

Abstract
Iron is an essential element for numerous fundamental biologic processes, but excess iron is toxic. Abnormalities in systemic iron balance are common in patients with chronic kidney disease (CKD) and iron administration is a mainstay of anemia management in many patients. This review provides an overview of the essential role of iron in biology, the regulation of systemic and cellular iron homeostasis, how imbalances in iron homeostasis contribute to disease, and the implications for CKD patients.
Keywords: Iron metabolism, iron deficiency, iron overload, anemia, chronic kidney disease
INTRODUCTION
Iron is one of the most abundant elements of the Earth’s crust. It is also a transition metal that can readily donate and accept electrons to participate in oxidation-reduction reactions that are essential for a number of fundamental biologic processes. It is therefore not surprising that iron has had an essential role in almost all living organisms, possibly dating back to very origins of life.1 In humans, iron is incorporated into proteins as a component of heme (e.g. hemoglobin, myoglobin, cytochrome proteins, myeloperoxidase, nitric oxide synthetases), iron sulfur clusters (e.g. respiratory complexes I-III, coenzyme Q10, mitochondrial aconitase, DNA primase), or other functional groups (e.g. hypoxia inducible factor prolyl hydroxylases).2 These iron-containing proteins are required for vital cellular and organismal functions including oxygen transport, mitochondrial respiration, intermediary and xenobiotic metabolism, nucleic acid replication and repair, host defense, and cell signaling.
With the oxygenation of the Earth’s atmosphere over 2 billion years ago, abundant soluble Fe2+ was oxidized to insoluble Fe3+, making bioavailable iron much more scarce.3 At the same time, iron became potentially more toxic since the redox cycling of iron in the presence of oxygen and hydrogen peroxide catalyzes the production of free radicals in the Fenton reaction that can damage DNA, protein, and lipids.4 Humans and other organisms therefore evolved specialized proteins and tightly regulated homeostatic mechanisms for the uptake, transport, storage, and export of iron to provide adequate iron for essential biologic process, but to limit the toxicity of iron excess.
SYSTEMIC IRON HOMEOSTASIS
Reflecting the scarcity of biologically available iron, humans efficiently conserve and recycle iron (Figure 1).5–6 The majority of iron is contained in red blood cell hemoglobin (~2 gm iron) and is recycled in the process of erythrophagocytosis by reticuloendothelial macrophages. These iron recycling macrophages are a major storage site for iron in addition to liver hepatocytes. All other cells in the body contain smaller amounts of iron for essential cellular processes as noted above. The circulating pool of iron is comparatively small (2–4 mg) and must be turned over every few hours to meet the daily requirement of iron to support erythropoiesis and other body needs (~20–25 mg). Approximately 1–2mg of iron is provided by dietary absorption in the duodenum, which is balanced by an unregulated loss of 1–2mg of iron, primarily through epithelial desquamation and blood loss. Urinary iron excretion appears to be minimal due to the largely protein bound form of circulating iron (bound to transferrin) and multiple mechanisms for iron reclamation in the kidney.7 Since body iron losses are not regulated, the major avenues for regulating systemic iron balance are in controlling dietary iron update, and iron release from recycling macrophages and hepatocytes.
Figure 1. Systemic iron homeostasis.
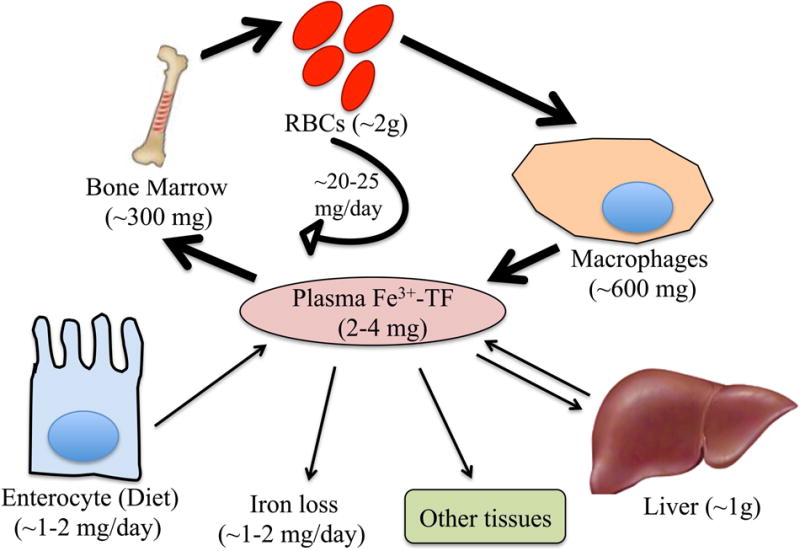
Iron (Fe) circulates in the bloodstream bound to transferrin (TF). The majority of iron is delivered to the bone marrow for red blood cell (RBC) production, with lesser amounts going to other tissues for fundamental cellular processes and the excess transported to the liver for storage. Systemic iron homeostasis is maintained predominantly by recycling iron from RBCs via reticuloendothelial macrophages. A smaller amount of iron is provided by dietary absorption via duodenal enterocytes, which is matched by an unregulated loss of iron through desquamation and blood loss.
IRON ABSORPTION AND RECYCLING
Dietary iron is absorbed in several forms: inorganic, heme, and ferritin. Inorganic dietary iron is mainly present in the oxidized form (Fe3+) and must be reduced to the Fe2+ form prior to intestinal uptake. This reduction is thought to be mediated by ferrireductases in the intestinal cell apical membrane such as duodenal cytochrome B (DCYTB), facilitated by ascorbic acid.8 Once reduced, Fe2+ is transported across the apical membrane by divalent metal transporter 1 (DMT1).9–11 This uptake is facilitated by the acidic microenvironment and the H+ gradient generated by the brush border Na+/H+ exchanger.12 Although the mechanisms for uptake of dietary heme and ferritin are less well understood, evidence suggests that iron is subsequently liberated and enters a common pathway as inorganic iron in the enterocyte.13–14
Iron taken up by enterocytes can be used directly for intrinsic cellular metabolic processes, stored, or exported across the basolateral membrane for systemic delivery. Iron is stored in enterocytes, like other cells, largely in the form of ferritin, which is comprised of a spherical nanocage of heavy (H) and light (L) chains surrounding a core of iron that is oxidized by H-ferritin.15 This allows iron to be stored in an inert form that limits the production of damaging redox reactive species. Ferritin storage allows for a more controlled delivery of iron to basolateral iron exporters and the ability to limit systemic iron delivery by intestinal cell desquamation, which occurs every few days.16
Systemic iron delivery is mediated by the basolateral iron exporter ferroportin.17–19 Although the molecular mechanisms of iron export by ferroportin are still not well understood, it is thought that iron is exported in the Fe2+ form, oxidized to Fe3+ by hephaestin and ceruloplasmin, and loaded onto transferrin, the main plasma iron carrier.20–23
A major source of daily iron is provided by iron recycling macrophages.5 This specialized macrophage population phagocytoses old and damaged red blood cells after a mean lifespan of 120 days. Red blood cells are lysed, and iron is released from hemoglobin by heme oxygenase-1. Iron can then be stored in ferritin and exported to the bloodstream by ferroportin via a similar process described above for duodenal enterocytes. Ferroportin, the only known mammalian iron exporter, therefore functions as a major gatekeeper controlling iron entry into the bloodstream.
CIRCULATING IRON
Transferrin-bound iron is the main form of iron present in the bloodstream under normal conditions. Transferrin can carry up to 2 iron molecules and maintains iron in a redox inert state. Approximately 20–40% of iron binding sites on transferrin are normally occupied by iron, corresponding to transferrin saturation. Transferrin delivers iron to tissues for uptake by the ubiquitously expressed transferrin receptor 1 (TFR1).6 A homologue with more restricted expression, transferrin receptor 2 (TFR2), may function as a sensor of transferrin-bound iron levels in other tissues including the liver and erythrocyte.5, 24 In conditions where iron exceeds the carrying capacity of transferrin, non-transferrin bound iron (NTBI) may circulate, including highly reactive labile plasma iron (LPI) that can be taken up in organs such as the liver, pancreas, and heart, where it leads to organ damage.25 Uptake mechanisms for NTBI differ from transferrin-bound iron, for example ZIP14 is a major mediator of NTBI uptake in hepatocytes.26 Ferritin is also measurable in the bloodstream. Circulating ferritin mainly originates from macrophages27 and generally correlates with body iron stores, although its levels can also be influenced by inflammation, infection, liver disease, and malignancy among other conditions.25 Circulating ferritin is relatively iron-poor, but has been hypothesized to act as another extracellular iron carrier in some conditions28. Additional circulating carriers of iron include the heme and hemoglobin scavengers hemopexin and haptoglobin. Neutrophil gelatinase associated lipocalin (NGAL)/lipocalin-2 has also been proposed to function as an extracellular iron carrier by binding to siderophores, small iron-binding compounds typically secreted by microorganisms, but which may also be produced in mammalian systems.29
CELLULAR IRON HOMEOSTASIS
Circulating iron is delivered to erythrocytes and other cells in the body via specific uptake mechanisms (Figure 2).6 A major common mechanism is uptake of transferrin-bound iron by TFR1 via receptor mediated endocytosis into clathrin coated pits. Iron is released in the acidic environment of the endosome, Fe3+ is reduced to Fe2+ by the ferriductase STEAP3, and Fe2+ is exported out of the endosome by DMT1, while TFR1 is recycled to the cell surface. Iron then enters the so-called chelatable or labile-iron pool and is either utilized directly, trafficked to the mitochondria for incorporation into the heme or iron sulfur cluster synthesis pathways, stored in an inert form in cytosolic or mitochondrial ferritin as described above, or exported out of the cell. Intracellular trafficking and transport across the mitochondrial membranes requires additional specialized proteins including the cytosolic iron chaperone poly (rC) binding proteins30, the mitochondrial chaperone frataxin31, and mitochondrial iron and heme transporters such as mitoferrins32 and feline leukemia virus type C receptor 1B (FLVCR1B).33 Iron stored in ferritin can be released for utilization or export by autophagic turnover (ferritinophagy) mediated by the cargo receptor nuclear receptor coactivator 4 (NCOA4).34–35 Additional transporters are involved in iron export from cells, such as ferroportin for elemental iron as described above and FLVCR1A for heme iron.36 Although much progress has been made in understanding cellular iron trafficking pathways, many of these pathways are still not fully understood.
