

Abstract
Many human genes have tandem promoters driving overlapping transcription, but the value of this distributed promoter configuration is generally unclear. Here we show that MICA, a gene encoding a ligand for the activating immune receptor NKG2D, contains a conserved upstream promoter that expresses a noncoding transcript. Transcription from the upstream promoter represses the downstream standard promoter activity in cis through transcriptional interference. The effect of transcriptional interference depends on the strength of transcription from the upstream promoter and can be described quantitatively by a simple reciprocal repressor function. Transcriptional interference coincides with recruitment at the standard downstream promoter of the FACT histone chaperone complex, which is involved in nucleosomal remodelling during transcription. The mechanism is invoked in the regulation of MICA expression by the physiological inputs interferon‐γ and interleukin‐4 that act on the upstream promoter. Genome‐wide analysis indicates that transcriptional interference between tandem intragenic promoters may constitute a general mechanism with widespread importance in human transcriptional regulation.
Keywords: FACT, MICA, NKG2D, tandem promoter, transcriptional interference
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Signal Transduction; Transcription
Introduction
Genomic analyses demonstrate that many human genes have tandem promoter gene structure (Djebali et al, 2012; Fantom Consortium and the RIKEN PMI and CLST, 2014). A transcript initiated from an upstream promoter can overlap that from a downstream promoter; the transcripts will differ in their first exons, but may share similar downstream sequence. Additional promoters have been studied mainly in the context of the protein coding function of the different transcripts generated (Carninci et al, 2006; Davuluri et al, 2008; Wang et al, 2016). Less explored are the consequences of transcription from additional upstream promoters on gene expression.
Transcription can have an in cis influence on promoters that lie in the path of transcriptional elongation through a process known as transcriptional interference (Shearwin et al, 2005; Palmer et al, 2011). Transcriptional interference is thought to occur when a traversing RNA polymerase elongation complex, arising from one promoter, encounters and displaces a transcription initiation complex, which has formed transiently at a different promoter (Shearwin et al, 2005; Palmer et al, 2011). In some cases, epigenetic changes of transcriptional elongation have been associated with transcriptional interference (Houseley et al, 2008; Hainer et al, 2011; Ard & Allshire, 2016). Transcriptional interference is well characterized in lower eukaryotes with compact genomes where the transcriptional path of one gene runs on into the promoter of another gene (Greger et al, 2000; Martens et al, 2005; Hongay et al, 2006; Petruk et al, 2006; Gummalla et al, 2012; Ard et al, 2014). In mammalian species, reports of transcriptional interference are limited to a few examples of developmentally regulated genes that are subject to relatively stable epigenetic regulation following the cell differentiation process (Abarrategui & Krangel, 2007; Racanelli et al, 2008; Latos et al, 2012; MacIsaac et al, 2012). A necessary condition for transcriptional interference is overlapping transcription. Tandem promoter gene structure, as found in many mammalian genes, strictly implies overlapping transcription within the gene, such that the upstream promoter drives transcription through the downstream promoter. This raises the possibility that for some of the human genes with additional upstream promoters, the tandem promoter arrangement may have evolved to regulate gene expression through transcriptional interference.
