

Abstract
In the USA, rare diseases are defined as those affecting fewer than 200,000 patients at any given time. Patients with rare diseases are frequently misdiagnosed or undiagnosed which may due to the lack of knowledge and experience of care providers. We hypothesize that patients’ phenotypic information available in electronic medical records (EMR) can be leveraged to accelerate disease diagnosis based on the intuition that providers need to document associated phenotypic information to support the diagnosis decision, especially for rare diseases. In this study, we proposed a collaborative filtering system enriched with natural language processing and semantic techniques to assist rare disease diagnosis based on phenotypic characterization. Specifically, we leveraged four similarity measurements with two neighborhood algorithms on 2010-2015 Mayo Clinic unstructured large patient cohort and evaluated different approaches. Preliminary results demonstrated that the use of collaborative filtering with phenotypic information is able to stratify patients with relatively similar rare diseases.
Introduction
In the USA, rare diseases are defined as those affecting fewer than 200,000 patients at any given time1. According to research conducted by Rare Disease Day2 and the National Organization for Rare Disorders (NORD)3, to date, there are over 7,000 known rare diseases inflicting 30 million Americans (nearly 1 in 10), more than half of whom are children2. However, due to the lack of scientific knowledge and clinical experience for rare diseases, most patients with rare diseases are frequently misdiagnosed or undiagnosed. In addition, only 5% of those diseases have corresponding treatment plans2. Therefore, there is an urgent need to accelerate the diagnosis of rare diseases as well as explore the science behind them.
Almost 80% of rare diseases are genetic2 and the first step towards rare disease research and diagnosis is to identify patients with similar phenotypes, where a phenotype refers to a clinical observable sign or symptom. Multiple studies have reported on studying rare disease phenotyping. To facilitate the discovery of genotype-phenotype association, the Human Phenotype Ontology (HPO)4 was developed to capture human phenotype information and is one of the most representative efforts that conducts phenotype-oriented analysis for rare disease differential diagnosis. The current version of HPO contains more than 116,000 annotations for over 7,000 rare diseases. To understand genotypes for rare diseases, Phen-Gen5 provides a method to combine phenotypes and patients’ sequencing data with domain knowledge in order to locate genotypes for rare disorders. Other efforts, PhenomeNET6 and PheWAS7 leveraged phenotypic information to discover genotype-phenotype associations that can be applied to prioritize genes for rare diseases.
Meanwhile, in the era of e-commerce, collaborative filtering techniques8 are popularly applied to recommend products to a customer based on customers with similar purchase preferences or other interests. The problem of diagnosing a patient with a disease based on patients’ phenotypic information is very similar to recommending a product to a customer, and therefore it is natural to propose the use of collaborating filtering for disease diagnosis. For example, given the hypothesis that disease prediction is achievable through analyzing phenotype and disease history, CARE provided an individualized healthcare framework by analyzing ICD-9-CM codes in patient’s medical history9-11. Similarly, Steinhaeuser and Chawla proposed a combined disease network and collaborative filtering to perform disease prediction on structured patient data12.
In this study, we proposed a patient based collaborative filtering system enriched with natural language processing (NLP) and semantic techniques to assist rare disease diagnosis based on phenotypic information extracted from free-text clinical notes. Specifically, we extracted the Unified Medical Language System (UMLS)13 concepts using MetaMap and used terms in the Human Phenotype Ontology (HPO)4, 14 and the Genetic and Rare Diseases Information Center (GARD)15 based on dictionary lookup to preprocess 2010-2015 clinical notes from Mayo Clinic Rochester campus. We then used a patient based collaborative filtering framework for rare disease diagnosis and compared four similarity measurements and two neighborhood algorithms.
In the following, we first introduce materials used in this study. Next, we describe the methods used to build the framework and conduct the evaluation. We then present the results followed by discussion. Lastly, we conclude and discuss potential future directions.
Materials
Clinical Data Collection
We collected all clinical notes during the years of 2010 to 2015 generated at Mayo Clinic Rochester campus. The resulting corpus contains 12.8 million clinical notes corresponding to 729,000 patients. We limited our annotation to sections containing problems and diagnoses.
