

Abstract
Clinical investigations have suggested that repression of the TGF-s type II receptor (TβRII) may be an important step in progression of lung adenocarcinoma from an indolent in situ state to a frank invasive carcinoma. To test this hypothesis, we compared the effects of deleting the murine homolog of this receptor (Tgfbr2) in a mouse model of mutant K-ras-induced lung carcinoma, that normally induces the formation of multifocal tumors of low invasive potential. In this model, loss of Tgfbr2 induced a highly invasive phenotype associated with lymph node metastasis and reduced survival. Tumor-associated stromal cells displayed an immunosuppressive profile marked by increased numbers of B and T cells. Moreover, tumor stromal cell profiling revealed a developmental TGF-β response profile that associated with a collagenized extracellular matrix and increased invasion of TGF-β non-responsive tumor cells. Together, these results suggest that our KrasTgfbr2 −/− mouse model of invasive lung carcinoma mirrors the genomic response and clinical progression of human lung adenocarcinoma, recapitulating changes in lung stromal pathways that occur in the tumor microenvironment during malignant progression in this disease.
Keywords: Lung adenocarcinoma, Lung cancer, Tumor microenvironment, TGF-β, Invasion
INTRODUCTION
The extent of the invasive component in lung adenocarcinoma is associated with clinical outcomes. Similar to malignancies in other organs, such as breast and cervix, tumors are defined as non-invasive (in-situ carcinoma), minimally-invasive (microscopic invasion), or as invasive carcinomas. The World Health Organization has subclassified lung adenocarcinoma based upon predominant cell morphology and growth pattern such as bronchioloalveolar carcinoma (BAC), adenocarcinoma with mixed subtypes (AC-mixed), and homogenously invasive tumors with a variety of histological patterns (1). BAC tumor cells are cuboidal to columnar, with or without mucin, and grow in a noninvasive, lepidic fashion along alveolar walls. Adenocarcinomas with mixed subtypes frequently contain regions of lepidic pattern tumor at the periphery of invasive tumor. Pure invasive adenocarcinomas are devoid of lepidic morphology.
The clinical importance of lung adenocarcinoma invasion is supported by several recent studies indicating that the risk of death in non-mucinous BAC is significantly lower than that of pure invasive tumors and in tumors with greater than 0.5 cm of fibrosis or linear invasion (2). These features have been incorporated into a proposed revision of lung adenocarcinoma classification that recognize the indolent nature of pure lepidic non-mucinous tumors as adenocarcinoma in situ (AIS) and of minimally invasive adenocarcinoma (MIA) (3). Together, these studies suggest that non-invasive lung adenocarcinomas are biologically indolent with five-year survival after resection approaching 100%, and that following the paradigm of other adenocarcinoma in other organs, these in situ tumors may acquire molecular alterations that promote invasion and thus increases the risk of metastatic disease and death.
To identify molecular pathways important for distinguishing adenocarcinoma subclasses and for mediating the acquisition and progression of invasion, we and others (4–8) performed microarray gene expression profiling of lung adenocarcinomas to identify signatures associated with histology and invasion. The results of unsupervised analyses show lung adenocarcinomas segregate into three major branches comprised predominantly of BAC/AIS, AC-Mixed subtype, and pure invasive tumors and provide biological plausibility to support the notion that these adenocarcinoma subtypes are distinct. Among the genes differentially expressed in the progression from BAC/AIS to invasive tumors was the transforming growth factor-β (TGF-β) type II receptor (TβRII)(4), which was expressed at reduced levels by AC-Mixed and solid invasive tumors compared with BAC/AIS. This finding suggests that TβRII repression is important for lung adenocarcinoma progression from BAC/AIS to invasive adenocarcinoma, and is supported by in vitro studies showing that loss of TGFβRII in lung cancer cells increases tumor cell invasion, as well as by constitutive genetic models of TGFβRII targeted deletion in other organ systems (9–11). This observation is also consistent with data demonstrating that TGF-β signaling in carcinoma cells appears to be a major regulator in the tumor microenvironment. Studies in murine models of oncogene-driven breast cancer in which the TGFβRII was deleted have shown that loss of response to TGF-β in mammary epithelial cells resulted in increased secretion of the chemokines Cxcl1, Cxcl5, and Cxcl12/Sdf1, followed by increased infiltration with myeloid cells that appeared to alter invasiveness and tumor migration (12). This paradigm has not yet been evaluated in murine models of lung cancer.
The goals of this paper were to determine: 1) biologic significance of our clinical observations relating loss of the TBRII to lung cancer invasiveness by creating a mouse model in which oncogenesis could be driven in the presence or absence of the TBRII in lung epithelial cells; 2) whether this model would resemble other mouse models of tumor cell loss of Tgfbr2; 3) whether this new model would more resemble human invasive lung adenocarcinomas than previously described mouse models; and 4) the downstream signaling events in the tumor microenvironment that are important for lung tumor invasion and progression.
MATERIALS AND METHODS
Mice
To generate orthotopic lung cancers, we used a model in which an oncogenic mutated Kras gene is expressed ubiquitously under the control of a Lox-stop-Lox promoter (13). The mutated protein is not expressed until the animals are administered an adenoviral vector encoding Cre-recombinase (Ad.Cre). Once the Ad.Cre virus is given intranasally or intra-tracheally, airway and alveolar epithelial cells begin to produce the mutated Kras product leading to epithelial multifocal tumors that resemble BAC.
Breeding pairs of Lox-Stop-Lox (LSL) KrasG12D mice (on a mixed 129Sv.J and C57BL/6 background) were generously provided by Dr. David Tuveson while he was at the University of Pennsylvania (14). Mice were genotyped by PCR amplification of genomic DNA obtained from tail samples (primer sequences available upon request). Breeding pairs of Tgfbr2 (Tgfbr2flox/flox) mice (on the C57/Bl6 background) were generously provided by Dr. Harold Moses of Vanderbilt University (15).
LSL-KrasG12D positive mice were simultaneously backcrossed to C57/BL6 mice and to the Tgfbr2flox/flox mice to allow comparisons with the same genetic background. Mice were backcrossed nine generations. Some experiments were conducted at the F5 generation, with similar results to the final F9 generation. Animals used for all experiments were between 6–10 weeks old and were housed in the animal facility at Wistar Institute (Philadelphia, PA). All protocols were approved by the Animal Use Committees of the Wistar Institute and University of Pennsylvania and were in compliance with the guide for care and use of animals.
Lung Tumor Model
To induce tumors, 100 μl of saline containing 109 pfu particles of an adenovirus containing the Cre recombinase (Ad.Cre) was administered to each LSL-KrasG12D mouse intra-nasally (for details see Wilderman et al (16)). Virus was suspended in serum-free DMEM medium (which contains 125 mg/L of NaH2PO4). Fifteen minutes prior to administration of adenovirus, 0.5 μl of 2M CaCl2 was added; calcium phosphate co-precipitation has been shown to improve lung gene transfer (14, 17). Animals were closely observed daily for signs of distress. When they appeared lethargic, had ruffled fur or increased breathing rates, the animals were sacrificed. Massive tumor infiltration of the lung was confirmed by histology. At sacrifice, one lung was excised and frozen in optimal embedding compound (OCT). The other lung and the mediastinum were fixed in formalin and paraffin embedded.