MICA is a transmembrane protein with structural similarity to MHC class I molecules and is encoded by the MICA gene within the human MHC complex (Bahram et al, 1994; Li et al, 2001). MICA is a ligand for NKG2D, an activating immune receptor expressed on CD8+ T cells, γδ T cells and natural killer cells (Bauer et al, 1999; Wu et al, 1999). Engagement of NKG2D on NK cells by MICA expressed on target cells promotes cytokine release and killing of the target cells and is implicated in cancer immunity, antiviral immunity and autoimmunity (Bauer et al, 1999; Ullrich et al, 2013; Lanier, 2015). Homologs of the MICA gene are present in all mammals studied, except rodents (Kasahara & Sutoh, 2015). The MICA gene is highly polymorphic, with over 80 coding alleles known to date (Robinson et al, 2015). Genetic studies have associated the MICA locus with inflammatory diseases, and with the response to virus infection (Kumar et al, 2011; Zhou et al, 2014; Zhang et al, 2016), although strong linkage disequilibrium between MICA and other genes within the MHC, especially HLA‐B, is a confounding factor in such studies (Le Clerc et al, 2014; Okada et al, 2014). MICA expression is upregulated in cancer tissues, and in response to a diverse range of stimuli including virus infection, heat shock, metabolic stress, cell proliferation and cytokines, such as TNFα and interferon‐γ (Groh et al, 1996; Zou et al, 2005; Cerboni et al, 2007; Schwinn et al, 2009; Lin et al, 2012; McCarthy et al, 2017). However, the molecular mechanisms that govern MICA regulation remain enigmatic. Many stimuli are known to affect MICA transcription, but only a few specific transcription factors, such as NF‐κB and HSF1, have been shown to directly regulate MICA transcription through binding to the MICA promoter (Venkataraman et al, 2007; Lin et al, 2012).
Here, we show that in MICA a conserved upstream additional promoter drives transcription of an unstable noncoding transcript and that this transcriptional activity inhibits transcription of the coding transcript from the standard downstream promoter in cis through transcriptional interference. This mode of transcriptional regulation is exemplified by the physiological signals interferon‐γ and interleukin‐4, which exert transcription factor‐mediated positive and negative influences, respectively, on the upstream promoter, resulting in opposite effects on production of the MICA coding transcript from the standard downstream promoter. Our results demonstrate for the first time that intragenic transcriptional interference occurs in humans and constitutes a potentially powerful form of real‐time transcriptional regulation. Evolutionary selection for upstream promoter components provides a means for reversing the polarity of an input signal on expression of a coding transcript, thus turning activators into repressors or vice versa.
Results
An alternative upstream MICA promoter drives expression of an unstable noncoding transcript
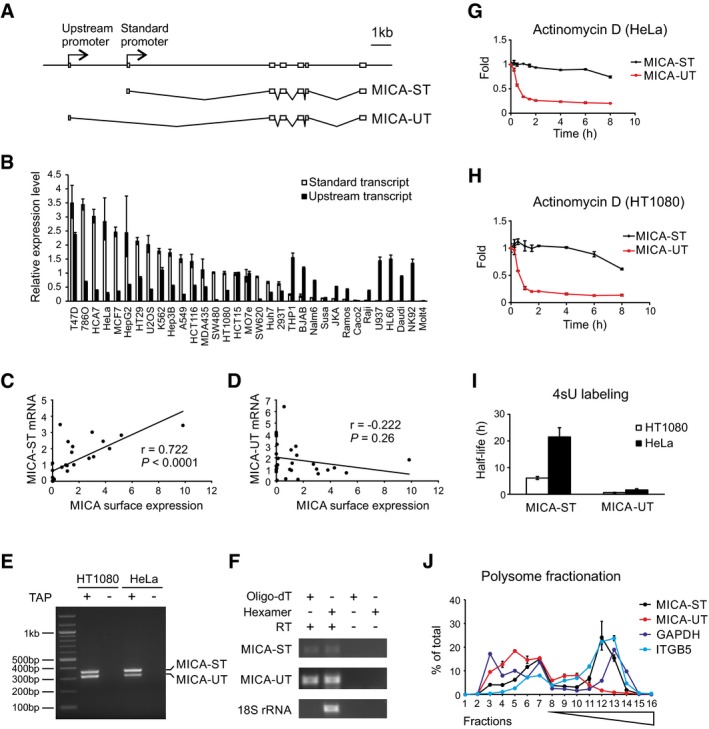
MICA encodes two major transcripts, a standard transcript (MICA‐ST) that initiates from the standard MICA promoter, and an alternative upstream transcript (MICA‐UT) that initiates from an alternative promoter 2.9 kb upstream (Fig 1A). The two transcripts have different first exons, but share common downstream exons. We found that both transcripts were widely expressed across a range of human cells and tissues, but with distinct expression patterns—in some cases there was abundant expression of the upstream transcript with little or no expression of the standard transcript (Figs 1B, and EV1A and B).