The Unified Medical Language System and MetaMap
The Unified Medical Language System (UMLS), developed at National Library of Medicine (NLM), is an integrated knowledge base that involves key medical terminologies and related resources. There are three components in the UMLS, the Metathesaurus, Semantic Network, and Specialist Lexicon. The Metathesaurus lists all clinical concepts integrated from over a hundred of terminological resources. A unique identifier, Concept Unique Identifier (CUI), is assigned to each medical concept. Each concept can be associated with one or more semantic types defined in Semantic Network. MetaMap16 is a configurable application for mapping biomedical text to the Metathesaurus. Here, we applied MetaMap with the UMLS version 2015 to extract UMLS concepts from clinical notes as a preprocessing step.
Human Phenotype Ontology
The Human Phenotype Ontology (HPO) has been developed as a controlled vocabulary for phenotypes by mining and integrating phenotype knowledge from medical literature and ontologies. HPO also provides associations with other biomedical resources such as Gene Ontology17. HPO contains four sub-ontologies focusing on different annotation areas: Phenotypic Abnormality, Mode of Inheritance, Clinical Modifier, and Mortality/Aging. HPO and the UMLS have been cross-referred and we limited phenotype concepts extracted from clinical notes only to UMLS concepts that are cross-referred with the HPO released on September 2016. We only considered concepts from HPO sub-ontology Phenotypic Abnormality (HP_0000118) and its descendants, consisting of 11,721 phenotypes.
Genetic and Rare Diseases Information Center
The Genetic and Rare Diseases (GARD) Information Center is a program initiated by the National Center for Advancing Translational Sciences (NCATS) and funded by the National Institutes of Health (NIH). GARD extracts information from NIH resources, medical textbooks/databases, literature and the Internet to aggregate knowledge of rare or genetic diseases and group them into 32 categories. In the current version, GARD contains 4,560 diseases. We limited rare diseases extracted from clinical notes only to UMLS concepts that can be mapped to GARD.
Methods
Figure 1 shows the overview of our system workflow which includes two modules: i) a preprocessing module leveraging NLP and semantic processing techniques to identify patients with rare diseases and collect their phenotypes; and ii) a collaborative filtering model to recommend similar patients and possible disease recommendations.
Figure 1.
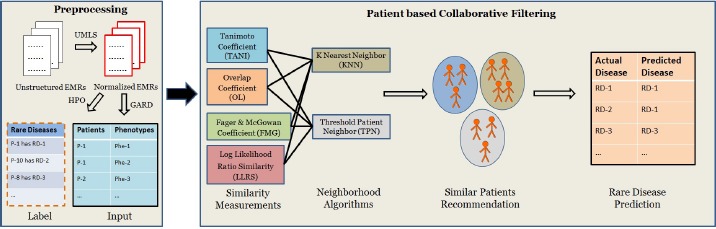
System workflow.
Preprocessing Module
We first extracted problems and diagnoses from clinical notes using MetaMap. We kept UMLS concepts from the following semantic types: Disease or Syndrome (dsyn), Neoplastic Process (neop), Mental or Behavioral Dysfunction (mobd), Anatomical Abnormality (anab), Congenital Abnormality (cgab), Injury or Poisoning (inpo), Finding (fndg) and Sign or Symptom (sosy). To limit our analysis to only patients’ phenotypes and rare diseases, we conducted another refinement round on preprocessed UMLS terms to only keep phenotype-related concepts in HPO and rare diseases in GARD. For patients with rare diseases, we collected their phenotypic information within the last twelve months of the first mention of rare diseases.
Patient based Collaborative Filtering Module
In traditional user-based collaborative filtering, user preference data with various features describe different angles of user interests. Similarly, in the clinical domain, we considered patients as users and leverage large clinical cohorts to extract phenotypes as items for each patient. In contrast to rating-based recommender systems in which preference score matters, we considered patient profile with phenotypes as binary inputs, that is, the relationship between patient and phenotype is either yes or no.
This module was designed on top of a user based collaborative filtering engine consisting of similarity measurements and neighborhood algorithms. Specifically, in this study, we applied Tanimoto coefficient similarity (TANI)18, Overlap coefficient similarity (OL)19, Fager & McGowan coefficient similarity (FMG)20, and Log likelihood ratio similarity (LLRS)21 as four similarity measurements for binary data to calculate patients’ similarity based on phenotypes. Each measurement is feasible for different recommendation tasks, and as we did not have any a priori reason to prefer one measurement over another, we tested all four methods.