Fluorescence-activated Cell Sorting
Lungs were removed from euthanized mice and minced into fine pieces in digestion buffer containing 0.1 mg/ml DNAse-I and 2.0 mg/ml collagenase type IV (Sigma, St. Louis, MO). Samples were incubated in digestion buffer at 37°C for 30 min, filtered through a 70 μm filter and washed 2 times with serum-free media. All flow cytometry was performed using a Becton-Dickinson FACSCalibur flow cytometer. Data analysis was performed utilizing FlowJo software (Ashland, OR). Antibodies were purchased from BD Biosciences Pharmingen (San Diego, CA).
Genomics (details in Supplementary Methods)
To enrich for tumor and stromal cells, we used the PALM MicroBeam LCM system (Zeiss, Thornwood, NY) system to acquire cells for mRNA extraction using the RNAqueous-Micro Scale RNA Isolation kit (Ambion, Austin, TX). From each specimen, approximately 600,000 μ2 of cells were dissected, yielding 20 ng RNA for genomics analysis using Affymetrix Mouse 430 2.0 Arrays. We identified genes that were differentially expressed between tumors of KrasTgfbr2 −/− and those of KrasTgfbr2 WT mice using a permuted random variance t-test with a two-fold change cut-off and the Significance of Microrarrays (SAM) method (3). Genes were considered to be statistically significant if their permuted t-test p value was less than 0.001 and the estimated false discovery rate was less than 0.01, see Supplemental Table 1. Genomic profiles were compared to human lung adenocarcinoma tumor cells acquired by microdissection in 40 cases (New York Lung Adenocarcinoma). Human studies were approved by the Columbia University Medical Center Institutional Review Board. Gene set Enrichment Analysis (GSEA) was used to determine enrichment of murine tumor gene sets (Supplemental Table 6) in the human New York Lung Adenocarcinoma gene profile and in the Japan Lung Adenocarcinoma (5) gene profiles. Microarray data have been deposited with the GEO repository, accession number GSE27719.
Immunohistochemistry
Immunohistochemistry for cytokeratin 18 (KRT18, Epitomics, Burlingame, CA, polyclonal rabbit antibody, dilution 1:100) was performed using Target retrieval buffer pH 9 (DAKO, Carpinteria CA), one hour primary antibody incubation at room temperature and the Vector Elite Rabbit ABC kit with 3,3′-diaminobenzidine (Vector Laboratories, Burlingame, CA) for 5 minutes. For mediastinal lymph node dissection, ten level sections were cut and numbered; and hematoxylin and eosin stains were performed on the odd sections and immunohistochemistry for KRT18 was performed on the even sections, using a technique similar to sentinel node histologic examination. All foci staining for KRT18 were confirmed by examination of adjacent morphologic tumor on the H&E section before being counted as a node metastasis. Mediastinal lymph nodes were evaluated in 19 KrasTgfbr2 −/− animals and 7 KrasTgfbr2WT animals.
Collagen quantitation
This was performed using a modification of the method of Ornberg et al (18). Briefly, five fields each of trichrome stained tumor at 100X were captured from KrasTgfbr2 −/− lungs (n=6) and KrasTgfbr2WT lungs (n=9). Using NIH Image Jv1.37c (19) images were converted to grayscale using RGB stack, generating a red, green and blue grayscale rendition. Using the blue stack, the image threshold was adjusted so that segmented pixels were highlighted, representing areas of the field not containing collagen. Fields did not include blank areas. The area and ratio of blue staining to collagen containing areas were calculated; measurements were averaged by animal and were evaluated using two-tailed T-test.
Statistical Analysis
Unless otherwise noted, data comparing differences between groups were assessed using one-way analysis of variance (ANOVA) with appropriate post-hoc testing. Survival studies were assessed using Kaplan-Meier survival curves and analyzed with the Mantel-Cox log rank test. Differences were considered significant when p-value was <0.05. Statistics were performed using SPSS software, version 16.0.
RESULTS
Loss of TBRII leads to increased invasiveness of Kras-induced tumors
To determine the direct role of TβRII repression on adenocarcinoma invasion in vivo, we generated a genetic model of murine lung adenocarcinoma with targeted deletion of Tgfbr2 (see Methods). In this model, intranasal instillation of Ad.Cre led to both activation of a mutated Kras gene and loss of the Tgfbr2 gene in the same epithelial cells. We wanted to test the hypothesis that in the oncogenic Kras murine model of lung cancer, loss of TβRII would increase tumor invasion and metastasis.
Intranasal instillation of Ad.Cre into the Tgfbr2flox/flox mice did not lead to any discernable tumors over a ten week period, indicating that loss of TβRII is not sufficient for tumor initiation. When we administered Ad.Cre to mice that were KrasTgfbr2 heterozygous, we did not identify differences in tumor morphology compared with the LSL-KrasTgfbr2WT mice (data not shown). All subsequent breeding was directed towards generation of KrasTgfbr2 KO homozygous mice (KrasTgfbr2 −/−). We confirmed recombination of the Tgfbr2 floxed allele, and loss of TβRII expression after administration of intranasal Adeno-Cre to KrasTgfbr2 flox/flox mice (Supplementary Figure 1).
In contrast to the heterozygous mice, instillation of Ad.Cre to homozygous animals resulted in striking differences in tumor morphology and tumorigenesis (Figure 1). Instillation of 109 pfu of Ad.Cre into KrasTgfbr2WT mice led to the expected development, over a six to ten week period, of non-invasive adenoma and adenocarcinoma characterized by epithelial cell proliferation without destruction of alveolar walls (lepidic growth) or stromal reaction (16). In contrast, lung tumors in KrasTgfbr2 −/− were histologically similar to human mixed subtype adenocarcinomas in that lepidic areas were associated with transitions to invasive adenocarcinoma involving vessels, airways, and pleura. Tumor cells were grouped as small glands, small nests and single cells amidst distinct areas of desmoplastic stroma. Immunohistochemistry for cytokeratin 18 (KRT18) confirmed the epithelial nature of these infiltrating tumor cells. Trichrome stain highlighted the increased collagen in invasive areas in KrasTgfbr2 −/− mouse lungs when compared to KrasTgfbr2WT, with significant difference (T-test, p< .0001) when analyzed quantitatively.
Figure 1. Mouse Lung Tumor Morphology in Kras-Tgfbr2−/− Model.