Figure 1. Properties of the MICA upstream transcript.
-
AExon structure of the MICA gene, standard transcript (MICA‐ST) and upstream transcript (MICA‐UT). In the upstream transcript, an alternative upstream first exon is spliced to exon 2 of the gene. Both transcripts share common downstream exons.
-
BThe upstream transcript and standard transcript levels measured by qPCR in different cells. Relative expression levels were normalized to that of the standard transcript in HT1080 cells and ranked according to expression of the standard transcript. Error bars represent standard deviations of three replicates.
-
CPositive correlation between the level of the standard transcript and cell surface MICA expression in 28 different cell types.
-
DNo significant correlation was found between the level of the upstream transcript and cell surface MICA expression in these different cells. The Pearson's correlation coefficient r and associated P‐values are shown.
-
ERLM‐RACE analysis of the upstream and standard MICA transcripts using a common primer in exon 2 demonstrates that both transcripts are 5′ capped. Samples prepared without tobacco acid pyrophosphatase (TAP) treatment were used as negative controls.
-
FRT–PCR using hexamer or oligo‐dT primed cDNA shows that both transcripts are 3′ polyadenylated. Nonpolyadenylated 18S rRNA and the RT–PCR without reverse transcriptase (RT) were used as negative controls.
-
G, HStability of the upstream and standard MICA transcripts measured following actinomycin D treatment in HeLa cells (G) or HT1080 cells (H). Error bars represent standard deviations of three replicates.
-
IStability of the upstream and standard MICA transcripts measured by 4sU metabolic labelling in HeLa or HT1080 cells. Error bars represent standard deviations of three replicates.
-
JPolysome profiling of the standard transcript (MICA‐ST), upstream transcript (MICA‐UT) and GAPDH and ITGB5 as controls in HT1080 cells. Distributions of mRNA across sucrose gradient fractions are shown. Numbers 1–6 represent ribosome‐free fractions, number 7 represents the monosome fraction, and numbers 8–16 represent polysome fractions with increasing number of ribosomes. Error bars represent standard deviations of three replicates.
Figure EV1. Properties of the MICA upstream transcript.
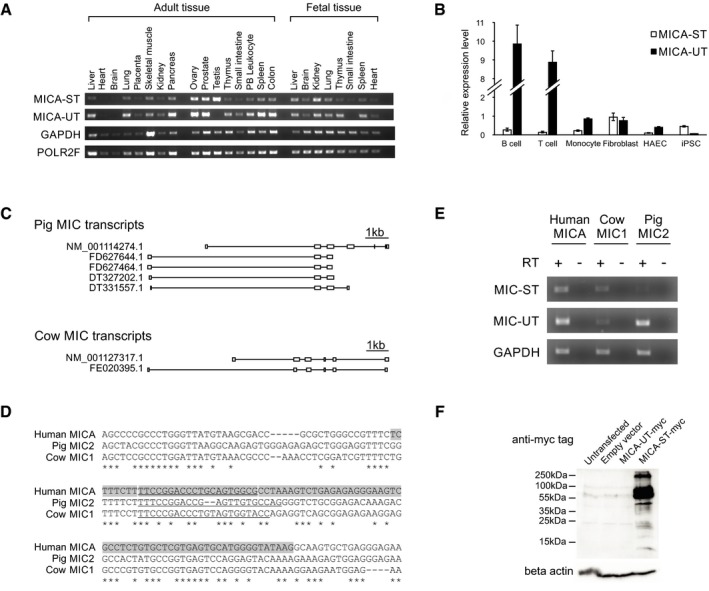
- Semi‐quantitative RT–PCR analysis of the transcript levels in adult and foetal tissues. GAPDH and POLR2F levels are shown as controls.
- The upstream transcript and standard transcript levels measured by qPCR in different primary human cells. Relative expression levels were normalized to that of the standard transcript in HT1080 cells. Error bars represent standard deviations of three replicates.