Let |Phei| and |Phej| be the total number of phenotypes, and |Phei∩Phej| be the number of common phenotypes between any two patients i and j. Tanimoto, Overlap and Fager & McGowan coefficient similarity are defined as shown in Equations 1, 2, and 3, respectively.
| (Eq 1) |
| (Eq 2) |
| (Eq 3) |
Log likelihood ratio similarity is built based on Shannon entropy22. Given an event X with its possible value xi, Shannon entropy is defined in Equation 4.
| (Eq 4) |
In Shannon entropy, let pij, , , denote probabilities of common phenotypes shared by both patient i and j, patient j but not i, patient i but not j, and neither patient i nor j, respectively. The Log likelihood ratio (LLR) between any two patients i and j can be defined as shown in Equation 5.
| (Eq 5) |
As a result, Log likelihood ratio similarity (LLRS) between patient i and j is defined in Equation 6.
| (Eq 6) |
We also applied two commonly used neighborhood algorithms in collaborative filtering for given similarity metrics to recommend patients. One is K Nearest Neighbor (KNN)23, where the neighbors included are the k nearest neighbors. The other is Threshold Patient Neighbor (TPN)24, where a similarity threshold t is used to select neighbors.
Evaluation
This proposed system was implemented in Java with Eclipse Standard/SDK version Luna 4.4.025 running on 64 bit Linux CentOS 6.8 servers hosted by Mayo Clinic. We used the Apache Mahout framework26 to establish the environment for implementing the collaborative filtering framework. We evaluated eight different experimental groups formed as: 1) TANI with KNN; 2) LLRS with KNN; 3) OL with KNN; 4) FMG KNN; 5) TANI with TPN; 6) LLRS with TPN; 7) OL with TPN; 8) FMG with TPN.
To determine the best k and t, we first conducted a 10-fold cross validation for each experimental group. For each round, we randomly selected 90% data for training and the rest 10% for testing. To deal with binary preference value (i.e. either yes or no to a phenotype), for a specific patient, the Apache Mahout framework considered phenotype preference values as the summation of all his/her neighbors who also have the same phenotype. To determine the best k for KNN and t for TPN, we compared those estimated preference values with patients’ actual phenotype binary values (0 or 1). Specifically, for each predicted value pi’ and its corresponding actual value pi, we applied root-mean-square error (RMSE)27 on the total n results across all patients as shown in Equation 7.
| (Eq 7) |
We then computed second derivative for each RMSE function to select the optimal k and t at the biggest second derivative value. For function y=f(x), second derivative sd can be used to detect the concavity of a graph as shown in Equation 8.
| (Eq 8) |
After the optimal k and t for each experiment was confirmed, the system returned a number of neighbors for each patient ranked in descending order by similarity scores.
To evaluate the performance of patient recommendation, we used information retrieval techniques to evaluate all ranked recommendations. We selected k recommended patients for KNN and all patients with similarity higher than t for TPN. We considered each patient as a query and their neighbors as a group of ranked recommendations. In addition, we used patients’ diseases as a gold standard to validate their similarity and considered a recommendation as an optimal one as long as the recommended patient affected similar rare disease(s). The confusion matrix in Table 1 generally depicts how to evaluate the system performance. According to Table 1, precision, recall and F measure can be computed as shown in Equations 9, 10 and 11. To analyze the performance and observe the trade-off between precision and recall, starting from top ranked patients, we divided them into ten portions and plotted precision-recall curves for each experiment. In addition, we calculated precision-recall area under curve (PRAUC)28 for each case. Moreover, we computed mean average precision (MAP)29 for recommendations of all patients. As shown in Equation 12, for each query q, we computed average precision AveP over all relevant answers and calculated summation of AveP for all queries then divided by the number of total queries |Q| to compute the MAP.
Table 1.
Confusion matrix for system performance evaluation.
| Recommended Rare Disease | Not Recommended Rare Disease | |
|---|---|---|
| Relevant Rare Disease | True Positive (TP) | False Negative (FN) |
| Not Relevant Rare Disease | False Positive (FP) | True Negative (TN) |
To further evaluate performance for individual rare disease prediction, we assigned similarity scores of recommended patients to their corresponding rare diseases and accumulated all similarity scores for each disease. We considered the rare disease with the highest accumulated similarity score as the final diagnosis to evaluate performance for rare disease prediction. We used the optimal algorithm to conduct the evaluation. Precision, recall and F measure were computed for each rare disease as evaluation outputs.