Figure 2. Cellular iron homeostasis.
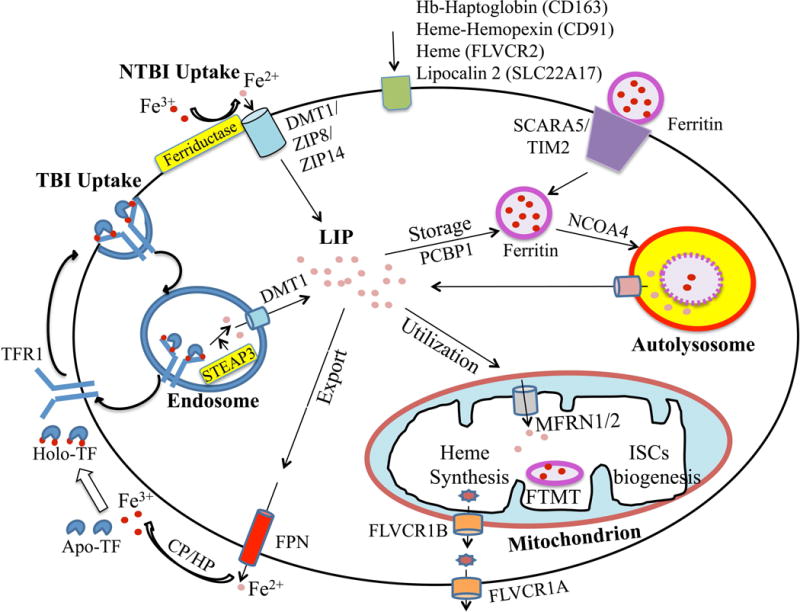
Depicted cell is an amalgam of many cell types; not all proteins/pathways are present in all cells. Iron enters into cells primarily by transferrin receptor 1 (TFR1)-mediated endocytosis. In endosomes, iron is freed from TF and reduced by a ferriductase (STEAP3) before exiting into the cytosol via divalent metal transporter (DMT1). TF and TFR1 are recycled back to the cell membrane for further cycles. DMT1 and other transporters (ZIP8, ZIP14) function in non-TF bound iron (NTBI) uptake pathways in some cell types. Other iron acquisition pathways in some cell types include uptake of hemoglobin(Hb)-haptoglobin, heme-hemopexin, heme, lipocalin 2, and ferritin via CD163, CD91, FLVCR2, SLC22A17, SCARA5, and TIM2 receptors respectively. In the cytosol, iron enters the labile iron pool (LIP), and is then utilized, stored, or exported out of the cell. Cytoplasmic iron transport is assisted in some cases by the chaperone poly (rC) binding protein 1 (PCBP1). Iron is mainly utilized by mitochondria for heme synthesis and iron-sulfur clusters (ISCs) biogenesis, with mitoferrins (MFRN1/2) playing a role in mitochondrial iron import and FLVCR1B playing a role in mitochondrial heme export. Excess iron in the cytosol is stored safely in ferritin. A mitochondrial form of ferritin (MTFT) is also expressed in some cell types. When the demand arises, ferritin can be targeted for autophagic turnover by nuclear receptor coactivator 4 (NCOA4) to release iron into the cytosolic LIP. Iron is exported out of the cell by ferroportin (FPN), assisted by ferroxidases ceruloplasmin (CP)/hephaestin (HP), followed by iron loading onto TF. FLVCR1A may play a role in heme export in some cell types.
REGULATION OF CELLULAR AND SYSTEMIC IRON HOMEOSTASIS
A major regulator of cellular iron homeostasis is the iron regulatory protein (IRP) system (Figure 3A). When cellular iron levels are low, IRPs regulate expression of numerous iron homeostasis proteins by binding to iron responsive elements (IREs) in the mRNA 5′ untranslated region (UTR) to inhibit translation (e.g for ferritin and ferroportin) or the 3′ UTR to prevent degradation (e.g. for TFR1).37 This leads to an increase in iron uptake and a decrease in iron storage and export when cellular iron levels are low, and the converse occurs when iron levels are high. IRP-IRE binding is inhibited in iron replete conditions by 2 mechanisms: 1) the binding of an iron-sulfur cluster in the RNA-binding cleft of IRP137, and 2) the targeting of IRP2 for proteasomal degradation by the E3-ubiqutin ligase F-box/leucine-rich repeat protein 5 FBXL538–39, whose stability is regulated by an iron-binding hemerythrin domain. Since IRPs target proteins that are also key mediators of systemic iron homeostasis, IRPs may also influence systemic iron balance.
Figure 3. Regulation of cellular iron homeostasis. A.
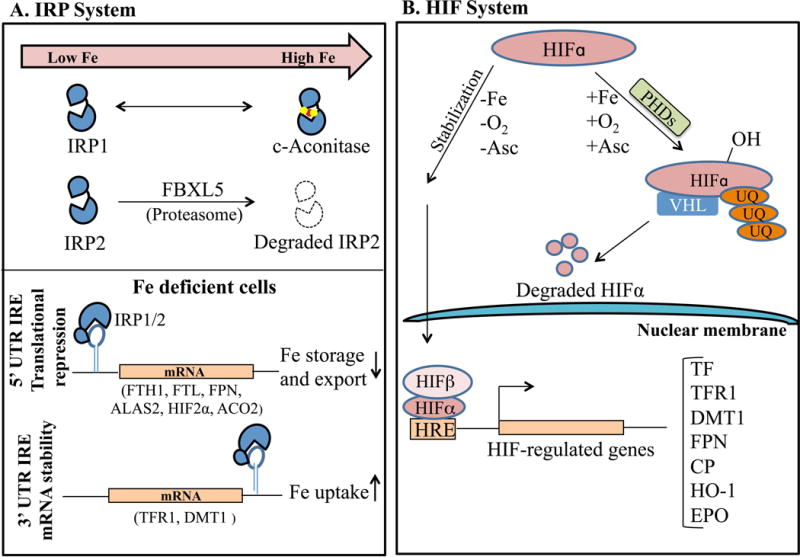
Under iron-deficient conditions, iron regulatory proteins (IRPs) bind to iron-responsive elements (IREs) in the 5′ untranslated regions (UTR) of iron homeostasis mRNAs such as ferritin (FTH1, FTL1) and ferroportin (FPN) to block their translation, while IRP binding in the 3′ UTR of TFR1 and DMT1 enhances mRNA stability. This leads to an increase in iron uptake and a decrease in iron storage and export. Under the iron-replete conditions, the IRP-IRE interaction is inhibited: IRP2 is targeted for proteasomal degradation by F-box/leucine-rich repeat protein 5 (FBXL5), and IRP1 gets converted into cytosolic aconitase. B. Under low iron and oxygen (O2) conditions, the regulatory hypoxia-inducible factor alpha subunit (HIFα) forms a complex with the ubiquitously expressed β subunit and translocates to the nucleus to regulate transcription of numerous iron homeostasis genes including TFR1, DMT1, FPN, CP, and erythropoietin (EPO). On the contrary, under iron-replete, normoxic conditions, HIFα subunits are targeted for proteasomal degradation by the oxygen- and iron-dependent prolyl hydroxylases (PHDs) and von Hippel-Lindau (VHL) protein. Abbreviations: ALAS, 5′-Aminolevulinate Synthase 2; Asc, ascorbate; UQ, ubiquitin; HRE, hypoxia response element; HO-1, heme oxygenase 1.
Another intracellular iron sensor is the hypoxia inducible factor (HIF) system (Figure 3B). Under low iron and oxygen conditions, the regulatory alpha subunits (HIF1α, HIF2α, and HIF3α) form a complex with the ubiquitously expressed β subunit (ARNT) and translocate to the nucleus to regulate gene transcription. Under iron-replete, normoxic conditions, HIFα subunits are targeted for degradation by the oxygen- and iron-dependent prolyl hydroxylases.40 HIFs target a number of genes involved in iron homeostasis to influence not only cellular iron homeostasis, but also systemic iron homeostasis and erythropoiesis. In particular, HIF2α is critical regulator of erythropoietin expression41 and contributes to the upregulation of iron transport proteins DMT1 and ferroportin in duodenal enterocytes under iron deficiency conditions42–43. Interestingly, HIF2α is also an IRP target, suggesting another mechanism by which cellular iron levels can influence this pathway.44
At the systemic level, the iron hormone hepcidin is a major regulator of body iron balance (Figure 4)5. Hepcidin controls iron entry into circulation from absorptive enterocytes, iron recycling macrophages, and hepatocytes by binding to ferroportin and inducing its internalization and degradation in lysosomes.45 Stimulating hepcidin expression thereby inhibits iron absorption from the diet and iron release from recycling macrophages and other body stores. In contrast, lowering hepcidin levels promotes iron availability.
Figure 4. Regulation of systemic iron homeostasis.