- Exon structure of the upstream and standard transcripts of the MICA homologs in pig and cow based on EST and RefSeq data. Transcript sequences were aligned to the pig (CT737281.12) or cow (AC_000180.1) genomes for visualization.
- Alignment of the region around the first exon of the upstream transcript (highlighted in grey) demonstrating sequence conservation. The sequence information was used to design species‐specific primers (underlined) for detection of MICA homolog transcripts in these species.
- RT–PCR demonstrates expression of upstream transcripts and standard transcripts for MICA homologs in adult kidney tissue from different species. Reactions without reverse transcriptase (RT) were used as negative controls.
- Western blot analysis of lysates from 293T cells transfected with C‐terminal in‐frame myc‐tagged upstream transcript (MICA‐UT‐myc) or standard transcript (MICA‐ST‐myc) expression constructs demonstrate that no protein was detectable for the upstream transcript.
There are MICA homologs in the genomes of almost all mammals studied except rodents (Kasahara & Sutoh, 2015). We identified homologs of both the upstream and standard transcripts for pigs and cows (Fig EV1C) and alignment with the corresponding genomes revealed conservation of the tandem promoter gene structure and local sequence around the upstream promoter in these species and humans (Fig EV1C and D). RT–PCR analysis confirmed the expression of both transcripts in adult pigs and cows (Fig EV1E). This conservation suggested that the upstream promoter or transcript could have a biological function.
Cell surface expression of human MICA protein correlated with expression of the standard transcript, but not the upstream transcript (Fig 1C and D), consistent with the standard transcript encoding the MICA protein. Both transcripts were 5′ capped, and poly‐A tailed (Fig 1E and F), indicating that the upstream transcript is transcribed by RNA polymerase II and processed in the same way as the standard transcript. Bioinformatic analysis of the upstream transcript sequence predicted that it would undergo nonsense‐mediated decay due to the presence of an upstream open reading frame encoding a premature termination codon (Hug et al, 2016). Consistent with this, it displayed rapid decay kinetics compared to the standard MICA transcript, as measured by global transcriptional inhibition with actinomycin D (Fig 1G and H) or by 4‐thiouridine metabolic labelling (Fig 1I). No protein product could be detected for the major open reading frame of this transcript (Fig EV1F). The standard transcript was enriched in RNA isolated from heavy polysome fractions similar to GAPDH and ITGB5, consistent with active translation of this transcript, but the upstream transcript was predominantly in the ribosome‐free and monosome fractions (Fig 1J). This indicates that the upstream transcript is poorly translated, which is consistent with the prediction that it is noncoding and undergoes nonsense‐mediated decay. Together, these data demonstrate that the upstream transcript of the MICA gene is an unstable noncoding transcript.
The upstream promoter represses expression of the standard coding MICA transcript in cis
Expression of the upstream transcript in trans or post‐transcriptional siRNA‐mediated downregulation of the upstream transcript had no effect on the expression of endogenous MICA (Fig 2A and B). These findings suggest that the transcript itself does not regulate MICA expression, leaving open the possibility that the alternative upstream promoter might itself regulate expression of the downstream coding transcript. To test this hypothesis, we used CRISPR genome editing tools to delete the upstream promoter in primary cells (Fig 2C). We mapped the core upstream and standard MICA promoters by serial deletion using reporter assays (Fig EV2A and B), and designed pairs of CRISPR guide RNAs for deletion of the core upstream promoter (Fig EV2C). In primary human fibroblasts and arterial endothelial cells, transient expression of the Cas9 nuclease and either of two different pairs of CRISPR guide RNAs for the upstream promoter caused significant upregulation of MICA expression compared to CRISPR guide RNAs targeting control loci and the effect was greater in the fibroblasts (Figs 2D, and EV2D and E). We also used a deactivated Cas9 (dCas9)‐based programmable transcriptional activation system to specifically activate the upstream promoter and study its effect on MICA expression. This synergistic activation mediator (SAM) system consists of dCas9 fused to a VP64 transactivation domain and a locus‐specific guide RNA that recruits multiple copies of a p65 and HSF1‐derived multidomain transactivator (Konermann et al, 2015; Fig 2E). dCas9‐based activation of the upstream promoter downregulated MICA expression from the standard downstream promoter, whereas activation of the standard promoter upregulated MICA expression (Fig 2F). These observations indicate that the upstream promoter negatively regulates the activity of the standard downstream promoter.