Kras-Tgfbr2−/− (panels A, B, C): lung tumor at low power (A) shows an area of alveolar/lepidic tumor cell proliferation (arrowheads) which transitions into an infiltrative and desmoplastic tumor that surrounds an adjacent vessel. The box in Panel A is magnified in Panel B. The bland alveolar epithelial proliferation in Panel B (subset of Panel A) transitions into a nest and gland forming tumor amidst a cellular stroma with the vessel lumen at the upper left. KRT18 immunohistochemistry (C) confirms the epithelial differentiation, gland formation and irregular nests of tumor. Kras-Tgfbr2WT (panels D, E, F): lung tumor at low power (D) shows proliferation without replacement of alveolar walls or stromal reaction near bronchovascular bundle. Higher magnification (E) shows a bland alveolar and focal aggregate of tumor cells without desmoplastic stroma. KRT18 immunohistochemistry stains an aggregate of tumor cells and alveolar epithelial proliferation (Panel F). Collagen deposition (blue trichrome stain) is markedly increased in Kras-Tgfbr2−/− tumors (G) vs. Kras-Tgfbr2WT tumors (H). Collagen deposition, as measured by image analysis of trichrome positive surface area, was increased three-fold in Kras-Tgfbr2−/− tumors (I). (A–D Hematoxylin and eosin, original magnification A, D × 25 and B, E ×150; C, F DAB immunohistochemistry × 150; G, H Trichrome × 50).
Further examination of the KrasTgfbr2 −/− lung tumors demonstrated nuclear TITF-1 and cytoplasmic pro-surfactant protein C immunoreactivity, while non-reactive for CCSP (data not shown), suggesting alveolar type II cell differentiation. In addition, the KrasTgfbr2 −/− lung tumors showed evidence of inflammatory cell recruitment and tumor microenvironment remodeling with neoangiogenesis that was not seen in the KrasTgfbr2WT tumors (Supplemental Figure 2).
Vascular Invasion and Metastasis
In addition to desmoplastic stroma deposition, KrasTgfbr2 −/− mouse tumors demonstrated transmural invasion of vessels and pleural invasion (Figure 2A–D). We also detected regional metastases to mediastinal lymph nodes in 10 of 19 (53%) of KrasTgfbr2 −/− mice, whereas no metastases were detected in the KrasTgfbr2WT (n=7) mice, despite examination of 10 level sections with alternating KRT18 stains (Fisher p = 0.02, Figure 2E–F). The metastases were comprised of small clusters of tumor cells, detected morphologically and confirmed by KRT18 immunohistochemistry. No metastases were seen in the liver, spleen, or other extrathoracic sites.
Figure 2. Vascular Invasion and Metastasis in Kras-Tgfbr2−/− Mice.

Kras-Tgfbr2−/− tumor cells (Panel A, B) that are KRT18 positive (inset Panel B) invade through the vessel wall into intima with an endothelial cell layer overlying the nest. This is confirmed by CD31 immunohistochemistry in endothelium (arrows) in Panel C over the non-staining tumor cells (asterisk). A small fibrin thrombus is seen between the arrows. Visceral pleural invasion is seen using elastic stain (Panel D) demonstrating intact pleural elastic tissue (left arrowhead) and tumor cell invasion through the visceral pleural elastic tissue, causing a defect (right 2 arrowheads) and tumor growth on the pleural surface. Discrete foci of tumor cells were detected within mediastinal lymph nodes (E, F). The epithelial origin of these cells was confirmed by KRT18 immunostaining (inset F). (Hematoxylin and eosin, Original magnification × 50, A, E; and × 150 B, F; Inset B and F DAB IHC Original magnification × 150; C, DAB IHC Original magnification × 150; D, Elastic von Gieson, Original magnification × 25).
In summary, the morphological examination indicates that the KrasTgfbr2 −/− mouse recapitulates the progression of a pure non-invasive adenocarcinoma to an adenocarcinoma with mixed invasive morphology and metastatic potential.
Loss of TBRII leads to decreased survival in mice with Kras-induced tumors
These changes in morphology led to changes in survival. We studied the effect of intranasal Ad.Cre on survival in the F5 and F9 generation LSL-K-rasG12D-positive mice that were homozygous for the Tgfbr2 flox/flox allele vs. littermate controls without the floxed Tgfbr2 allele (n= 10 in each group). In the F9 mice, the KrasTgfbr2WT mice had a median survival of 79 days while the median survival in the KrasTgfbr2 −/− group was reduced to only 43 days (logrank p<0.001) (Figure 3). The survival data from the F5 mice was similar (data not shown).
Figure 3. Survival is Shortened in Kras-Tgfbr2−/− Mice.
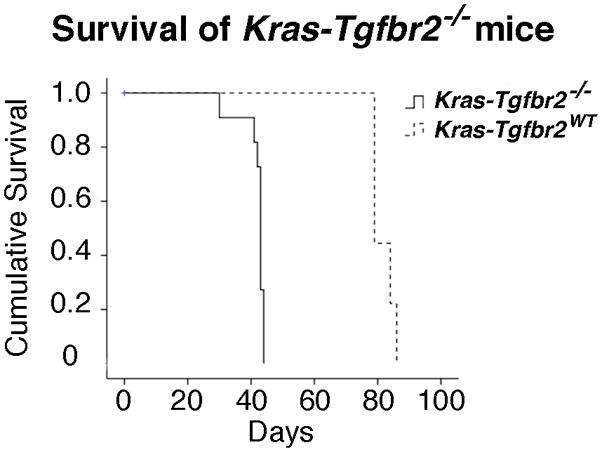
After administration of Ad.Cre, animals were followed until death or moribund condition. Kaplan Meier survival curve indicates median survival of Kras-Tgfbr2−/− mice was 43 days vs. 79 days in the Kras-Tgfbr2WT animals, P < .001.
Alterations in gene profiles in KrasTgfbr2 −/− versus KrasTgfbr2WT mice
To determine the key activated molecular signaling pathways in these invasive murine lung adenocarcinomas and to determine if the murine model transcriptional profiles were similar to those of human adenocarcinoma, we examined gene expression profiles of the microdissected murine tumors using microarray analysis of tumor cell mRNA. We generated a signature of KrasTgfbr2 −/− tumors by combining results of a permuted T- test, P.001 with those from Significance Analysis of Microarrays algorithm, with a false discovery rate (FDR) of 1%. The intersection of these lists consisted of 243 upregulated and 707 downregulated genes (Supplemental Table 1).
Comparative Genomics
A primary goal of this work was to determine if the KrasTgfbr2 −/− signature of upregulated genes was enriched in human invasive mixed-subtype lung adenocarcinoma tumor cells compared with non-invasive non-mucinous bronchioloalveolar carcinoma. In order to accomplish this, we first generated a gene expression dataset of 40 BAC and AC-mixed human tumors from which we acquired mRNA from laser capture microdissected tumor cells (Supplemental Table 2). Unsupervised hierarchical clustering identified two main clusters (Figure 4A). The mixed-subtype adenocarcinomas were grouped in cluster 1 (15/23) while BACs were grouped in cluster 2 (13/17); Fisher p=0.01. DNA direct sequencing analysis indicated the prevalence of EGFR and Kras mutations was 38% and 7%, respectively in these non-solid adenocarcinomas, which is in the range of those reported by others. Similarly, the clusters of adenocarcinoma histological subtype classes were independent of EGFR and Kras mutation status (6, 20). Taken together, these results indicate that tumor cell molecular signatures of non-invasive and minimally invasive lung adenocarcinomas are distinct from other mixed-subtype invasive adenocarcinoma.
