce, Rockford, IL) to monitor yield and quality.
Affinity Chromatography
Twenty-four hours before transfection, the C2C12 cells were seeded at 5 × 105 cells per 100-mm plates in complete medium. Transient transfections were performed for 24 h in complete medium with 4 μg of pCI-c-Jun expression vector in the presence of 12 μl of Fugene 6 (Roche). C2C12-transfected cells were lysed by sonication in 0.2 M NaCl column buffer (20 mM Tris-Cl, pH 8, 200 mM NaCl, 0.1 mM EDTA, 0.1% Triton X-100) in the presence of 1 μg/ml anti-proteases (leupeptin, pepstatin, aprotinin) and 1 mM PMSF. Total C2C12 cell extracts were precleared onto a chitin column (NEB) to reduce unspecific binding. Bacterial extracts, obtained from E. coli cells transformed with pTYB2 and pTYB2-NAC plasmids, and containing an equivalent quantity of proteins (data not shown), were immobilized on the chitin columns. After washes, 1 ml of precleared total extracts from pCI-c-Jun-transfected C2C12 cells was loaded on the αNAC-Chitin affinity columns and incubated overnight at 4°C. After extensive washes with column buffer, the bound proteins were eluted with elution buffer (20 mM Tris-Cl, pH 8, 700 mM NaCl, 0.1 mM EDTA). The eluted proteins were loaded onto a 10% SDS-polyacrylamide gel and transferred to nitrocellulose membranes (Amersham-Pharmacia). The specific signal was detected with the anti-phospho-c-Jun (S63) antibody (NEB) and revealed with the Supersignal West femto maximum sensitivity substrate kit (Pierce).
Immunocytochemistry
The COS-7 cells were plated at 1.2 × 105 cells/35-mm plate, on gelatin-coated coverslips, and transiently transfected with 0.4 μg of pSI-NAC-Flag, or mutant, and 1.6 μg of the pBluescript plasmid (Stratagene, La Jolla, CA). Twenty-four hours post-transfection, the cells were fixed in 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and the endogenous peroxidase activity was quenched with 1% H2O2. Following blocking with 1% Blocking Reagent (Roche) supplemented with 0.2% Tween-20, the cells were incubated with the anti-Flag M2 antibody (Sigma, Oakville, ON), then with biotinylated secondary anti-mouse IgG antibody (Vector Lab. Inc., Burlingame, CA). After washes, the cells were immersed in the Avidin Biotin peroxidase reagent (Vector Lab. Inc.). The peroxidase staining was revealed with DAB reagent and visualized on a Leica DM-R microscope at 200×.
Luciferase Assays
The COS-7 cells were transiently cotransfected with 500 ng of wild-type αNAC or m4 mutant, 500 ng of a constitutively active form of the Integrin-Linked Kinase (pcDNA3.1/V5-His-ILK S343D) (an activator of the AP-1 pathway) (23), and plasmids from the PathDetect c-Jun trans-reporting system (Stratagene). The PathDetect plasmids were as followed: 500 ng of pFR-Luc reporter plasmid containing 5xGal4 binding sites fused to the luciferase gene and 50 ng of pFA2-cJun expression vector (c-Jun-GAL4 DNA binding domain fusion, Gal4-c-Jun) or pFC2-dbd as a negative control (Gal4-DNA binding domain). Corresponding empty vectors served as controls. Transient transfections were carried out in COS-7 cells for 48 h in 0.5% FBS containing DMEM. Subsequently, the cells were lysed 20 min in the reporter gene assay lysis buffer (Roche). Twenty microliters of cell lysate was used for single luciferase reporter assays following the manufacturer’s procedure (Promega) and analyzed with a Monolight 2010 luminometer (Analytical Luminescence Laboratory, San Diego, CA). The presence of the transfected αNAC-Flag proteins was controlled by immunoprecipitation with anti-Flag M2 beads (Sigma) followed by immunoblotting with the anti-NAC antibody (27). The Gal4-c-Jun proteins were detected by immunoblotting with the anti-Gal4 antibody, recognizing the DNA binding domain of Gal4 (Santa Cruz Biotechnology Inc., Santa Cruz, CA).