Here we applied three level matching criteria to determine if two rare diseases are similar or not. The first one is string matching. That is, we compared two diseases directly by checking their exact names. Considering each physician might use different terms or concepts to make a diagnosis, the use of strict string matching would probably miss some semantically similar diseases. The Systematized Nomenclature of Medicine – Clinical Terms (SNOMED-CT)30 groups comprehensive medical terminologies with a semantic hierarchy in a standard manner. Therefore, as a second level matching, we mapped diseases to SNOMED-CT and considered two diseases to be related if they contributed to a common ancestor node within 3 hierarchical generations31. In addition, to have broader rare disease similarity checking in terms of categorization, we used the GARD dictionary and considered two diseases have a close relationship if they were listed in the same rare disease category. Therefore, for different matching criteria, definitions of relevant or not in confusion matrix are different.
| (Eq 9) |
| (Eq 10) |
| (Eq 11) |
| (Eq 12) |
Results
After pre-processing, there are about 31,000 patients with at least one rare disease diagnosis. After removing rare diseases affecting only one patient, the final data set includes about 29,000 patients with 437 rare diseases (29 out of 32 GARD categories) and 2,400 phenotypes. In addition, 24,000 patients were diagnosed with only 1 rare disease and 5,000 patients were diagnosed with 2 rare diseases.
Figure 2 shows statistics of rare diseases in our clinical notes with GARD categories. Unique count indicates the number of unique rare diseases in each GARD category, and category coverage shows the percentile of rare diseases out of the total number of rare disease in each GARD category. The category with the biggest unique count is Rare Cancer consisting of 155 diseases, for instance, lentigo maligna melanoma and papillary thyroid carcinoma. In terms of category coverage, Autoimmune Autoinflammatory Diseases is the highest one with 45.45% (5 of 11), which includes autoimmune hepatitis, crest syndrome, addison’s disease and so on.
Figure 2.
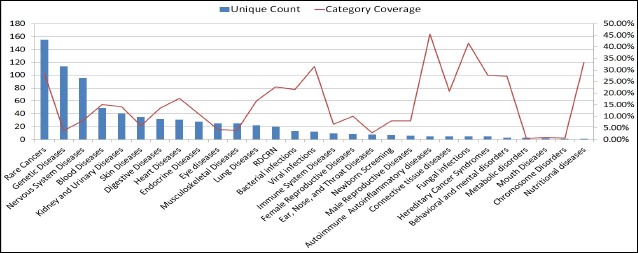
Statistics of rare disease in clinical notes with GARD categories.
Root-Mean-Square Error for Optimal Threshold Selection
For different similarity measurements with KNN, Figure 3(a) plots the relationship between RMSE and the increasing number of neighbors from 2 to 30. Since KNN selected the closest k nodes incrementally, more neighbors will be included with k increased even though they do not locate within an absolutely close distance, which should increase the error. We found that with k increased, RMSE for LLRS had a fastest increase rate after optimal k, indicating that LLRS is too sensitive to the change of k. RMSE didn’t change obviously for OL and FMG, which shows that these two similarities with KNN are not capable of differentiating neighbors and non-neighbors. RMSE for TANI slightly increased RMSE after its optimal k, suggesting that TANI+KNN is able to group neighbors in a moderate way.
Figure 3.
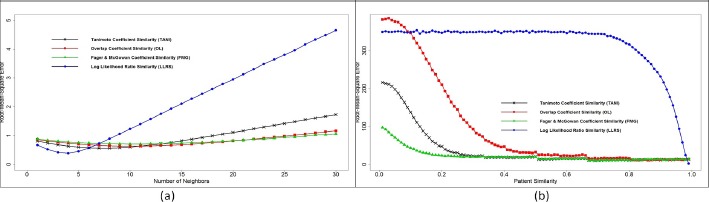
RMSE evaluation for KNN (a) and TPN (b) with four similarity measurements.
Similarly, Figure 3(b) depicts the relationship between RMSE and patient similarity threshold varying from 0.01 to 0.99 for different similarity measurements with TPN. Generally, RMSE decreased as we increased patient similarity. The reason is that criteria for neighbor selection became higher when we increased the user similarity, which resulted in more precise neighbor detection and thus decreased the error. We found that RMSE for LLRS stayed stable until similarity threshold t became relatively higher, which indicates that LLRS is less sensitive to similarity threshold. We also found that curves for TANI, OL, and FMG had the similar trends but OL started with the highest RMSE and FMG started with the lowest RMSE. TANI had the moderate RMSE at the beginning and reached its optimal threshold t before OL and after FMG.
As a result, the best k and t for each combination are given in Table 2.
Table 2.