Figure 2. The upstream promoter represses MICA expression in cis .
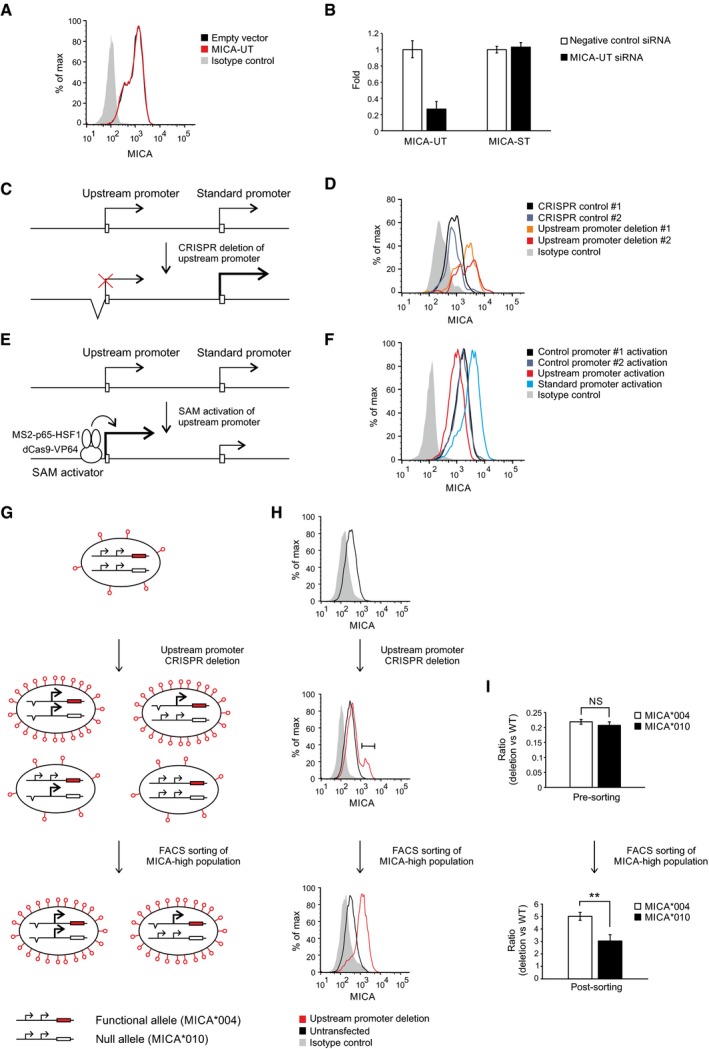
- Flow cytometric analysis of MICA surface expression in 293T cells transfected with plasmids expressing the full‐length upstream transcript or empty vector control and the pEGFP‐N1 plasmid as transfection control. Cells were gated for the GFP‐positive population.
- qPCR analysis of the upstream transcript and standard transcript in 293T cells transfected with siRNA targeting the upstream transcript or with control siRNA. Error bars represent standard deviations of three replicates.
- Diagram of CRISPR deletion of the core upstream promoter.
- Flow cytometry of cell surface MICA expression in primary human fibroblasts following transfection with CRISPR plasmids targeting deletions of the MICA upstream promoter or control genes (HLA‐B or PDPN). Cells were gated for the CRISPR Cas9 nuclease‐transfected GFP‐positive population.
- Diagram of dCas9‐based transcriptional activation of the upstream promoter. The SAM transcription activator consists of dCas9 fused to a VP64 transcription activator and a guide RNA containing RNA aptamers that recruit multiple copies of the MS2 bacteriophage coat protein fused to a multidomain transcription activator derived from the p65 and HSF1 transcription factors.