RESULTS
αNAC is a small protein of 215 amino acids, with an apparent molecular weight of 37 kDa. We have engineered N-terminal and C-terminal deletion mutant cDNAs by PCR (primer sequences available upon request) and introduced them in bacterial and eukaryotic expression vectors. Figure 1A shows a schematic representation of αNAC (WT) and the deletion mutant proteins that were used in this study. One microgram of each recombinant protein (wild-type αNAC and mutants) was loaded onto a 12% denaturing polyacrylamide gel and the purified proteins were revealed with the Gel code blue staining reagent. These data showed that all the proteins were produced. They were of good quality and purity, and had the predicted sizes (Fig. 1B). Mutant m4 always exhibited a diffuse pattern when revealed by staining of the SDS-PAGE gels (Fig. 1B, lane 4).
To localize the domain of αNAC interacting with c-Jun, we used recombinant αNAC proteins for affinity chromatography. αNAC WT and mutants, as well as ICBD moiety as a negative control, were immobilized onto chitin beads to prepare affinity columns. C2C12 cells were transiently transfected with c-Jun expression vector before retrieving the cellular extracts. The c-Jun-containing cell extracts were precleared on chitin columns, then loaded on the αNAC affinity columns. After overnight incubation, the proteins interacting with αNAC were eluted with high salt buffer and controlled for the presence of phosphorylated c-Jun by immunoblotting with the anti-phospho-c-Jun antibody.
The immunoblotting revealed that αNAC interacted with the activated, phosphorylated form of c-Jun (Fig. 2, lane 8). Deleting residues 4 to 45 (Δ4–45), 46 to 69 (Δ46–69), or 12 to 69 (Δ12–69) did not affect the interaction of αNAC with the phosphorylated form of c-Jun (Fig. 2, lanes 2, 3, and 4, respectively). We observed differences in the interaction between c-Jun and αNAC when the C-terminal region of the protein was deleted. While deleting residues 130 to 149 (Δ130–149) or 150 to 215 (m2) did not affect interaction with c-Jun (Fig. 2, lanes 5 and 6), a further deletion to amino acid 88 (deletion 89 to 215, mutant m4) almost completely abolished interaction with phospho-c-Jun (Fig. 2, lane 7). Because deleting up to residue 130 was neutral (Δ130–149 and m2, Fig. 2, lanes 5 and 6) but further deleting to residue 88 abolished interaction (m4, Fig. 2, lane 7), we concluded that residus 89 to 129 of the αNAC protein are essential for its interaction with the c-Jun activator.
Figure 2.
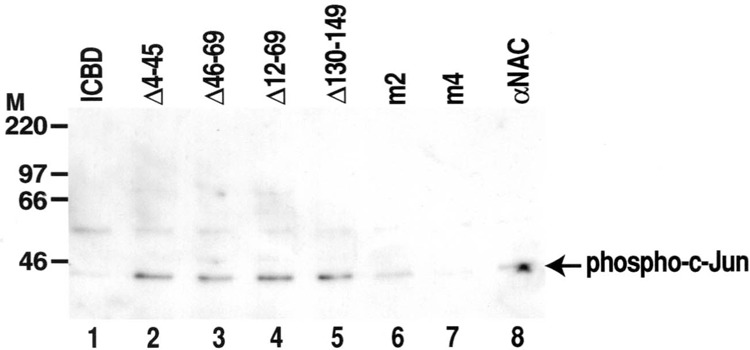
Identification of the c-Jun-interacting domain. αNAC fusion proteins (WT or mutants), or ICBD as a negative control, were immobilized onto chitin beads columns. Total extracts of C2C12 cells transfected with a c-Jun expression vector were loaded on the affinity columns. The proteins interacting with αNAC were eluted with high salt concentration buffer. Immunoblotting of the eluates with the anti-phospho-c-Jun antibody revealed a specific interaction between αNAC proteins and the active, phosphorylated form of c-Jun. A specific c-Jun signal was detected with αNAC WT (lane 8), C-terminal and N-terminal deletion mutant proteins (lanes 2 to 6). No significant binding was observed with the control intein chitin binding domain (ICBD, lane 1). A highly significant decrease was observed with the m4 mutant (lane 7).
We further checked whether the loss of interaction between αNAC and c-Jun could interfere with the subcellular localization of αNAC. For that purpose, we performed immunocytochemistry in COS-7 cells transfected with pSI-NAC-Flag WT and the m4 construct (Fig. 3). Specific signals were detected with the anti-Flag M2 antibody. As shown in Figure 3a, background signal from cells transfected with the inert vector was barely detectable. The subcellular localization of WT αNAC was cytosolic and mostly perinuclear (Fig. 3b), whereas the m4 mutant had an exclusively nuclear staining pattern (Fig. 3c). The m4 mutant, despite its loss of c-Jun interaction, can nevertheless enter the nucleus of cells.