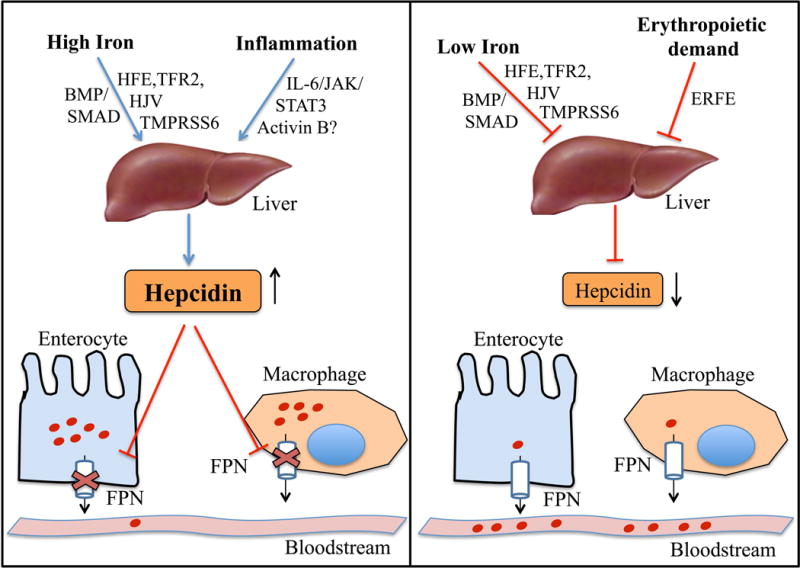
Secreted by the liver, hepcidin is a key iron hormone controls iron entry into circulation from absorptive enterocytes and iron-recycling macrophages by inducing FPN degradation. Iron loading and inflammation stimulate hepcidin transcription to prevent iron overload and sequester iron from pathogenic microorganisms (left panel). Iron deficiency and erythropoietic drive inhibit hepcidin transcription to provide adequate iron for erythropoiesis and other body requirements (right panel). Mediators of hepcidin regulation by iron, inflammation, and erythropoietic drive are indicated. Abbreviations: BMP, bone morphogenetic protein; HFE, hemochromatosis protein; TFR2, transferrin receptor 2; HJV, hemojuvelin; TMPRSS6 transmembrane protease serine 6; IL-6, interleukin 6; JAK, janus kinase; STAT3, signal transducer and activator of transcription 3; ERFE, erythroferrone.
The liver is the major source of circulating hepcidin that regulates systemic iron balance.46 Hepatocyte hepcidin expression is regulated by a number of different signals that communicate the body’s need for more or less iron (Figure 4).47 Major hepcidin regulators include iron, erythropoietic drive, and inflammation. In particular, iron loading stimulates hepcidin expression, whereas iron deficiency inhibits hepcidin as a feedback mechanism to maintain normal body iron levels. Enhancers of erythropoietic drive, such as anemia, erythropoietin administration, and hypoxia suppress hepcidin expression to increase the availability of iron for red blood cell production. Inflammatory cytokines stimulate hepcidin expression to limit the supply of iron to pathogenic microorganisms in the context of infection, but this also leads to hypoferremia and iron restricted erythropoiesis in chronic inflammatory diseases including CKD.
IRON HOMEOSTASIS DISORDERS
Given the essential functions of iron but the toxicity associated with iron excess, abnormalities in iron homeostasis are associated with a number of diseases.
IRON DEFICIENCY
The most widely recognized clinical manifestation of iron deficiency is anemia. According the the World Health Organization, anemia affects nearly one quarter of the world’s population, with 50% of cases attributable to iron deficiency.48 Major health consequences of anemia include an increased risk of maternal and child mortality, impaired cognitive and physical development in children, reduced physical performance and work productivity in adults, and cognitive decline in the elderly.48–49 In patients with CKD, anemia is associated with a lower quality of life and a higher risk of numerous adverse outcomes including hospitalization, cardiovascular disease, cognitive impairment, and death.50
Iron deficiency can be due to insufficient dietary iron absorption to meet the body iron needs from nutritional deficiency, malabsorption (e.g. due to celiac disease, gastric/gut resection, Helicobacter pylori colonization, protein pump inhibitors/H2 antagonists), increased iron requirements during pregnancy and rapid growth in children, or increased blood loss (e.g. due to gynecological losses or gastrointestinal losses from parasites, ulcers, malignancy, aspirin/nonsteroidal anti-inflammatory drugs).49 Patients with CKD are predisposed to iron deficiency due to nutritional deficits, medications that impair enterocyte iron uptake (phosphate binders, antacids), increased iron utilization induced by erythropoiesis stimulating agents, and increased blood losses from hemodialysis, frequent phlebotomy, and uremic platelet dysfunction.51
In addition to true iron deficiency, where total body iron levels are reduced, anemia can also be caused by functional iron deficiency, characterized by reduced levels of circulating iron that limit erythropoiesis despite adequate or high stores of total body iron. This is a characteristic feature of anemia of chronic disease or anemia of inflammation associated with a number of chronic diseases including autoimmune disorders, malignancy, and CKD.47 In these disorders, inflammatory cytokines such as IL6 activate the JAK/STAT3 pathway to induce the transcription of hepcidin, which downregulates ferroportin expression to inhibit dietary iron absorption and macrophage iron recycling (Figure 4). Hepcidin excess and functional iron deficiency in CKD can also be influenced by reduced renal clearance of this small peptide hormone.51
More rarely, iron deficiency anemia can be a consequence of mutations in genes involved in duodenal iron uptake, iron mobilization from body stores, or erythroid iron uptake or utilization, including DMT1 (SLC11A2), ceruloplasmin (CP), transferrin (TF).52–54 In many of these cases, the iron deficiency anemia is associated with tissue iron overload. Mutations in the hepcidin regulatory gene, TMPRSS6, lead to iron refractory iron deficiency anemia due to inability to suppress hepcidin production in the liver (Figure 4).55–56 These patients do not respond to oral iron supplementation and only partially respond to parenteral iron supplementation due to hepcidin-mediated ferroportin degradation that limits dietary iron absorption and inhibits iron release from macrophages that mobilize iron from parenteral preparations.
IRON OVERLOAD
Major causes of systemic iron overload are hereditary hemochromatosis, iron loading anemias (thalassemias, congenital dyserythropoietic anemias, sideroblastic anemias, myelodysplastic syndromes), and transfusional or other secondary forms of iron overload.57–58 In these disorders, iron exceeds the buffering capacity of transferrin leading to highly reactive forms of NTBI that are taken up in the liver, heart, and endocrine glands, where the excess iron fuels oxidative damage and organ dysfunction, leading to cirrhosis, cardiomyopathy, diabetes mellitus, and other endocrinopathies.
Hereditary hemochromatosis is due to mutations in genes encoding hepcidin itself (HAMP) or mutations in the genes that are the major inducers of hepcidin expression in response to iron: HFE, transferrin receptor 2 (TFR2), or hemojuvelin (HFE2, also known as HJV) (Figure 4).59–62 Mutations in these genes impair the ability of hepatocytes to detect increasing iron levels and activate signaling via the bone morphogenetic protein (BMP)-SMAD cascade to stimulate hepcidin transcription.51 This results in inappropriately low hepcidin levels, unregulated ferroportin activity, and inability to suppress dietary iron absorption and macrophage iron release, leading to iron accumulation in other tissues. Mutations in the hepcidin-binding site of ferroportin that render it resistant to hepcidin-mediated degradation cause a similar phenotype.63 Loss of function mutations in ferroportin lead to a distinct disorder (ferroportin disease) characterized by iron accumulation in macrophages.57, 64
Iron loading anemias are disorders characterized by ineffective erythropoiesis, resulting in hepcidin suppression, dietary hyperabsoprtion of iron and secondary iron overload.58 A prototypical example is β-thalassemia, where abnormalities in β-globin synthesis lead to toxicity and apoptosis of mature erythroblasts, resulting in anemia. This stimulates erythropoietin production and expansion of immature erythroid precursors, but erythropoiesis remains ineffective and anemia persists. The expanded erythroid precursor population secretes an excess of the erythroid regulator(s) that normally suppress hepcidin to increase iron availability for red blood cell production, thereby resulting in excessive iron absorption and iron overload. One such erythroid regulator of hepcidin was recently identified as erythroferrone (Figure 4).65 Iron overload is exacerbated in many of these conditions by blood transfusions.
Transfusional iron overload was previously common in the dialysis patient population before the advent of treatment with erythropoiesis stimulating agents. In the wake of studies raising safety concerns for ESAs when used to target higher hemoglobin levels and changes for dialysis reimbursement, intravenous iron supplementation transiently increased and average ferritin levels remain persistently higher in dialysis patients in the United States.66 Although the degree of iron loading in CKD patients is lower than in patients with hemochromatosis and thalassemia, where clear end organ toxicity from iron has been well described, there is evidence that iron may also be associated with disease outside the context of the massive iron overload seen in these genetic disorders.