- Flow cytometry of cell surface MICA expression in 293T cells following transfection with SAM plasmids activating the MICA upstream promoter, standard promoter or control promoters (CD43 and CD36). Cells were gated for the transfected GFP‐positive population.
- Schematic diagram representing the different possible genotypes that may arise and the associated phenotypes predicted by the in cis transcriptional interference hypothesis. Primary human fibroblasts were used which are heterozygous for the MICA*004 and MICA*010 alleles, of which only MICA*004 (red) reaches the cell surface. CRISPR deletion of the upstream promoter can result in three additional genotypes depending on whether one or both MICA alleles are affected. If, as we hypothesized, there is intragenic transcriptional interference, then deletion of the upstream promoter of the MICA*004 allele will result in upregulation of cell surface MICA expression, whereas deletion of the upstream promoter of the MICA*010 null allele will have no effect on cell surface MICA expression compared to wild‐type cells. Accordingly, cells sorted for upregulated MICA surface expression should be enriched for cells with deletion of the MICA*004 allele if transcriptional interference occurs in cis.
- MICA surface expression of cells at different stages of the experiment. After CRISPR deletion of the upstream promoter, there is a population of cells with upregulation of MICA expression (middle panel) and this population was sorted for further analysis (lower panel).
- PCR analysis of the CRISPR‐deletion genotype for each allele before and after sorting of cells with upregulated cell surface MICA expression. One of the differences between the MICA*004 and MICA*010 alleles is at SNP rs2596539 within the upstream promoter outside the deleted region, and this allows differentiation between the two alleles using restriction digestion prior to PCR amplification. As indicated by the genotype before sorting, CRISPR‐mediated deletion of the upstream promoter arises with equal frequency for both alleles (upper panel). However, consistent with the in cis transcriptional interference hypothesis, the cells that have high MICA*004 surface expression are enriched for deletion of the in cis MICA*004 upstream promoter compared to the MICA*010 upstream promoter (lower panel). The difference in the enrichment of MICA*004 compared to MICA*010 upstream promoter deletion is modest as the majority of the sorted cells have biallelic deletion. Error bars represent standard deviations of four replicates. NS not significant; **P < 0.01, Student's t‐test.
Figure EV2. Transcription from the upstream promoter represses MICA expression.
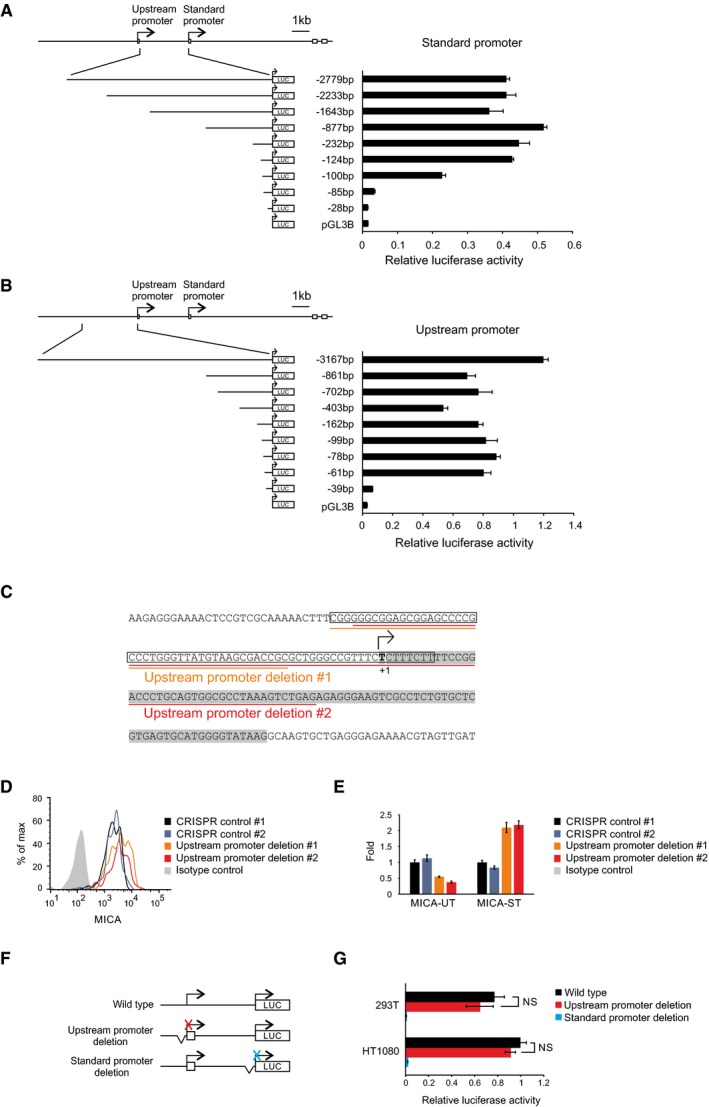
-
A, BMapping of the core standard promoter (A) or upstream promoter (B) using luciferase reporter assays with constructs carrying serial promoter truncations in HT1080 cells. Error bars represent standard deviations of three biological replicates.