Figure 4. Comparative Genomics.

Human Lung Adenocarcinoma Unsupervised Clustering (Panel A). Tumors segregate into two distinct clusters associated with invasion (BAC-cluster 2 vs. AC-Mixed-cluster 1, P= .01). We performed reproducibility analysis on the two major clusters identified in the dendrogram. The Robustness index (R-index) measures the proportion of pairs of specimens within a cluster for which the members of the pair remain together in the reclustered perturbed data. The Discrepancy index (D-index) measures the number of discrepancies (additions or omissions) comparing an original cluster to a best-matching cluster in the re-clustered perturbed data. Using 100 permutations, the R-index was .95 and the D-index was .64, indicating that the clustering results were robust. BAC Tumor histology indicated in blue and Invasive AC histology indicated in yellow. Kras mutation status for exon 2 and exon 3 are indicated in red. EGFR exon 18 –21 status is indicated in green.
Kras-Tgfbr2−/− Tumor Microenvironment Gene Expression (Panel B). Heat map of top 25% differentially expressed genes in laser capture microdissected tumor and tumor associated stromal tissue from five-week mouse Kras-Tgfbr2−/− (yellow) and nine-week Kras-Tgfbr2WT (blue) lungs, clustered by gene expression. . Box 1 indicates genes upregulated in tumor cells (black) compared to non-tumoral stroma (gray) tissues. Box 2 indicates genes upregulated in Kras-Tgfbr2−/− genotype tumor cells. Box 3 indicates genes upregulated in non-tumor stroma compared to tumor cells. Box 4 indicates genes upregulated in Kras-Tgfbr2−/− genotype tumor associated stroma.
With this human data in hand, we used Gene Set Enrichment Analysis to: 1) determine if the gene signature of KrasTgfbr2 −/− tumors was enriched in human AC-mixed tumors compared to BAC tumors and 2) if the KrasTgfbr2 −/− model was more similar to human invasive lung adenocarcinoma than other previously described mouse lung cancer models. To determine the relative similarity of the KrasTgfbr2 −/− model with other murine lung adenocarcinoma models, we tested gene signatures from: 1. the KrasTgfbr2 −/−- model (this paper); 2. the non-invasive Kras-LA model (14, 21); 3. the KrasP53mut model (22); 4. the KrasLkb1−/−, model (23); and 5. the KrasHif2a models of advanced lung carcinoma (24) (Table 1). For each murine model, we calculated a normalized enrichment score (NES), which indicates the degree to which the gene set is overrepresented at the top or bottom of the ranked list of genes in the human AC expression dataset (New York Lung Adenocarcinoma), normalized across the analyzed gene sets. In human AC invasive tumors, three murine gene sets were enriched, with NES of 1.29, 1.03 and 0.87 for the KrasTgfbr2 −/−, KrasLkb1−/−, and KrasP53mut gene sets, respectively; but only the KrasTgfbr2 −/− enrichment was significant with nominal P-value <.05 and False discovery rate < 25%. To examine the generalizability of these findings and to exclude laser capture microdissection as a confounding variable for gene set enrichment, we repeated the procedure in a human adenocarcinoma gene expression dataset acquired from 163 lung specimens (Japan invasive AC). We examined gene set enrichment in the adenocarcinomas with Terminal respiratory (TRU-a) morphology that is similar to AC-Mixed tumors in comparison to TRU-B, BAC-like tumors. Again, we detected enrichment of the KrasTgfbr2 −/− gene set with a NES of 1.35 (P .025, FDR .045). The KrasP53mut and Kras-LA gene sets were also enriched but the NES was not significant. Taken together, the genomics results in two human lung adenocarcinoma datasets suggest that the murine KrasTgfbr2 −/− model closely recapitulates the gene expression alterations detected in the progression of human BAC to AC-Mixed subtypes.
Table 1.
Geneset Enrichment Analysis of Mouse Model Genes in Human Lung Adenocarcinoma Tumors.
| NY Lung Adenocarcinoma | |||
|---|---|---|---|
| Gene Set | Normalized Enrichment Score | Nominal p value | FDR q value |
| KrasTgfbr2 −/−- | 1.29 | 0.05 | 0.08 |
| KrasLkb1−/−, | 1.03 | 0.35 | 0.53 |
| KrasP53mut | 0.87 | 0.92 | 0.86 |
| Kras-LA | −1.14 | 0.10 | 0.14 |
| KrasHif2a | −1.69 | 0.00 | 0.00 |
| Japan Lung Adenocarcinoma | |||
| Gene Set | Normalized Enrichment Score | Nominal p value | FDR q value |
| KrasTgfbr2 −/−- | 1.35 | 0.02 | 0.05 |
| KrasP53mut | 1.11 | 0.16 | 0.27 |
| Kras-LA | 0.95 | 0.67 | 0.64 |
| KrasLkb1−/−, | −1.13 | 0.17 | 0.17 |
| KrasHif2a | −1.21 | 0.13 | 0.17 |
For each murine model, we calculated a normalized enrichment score (NES), which indicates the degree to which the gene set is overrepresented at the top or bottom of the ranked list of genes in the human AC expression dataset (New York Lung Adenocarcinoma invasive vs. BAC/AIS and Japan Lung Adenocarcinoma TRU-A vs. TRU-B tumors), normalized across the analyzed gene sets. The significance was indicated by a nominal p value and by a permuted false discovery rate.
Tumor Microenvironment
The biologic process ontology of the KrasTgfbr2 −/− mouse tumor model genes suggested activation of pathways important for immune response and development, including lung branching morphogenesis. (Supplemental Table 3). These processes require interactions between epithelial and stromal cells within the tumor microenvironment. To examine the stromal response to Tgfbr2 deficient tumor cells, we compared stromal cell transcriptional profiles from five-week KrasTgfbr2 −/− tumors and nine-week KrasTgfbr2 WT. We used mice at these time points to allow comparison of the stromal compartment of similarly advanced tumors. However, even with this adjustment, the amount of tumor-associated stroma in advanced nine-week KrasTgfbr2 WT mice was less than in the five-week KrasTgfbr2 −/− mice. The gene expression heatmap revealed clusters of genes that were upregulated in stroma and not tumor (indicating successful microdissection) as well as genes differentially expressed in KrasTgfbr2 −/− stroma compared to KrasTgfbr2 WT stroma (Figure 4B, Supplemental Table 4). Ontology analysis of KrasTgfbr2 −/− stroma profile showed upregulation of genes with functions important in wound healing response, development, and immune responses (Supplemental Table 5), similar to ontologies of genes acquired from tumor cells.