IRON AND LIVER DISEASE
The liver is a main storage depot for iron and is the primary organ that clears excess circulating NTBI in conditions of iron overload. When the iron storage and anti-oxidant capacity of the liver is exceeded, iron overload can lead to oxidant-mediated liver injury, cirrhosis, and hepatocellular carcinoma, as seen in hereditary hemochromatosis and β-thalassemia.57–58 A number of other chronic liver diseases including alcoholic liver disease, nonalcoholic fatty liver disease, and viral hepatitis, are also associated with liver iron loading.57 In some cases, the pathogenic factors associated with these diseases and the liver damage itself may alter hepcidin expression to contribute to the iron overload.57 Although liver iron loading is typically lower grade in these conditions, iron-induced oxidative stress is thought to contribute to liver disease progression.57
IRON AND DIABETES MELLITUS
Diabetes mellitus is a common complication of iron overload disorders such as hemochromatosis, which was originally termed “bronze diabetes”, and β-thalassemia.67–68 Even in apparently healthy populations, increased dietary heme iron and high body iron stores, as measured by serum ferritin, are associated with an increased risk of type 2 diabetes and other insulin resistance states.69–72 A causative role for iron in the development of diabetes mellitus is supported by the fact that body iron reduction by phlebotomy or iron chelation improves glycemic control in these patients.73–75 The mechanisms by which iron contribute to diabetes pathogenesis are still not fully understood, but appear to be multifactorial, and may differ depending on the underlying cause of iron overload and tissue iron distribution.76 In hemochromatosis, iron accumulates in pancreatic islets, where it is thought to have a direct toxic effect on β cells by inducing oxidative stress and apoptosis, thereby impairing insulin secretion.77 Iron overload also causes insulin resistance by toxic effects on the liver. In secondary iron overload where hepcidin is upregulated and ferroportin expression is reduced, iron accumulates in adipocytes and contributes to insulin resistance by reducing production of the insulin-sensitizing hormone adiponectin.78
IRON AND CARDIOVASCULAR DISEASE
The heart is a major organ where iron accumulates in hereditary hemochromatosis and β-thalassemia. Iron accumulation is associated with cardiomyopathy, which is a major cause of morbidity and mortality in these patients.79 Clinically, this presents as a restrictive cardiomyopathy with prominent early diastolic dysfunction, arrhythmias, and progression to an end-stage dilated cardiomyopathy.79 The pathophysiology of iron-overload cardiomyopathy is multifactorial including oxidant-mediated injury, interference with cardiac electrical function, and promotion of fibrosis.79–80
Iron deficiency also has adverse consequences on the heart, an organ with high energy demands. Approximately 30–50% of patients with chronic heart failure are iron deficient81, which is associated with more severe heart failure symptoms and worse cardiovascular outcomes.82 Intravenous iron supplementation has been shown to improve heart failure symptoms, exercise capacity, and quality of life in this patient population83–84 and is therefore a recommended treatment for heart failure patients with iron deficiency.
An unresolved question is whether iron also has a role in promoting atherosclerosis, which was originally postulated over 30 years ago.85 Some epidemiologic studies in the general population have demonstrated an association between increased heme iron intake, body iron stores, and cardiovascular risk86–88, although other studies have found conflicting results.89 Pathogenic mechanisms suggested by some studies include promoting endothelial cell dysfunction, monocyte adhesion, and/or oxidative stress and plaque instability90–91; however, other studies do not support a pathogenic role.92 In CKD patients, some observational studies have suggested an association between higher doses of IV iron and cardiovascular death93, but no association has been seen in other studies.94–95 Limitations in serologic markers of iron status, the observational nature and/or limited size and duration of most studies, and different methods and location of iron accumulation may account for some differences between studies. Large prospective randomized trials are awaited to provide further clarification.
IRON AND NEURODEGENERATIVE DISEASES
Iron is the most abundant transition metal in the brain, where it is required to sustain the brain’s high respiratory activity, for myelinogenesis, and for the production of several neurotransmitters including dopamine and norepinephrine.96 Iron deficiency is well-established to impair brain development and cognition.97–98 On the contrary, iron overload or pathological deposition of this redox-active metal in the brain has been associated with many neurodegenerative disorders.99–101 Certain areas of the brain are particularly iron rich, including the basal ganglia, and it is these regions that tend to be impacted most in a group of disorders known as neurodegeneration with brain iron accumulation (NBIA) diseases, characterized by movement disorders and in some cases dementia and other neuropsychiatric symptoms.99–101 The clearest causal association is seen in a subset of NBIA diseases due to mutations in the iron homeostasis genes ceruloplasmin or ferritin light chain. In aceruloplasminemia, the loss of ceruloplasmin ferroxidase activity impairs astrocyte iron export and incorporation into extracellular transferrin for uptake by neurons. This leads to a combination of oxidative damage from iron overload in astrocytes and possibly also toxicity from neuronal iron deficiency.99–101 In neuroferritinopathy, mutations in ferritin light chain impair safe iron storage leading to iron-mediated oxidative damage.99–101 Mutations in frataxin, a mitochondrial iron chaperone important for iron-sulfur cluster assembly, heme biosynthesis, and mitochondrial iron storage, cause Friedrich’s ataxia, characterized by a loss of nerve cells in the spinal cord, cerebellum, and dorsal root ganglia, which is thought to be a consequence of mitochondrial dysfunction, oxidative stress, and possibly induction of sphingolipid synthesis and other signaling pathways induced by iron accumulation.99, 101–102
Iron accumulation is also associated with more common neurodegenerative diseases including Parkinson’s disease and Alzheimer’s disease.99 Whether iron accumulation contributes to the pathogenesis of these neurological disorders or whether iron accumulation occurs as a result of the pathogenic condition is still a matter of debate, but there is some evidence of a mechanistic role. A functional iron-responsive element (IRE) has been described in the 5-UTR of amyloid precursor protein (APP) mRNA and putative IRE-like sequence in the 5′-UTR of alpha-synuclein mRNA103–104, suggesting that these proteins associated with Alzheimer’s and Parkison’s disease pathogenesis could be upregulated as a consequence of iron loading. APP has been reported to stabilize the iron exporter ferroportin to facilitate iron egress from neurons.105–106 Similarly, elevated expression of DMT1 isoform in substantia nigra107 and alterations of other iron homeostasis proteins that may lead to iron accumulation have been reported in Parkinson’s disease patients.108 Neuronal iron accumulation may then exacerbate oxidative stress and neuotoxicity.
IRON AND CANCER
Iron overload is associated with an increased risk of hepatocellular carcinoma and possibly other cancers in patients with hereditary hemochromatosis and β-thalassemia.109–111 Iron excess is thought to contribute to cancer development by two main mechanisms.112–113 First, the pro-oxidant effects of iron can damage DNA and thereby promote oncogenesis. Second, cancer cells have an enhanced dependence on iron to maintain their rapid growth rate. Interestingly, multiple cancer cell types exhibit altered expression of iron homeostasis proteins that favor iron accumulation, including increased expression of TFR1 (to increase iron uptake), decreased expression of ferroportin (to reduce iron export), and increased hepcidin production (for autocrine downregulaton ferroportin expression to reduce iron export).112–113 Moreover, some data suggests that altered expression of these proteins may have a functional effect on tumor growth and survival. For example, in breast cancer, low tumor expression of ferroportin is associated with metastatic progression and reduced survival, while high tumor expression of ferroportin and low expression of hepcidin predicts a favorable prognosis.114 Iron chelators are being studied for use in cancer treatment by iron depleting cancer cells and/or promoting reactive oxidative stress to which cancer cells are particularly vulnerable.113 Erythropoiesis stimulating agents have been demonstrated to increase the risk of malignancy-related adverse outcomes115–116, and it will be important to study similar outcomes related to iron usage in larger prospective randomized controlled trials in CKD patients.
IRON AND KIDNEY DISEASE
Iron and iron-induced reactive oxygen species have been implicated in the pathogenesis of multiple models of acute kidney injury (AKI).117–118 Kidney iron accumulation has also been noted in chronic kidney disease models.119–121 It is thought that ischemic or toxic insults to the kidney increase the intracellular release of labile iron (also known as catalytic iron) that result in oxidative damage. More recently, a specific iron-dependent type of regulated cell death due to lipid peroxide accumulation, termed ferroptosis, has been identified and is implicated the pathogenesis of renal ischemia-reperfusion injury.122 Systemic iron mobilization from liver and spleen may also contribute to kidney iron accumulation and toxicity in renal-ischemia reperfusion injury.123 A functional role for iron in AKI pathogenesis is supported by the protective effects of treatment with iron chelators in many different AKI models.118, 124 Iron metabolism proteins heme-oxygenase 1 (HO-1), ferritin, and hepcidin have all been demonstrated to play protective roles in AKI by enhancing antioxidant defense, sequestering iron, and altering iron trafficking.123, 125–126 Interestingly, some evidence suggest that intravenous iron administration in combination with a protoporphyrin may have a protective effect in AKI, possibly by inducing some of these endogenous protective pathways.127 Further studies are needed to better understand the role of iron in the pathogenesis of kidney disease and the potentially harmful or beneficial effects of intravenous iron administration on kidney disease.