Figure 3.
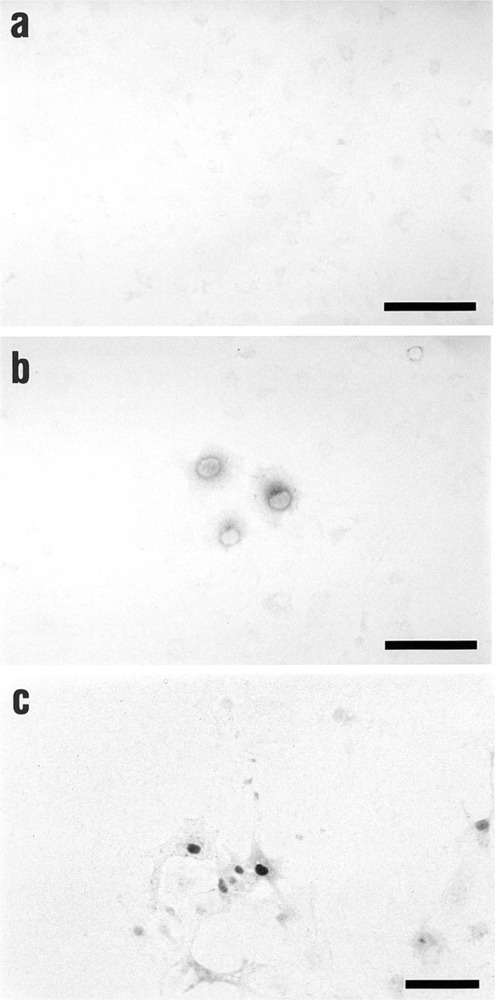
Nuclear localization of the m4 mutant. Immunocytochemistry was performed on COS-7 cells transiently transfected with Flag-tagged αNAC WT or m4 mutant expression vectors, or inert vector. The detection was performed using the anti-Flag M2 antibody and revealed by DAB staining. The cells transfected with the inert vector presented very low background (a). The WT αNAC protein had a cytosolic and perinuclear pattern (b), whereas the m4 mutant localized to the nucleus of cells (c). Bars = 100 μm.
αNAC was previously characterized as a coactivator of c-Jun homodimers (21). We showed above that the m4 mutant was not able to directly interact with c-Jun, but could still enter the nucleus. To study the coactivation potency of the m4 deletion mutant, we performed transient transfections in COS-7 cells. The c-Jun expression vector contained the N-terminal part of c-Jun fused to the Gal4-DNA binding domain, while the luciferase reporter gene was under the control of Gal4 binding sites. The 1–89 N-terminal domain of c-Jun was previously shown to be sufficient for transactivation (15) and for interaction with αNAC (21). The c-Jun pathway was activated by overexpression of ILK (integrin linked kinase) (23). Luciferase activity was measured after 2 days of culture in 0.5% serum to avoid the activation of the endogenous AP-1 cascade or other pathways. The luciferase signal detected was dependent upon the transfected expression vectors. In this experimental system, αNAC moderately but reproducibly potentiated the Gal4-c-Jun response (Fig. 4A). The m4 mutant was not able to potentiate the c-Jun response, demonstrating the importance of a direct interaction between the transcription factor and its coactivator. Immunoblotting with anti-Gal4 and anti-NAC antibodies demonstrated that the lack of response with the m4 mutant was not due to an absence of one of the two partners Gal4-c-Jun and m4, respectively (Fig. 4B).
Figure 4.