Tumor Microenvironment - Lung development and TGF-β
In light of the gene ontologies related to development in both the tumor and stromal analysis (Supplemental Tables 5 and 6) and of prior work suggesting that tumor cell signatures recapitulate key developmental pathways (25), we compared our KrasTgfbr2 −/− gene expression profiles to a published study of lung branching genomics (26). Lu and colleagues showed that expression of TGF-β responsive genes in the distal lung mesenchyme was associated with initiation of branching. Gene set analysis of our KrasTgfbr2 −/− stromal signature showed enrichment in the Lu branching morphogenesis gene expression profile (Efron-Tibshirani GSA p value <.005). To determine if this stromal signature was enriched for TGF-β responsive genes, we performed gene set analysis for the Gene ontology of cellular response to transforming growth factor beta stimulus (GO:0071560) and determined significant enrichment (Efron-Tibshirani GSA p value <.005).
As a result of these findings, we examined our microarray data for evidence suggestive of TGF-β effect in the stromal dissected compartment. Despite prior reports that TGF-β levels were increased in mice with tumor lacking TβRII (reviewed in Yang et al., (27)), we saw no clear differences in expression levels of Smads or TGF-β isoform mRNA in the stroma microarrays, although TGF-β2 was increased in the tumor microdissection microarrays. We did see a large increase in Thrombospondin 1 message (5 fold, p= 3 × 10−5), a matrix protein known to be important in activating latent TGF-β. Consistent with evidence of increased TGF-β activity, we saw: 1) increases in TGF-β responsive genes in the KrasTgfbr2−/− tumor stroma including Arginase 1 (63 fold, p<.0001), Transforming growth factor beta induced (2.85, p< .02), and Fibroblast activation protein (4.2 fold, p= .045) and 2) increases in matrix proteins known be responsive to TGFβ including Tenascin C (4.5 fold, p=0.006), Fibronectin 1 (5.88 fold, p=0.01), Collagen type I, alpha 1 (30.3 fold, p=0.03), Collagen type VIII, alpha 1 (2.63 fold, p= 0.018), Collagen XV, alpha 1 (3.22 fold, p= .045), and Cysteine rich protein 61 (15.2 fold increase, p= 0.02), Thus, our array data suggest a tumor microenvironment characterized by enhanced TGF-β activity in the stromal compartment. We confirmed expression of a number of these TGF-β related genes and calculated the ratio of expression levels of mRNAs by quantitative PCR between needle dissected (tumor plus tumor associated stroma) for KrasTgfbr2 −/− lungs and KrasTgfbr2WT lungs. We observed an increase in Arg-1 (2 fold, p.006) Fibroblast activation protein (1.8 fold, p=.02), and Fibronectin 1 (1.9 fold, p=.003), all validating the array results.
Tumor Microenvironment – Immune response
We then compared gene expression between the two types of mice with regard to inflammatory and immune modulatory programs (again, comparing the KrasTgfbr2−/− lungs to the KrasTgfbr2WT). Unlike the Yang breast cancer study (12), we did not see increases in Cccl5 (RANTES) or Cxcl12 in the KrasTgfbr2−/− lungs in arrays or using qRT-PCR. Ccl5 message was increased by 3 fold on the array, but we could not confirm this change using qRT-PCR. The interferon-induced chemokines that bind to Cxcr3 and attract activated Cd8 T cells, Cxcl9 (MIG), Cxcl10 (IP10), and Cxcl11 (i-TAC) were all highly down-regulated on the array (0.1 fold expression, p=0.001 for Cxcl9; 0.2 fold expression, p=0.006 for Cxcl10; and 0.3 fold expression, p=0.009 for Cxcl11). We validated a similar change for Cxcl10 using qRT-PCR (0.4 fold expression, p<0.001). The neutrophil chemokine, Cxcl3 (Gro3, MIP2β) was increased 2.1 fold by qRT-PCR (p=0.04), and 1.6 fold (p=0.2) on the array. Cytokine message levels were generally quite low making it difficult to assess changes. Finally, we did note some other changes consistent with a more immunosuppressive microenvironment in the KrasTgfbr2−/− tumors including upregulation of prostaglandin E2 synthase (increased 6–8 fold on the array; p=0.0001), arginase 2 (upregulated 2 fold in RT-PCR; p=0.006), and downregulation of Stat1 (0.4 fold; p<0.001).
In addition to mRNA expression data, we also evaluated the cellular composition of whole lungs from normal mice, mice bearing “five-week” KrasTgfbr2−/− tumors and mice with advanced “nine-week” KrasTgfbr2WT tumors. Again, we chose time points where tumor burden was approximately equal. After harvesting, the lungs were digested and subjected to flow cytometry. Two important caveats should be noted. First, digestion of lungs preferentially releases leukocytes (defined as CD45+ cells), so the overall percentage of CD45 cells recovered is less informative than analysis of the CD45 subsets. Second, we analyzed the entire lung, since we could not microdissect the tumor tissues. Our results thus reflect the admixture of cells within the lung tumors and the non-tumor-bearing lung (Figure 5).
Figure 5. Cellular Composition of Lungs.
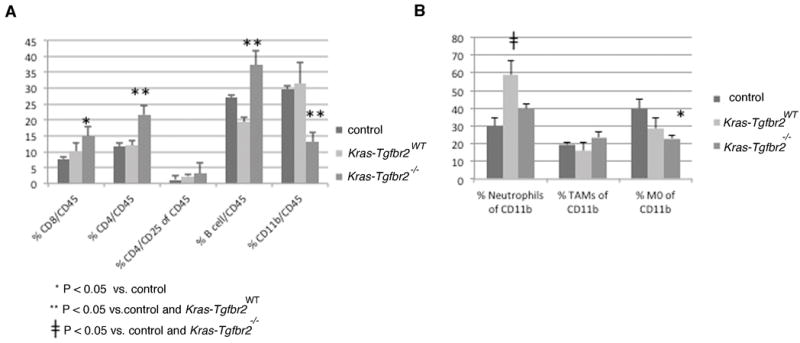
We harvested whole lungs from five-week mouse Kras-Tgfbr2−/−, nine-week Kras-Tgfbr2WT, and wildtype mice for analysis by flow cytometry. Lymphocyte populations (A) showed a significant increase of Cd4 T and B cells of the total Cd45 cells in Kras-Tgfbr2−/− compared to Kras-Tgfbr2WT lungs while myeloid populations (B) showed a decrease of Cd11b cells and undifferentiated myeloid M0 cells.
We noted a number of significant (p<0.05) changes in the lymphocyte populations in the KrasTgfbr2−/− lungs: 1) the % of Cd4 T cells of Cd45 cells and the % of B cells of Cd45 cells was increased compared to normal lungs and KrasTgfbr2WT tumor-bearing lungs, and 2) the % of Cd8 T cells of Cd45 cells was increased compared to control lungs. With regard to the Cd4 cells, the percentage of Cd45 cells that could be considered T-regulatory cells (defined as Cd4+/Cd25+) was increased (3.2%) in the KrasTgfbr2−/− lungs compared to the KrasTgfbr2WTlungs (2.3%), but this change did not reach significance.