IRON AND MINERAL AND BONE DISORDERS
Osteoporosis is prevalent in patients with hemochromatosis and β-thalassemia.128–129 The etiology is likely multifactorial, but there is evidence that iron deposits in bone and can directly impair bone formation or remodeling.130 Iron deficiency may also impact bone health by inducing production of the bone-derived hormone fibroblast growth factor 23 (FGF23) that regulates phosphorous and vitamin D homeostasis.131–132 In normal patients, the increased FGF23 production is coupled with an increase in cleavage so that there is minimal change in bioactive intact FGF23 levels. However, in patients with autosomal dominant hypophosphatemic rickets with mutations that impair FGF23 cleavage, iron deficiency increases intact FGF23 levels and exacerbates clinical manifestations of renal phosphate wasting, inappropriately low vitamin D levels, and rickets/osteomalacia.133–134 FGF23 levels are markedly elevated in CKD patients, and this is an important causative factor in the mineral and bone disorders (CKD-MBD) common in this patient population.131 Notably, FGF23 cleavage is also impaired in CKD for reasons that are still poorly understood, and iron deficiency may therefore may contribute to elevated FGF23 levels, CKD-MBD, and other adverse consequences associated with excess FGF23 in CKD patients.131–132
IRON, IMMUNITY AND INFECTION
Pathogenic microorganisms, like humans, require iron for survival and proliferation, and have evolved a number of mechanisms for obtaining iron from their human hosts. It has been hypothesized that one function of the hepcidin-ferroportin system in humans is to restrict iron availability in the context of infection by sequestering iron in macrophages and reducing circulating iron levels.135 This is most clearly established for siderophilic bacteria such as Vibrio vulnificus and Yersinia enterolitica, since these infections have increased pathogenicity in patients with hemochromatosis or β-thalassemia135 and hepcidin knockout mice.136 The protective effects of iron deficiency for malaria, diarrheal illnesses, and tuberculosis in the developing world is supported by the increased rate of these infections and their associated morbidity and mortality in some iron supplementation studies.137–139 Notably, hepcidin upregulation and macrophage iron sequestration is not always protective and may depend on the organism and its niche. For example, intracellular organisms that resides in macrophages, such as Salmonella, may have increased pathogenicity in this context.140–141 In addition to impacting iron availability to pathogens, iron may also influence the host immune response by altering macrophage cytokine production and function.142–144 The impact of altered iron homeostasis and iron supplementation strategies on infectious complications in CKD patients is still not well understood.145
CONCLUSIONS
CKD patients are prone to iron deficiency anemia and functional iron deficiency as a consequence of hepcidin excess, and intravenous iron administration is a mainstay of anemia management in this patient population. Although both iron deficiency and iron excess are associated with numerous adverse health consequences in many different organ systems, data are limited to understand the optimal iron treatment strategy for CKD patients. There are many key areas for future research. We need better diagnostic parameters for accurately gauging iron status and a better understanding of the risks versus benefits of intravenous iron administration, differences among available iron preparations, and optimal dosing regimens and treatment targets for iron administration. Fundamental research, animal studies, and larger prospective randomized controlled trials in human patients with hard clinical outcomes are needed to improve our knowledge and evidence based practice moving forward.
Acknowledgments
FUNDING DISCLOSURE
JLB is supported by a Howard Goodman Fellowship Award from the Massachusetts General Hospital and NIH grants RO1-DK087727 and R01-DK102815.
Footnotes
CONFLICT OF INTEREST STATEMENT
JLB has ownership interest in Ferrumax Pharmaceuticals, Inc. SD has nothing to declare.
References
- 1.Martin W, Russell MJ. On the origins of cells: a hypothesis for the evolutionary transitions from abiotic geochemistry to chemoautotrophic prokaryotes, and from prokaryotes to nucleated cells. Philos Trans R Soc Lond B Biol Sci. 2003;358(1429):59–83. doi: 10.1098/rstb.2002.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Evstatiev R, Gasche C. Iron sensing and signalling. Gut. 2012;61(6):933–952. doi: 10.1136/gut.2010.214312. [DOI] [PubMed] [Google Scholar]
- 3.Pietrangelo A. Pathogens, Metabolic Adaptation, and Human Diseases–An Iron-Thrifty Genetic Model. Gastroenterology. 2015;149(4):834–838. doi: 10.1053/j.gastro.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 4.Jomova K, Valko M. Advances in metal-induced oxidative stress and human disease. Toxicology. 2011;283(2–3):65–87. doi: 10.1016/j.tox.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 5.Ganz T. Systemic iron homeostasis. Physiol Rev. 2013;93(4):1721–1741. doi: 10.1152/physrev.00008.2013. [DOI] [PubMed] [Google Scholar]
- 6.Pantopoulos K, Porwal SK, Tartakoff A, Devireddy L. Mechanisms of mammalian iron homeostasis. Biochemistry. 2012;51(29):5705–5724. doi: 10.1021/bi300752r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang J, Mori K, Li JY, Barasch J. Iron, lipocalin, and kidney epithelia. Am J Physiol Renal Physiol. 2003;285(1):F9–18. doi: 10.1152/ajprenal.00008.2003. [DOI] [PubMed] [Google Scholar]
- 8.McKie AT, Barrow D, Latunde-Dada GO, et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science. 2001;291:1755–1759. doi: 10.1126/science.1057206. [DOI] [PubMed] [Google Scholar]
- 9.Fleming MD, Trenor CC, III, Su MA, et al. Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat Genet. 1997;16:383–386. doi: 10.1038/ng0897-383. [DOI] [PubMed] [Google Scholar]
- 10.Gunshin H, Mackenzie B, Berger UV, et al. Cloning and characterization of a mammalian proton-ion transporter. Nature. 1997;388:482–488. doi: 10.1038/41343. [DOI] [PubMed] [Google Scholar]
- 11.Gunshin H, Fujiwara Y, Custodio AO, et al. Slc11a2 is required for intestinal iron absorption and erythropoiesis but dispensable in placenta and liver. J Clin Invest. 2005;115:1258–1266. doi: 10.1172/JCI24356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shawki A, Engevik MA, Kim RS, et al. Intestinal brush-border Na+/H+ exchanger-3 drives H+-coupled iron absorption in the mouse. Am J Physiol Gastrointest Liver Physiol. 2016;311(3):G423–430. doi: 10.1152/ajpgi.00167.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weintraub LR, Weinstein MB, Huser HJ, et al. Absorption of hemoglobin iron: the role of a heme-splitting substance in the intestinal mucosa. J Clin Invest. 1968;47:531–539. doi: 10.1172/JCI105749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Theil EC, Chen H, Miranda C, et al. Absorption of iron from ferritin is independent of heme iron and ferrous salts in women and rat intestinal segments. J Nutr. 2012;142(3):478–483. doi: 10.3945/jn.111.145854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arosio P, Carmona F, Gozzelino R, Maccarinelli F, Poli M. The importance of eukaryotic ferritins in iron handling and cytoprotection. Biochem J. 2015;472(1):1–15. doi: 10.1042/BJ20150787. [DOI] [PubMed] [Google Scholar]
- 16.Vanoaica L, Darshan D, Richman L, Schümann K, Kühn LC. Intestinal ferritin H is required for an accurate control of iron absorption. Cell Metab. 2010;12(3):273–282. doi: 10.1016/j.cmet.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 17.Donovan A, Brownlie A, Zhou Y, et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature. 2000;403:776–781. doi: 10.1038/35001596. [DOI] [PubMed] [Google Scholar]
- 18.Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem. 2000;275:19906–19912. doi: 10.1074/jbc.M000713200. [DOI] [PubMed] [Google Scholar]
- 19.McKie AT, Marciani P, Rolfs A, et al. A novel duodenal ironregulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell. 2000;5:299–309. doi: 10.1016/s1097-2765(00)80425-6. [DOI] [PubMed] [Google Scholar]
- 20.Drakesmith H, Nemeth E, Ganz T. Ironing out Ferroportin. Cell Metab. 2015;22(5):777–87. doi: 10.1016/j.cmet.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vulpe CD, Kuo YM, Murphy TL, et al. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet. 1999;21:195–199. doi: 10.1038/5979. [DOI] [PubMed] [Google Scholar]
- 22.Harris ZL, Durley AP, Man TK, Gitlin JD. Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc Natl Acad Sci U S A. 1999;96(19):10812–10817. doi: 10.1073/pnas.96.19.10812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cherukuri S, Potla R, Sarkar J, Nurko S, Harris ZL, Fox PL. Unexpected role of ceruloplasmin in intestinal iron absorption. Cell Metab. 2005;2(5):309–319. doi: 10.1016/j.cmet.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 24.Nai A, Lidonnici MR, Rausa M, et al. The second transferrin receptor regulates red blood cell production in mice. Blood. 2015;125(7):1170–1179. doi: 10.1182/blood-2014-08-596254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wood JC. Guidelines for quantifying iron overload. Hematology Am Soc Hematol Educ Program. 2014;2014(1):210–215. doi: 10.1182/asheducation-2014.1.210. [DOI] [PubMed] [Google Scholar]