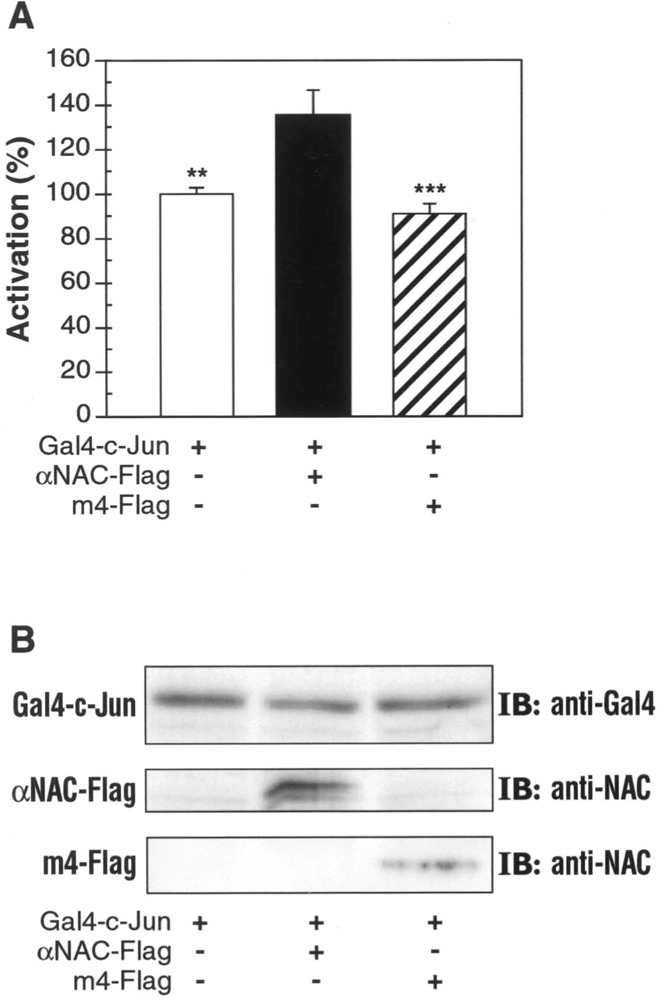
Decreased coactivation activity of the m4 mutant. (A) COS-7 cells were transiently transfected with expression vectors for αNAC WT or m4 mutant, Gal4-c-Jun and ILK, and a luciferase reporter gene. After 48 h in 0.5% serum, single luciferase assays were performed. WT αNAC protein coactivates c-Jun transcriptional activity (black column) compared with activated c-Jun transcriptional level (white column). Deletion of the c-Jun interacting domain (m4 mutant) blocks the coactivation capacity of αNAC (striped column). The results shown (mean ± SEM) are representative of three independent experiments performed in triplicate. The results were statistically analyzed using ANOVA and the Tukey posttest (**p < 0.01, ***p < 0.001). (B) Total cell extracts obtained from tranfected COS-7 cells were probed with the anti-Gal4 antibody to detect the expression of Gal4-c-Jun (upper panel). After immunoprecipitation with anti-Flag M2 beads, the expression of αNAC WT or m4 mutant were revealed with the anti-NAC antibody (middle and lower panels, respectively).
DISCUSSION
It has been established that αNAC is a transcriptional coactivator (21,26) that interacts with the c-Jun transcription factor, a member of the AP-1 family (21). But the structural motifs mediating the αNAC interaction remained to be identified. In this study, we have identified the amino acid residues that are essential for allowing αNAC to bind c-Jun, and addressed the role of the c-Jun–αNAC interaction in the function of αNAC as a transcriptional coactivator.
Using internal, N-terminal, and C-terminal deletion mutants, the c-Jun interaction domain was localized to a short region of 40 amino acids corresponding to the middle part of the αNAC protein. This region was necessary for the interaction with the activated form of c-Jun. c-Jun is activated by phosphorylation in its N-terminal domain by the Jun N-terminal kinases (JNKs) on serines 63 and 73. This phosphorylation enhances c-Jun transactivation properties by recruiting coactivator proteins such as CBP (1,22).
c-Jun coactivators, such as CBP and JAB-1, also contact the c-Jun N-terminal activation domain (1,3,7). CBP is a coactivator of c-Jun that stimulates the activity of c-Jun in vivo, and a reduced binding between CBP and c-Jun abolishes the increase of transcription in vivo (2). It is interesting to note the similarities between the mechanism of action of these coactivators and that of αNAC, despite the absence of sequence similarity. Both JAB-1 and αNAC proteins interact with the N-terminal part of c-Jun and can bind the phosphorylated as well as the unphosphorylated forms of c-Jun [(7,21); our data]. The CBP–c-Jun interaction, however, is observed only with the phosphorylated form of the transcription factor (2). The αNAC coactivator interacts with the c-Jun N-terminal activation domain and therefore co-activates its response. As for CBP, the loss of binding between c-Jun and αNAC was detrimental for the further augmentation of AP-1 response. These data are consistent with a mechanism by which αNAC co-activates c-Jun activity by interacting with the c-Jun activation domain.