There were also changes in the myeloid cell populations (defined as Cd45+ cells that express the adhesion molecule Cd11b). First, the myeloid (Cd11b+) cell population was significantly lower in the KrasTgfbr2−/− lungs than in the normal or KrasTgfbr2WT lungs. The types of myeloid cells were also different. Whereas the % of neutrophils (defined as Cd11b+/F4/80-/Ly6G+) of the total myeloid population was significantly (p<0.05) increased in the KrasTgfbr2WT lungs (58%) versus normal lungs (30%), this increase was not seen in the in the KrasTgfbr2−/− lungs. The percentage of tumor-associated macrophages (TAM, defined as Cd11b+/F4/80+/Ly6G-) of total myeloid cells were similar among the three groups, however, given the smaller percentage of myeloid cells in the KrasTgfbr2−/− lungs, the total number of lung TAMs would be lower. Finally, there seemed to be a decreased proportion of undifferentiated myeloid cells (M0 cells, defined as CD11b+/F4/80-/Ly6G-) in the KrasTgfbr2−/− lungs. In summary, compared to the KrasTgfbr2WT lungs, the KrasTgfbr2−/− lungs had more T and B cells and less myeloid cells, somewhat different than the results reported in the breast cancer models.
Expression of the TGF-β type II receptor in Human Lung Adenocarcinoma
TBRII protein expression is inversely correlated with extent of invasion in early stage lung adenocarcinoma (4). We extended this observation by examining the correlation of TBRII protein expression with Tumor stage in Clinical Stage I patients within a larger cohort of 183 lung adenocarcinoma patients (2). Reduced expression (TBRII intensity score 0, 1) was detected in 46% of Stage T2 tumors and 22% of Stage T1 tumors, p <0.05 vs TBRII intensity score 2. We examined the prognostic significance of TGFBRII mRNA repression in human lung adenocarcinoma using the Japanese Lung Adenocarcinoma dataset. Low expression of TGFBRII (below median level of expression of probe A_23_P211957) was associated with lymph node metastasis (N1–N2 vs. N0) and with increased risk of death, p <.001 and p. <.05, respectively. Taken together, these results confirm the clincopathologic significance of TGF-β type II receptor expression in human lung adenocarcinoma during malignant progression of this disease.
DISCUSSION
Experience over time demonstrates that human lung adenocarcinoma progresses from a non-invasive lesion, usually manifest as a ground-glass opacity, to an invasive carcinoma with capacity for metastasis (28). The purpose of this study is to model this progression in the mouse and to determine the importance of the Type II TGF Beta Receptor in mediating lung adenocarcinoma invasion and metastasis, in vivo. We show that loss of Tgfbr2 in lung epithelial cells induces invasive morphology in non-invasive Kras mutant tumors, promotes lymph node metastases, and shortens survival. Further, we demonstrate that this murine model recapitulates both the histological and genomic changes that accompany invasion and metastasis in human lung adenocarcinoma tumors.
TGF-β, the ligand for the TGF-β type II receptor is a pleiotropic cytokine comprised of family members TGF-β 1, 2, 3 that regulate tissue homeostasis and prevent tumor initiation by inhibiting cellular proliferation, differentiation, and survival (29). It is secreted as a latent molecule and is activated by cleavage by proteases and other molecules. Signaling primarily occurs through SMAD protein dependent pathways whereby ligand binding to TβRII induces phosphorylation and activation of TGF-β type I receptor (TβRI). TGF-β signaling may also proceed via less well understood SMAD independent pathways (30). Depending upon context, TGF-β signaling may alternatively function to suppress tumor growth in early cancers or to promote tumor cell invasion and metastasis in late cancers (31–34). Rojas and colleagues have shown that different levels of repression of the TGF-β receptor are associated with variable activation of the SMAD and MAPK pathways, such that at lower levels of TGF-β receptor activation, the pro-tumorigenic non-SMAD signaling pathways dominate (35).
Type II receptor genetic alterations are well characterized in human gastrointestinal tumors, in which 25% of colorectal carcinomas have missense mutations associated with microsatellite instability. Somatic mutations are rare in other human neoplasms. In human lung tumor specimens, type II receptor repression is evident in ~40% of lung adenocarcinomas overall and in up to 100% of poorly differentiated adenocarcinomas (4, 36). Potential mechanisms of repression include promoter hypermethylation (37), miRNA (38), microsatellite instability, and frameshift mutations involving the poly(A) tract (39).
In advanced cancers, induction of TGF-β signaling promotes epithelial to mesenchymal transition, a characteristic of invasive and metastatic cells (40, 41), with constitutive activation of TGF-β or TβRI leading to increased metastases in animal models of breast cancer (42–44). Systemic inhibition of TGF-β has been shown to suppress metastasis (45–48) and clinical cancer trials are in progress. In animal models of early tumor progression, TGF-β signaling is tumor suppressive, with animal models of targeted repression of TβRII consistently resulting in tumor cell invasion, progression and metastasis. However, the signaling pathways that are activated in the setting of TGF-β signaling inhibition are varied. Yang and colleagues showed that targeted deletion of TβRII in the mammary epithelium promoted breast cancer metastases through the Cxcl5/Cxcr2 chemokine axis mediated recruitment of Gr-1+/Cd11b+ myeloid-derived suppressor cells. Increased stromal TGF-β levels at the invasive front of tumors was shown to be important for tumor progression and for inhibition of tumor immunosurveillance (12). Our lung model gene expression data support a role for TβRII repression in producing an immunosuppressed tumor microenvironment. Our FACS data do not show a significant increase of Gr-1+/Cd11b+ cells but rather an increase in Cd4 positive T cells and B cells that may be important in facilitating tumor progression and invasion in the lung as has been shown in the prostate and breast (49, 50). The differences in inflammatory cell recruitment and chemokine profile between our inducible model and those reported by Yang and colleagues may be attributable to organ specific microenvironment influences or to developmental compensation in a constitutive model of Tgfbr2 deficiency.
The identification of developmental gene ontologies and specifically the enrichment of genes involved in lung branching morphogenesis in our mouse lung tumor and stroma gene expression profiles supports the concept that key events in tumor progression recapitulate fundamental developmental pathways. Similar to the distal branching lung, TGF-β non-responsiveness in the KrasTgfbr2−/− adenocarcinoma model determines epithelial fate in terms of cell motility, adhesion and invasion. Unlike normal lung development (26), the tumor model has continuous activation of Kras with TGF-β responsiveness only in the non-tumoral stroma, resulting in a TGF-β response-associated stromal matrix profile, but not growth stabilization. The resulting tumor manifestation is disorganized collagenized matrix and invasion rather than an organized epithelial-mesenchymal structure. Recent work using cell lines with repressed TBRII expression demonstrate tumor cell autonomous effects of TBRII repression on tumor invasion (4). Thus, it is unclear whether the tumor invasion and progression phenotype requires an activated TGF- β stromal response or is independent of stromal influence; this will require further testing of systemic and targeted TGF-B signaling blockade.