The optimal k and t for different experiments.
| TANI | OL | FMG | LLRS | |
|---|---|---|---|---|
| Best k for KNN | 8 | 11 | 10 | 4 |
| Best t for TPN | 0.21 | 0.34 | 0.17 | 0.84 |
Performance for Patient Recommendation
Figure 4 shows precision-recall curves for 8 experiments and PRAUC for each matching criterion. We found that GARD matching yielded the best performance overall, and SNOMED-CT semantic matching always performed better than string matching. There is no significant difference between TANI+KNN and TANI+TPN for each of the three matching criteria. Specifically, TANI+TPN contributed to the best PRAUC for string matching while TANI+KNN led to the best PRAUC for SNOMED-CT and GARD matching. LLRS was suboptimal and LLRS+KNN outperformed LLRS+TPN with all three matching criteria. In contrast, PRAUC indicated that FMG+TPN performed better than FMG+KNN. OL contributed to relatively lower PRAUC than other three similarity measurements, OL+KNN performed slightly better for SNOMED-CT and GARD matching while OL+TPN was more suitable for string matching.
Figure 4.
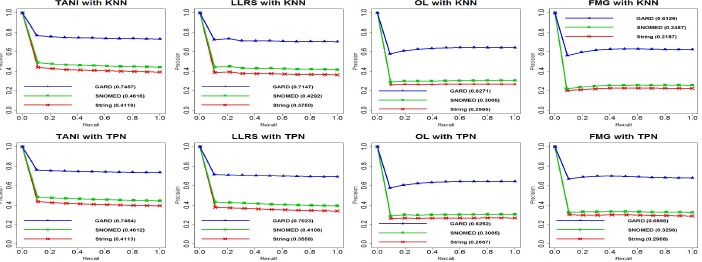
Precision-Recall curves for 8 experiments with 3 matching criteria (number in bracket indicates PRAUC).
Table 3 shows mean average precision for patients with all their recommendations. MAP scores showed consistent performance with what PRAUC evaluated. But the only difference is that MAP scores indicated that TANI with KNN performed slightly better for string matching while TANI with TPN was slightly better for SNOMED-CT and GARD matching.
Table 3.
Mean average precision for 8 experiments with 3 matching criteria (highest value in bold).
| TANI | LLRS | OL | FMG | |||||
|---|---|---|---|---|---|---|---|---|
| KNN | TPN | KNN | TPN | KNN | TPN | KNN | TPN | |
| String | 0.4229 | 0.4228 | 0.3736 | 0.3672 | 0.2671 | 0.2672 | 0.2124 | 0.3007 |
| SNOMED | 0.4727 | 0.4729 | 0.4297 | 0.4215 | 0.2989 | 0.2988 | 0.2387 | 0.3303 |
| GARD | 0.7525 | 0.7530 | 0.7116 | 0.7084 | 0.6114 | 0.6094 | 0.5964 | 0.6841 |
Performance for Rare Disease Prediction
Here we chose TANI with KNN as the optimal algorithm and applied it on 24,000 patients with only one rare disease. Scatter plots in Figure 5 describe prediction performance for different matching criteria. There are 417 rare diseases in total. For these diseases, string, SNOMED-CT and GARD matching found 49.4%, 56.6%, and 92.1% correct predicted rare diseases, and weighted micro-average F measure for them are 0.4, 0.44, and 0.71 respectively.
Figure 5.
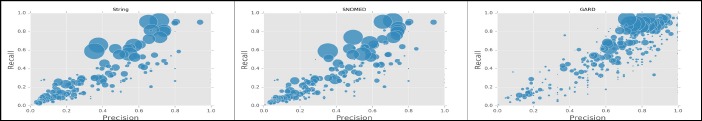
Scatter plot of precision-recall for rare disease prediction (circle size is proportional to the number of affected patients).
Table 4 shows the top five diseases with the best F measures for each matching criterion. To protect patients’ privacy, any rare disease with affected cases less than 10 were marked as <10. We found for some diseases, the unique features affecting very few patients may contribute to a high prediction performance. For example, there were less than 10 patients with ichthyosis bullosa of siemens, but the prediction F measure with string matching is 0.8. All these patients have symptom ichthyosis, some of them have ichthyosis and hyperkeratosis, and some others have ichthyosis and congenital bullous ichthyosiform erythroderma. These phenotypes grouped as unique combinations that can help diagnose ichthyosis bullosa of siemens. Meanwhile, with 1,380 affected patients, abdominal aortic aneurysm had a high F measure with string matching. However, not all rare diseases with a relatively large number of affected patients yielded the same performance. For instance, meningioma had 1,476 affected patients, but prediction F measures for it is only 0.44.