- 26.Jenkitkasemwong S, Wang CY, Coffey R, et al. SLC39A14 Is Required for the Development of Hepatocellular Iron Overload in Murine Models of Hereditary Hemochromatosis. Cell Metab. 2015;22(1):138–150. doi: 10.1016/j.cmet.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cohen LA, Gutierrez L, Weiss A, et al. Serum ferritin is derived primarily from macrophages through a nonclassical secretory pathway. Blood. 2010;116(9):1574–1584. doi: 10.1182/blood-2009-11-253815. [DOI] [PubMed] [Google Scholar]
- 28.Meyron-Holtz EG, Moshe-Belizowski S, Cohen LA. A possible role for secreted ferritin in tissue iron distribution. J Neural Transm (Vienna) 2011;118(3):337–347. doi: 10.1007/s00702-011-0582-0. [DOI] [PubMed] [Google Scholar]
- 29.Bao G, Clifton M, Hoette TM, et al. Iron traffics in circulation bound to a siderocalin (Ngal)-catechol complex. Nat Chem Biol. 2010;6(8):602–609. doi: 10.1038/nchembio.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi H, Bencze KZ, Stemmler TL, Philpott CC. A cytosolic iron chaperone that delivers iron to ferritin. Science. 2008;320(5880):1207–1210. doi: 10.1126/science.1157643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Babcock M, de Silva D, Oaks R, et al. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science. 1997;276(5319):1709–1712. doi: 10.1126/science.276.5319.1709. [DOI] [PubMed] [Google Scholar]
- 32.Shaw GC, Cope JJ, Li L, et al. Mitoferrin is essential for erythroid iron assimilation. Nature. 2006;440(7080):96–100. doi: 10.1038/nature04512. [DOI] [PubMed] [Google Scholar]
- 33.Chiabrando D, Marro S, Mercurio S, et al. The mitochondrial heme exporter FLVCR1b mediates erythroid differentiation. J Clin Invest. 2012;122(12):4569–4579. doi: 10.1172/JCI62422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509(7498):105–109. doi: 10.1038/nature13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mancias JD, Pontano Vaites L, Nissim S, et al. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife. 2015;4 doi: 10.7554/eLife.10308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keel SB, Doty RT, Yang Z, et al. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science. 2008;319(5864):825–828. doi: 10.1126/science.1151133. [DOI] [PubMed] [Google Scholar]
- 37.Muckenthaler MU, Galy B, Hentze MW. Systemic iron homeostasis and the iron-responsive element/ironregulatory protein (IRE/IRP) regulatory network. Annu Rev Nutr. 2008;28:197–213. doi: 10.1146/annurev.nutr.28.061807.155521. [DOI] [PubMed] [Google Scholar]
- 38.Vashisht AA, Zumbrennen KB, Huang X, et al. Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science. 2009;326(5953):718–721. doi: 10.1126/science.1176333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salahudeen AA, Thompson JW, Ruiz JC, et al. An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science. 2009;326(5953):722–726. doi: 10.1126/science.1176326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peyssonnaux C, Nizet V, Johnson RS. Role of the hypoxia inducible factors HIF in iron metabolism. Cell Cycle. 2008;7(1):28–32. doi: 10.4161/cc.7.1.5145. [DOI] [PubMed] [Google Scholar]
- 41.Gruber M, Hu CJ, Johnson RS, Brown EJ, Keith B, Simon MC. Acute postnatal ablation of Hif-2α results in anemia. Proc Natl Acad Sci U S A. 2007;104(7):2301–2306. doi: 10.1073/pnas.0608382104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shah YM, Matsubara T, Ito S, Yim SH, Gonzalez FJ. Intestinal hypoxia-inducible transcription factors are essential for iron absorption following iron deficiency. Cell Metab. 2009;9(2):152–164. doi: 10.1016/j.cmet.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taylor M, Qu A, Anderson ER, et al. Hypoxia-inducible factor-2α mediates the adaptive increase of intestinal ferroportin during iron deficiency in mice. Gastroenterology. 2011;140(7):2044–2055. doi: 10.1053/j.gastro.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anderson SA, Nizzi CP, Chang YI, et al. The IRP1-HIF-2α axis coordinates iron and oxygen sensing with erythropoiesis and iron absorption. Cell Metab. 2013;17(2):282–290. doi: 10.1016/j.cmet.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 46.Zumerle S, Mathieu JR, Delga S, et al. Targeted disruption of hepcidin in the liver recapitulates the hemochromatotic phenotype. Blood. 2014;123(23):3646–3650. doi: 10.1182/blood-2014-01-550467. [DOI] [PubMed] [Google Scholar]
- 47.Wang CY, Babitt JL. Hepcidin regulation in the anemia of inflammation. Curr Opin Hematol. 2016;23(3):189–197. doi: 10.1097/MOH.0000000000000236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.WHO Global Database on Anaemia. de Benoist Bruno, McLean Erin, Egli Ines, Cogswell Mary., editors. World prevalence of anaemia 1993–2005. Geneva: World Health Organization; 2008. [Google Scholar]
- 49.Lopez A, Cacoub P, Macdougall IC, Peyrin-Biroulet L. Iron deficiency anaemia. Lancet. 2016;387(10021):907–916. doi: 10.1016/S0140-6736(15)60865-0. [DOI] [PubMed] [Google Scholar]
- 50.National Kidney Foundation. KDOQI Clinical Practice Guidelines and Clinical Practice Recommendations for Anemia in Chronic Kidney Disease. Am J Kidney Dis. 2006;47(5 suppl 3):S1–145. doi: 10.1053/j.ajkd.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 51.Zumbrennen-Bullough K, Babitt JL. The iron cycle in chronic kidney disease (CKD): from genetics and experimental models to CKD patients. Nephrol Dial Transplant. 2014;29(2):263–273. doi: 10.1093/ndt/gft443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mims MP, Guan Y, Pospisilova D, et al. Identification of a human mutation of DMT1 in a patient with microcytic anemia and iron overload. Blood. 2005;105(3):1337–1342. doi: 10.1182/blood-2004-07-2966. [DOI] [PubMed] [Google Scholar]
- 53.Beutler E, Gelbart T, Lee P, Trevino R, Fernandez MA, Fairbanks VF. Molecular characterization of a case of atransferrinemia. Blood. 2000;96(13):4071–4074. [PubMed] [Google Scholar]
- 54.Harris ZL, Takahashi Y, Miyajima H, Serizawa M, MacGillivray RT, Gitlin JD. Aceruloplasminemia: molecular characterization of this disorder of iron metabolism. Proc Natl Acad Sci U S A. 1995;92(7):2539–2543. doi: 10.1073/pnas.92.7.2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Finberg KE, Heeney MM, Campagna DR, et al. Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA) Nat Genet. 2008;40(5):569–571. doi: 10.1038/ng.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Du X, She E, Gelbart T, et al. The serine protease TMPRSS6 is required to sense iron deficiency. Science. 2008;320(5879):1088–1092. doi: 10.1126/science.1157121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pietrangelo A. Iron and the liver. Liver Int. 2016;36(Suppl 1):116–123. doi: 10.1111/liv.13020. [DOI] [PubMed] [Google Scholar]
- 58.Camaschella C, Nai A. Ineffective erythropoiesis and regulation of iron status in iron loading anaemias. Br J Haematol. 2016;172(4):512–523. doi: 10.1111/bjh.13820. [DOI] [PubMed] [Google Scholar]
- 59.Roetto A, Papanikolaou G, Politou M, et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet. 2003;33(1):21–22. doi: 10.1038/ng1053. [DOI] [PubMed] [Google Scholar]
- 60.Feder JN, Gnirke A, Thomas W, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13(4):399–408. doi: 10.1038/ng0896-399. [DOI] [PubMed] [Google Scholar]
- 61.Camaschella C, Roetto A, Calì A, et al. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat Genet. 2000;25(1):14–15. doi: 10.1038/75534. [DOI] [PubMed] [Google Scholar]
- 62.Papanikolaou G, Samuels ME, Ludwig EH, et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat Genet. 2004;36(1):77–82. doi: 10.1038/ng1274. [DOI] [PubMed] [Google Scholar]
- 63.Drakesmith H, Schimanski LM, Ormerod E, et al. Resistance to hepcidin is conferred by hemochromatosis-associated mutations of ferroportin. Blood. 2005;106(3):1092–1097. doi: 10.1182/blood-2005-02-0561. [DOI] [PubMed] [Google Scholar]
- 64.Montosi G, Donovan A, Totaro A, Garuti C, Pignatti E, Cassanelli S, Trenor CC, Gasparini P, Andrews NC, Pietrangelo A. Autosomal-dominant hemochromatosis is associated with a mutation in the ferroportin (SLC11A3) gene. J Clin Invest. 2001;108(4):619–23. doi: 10.1172/JCI13468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kautz L, Jung G, Valore EV, Rivella S, Nemeth E, Ganz T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet. 2014;46(7):678–684. doi: 10.1038/ng.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Karaboyas A, Zee J, Morgenstern H, et al. Understanding the Recent Increase in Ferritin Levels in United States Dialysis Patients: Potential Impact of Changes in Intravenous Iron and Erythropoiesis-Stimulating Agent Dosing. Clin J Am Soc Nephrol. 2015;10:1814–1821. doi: 10.2215/CJN.02600315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McClain DA, Abraham D, Rogers J, et al. High prevalence of abnormal glucose homeostasis secondary to decreased insulin secretion in individuals with hereditary haemochromatosis. Diabetologia. 2006;49(7):1661–1669. doi: 10.1007/s00125-006-0200-0. [DOI] [PubMed] [Google Scholar]
- 68.Vogiatzi MG, Macklin EA, Trachtenberg FL, et al. Thalassemia Clinical Research Network. Differences in the prevalence of growth, endocrine and vitamin D abnormalities among the various thalassaemia syndromes in North America. Br J Haematol. 2009;146(5):546–556. doi: 10.1111/j.1365-2141.2009.07793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ford ES, Cogswell ME. Diabetes and serum ferritin concentration among U.S. adults. Diabetes Care. 1999;22(12):1978–1983. doi: 10.2337/diacare.22.12.1978. [DOI] [PubMed] [Google Scholar]