The oncogenic Kras lung adenocarcinoma mouse has proven to be a reproducible model of non-invasive lung adenocarcinoma that is more similar genomically to human lung adenocarcinoma than the carcinogen-induced models, which also induce non-invasive adenoma lesions. Combination of Kras induction with alterations in Lkb1, p53, and Hif2a has generated lung tumor models with accelerated progression, dedifferentiation, and angiogenesis and provided important insights into mechanisms related to advanced disease. The unique features of our model are: the retention of non-invasive morphology in the background of invasive tumor with extensive desmoplastic stromal reaction; the relation of TβRII expression to lymph node metastasis and survival; and the similarity of gene expression profiles in Tgfbr2 −/− murine lung tumors to human invasive lung adenocarcinoma. Although not tested yet, it is possible that using smaller amounts of Ad.Cre, would lead to the slower development of tumors and allow for the development of distant metastasis. Of human lung cancer, adenocarcinoma is the most common histology and the incidence is expected to increase with the anticipated increased implementation of lung cancer clinical screening by computed tomography. Screening scans typically detect adenocarcinomas with approximately 25% of tumors of non-invasive BAC/AIS morphology. Thus, the delineation of genomic and molecular alterations that capture the spectrum of non-invasive and invasive adenocarcinoma will have significant impact on this important clinical problem. The model of lung specific Tgfbr2 loss in the oncogenic Kras mouse faithfully recapitulates the morphology, genomics and clinical progression of lung adenocarcinoma and promises to serve as an important tool for the evaluation of diagnostic and therapeutic advances in the management of human lung adenocarcinoma.
Supplementary Material
Acknowledgments
Funding: This work is supported by NIH Grant CA120174 (C.A.P.).
Footnotes
The authors declare no conflicts of interest
References
- 1.Brambilla E, Travis WD, Colby TV, Corrin B, Shimosato Y. The new World Health Organization classification of lung tumours. Eur Respir J. 2001;18:1059–68. doi: 10.1183/09031936.01.00275301. [DOI] [PubMed] [Google Scholar]
- 2.Borczuk AC, Qian F, Kazeros A, Eleazar J, Assaad A, Sonett JR, et al. Invasive size is an independent predictor of survival in pulmonary adenocarcinoma. Am J Surg Pathol. 2009;33:462–9. doi: 10.1097/PAS.0b013e318190157c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Travis WD, Brambilla E, Noguchi M, Nicholson AG, Geisinger KR, Yatabe Y, et al. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol. 2011;6:244–85. doi: 10.1097/JTO.0b013e318206a221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borczuk AC, Kim HK, Yegen HA, Friedman RA, Powell CA. Lung adenocarcinoma global profiling identifies type II transforming growth factor-beta receptor as a repressor of invasiveness. Am J Respir Crit Care Med. 2005;172:729–37. doi: 10.1164/rccm.200504-615OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takeuchi T, Tomida S, Yatabe Y, Kosaka T, Osada H, Yanagisawa K, et al. Expression Profile-Defined Classification of Lung Adenocarcinoma Shows Close Relationship With Underlying Major Genetic Changes and Clinicopathologic Behaviors. Journal of Clinical Oncology. 2006;24:1679–88. doi: 10.1200/JCO.2005.03.8224. [DOI] [PubMed] [Google Scholar]
- 6.Motoi N, Szoke J, Riely GJ, Seshan VE, Kris MG, Rusch VW, et al. Lung adenocarcinoma: modification of the 2004 WHO mixed subtype to include the major histologic subtype suggests correlations between papillary and micropapillary adenocarcinoma subtypes, EGFR mutations and gene expression analysis. Am J Surg Pathol. 2008;32:810–27. doi: 10.1097/PAS.0b013e31815cb162. [DOI] [PubMed] [Google Scholar]
- 7.Beer DG, Kardia SL, Huang CC, Giordano TJ, Levin AM, Misek DE, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;8:816–24. doi: 10.1038/nm733. [DOI] [PubMed] [Google Scholar]
- 8.Director’s Challenge Consortium for the Molecular Classification of Lung A. Shedden K, Taylor JM, Enkemann SA, Tsao MS, Yeatman TJ, et al. Gene expression-based survival prediction in lung adenocarcinoma: a multi-site, blinded validation study. Nat Med. 2008;14:822–7. doi: 10.1038/nm.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ijichi H, Chytil A, Gorska AE, Aakre ME, Fujitani Y, Fujitani S, et al. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev. 2006;20:3147–60. doi: 10.1101/gad.1475506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu SL, Herrington H, Reh D, Weber S, Bornstein S, Wang D, et al. Loss of transforming growth factor-beta type II receptor promotes metastatic head-and-neck squamous cell carcinoma. Genes Dev. 2006;20:1331–42. doi: 10.1101/gad.1413306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Munoz NM, Upton M, Rojas A, Washington MK, Lin L, Chytil A, et al. Transforming growth factor beta receptor type II inactivation induces the malignant transformation of intestinal neoplasms initiated by Apc mutation. Cancer Res. 2006;66:9837–44. doi: 10.1158/0008-5472.CAN-06-0890. [DOI] [PubMed] [Google Scholar]
- 12.Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13:23–35. doi: 10.1016/j.ccr.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–6. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 14.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes & Development. 2001;15:3243–8. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chytil A, Magnuson MA, Wright CV, Moses HL. Conditional inactivation of the TGF-beta type II receptor using Cre:Lox. Genesis. 2002;32:73–5. doi: 10.1002/gene.10046. [DOI] [PubMed] [Google Scholar]
- 16.Wilderman MJ, Sun J, Jassar AS, Kapoor V, Khan M, Vachani A, et al. Intrapulmonary IFN-beta gene therapy using an adenoviral vector is highly effective in a murine orthotopic model of bronchogenic adenocarcinoma of the lung. Cancer Res. 2005;65:8379–87. doi: 10.1158/0008-5472.CAN-05-0920. [DOI] [PubMed] [Google Scholar]
- 17.Fasbender A, Lee JH, Walters RW, Moninger TO, Zabner J, Welsh MJ. Incorporation of adenovirus in calcium phosphate precipitates enhances gene transfer to airway epithelia in vitro and in vivo. J Clin Invest. 1998;102:184–93. doi: 10.1172/JCI2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ornberg RL, Woerner BM, Edwards DA. Analysis of stained objects in histological sections by spectral imaging and differential absorption. J Histochem Cytochem. 1999;47:1307–14. doi: 10.1177/002215549904701010. [DOI] [PubMed] [Google Scholar]
- 19.Rasband WS. Image J. 1997–2011 [cited 2011; Available from: http://rsb.info.nih.gov/ij/
- 20.Takeuchi T, Tomida S, Yatabe Y, Kosaka T, Osada H, Yanagisawa K, et al. Expression profile-defined classification of lung adenocarcinoma shows close relationship with underlying major genetic changes and clinicopathologic behaviors. J Clin Oncol. 2006;24:1679–88. doi: 10.1200/JCO.2005.03.8224. [DOI] [PubMed] [Google Scholar]