Table 4.
Recommendation performance for selected rare diseases.
| Approaches | Top Diseases | Number of Affections | Precision | Recall | F Measure |
|---|---|---|---|---|---|
| Tani+KNN with String Matching | osteochondritis dissecans | 158 | 0.94 | 0.9 | 0.92 |
| frontotemporal dementia | 221 | 0.81 | 0.91 | 0.85 | |
| spasmodic dysphonia | 173 | 0.8 | 0.9 | 0.84 | |
| abdominal aortic aneurysm | 1,380 | 0.71 | 0.92 | 0.8 | |
| ichthyosis bullosa of siemens | <10 | 0.8 | 0.8 | 0.8 | |
| Tani+KNN with SNOMED Matching | acute myelomonocytic leukemia | 11 | 1 | 1 | 1 |
| osteochondritis dissecans | 158 | 0.94 | 0.9 | 0.92 | |
| frontotemporal dementia | 221 | 0.81 | 0.91 | 0.85 | |
| spasmodic dysphonia | 173 | 0.8 | 0.89 | 0.84 | |
| abdominal aortic aneurysm | 1,380 | 0.71 | 0.92 | 0.8 | |
| Tani+KNN with GARD Matching | acute myelomonocytic leukemia | 11 | 1 | 1 | 1 |
| angioimmunoblastic t-cell lymphoma | 18 | 1 | 1 | 1 | |
| ataxia telangiectasia | <10 | 1 | 1 | 1 | |
| chronic myelomonocytic leukemia | <10 | 1 | 1 | 1 | |
| congenital heart block | <10 | 1 | 1 | 1 |
For string matching, some rare diseases didn’t get any correct predictions, such as cadasil (<10 patients) and myotonic dystrophy (25 patients), the reason is that those diseases not only had a small group of affected patients but also didn’t show unique groups of symptoms and signs. Due to the semantic hierarchical processing, SNOMED-CT matching had a slightly better performance than string matching. It was able to predict some diseases that string matching considered as non-relevant, such as acquired von willebrand syndrome (<10 patients) and acute disseminated encephalomyelitis (<10 patients). What is more, GARD gave predictions based on category and all top 5 predictions are 100% predicted. This indicates that phenotypes can help to infer similar type of rare diseases. Although such prediction cannot identify the exact rare diseases, similar rare diseases within the same category can still give clues for physicians to make diagnoses.
Discussion
Our study utilized similarity measurements that do not take individual preference scores, therefore, other methods provided by the Mahout collaborative filtering engine that either consider preference values or preference rankings were not suitable for our preprocessed clinical notes (e.g., Euclidean distance similarity, Pearson correlation similarity, Uncentered cosine similarity, and Spearman correlation similarity)26. TANI is similar to LLRS, with the only difference that the latter gives more weight to dissimilar patients if they have common phenotypes and assigns less weight to similar patients even they share exactly the same phenotypes. This weighting difference slightly depressed LLRS’s performance relative to TANI. Similar to LLRS, FMG gives weight to similarity, but not as much as LLRS. In addition, OL gives too much weight to patients’ similarities even with few shared phenotypes, which lacks the ability to stratify patients well. What is more, FMG is the only one that is sensitive to the selection of KNN or TPN, and significantly outperformed TPN. The reason is that setting threshold as 0.17 is more suitable than selecting 10 neighbors according to the neighborhood density formed by FMG in our clinical notes. Therefore, making a good balance between KNN and TPN has a potential ability to optimize the overall performance with idealized neighbors and similarity at the same time.
For overall performance across all recommendations, considering physicians have different descriptions of specific diagnosis, string matching may overlook some associations, so we enriched disease matching with semantic similarity. There exist some ontologies that describe diseases, such as Disease Ontology (DO)32 and Orphanet Rare Disease Ontology (ORDO)33. However, for the detected rare diseases from clinical notes, only 56% and 52% are covered by DO and ORDO, respectively. Therefore, we used SNOMED-CT, a more comprehensive ontology that comprises 60% of our extracted rare diseases and maintains a more complicated hierarchical semantic relationship for discovering disease associations. Nevertheless, due to the limited coverage, semantic disease checking didn’t significantly improve the performance. To address this, some associations among rare diseases can be mined from literature for evaluation.