We also observed that deletion of the c-Jun interaction domain resulted in nuclear translocation of the αNAC mutant protein. This nuclear patterning could result from the loss of interaction between αNAC and c-Jun, although this interpretation appears unlikely. We hypothesize that the nuclear patterning of the m4 mutant was due to the large deletion within the molecule. The resulting mutant protein was short (104 amino acids including Flag tag epitope) and small enough to passively diffuse from the cytosol to the nucleus. Indeed, the cutoff for efficient passive diffusion into the nucleus is 20–30 kDa (13).
Despite the apparent passive diffusion of the m4 deletion mutant, we do not believe that the wild-type αNAC protein could localize to the nucleus by the same mechanism. Structure–function analysis of the αNAC molecule did not reveal the presence of a functional nuclear localization signal (data not shown). We favor the model that the interaction of αNAC with c-Jun is the signal for a controlled nuclear entry of the coactivator, as c-Jun nuclear import itself is regulated (5,11).
In conclusion, we identified the c-Jun interaction domain within the αNAC coactivator. We demonstrated that nuclear localization of αNAC and coactivation can be functionally separated from one another; however, interaction between c-Jun and its coactivator is fundamental for the potentiation of the c-Jun transcriptional response in cells.
ACKNOWLEDGMENTS
Dr. Shoukat Dedhar (Vancouver, BC) kindly provided the pcDNA3.1/V5-His-ILK S343D plasmid. We thank Guylaine Bédard for preparing the figures. This work was supported by a grant from the Shriners of North America. R. St-Arnaud is a Chercheur-Boursier from the Fonds de la Recherche en Santé du Québec.
REFERENCES
- 1. Arias J.; Alberts A. S.; Brindle P.; Claret F. X.; Smeal T.; Karin M.; Feramisco J.; Montminy M. Activation of cAMP and mitogen responsive genes relies on a common nuclear factor. Nature 370:226–229; 1994. [DOI] [PubMed] [Google Scholar]
- 2. Bannister A. J.; Oehler T.; Wilhem D.; Angel P.; Kouzarides T. Stimulation of c-Jun activity by CBP: c-Jun residues Ser63/73 are required for CBP induced stimulation in vivo and CBP binding in vitro. Oncogene 11:2509–2514; 1995. [PubMed] [Google Scholar]
- 3. Bannister A. J.; Kouzarides T. CBP-induced stimulation of c-Fos activity is abrogated by E1A. EMBO J. 14:4758–4762; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brownell J. E.; Zhou J.; Ranalli T.; Kobayashi R.; Edmondson D. G.; Roth S. Y.; Allis C. Tetrahymena histone acetyltransferase A: A homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell 84:843–851; 1996. [DOI] [PubMed] [Google Scholar]
- 5. Chida K.; Vogt P. K. Nuclear translocation of viral Jun but not of cellular Jun is cell cycle dependent. Proc. Natl. Acad. Sci. USA 89:4290–4294; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chinenov Y.; Kerppola T. K. Close encounters of many kinds: Fos–Jun interactions that mediate transcription regulatory specificity. Oncogene 20:2438–2452; 2001. [DOI] [PubMed] [Google Scholar]
- 7. Claret F. X.; Hibi M.; Dhut S.; Toda T.; Karin M. A new group of conserved coactivators that increase the specificity of AP-1 transcription factors. Nature 383:453–457; 1996. [DOI] [PubMed] [Google Scholar]
- 8. Curran T.; Franza B. R. Fos and Jun: The AP-1 connection. Cell 55:395–397; 1988. [DOI] [PubMed] [Google Scholar]
- 9. Curran T.; Teich N. M. Candidate product of the FBJ murine osteosarcoma virus oncogene: Characterization of a 55,000-dalton phosphoprotein. J. Virol. 42:114–122; 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dikstein R.; Ruppert S.; Tjian R. TAFII250 is a bipartite protein kinase that phosphorylates the basal transcription factor RAP74. Cell 84:781–790; 1996. [DOI] [PubMed] [Google Scholar]