- 70.Jiang R, Ma J, Ascherio A, Stampfer MJ, Willett WC, Hu FB. Dietary iron intake and blood donations in relation to risk of type 2 diabetes in men: a prospective cohort study. Am J Clin Nutr. 2004;79(1):70–75. doi: 10.1093/ajcn/79.1.70. [DOI] [PubMed] [Google Scholar]
- 71.Song Y, Manson JE, Buring JE, Liu S. A prospective study of red meat consumption and type 2 diabetes in middle-aged and elderly women: the women’s health study. Diabetes Care. 2004;27(9):2108–2115. doi: 10.2337/diacare.27.9.2108. [DOI] [PubMed] [Google Scholar]
- 72.Jiang R, Manson JE, Meigs JB, Ma J, Rifai N, Hu FB. Body iron stores in relation to risk of type 2 diabetes in apparently healthy women. JAMA. 2004;291(6):711–717. doi: 10.1001/jama.291.6.711. [DOI] [PubMed] [Google Scholar]
- 73.Dymock IW, Cassar J, Pyke DA, Oakley WG, Williams R. Observations on the pathogenesis, complications and treatment of diabetes in 115 cases of haemochromatosis. Am J Med. 1972;52(2):203–210. doi: 10.1016/0002-9343(72)90070-8. [DOI] [PubMed] [Google Scholar]
- 74.Platis O1, Anagnostopoulos G, Farmaki K, Posantzis M, Gotsis E, Tolis G. Glucose metabolism disorders improvement in patients with thalassaemia major after 24–36 months of intensive chelation therapy. Pediatr Endocrinol Rev. 2004;2(Suppl 2):279–281. [PubMed] [Google Scholar]
- 75.Fernández-Real JM, Peñarroja G, Castro A, García-Bragado F, Hernández-Aguado I, Ricart W. Blood letting in high-ferritin type 2 diabetes: effects on insulin sensitivity and beta-cell function. Diabetes. 2002;51(4):1000–1004. doi: 10.2337/diabetes.51.4.1000. [DOI] [PubMed] [Google Scholar]
- 76.Simcox JA, McClain DA. Iron and diabetes risk. Cell Metab. 2013;17(3):329–341. doi: 10.1016/j.cmet.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cooksey RC, Jouihan HA, Ajioka RS, et al. Oxidative stress, beta-cell apoptosis, and decreased insulin secretory capacity in mouse models of hemochromatosis. Endocrinology. 2004;145(11):5305–5312. doi: 10.1210/en.2004-0392. [DOI] [PubMed] [Google Scholar]
- 78.Gabrielsen JS, Gao Y, Simcox JA, et al. Adipocyte iron regulates adiponectin and insulin sensitivity. J Clin Invest. 2012;122(10):3529–3540. doi: 10.1172/JCI44421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pennell DJ, Udelson JE, Arai AE, et al. American Heart Association Committee on Heart Failure and Transplantation of the Council on Clinical Cardiology and Council on Cardiovascular Radiology and Imaging. Cardiovascular function and treatment in β-thalassemia major: a consensus statement from the American Heart Association. Circulation. 2013;128(3):281–308. doi: 10.1161/CIR.0b013e31829b2be6. [DOI] [PubMed] [Google Scholar]
- 80.Murphy CJ, Oudit GY. Iron-overload cardiomyopathy: pathophysiology, diagnosis, and treatment. J Card Fail. 2010;16(11):888–900. doi: 10.1016/j.cardfail.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 81.Klip IT, Comin-Colet J, Voors AA, et al. Iron deficiency in chronic heart failure: an international pooled analysis. Am Heart J. 2013;165:575–582 e573. doi: 10.1016/j.ahj.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 82.Comin-Colet J, Enjuanes C, Gonzalez G, et al. Iron deficiency is a key determinant of health-related quality of life in patients with chronic heart failure regardless of anaemia status. Eur J Heart Fail. 2013;15:1164–1172. doi: 10.1093/eurjhf/hft083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Anker SD, Comin Colet J, Filippatos G, et al. Ferric carboxymaltose in patients with heart failure and iron deficiency. N Engl J Med. 2009;361:2436–2448. doi: 10.1056/NEJMoa0908355. [DOI] [PubMed] [Google Scholar]
- 84.Ponikowski P, van Veldhuisen DJ, Comin-Colet J, et al. Beneficial effects of long-term intravenous iron therapy with ferric carboxymaltose in patients with symptomatic heart failure and iron deficiency. Eur Heart J. 2015;36(11):657–668. doi: 10.1093/eurheartj/ehu385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sullivan JL. Iron and the sex difference in heart disease risk. Lancet. 1981;1(8233):1293–1294. doi: 10.1016/s0140-6736(81)92463-6. [DOI] [PubMed] [Google Scholar]
- 86.Ascherio A, Willett WC, Rimm EB, Giovannucci EL, Stampfer MJ. Dietary iron intake and risk of coronary disease among men. Circulation. 1994;89(3):969–974. doi: 10.1161/01.cir.89.3.969. [DOI] [PubMed] [Google Scholar]
- 87.van der ADL, Peeters PH, Grobbee DE, Marx JJ, van der Schouw YT. Dietary haem iron and coronary heart disease in women. Eur Heart J. 2005;26(3):257–262. doi: 10.1093/eurheartj/ehi027. [DOI] [PubMed] [Google Scholar]
- 88.Kiechl S, Willeit J, Egger G, Poewe W, Oberhollenzer F. Body iron stores and the risk of carotid atherosclerosis: prospective results from the Bruneck study. Circulation. 1997;96(10):3300–3307. doi: 10.1161/01.cir.96.10.3300. [DOI] [PubMed] [Google Scholar]
- 89.Danesh J, Appleby P. Coronary heart disease and iron status: meta-analyses of prospective studies. Circulation. 1999;99(7):852–854. doi: 10.1161/01.cir.99.7.852. [DOI] [PubMed] [Google Scholar]
- 90.Kuo KL, Hung SC, Lee TS, Tarng DC. Iron sucrose accelerates early atherogenesis by increasing superoxide production and upregulating adhesion molecules in CKD. J Am Soc Nephrol. 2014;25(11):2596–2606. doi: 10.1681/ASN.2013080838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sullivan JL. Iron in arterial plaque: modifiable risk factor for atherosclerosis. Biochim Biophys Acta. 2009;1790(7):718–723. doi: 10.1016/j.bbagen.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 92.Kautz L, Gabayan V, Wang X, et al. Testing the iron hypothesis in a mouse model of atherosclerosis. Cell Rep. 2013;5(5):1436–1442. doi: 10.1016/j.celrep.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bailie GR, Larkina M, Goodkin DA, et al. Data from the Dialysis Outcomes and Practice Patterns Study validate an association between high intravenous iron doses and mortality. Kidney Int. 2015;87(1):162–168. doi: 10.1038/ki.2014.275. [DOI] [PubMed] [Google Scholar]
- 94.Kshirsagar AV, Freburger JK, Ellis AR, Wang L, Winkelmayer WC, Brookhart MA. Intravenous iron supplementation practices and short-term risk of cardiovascular events in hemodialysis patients. PLoS One. 2013;8(11):e78930. doi: 10.1371/journal.pone.0078930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Miskulin DC, Tangri N, Bandeen-Roche K, et al. Developing Evidence to Inform Decisions about Effectiveness (DEcIDE) Network Patient Outcomes in End Stage Renal Disease Study Investigators. Intravenous iron exposure and mortality in patients on hemodialysis. Clin J Am Soc Nephrol. 2014;9(11):1930–1939. doi: 10.2215/CJN.03370414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Moos T, Rosengren Nielsen T, Skjørringe T, Morgan EH. Iron trafficking inside the brain. J Neurochem. 2007;103(5):1730–1740. doi: 10.1111/j.1471-4159.2007.04976.x. [DOI] [PubMed] [Google Scholar]
- 97.Beard J. Iron deficiency alters brain development and functioning. J Nutr. 2003;133(5 Suppl 1):1468S–1472S. doi: 10.1093/jn/133.5.1468S. [DOI] [PubMed] [Google Scholar]
- 98.Youdim MB. Brain iron deficiency and excess; cognitive impairment and neurodegeneration with involvement of striatum and hippocampus. Neurotox Res. 2008;14(1):45–56. doi: 10.1007/BF03033574. [DOI] [PubMed] [Google Scholar]
- 99.Singh N, Haldar S, Tripathi AK, et al. Brain iron homeostasis: from molecular mechanisms to clinical significance and therapeutic opportunities. Antioxid Redox Signal. 2014;20(8):1324–1363. doi: 10.1089/ars.2012.4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Poujois A, Devedjian JC, Moreau C, et al. Bioavailable Trace Metals in Neurological Diseases. Curr Treat Options Neurol. 2016;18(10):46. doi: 10.1007/s11940-016-0426-1. [DOI] [PubMed] [Google Scholar]
- 101.Rouault TA. Iron metabolism in the CNS: implications for neurodegenerative diseases. Nat Rev Neurosci. 2013;14(8):551–564. doi: 10.1038/nrn3453. [DOI] [PubMed] [Google Scholar]
- 102.Chen K, Lin G, Haelterman NA, et al. Loss of Frataxin induces iron toxicity, sphingolipid synthesis, and Pdk1/Mef2 activation, leading to neurodegeneration. Elife. 2016;5 doi: 10.7554/eLife.16043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rogers JT, Randall JD, Cahill CM, et al. An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J Biol Chem. 2002;277(47):45518–45528. doi: 10.1074/jbc.M207435200. [DOI] [PubMed] [Google Scholar]
- 104.Friedlich AL, Tanzi RE, Rogers JT. The 5′-untranslated region of Parkinson’s disease alpha-synuclein messengerRNA contains a predicted iron responsive element. Mol Psychiatry. 2007;12(3):222–223. doi: 10.1038/sj.mp.4001937. [DOI] [PubMed] [Google Scholar]