For rare disease prediction with string matching, some phenotypes cannot uniquely characterize a certain type of rare disease in our clinical notes. For example, hypopituitarism has 51 affected patients and prediction F measure is 0. Top frequent phenotypes for hypopituitarism are hypothyroidism, neoplasm, hypertension, apnea and hyperlipidemia, which are very common symptoms shared with other rare diseases and are not useful to make correct diagnose. Therefore, common comorbidities may create noises for decision making. In this preliminary study, we only focused on patients with rare diseases and filtered out phenotypes that didn’t happen within 12 months of their rare disease diagnosis encounter time. To better characterize rare diseases and comorbidities, in the future, we will pass all patients’ data to our system and target on giving recommendations for misdiagnosed and undiagnosed cases. In addition, it would be interesting to investigate cross-institutional rare diseases to acquire diagnosis experiences and intelligence from different hospitals and healthcare systems to build a more generic rare disease diagnosis system.
Conclusion and Future Work
In this study, we have investigated patient based collaborative filtering with NLP and semantic techniques on large patient cohort to assist rare disease diagnosis. We demonstrated its potential in facilitating rare disease prediction.
In the future, we plan to incorporate HPO phenotypic information content14, topic modeling34, word embedding35 and deep learning temporal sequence analysis36 to give more degrees of similarity measurement for improving prediction performance. In addition to phenotype based analysis, we plan to involve rare disease genotype from PheWAS7 and literature to assist rare disease diagnose as well.
Acknowledgements
The authors gratefully acknowledge the support from the National Institute of Health (NIH) grants R01GM102282, R01LM011934, R01EB19403, and OT3TR002019.
References
- 1.The orphan drug act implementation and impact. [Accessed 12 February 2017]. Available: https://oig.hhs.gov/oei/reports/oei-09-00-00380.pdf.
- 2.Rare disease day. [Accessed 12 February 2017]. Available: ht tp://rarediseaseday.us/
- 3.National organization for rare disorders. [Accessed 12 February 2017]. Available: https://rarediseases.org/
- 4.Robinson PN, Köhler S, Bauer S, Seelow D, Horn D, Mundlos S. The Human Phenotype Ontology: a tool for annotating and analyzing human hereditary disease. The American Journal of Human Genetics. 2008 Nov 17;83(5):610–5. doi: 10.1016/j.ajhg.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Javed A, Agrawal S, Ng PC. Phen-Gen: combining phenotype and genotype to analyze rare disorders. Nature methods. 2014 Sep 1;11(9):935–7. doi: 10.1038/nmeth.3046. [DOI] [PubMed] [Google Scholar]
- 6.Hoehndorf R, Schofield PN, Gkoutos GV. PhenomeNET: a whole-phenome approach to disease gene discovery. Nucleic acids research. 2011 Oct 1;39(18):e119. doi: 10.1093/nar/gkr538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denny JC, Ritchie MD, Basford MA, Pulley JM, Bastarache L, Brown-Gentry K, Wang D, Masys DR, Roden DM. Crawford DC. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics. 2010 May 1;26(9):1205–10. doi: 10.1093/bioinformatics/btq126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.CarlKadie JB. Empirical Analysis of Predictive Algorithms for Collaborative Filtering. Microsoft Research Microsoft Corporation One Microsoft Way Redmond, WA. 1998 May;:98052. [Google Scholar]
- 9.Davis DA, Chawla NV, Blumm N, Christakis N, Barabási AL. Predicting individual disease risk based on medical history. In Proceedings of the 17th ACM conference on Information and knowledge management; 2008 Oct 26; ACM; pp. 769–778. [Google Scholar]
- 10.Davis DA, Chawla NV, Christakis NA, Barabási AL. Time to CARE: a collaborative engine for practical disease prediction. Data Mining and Knowledge Discovery. 2010 May 1;20(3):388–415. [Google Scholar]
- 11.Chawla NV, Davis DA. Bringing big data to personalized healthcare: a patient-centered framework. Journal of general internal medicine. 2013 Sep 1;28(3):660–5. doi: 10.1007/s11606-013-2455-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steinhaeuser K, Chawla NV. InSocial computing and behavioral modeling. US: Springer; 2009. A network-based approach to understanding and predicting diseases; pp. 1–8. [Google Scholar]