- 21.Sweet-Cordero A, Mukherjee S, Subramanian A, You H, Roix JJ, Ladd-Acosta C, et al. An oncogenic KRAS2 expression signature identified by cross-species gene-expression analysis. Nat Genet. 2005;37:48–55. doi: 10.1038/ng1490. [DOI] [PubMed] [Google Scholar]
- 22.Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, et al. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 2005;65:10280–8. doi: 10.1158/0008-5472.CAN-05-2193. [DOI] [PubMed] [Google Scholar]
- 23.Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007;448:807–10. doi: 10.1038/nature06030. [DOI] [PubMed] [Google Scholar]
- 24.Kim WY, Perera S, Zhou B, Carretero J, Yeh JJ, Heathcote SA, et al. HIF2alpha cooperates with RAS to promote lung tumorigenesis in mice. J Clin Invest. 2009;119:2160–70. doi: 10.1172/JCI38443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Borczuk AC, Gorenstein L, Walter KL, Assaad AA, Wang L, Powell CA. Non-small-cell lung cancer molecular signatures recapitulate lung developmental pathways. Am J Pathol. 2003;163:1949–60. doi: 10.1016/S0002-9440(10)63553-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu J, Qian J, Izvolsky KI, Cardoso WV. Global analysis of genes differentially expressed in branching and non-branching regions of the mouse embryonic lung. Dev Biol. 2004;273:418–35. doi: 10.1016/j.ydbio.2004.05.035. [DOI] [PubMed] [Google Scholar]
- 27.Yang L, Pang Y, Moses HL. TGF-beta and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010;31:220–7. doi: 10.1016/j.it.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takashima S, Maruyama Y, Hasegawa M, Yamanda T, Honda T, Kadoya M, et al. CT findings and progression of small peripheral lung neoplasms having a replacement growth pattern. AJR Am J Roentgenol. 2003;180:817–26. doi: 10.2214/ajr.180.3.1800817. [DOI] [PubMed] [Google Scholar]
- 29.Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3:807–21. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- 30.Massague J. How cells read TGF-beta signals. Nat Rev Mol Cell Biol. 2000;1:169–78. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 31.Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–29. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 32.Elliott RL, Blobe GC. Role of Transforming Growth Factor Beta in Human Cancer. J Clin Oncol. 2005;23:2078–93. doi: 10.1200/JCO.2005.02.047. [DOI] [PubMed] [Google Scholar]
- 33.Tang B, Vu M, Booker T, Santner SJ, Miller FR, Anver MR, et al. TGF-beta switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. J Clin Invest. 2003;112:1116–24. doi: 10.1172/JCI18899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roberts AB, Wakefield LM. The two faces of transforming growth factor beta in carcinogenesis. Proc Natl Acad Sci U S A. 2003;100:8621–3. doi: 10.1073/pnas.1633291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rojas A, Padidam M, Cress D, Grady WM. TGF-beta receptor levels regulate the specificity of signaling pathway activation and biological effects of TGF-beta. Biochim Biophys Acta. 2009;1793:1165–73. doi: 10.1016/j.bbamcr.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang Y, Prentice MA, Mariano JM, Davarya S, Linnoila RI, Moody TW, et al. Transforming growth factor-beta 1 and its receptors in human lung cancer and mouse lung carcinogenesis. Exp Lung Res. 2000;26:685–707. doi: 10.1080/01902140150216765. [DOI] [PubMed] [Google Scholar]
- 37.Zhang HT, Chen XF, Wang MH, Wang JC, Qi QY, Zhang RM, et al. Defective expression of transforming growth factor beta receptor type II is associated with CpG methylated promoter in primary non-small cell lung cancer. Clin Cancer Res. 2004;10:2359–67. doi: 10.1158/1078-0432.ccr-0959-3. [DOI] [PubMed] [Google Scholar]
- 38.Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103:2257–61. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim WS, Park C, Hong SK, Park BK, Kim HS, Park K. Microsatellite instability(MSI) in non-small cell lung cancer(NSCLC) is highly associated with transforming growth factor-beta type II receptor(TGF-beta RII) frameshift mutation. Anticancer Res. 2000;20:1499–502. [PubMed] [Google Scholar]
- 40.Oft M, Heider KH, Beug H. TGFbeta signaling is necessary for carcinoma cell invasiveness and metastasis. Curr Biol. 1998;8:1243–52. doi: 10.1016/s0960-9822(07)00533-7. [DOI] [PubMed] [Google Scholar]
- 41.Deckers M, van Dinther M, Buijs J, Que I, Lowik C, van der Pluijm G, et al. The tumor suppressor Smad4 is required for transforming growth factor beta-induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells. Cancer Res. 2006;66:2202–9. doi: 10.1158/0008-5472.CAN-05-3560. [DOI] [PubMed] [Google Scholar]
- 42.Muraoka-Cook RS, Kurokawa H, Koh Y, Forbes JT, Roebuck LR, Barcellos-Hoff MH, et al. Conditional overexpression of active transforming growth factor beta1 in vivo accelerates metastases of transgenic mammary tumors. Cancer Res. 2004;64:9002–11. doi: 10.1158/0008-5472.CAN-04-2111. [DOI] [PubMed] [Google Scholar]
- 43.Muraoka-Cook RS, Shin I, Yi JY, Easterly E, Barcellos-Hoff MH, Yingling JM, et al. Activated type I TGFbeta receptor kinase enhances the survival of mammary epithelial cells and accelerates tumor progression. Oncogene. 2006;25:3408–23. doi: 10.1038/sj.onc.1208964. [DOI] [PubMed] [Google Scholar]
- 44.Siegel PM, Shu W, Cardiff RD, Muller WJ, Massague J. Transforming growth factor beta signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proc Natl Acad Sci U S A. 2003;100:8430–5. doi: 10.1073/pnas.0932636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bandyopadhyay A, Agyin JK, Wang L, Tang Y, Lei X, Story BM, et al. Inhibition of pulmonary and skeletal metastasis by a transforming growth factor-beta type I receptor kinase inhibitor. Cancer Res. 2006;66:6714–21. doi: 10.1158/0008-5472.CAN-05-3565. [DOI] [PubMed] [Google Scholar]
- 46.Yang YA, Dukhanina O, Tang B, Mamura M, Letterio JJ, MacGregor J, et al. Lifetime exposure to a soluble TGF-beta antagonist protects mice against metastasis without adverse side effects. J Clin Invest. 2002;109:1607–15. doi: 10.1172/JCI15333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Biswas S, Guix M, Rinehart C, Dugger TC, Chytil A, Moses HL, et al. Inhibition of TGF-beta with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression. J Clin Invest. 2007;117:1305–13. doi: 10.1172/JCI30740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nam JS, Terabe M, Mamura M, Kang MJ, Chae H, Stuelten C, et al. An anti-transforming growth factor beta antibody suppresses metastasis via cooperative effects on multiple cell compartments. Cancer Res. 2008;68:3835–43. doi: 10.1158/0008-5472.CAN-08-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, et al. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell. 2009;16:91–102. doi: 10.1016/j.ccr.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ammirante M, Luo JL, Grivennikov S, Nedospasov S, Karin M. B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature. 2010;464:302–5. doi: 10.1038/nature08782. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.