- 105.Duce JA, Tsatsanis A, Cater MA, et al. Iron-export ferroxidase activity of β-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell. 2010;142(6):857–867. doi: 10.1016/j.cell.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wong BX, Tsatsanis A, Lim LQ, Adlard PA, Bush AI, Duce JA. β-Amyloid precursor protein does not possess ferroxidase activity but does stabilize the cell surface ferrous iron exporter ferroportin. PLoS One. 2014;9(12):e114174. doi: 10.1371/journal.pone.0114174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Salazar J, Mena N, Hunot S, et al. Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson’s disease. Proc Natl Acad Sci U S A. 2008;105(47):18578–18583. doi: 10.1073/pnas.0804373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jin L, Wang J, Zhao L, et al. Decreased serum ceruloplasmin levels characteristically aggravate nigral iron deposition in Parkinson’s disease. Brain. 2011;134(Pt 1):50–8. doi: 10.1093/brain/awq319. [DOI] [PubMed] [Google Scholar]
- 109.Fracanzani AL, Conte D, Fraquelli M, et al. Increased cancer risk in a cohort of 230 patients with hereditary hemochromatosis in comparison to matched control patients with non-iron-related chronic liver disease. Hepatology. 2001;33(3):647–651. doi: 10.1053/jhep.2001.22506. [DOI] [PubMed] [Google Scholar]
- 110.Osborne NJ, Gurrin LC, Allen KJ, et al. HFE C282Y homozygotes are at increased risk of breast and colorectal cancer. Hepatology. 2010;51(4):1311–1318. doi: 10.1002/hep.23448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Borgna-Pignatti C, Vergine G, Lombardo T, et al. Hepatocellular carcinoma in the thalassaemia syndromes. Br J Haematol. 2004;124(1):114–117. doi: 10.1046/j.1365-2141.2003.04732.x. [DOI] [PubMed] [Google Scholar]
- 112.Manz DH, Blanchette NL, Paul BT, Torti FM, Torti SV. Iron and cancer: recent insights. Ann N Y Acad Sci. 2016;1368(1):149–161. doi: 10.1111/nyas.13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bystrom LM, Rivella S. Cancer cells with irons in the fire. Free Radic Biol Med. 2015;79:337–342. doi: 10.1016/j.freeradbiomed.2014.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pinnix ZK, Miller LD, Wang W, et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Transl Med. 2010;2(43):43ra56. doi: 10.1126/scisignal.3001127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bohlius J, Schmidlin K, Brillant C, et al. Recombinant human erythropoiesis-stimulating agents and mortality in patients with cancer: a meta-analysis of randomised trials. Lancet. 2009;373(9674):1532–1542. doi: 10.1016/S0140-6736(09)60502-X. [DOI] [PubMed] [Google Scholar]
- 116.Pfeffer MA, Burdmann EA, Chen CY, et al. TREAT Investigators A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med. 2009;361(21):2019–2032. doi: 10.1056/NEJMoa0907845. [DOI] [PubMed] [Google Scholar]
- 117.Baliga R, Ueda N, Shah SV. Increase in bleomycin-detectable iron in ischaemia/reperfusion injury to rat kidneys. Biochem J. 1993;291(Pt 3):901–905. doi: 10.1042/bj2910901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Baliga R, Zhang Z, Baliga M, Ueda N, Shah SV. In vitro and in vivo evidence suggesting a role for iron in cisplatin-induced nephrotoxicity. Kidney Int. 1998;53(2):394–401. doi: 10.1046/j.1523-1755.1998.00767.x. [DOI] [PubMed] [Google Scholar]
- 119.Nankivell BJ, Boadle RA, Harris DC. Iron accumulation in human chronic renal disease. Am J Kidney Dis. 1992;20:580–584. doi: 10.1016/s0272-6386(12)70222-6. [DOI] [PubMed] [Google Scholar]
- 120.Wang H, Nishiya K, Ito H, Hosokawa T, Hashimoto K, Moriki T. Iron deposition in renal biopsy specimens from patients with kidney diseases. Am J Kidney Dis. 2001;38:1038–1044. doi: 10.1053/ajkd.2001.28593. [DOI] [PubMed] [Google Scholar]
- 121.Naito Y, Sawada H, Oboshi M, et al. Increased renal iron accumulation in hypertensive nephropathy of salt-loaded hypertensive rats. PLoS One. 2013;8:e75906. doi: 10.1371/journal.pone.0075906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Linkermann A, Skouta R, Himmerkus N, et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci U S A. 2014;111(47):16836–16841. doi: 10.1073/pnas.1415518111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Scindia Y, Dey P, Thirunagari A, et al. Hepcidin Mitigates Renal Ischemia-Reperfusion Injury by Modulating Systemic Iron Homeostasis. J Am Soc Nephrol. 2015;26(11):2800–2814. doi: 10.1681/ASN.2014101037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Paller MS, Hedlund BE. Role of iron in postischemic renal injury in the rat. Kidney Int. 1988;34(4):474–480. doi: 10.1038/ki.1988.205. [DOI] [PubMed] [Google Scholar]
- 125.Zarjou A, Bolisetty S, Joseph R, et al. Proximal tubule H-ferritin mediates iron trafficking in acute kidney injury. J Clin Invest. 2013;123(10):4423–4434. doi: 10.1172/JCI67867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nath KA. Heme oxygenase-1: a provenance for cytoprotective pathways in the kidney and other tissues. Kidney Int. 2006;70(3):432–443. doi: 10.1038/sj.ki.5001565. [DOI] [PubMed] [Google Scholar]
- 127.Zager RA, Johnson AC, Frostad KB. Combined iron sucrose and protoporphyrin treatment protects against ischemic and toxin-mediated acute renal failure. Kidney Int. 2016;90(1):67–76. doi: 10.1016/j.kint.2016.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Guggenbuhl P, Deugnier Y, Boisdet JF, et al. Bone mineral density in men with genetic hemochromatosis and HFE gene mutation. Osteoporos Int. 2005;16(12):1809–1814. doi: 10.1007/s00198-005-1934-0. [DOI] [PubMed] [Google Scholar]
- 129.Vogiatzi MA, Macklin EA, Fung EB, et al. Bone disease in thalassemia: a frequent and still unresolved problem. J Bone Miner Res. 2009;24(3):543–557. doi: 10.1359/jbmr.080505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mahachoklertwattana P, Sirikulchayanonta V, Chuansumrit A, et al. Bone histomorphometry in children and adolescents with beta-thalassemia disease: iron-associated focal osteomalacia. J Clin Endocrinol Metab. 2003;88(8):3966–3972. doi: 10.1210/jc.2002-021548. [DOI] [PubMed] [Google Scholar]
- 131.David V, Francis C, Babitt JL. Ironing out the cross talk between FGF23 and inflammation. Am J Physiol Renal Physiol. 2017;312(1):F1–F8. doi: 10.1152/ajprenal.00359.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.David V, Martin A, Isakova T, et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 2016;89(1):135–146. doi: 10.1038/ki.2015.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Farrow EG, Yu X, Summers LJ, et al. Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor-23 (Fgf23) knock-in mice. Proc Natl Acad Sci USA. 2011;108:E1146–E1155. doi: 10.1073/pnas.1110905108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Imel EA, Peacock M, Gray AK, et al. Iron modifies plasma FGF23 differently in autosomal dominant hypophosphatemic rickets and healthy humans. J Clin Endocrinol Metab. 2011;96:3541–3549. doi: 10.1210/jc.2011-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ganz T, Nemeth E. Iron homeostasis in host defence and inflammation. Nat Rev Immunol. 2015;15:500–510. doi: 10.1038/nri3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Arezes J, Jung G, Gabayan V, et al. Hepcidin-induced hypoferremia is a critical host defense mechanism against the siderophilic bacterium Vibrio vulnificus. Cell Host Microbe. 2015;17:47–57. doi: 10.1016/j.chom.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sazawal S, Black RE, Ramsan M, et al. Effects of routine prophylactic supplementation with iron and folic acid on admission to hospital and mortality in preschool children in a high malaria transmission setting: community-based, randomised, placebo-controlled trial. Lancet. 2006;367(9505):133–143. doi: 10.1016/S0140-6736(06)67962-2. [DOI] [PubMed] [Google Scholar]
- 138.Soofi S, Cousens S, Iqbal SP, et al. Effect of provision of daily zinc and iron with several micronutrients on growth and morbidity among young children in Pakistan: a cluster-randomised trial. Lancet. 2013;382(9886):29–40. doi: 10.1016/S0140-6736(13)60437-7. [DOI] [PubMed] [Google Scholar]
- 139.Murray MJ, Murray AB, Murray MB, Murray CJ. The adverse effect of iron repletion on the course of certain infections. Br Med J. 1978;2(6145):1113–1135. doi: 10.1136/bmj.2.6145.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Darton TC, Blohmke CJ, Giannoulatou E, et al. Rapidly Escalating Hepcidin and Associated Serum Iron Starvation Are Features of the Acute Response to Typhoid Infection in Humans. PLoS Negl Trop Dis. 2015;9:e0004029. doi: 10.1371/journal.pntd.0004029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kim DK, Jeong JH, Lee JM, et al. Inverse agonist of estrogen-related receptor γ controls Salmonella typhimurium infection by modulating host iron homeostasis. Nat Med. 2014;20:419–424. doi: 10.1038/nm.3483. [DOI] [PubMed] [Google Scholar]
- 142.Wang L, Harrington L, Trebicka E, et al. Selective modulation of TLR4-activated inflammatory responses by altered iron homeostasis in mice. J Clin Invest. 2009;119(11):3322–3328. doi: 10.1172/JCI39939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Pagani A, Nai A, Corna G, et al. Low hepcidin accounts for the proinflammatory status associated with iron deficiency. Blood. 2011;118(3):736–746. doi: 10.1182/blood-2011-02-337212. [DOI] [PubMed] [Google Scholar]
- 144.Nairz M, Haschka D, Demetz E, Weiss G. Iron at the interface of immunity and infection. Front Pharmacol. 2014;5:152. doi: 10.3389/fphar.2014.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Macdougall IC, Bircher AJ, Eckardt KU, et al. Conference Participants. Iron management in chronic kidney disease: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. 2016;89(1):28–39. doi: 10.1016/j.kint.2015.10.002. [DOI] [PubMed] [Google Scholar]