- 11. Forwood J. K.; Lam M. H.; Jans D. A. Nuclear import of CREB and AP-1 transcription factors requires importin-beta 1 and Ran but is independent of importin-alpha. Biochemistry 40:5208–5217; 2001. [DOI] [PubMed] [Google Scholar]
- 12. Franke J.; Reimann B.; Hartmann E.; Köhler M.; Wiedmann B. Evidence for a nuclear passage of nascent polypeptide-associated complex subunits in yeast. J. Cell Sci. 114:2641–2648; 2001. [DOI] [PubMed] [Google Scholar]
- 13. Görlich D.; Kutay U. Transport between the cell nucleus and the cytoplasm. Annu. Rev. Cell Dev. Biol. 15:607–660; 1999. [DOI] [PubMed] [Google Scholar]
- 14. Hai T.; Curran T. Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proc. Natl. Acad. Sci. USA 88:3720–3724; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hirai S.; Bourachot B.; Yaniv M. Both Jun and Fos contribute to transactivation by the heterodimer. Oncogene 5:39–46; 1990. [PubMed] [Google Scholar]
- 16. Hu G.-Z.; Ronne H. Yeast BTF3 protein is encoded by duplicated genes and inhibits the expression of some genes in vivo. Nucleic Acids Res. 22:2740–2743; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kerppola T. K.; Curran T. Selective DNA bending by a variety of bZIP proteins. Mol. Cell. Biol. 13:5479–5489; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee S. K.; Kim H. J.; Na S. Y.; Kim T. S.; Choi H. S.; Im S. Y.; Lee J. W. Steroid receptor coactivator-1 coactivates activating protein-1-mediated transactivations through interaction with the c-Jun and c-Fos subunits. J. Biol. Chem. 273:16651–16654; 1998. [DOI] [PubMed] [Google Scholar]
- 19. Lee S. K.; Na S. Y.; Jung S. Y.; Choi J. E.; Jhun B. H.; Cheong J.; Meltzer P. S.; Lee Y. C.; Lee J. W. Activating protein-1, nuclear factor-kappaB, and serum response factor as novel target molecules of the cancer-amplified transcription coactivator ASC-2. Mol. Endocrinol. 14:915–925; 2000. [DOI] [PubMed] [Google Scholar]
- 20. Maki Y.; Bos T. J.; Davis C.; Starbuck M.; Vogt P. K. Avian sarcoma virus 17 carries the jun oncogene. Proc. Natl. Acad. Sci. USA 84:2848–2852; 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Moreau A.; Yotov W. V.; Glorieux F. H.; St-Arnaud R. Bone-specific expression of the alpha chain of the nascent polypeptide-associated complex, a coactivator potentiating c-Jun-mediated transcription. Mol. Cell. Biol. 18:1312–1321; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Smeal T.; Binetruy B.; Mercola D. A.; Birrer M.; Karin M. Oncogenic and transcriptional cooperation with Ha-Ras requires phosphorylation of c-Jun on serines 63 and 73. Nature 354:494–496; 1991. [DOI] [PubMed] [Google Scholar]
- 23. Troussard A. A.; Tan C.; Yoganathan T. N.; Dedhar S. Cell–extracellular matrix interactions stimulate the AP-1 transcription factor in an integrin-linked kinase- and glycogen synthase kinase 3-dependent manner. Mol. Cell. Biol. 19:7420–7427; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Verrijzer C. P.; Yokomori K.; Chen J. L.; Tjian R. Drosophila TAFII150: Similarity to yeast gene TSM-1 and specific binding to core promoter DNA. Science 264:933–941; 1994. [DOI] [PubMed] [Google Scholar]
- 25. Wiedmann B.; Sakai H.; Davis P. A.; Wiedmann M. A protein complex required for signal-sequence-specific sorting and translocation. Nature 370:434–440; 1994. [DOI] [PubMed] [Google Scholar]
- 26. Yotov W. V.; Moreau A.; St-Arnaud R. The alpha chain of the nascent polypeptide-associated complex functions as a transcriptional coactivator. Mol. Cell. Biol. 18:1303–1311; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yotov W. V.; St-Arnaud R. Differential splicing-in of a proline-rich exon converts alphαNAC into a muscle-specific transcription factor. Genes Dev. 10:1763–1772; 1996. [DOI] [PubMed] [Google Scholar]
- 28. Zheng X. M.; Black D.; Chambon P.; Egly J. M. Sequencing and expression of complementary DNA for the general transcription factor BTF3. Nature 344:556–559; 1990. [DOI] [PubMed] [Google Scholar]