- 13.Bodenreider O. The unified medical language system (UMLS): integrating biomedical terminology. Nucleic acids research. 2004 Jan 1;32(suppl 1):D267–70. doi: 10.1093/nar/gkh061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Groza T, Köhler S, Moldenhauer D, Vasilevsky N, Baynam G, Zemojtel T, Schriml LM, Kibbe WA, Schofield PN, Beck T, Vasant D. The human phenotype ontology: semantic unification of common and rare disease. The American Journal of Human Genetics. 2015 Jul 2;97(1):111–24. doi: 10.1016/j.ajhg.2015.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Genetic and rare diseases information center. [Accessed 12 February 2017]. Available: https://rarediseases.info.nih.gov/
- 16.Aronson AR. InProceedings of the AMIA Symposium. American Medical Informatics Association; 2001. Effective mapping of biomedical text to the UMLS Metathesaurus: the MetaMap program; p. 17. [PMC free article] [PubMed] [Google Scholar]
- 17.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA. Gene Ontology: tool for the unification of biology. Nature genetics. 2000 May 1;25(1):25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaccard P. Distribution de la Flore Alpine: dans le Bassin des dranses et dans quelques régions voisines. Rouge. 1901 [Google Scholar]
- 19.Szymkiewicz D. Une contribution statistique a la géographie floristique. Polskie Towarzystwo Botaniczne. 1934 [Google Scholar]
- 20.Fager EW, McGowan JA. Zooplankton Species Groups in the North Pacific: Co-occurrences of species can be used to derive groups whose members react similarly to water-mass types. Science. 1963 May 3;140(3566):453–60. doi: 10.1126/science.140.3566.453. [DOI] [PubMed] [Google Scholar]
- 21.Dunning T. Accurate methods for the statistics of surprise and coincidence. Computational linguistics. 1993 Mar 1;19(1):61–74. [Google Scholar]
- 22.Shannon CE. A mathematical theory of communication. ACM SIGMOBILE Mobile Computing and Communications Review. 2001 Jan 1;5(1):3–55. [Google Scholar]
- 23.Altman NS. An introduction to kernel and nearest-neighbor nonparametric regression. The American Statistician. 1992 Aug 1;46(3):175–85. [Google Scholar]
- 24.Bellogín A, Castells P, Cantador I. Neighbor selection and weighting in user-based collaborative filtering: a performance prediction approach. ACM Transactions on the Web (TWEB) 2014 Mar 1;8(2):12. [Google Scholar]
- 25.Eclipse luna. [Accessed 12 February 2017]. Available: https://eclipse.org/luna/
- 26.Schelter S, Owen S. Collaborative filtering with apache mahout. Proc. of ACM RecSys Challenge. 2012 [Google Scholar]
- 27.Hyndman RJ, Koehler AB. Another look at measures of forecast accuracy. International journal of forecasting. 2006 Dec 31;22(4):679–88. [Google Scholar]
- 28.Davis J, Goadrich M. The relationship between Precision-Recall and ROC curves. InProceedings of the 23rd international conference on Machine learning; 2006 Jun 25; ACM; pp. 233–240. [Google Scholar]
- 29.Manning CD, Raghavan P, Schütze H. Cambridge: Cambridge university press; 2008. Jul 12, Introduction to information retrieval. [Google Scholar]
- 30.Donnelly K. SNOMED-CT: The advanced terminology and coding system for eHealth. Studies in health technology and informatics. 2006 Jan;121:279. [PubMed] [Google Scholar]
- 31.Frick JM, Guha R, Peryea T, Southall NT. Evaluating disease similarity using latent Dirichlet allocation. bioRxiv. 2015 Jan 1;:030593. [Google Scholar]
- 32.Schriml LM, Arze C, Nadendla S, Chang YW, Mazaitis M, Felix V, Feng G, Kibbe WA. Disease Ontology: a backbone for disease semantic integration. Nucleic acids research. 2012 Jan 1;40(D1):D940–6. doi: 10.1093/nar/gkr972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orphanet Rare Disease Ontology. [Accessed 12 February 2017]. Available: http://www.orphadata.org/cgi-bin/inc/ordo_orphanet.inc.php.
- 34.Blei DM. Probabilistic topic models. Communications of the ACM. 2012 Apr 1;55(4):77–84. [Google Scholar]
- 35.Mikolov T, Sutskever I, Chen K, Corrado GS, Dean J. Distributed representations of words and phrases and their compositionality. InAdvances in neural information processing systems. 2013:3111–3119. [Google Scholar]
- 36.Farhan W, Wang Z, Huang Y, Wang S, Wang F, Jiang X. A Predictive Model for Medical Events Based on Contextual Embedding of Temporal Sequences. JMIR Medical Informatics. 2016 Oct;4(4) doi: 10.2196/medinform.5977. [DOI] [PMC free article] [PubMed] [Google Scholar]