-
CSequence of the upstream promoter region subjected to CRISPR‐mediated deletion. The core promoter mapped by reporter assays is boxed, the transcription start site mapped by 5′‐RACE is in bold, and the upstream first exon is highlighted in grey. The deletions produced with the two pairs of CRISPR guide RNAs are underlined in orange and red, respectively.
-
DFlow cytometry of cell surface MICA expression in primary human arterial endothelial cells following transfection with CRISPR plasmids targeting deletions of the MICA upstream promoter or control genes (HLA‐B or PDPN). Cells were gated for the CRISPR Cas9 nuclease‐transfected GFP‐positive population.
-
EqPCR analysis of MICA upstream (MICA‐UT) or standard transcript (MICA‐ST) expression in primary human fibroblasts transfected with CRISPR plasmids targeting deletions of the MICA upstream promoter or control genes (HLA‐B or PDPN). GFP‐positive cells were sorted 3 days post‐transfection for analysis. Error bars represent standard deviations of three replicates.
-
FDiagram of constructs for luciferase reporter assays with deletion of the upstream (red) or standard (blue) core promoters.
-
GReporter assays using the above constructs in 293T and HT1080 cells. Deletion of the core standard promoter completely abolished luciferase activity, confirming that the activity measured from the wild‐type construct is derived exclusively from the standard promoter. Deletion of the core upstream promoter has no effect. Error bars represent standard deviations of three biological replicates. NS, not significant: Student's t‐test.
In one case, the CRISPR‐deletion experiment was undertaken in primary human fibroblasts carrying one functional MICA allele (MICA*004) and one null allele (MICA*010) that does not reach the cell surface (Li et al, 2000). Exploiting this feature, we used a forward genetics approach to test whether the core upstream promoter regulates MICA expression in cis (Fig 2G). CRISPR deletion of the upstream promoter can result in four major genotypes: wild type, monoallelic deletion of either allele and biallelic deletion of the upstream promoter. If the upstream promoter only represses MICA expression in cis, then deletion of the upstream promoter of the functional MICA*004 allele will upregulate cell surface MICA expression, but deletion of the upstream promoter of the MICA*010 null allele will not. Therefore, cells sorted for upregulated MICA surface expression would be enriched for deletion of the promoter of the functional MICA*004 allele (Fig 2G). In contrast, with in trans regulation, no allele‐specific enrichment would be observed in cells sorted for upregulated MICA surface expression. As demonstrated in Fig 2H, deletion of the upstream promoter produced a population of cells with upregulation of MICA expression which is stable following cell sorting of this population. Although the upstream promoters of both alleles were deleted with equal efficiency in the pre‐sorted population, cells with upregulation of cell surface MICA expression were preferentially enriched for deletion of the functional MICA*004 allele compared to deletion of the null MICA*010 allele (Fig 2I). This confirms that the upstream promoter represses MICA expression from the standard promoter in cis.
The upstream promoter represses MICA expression through transcriptional interference
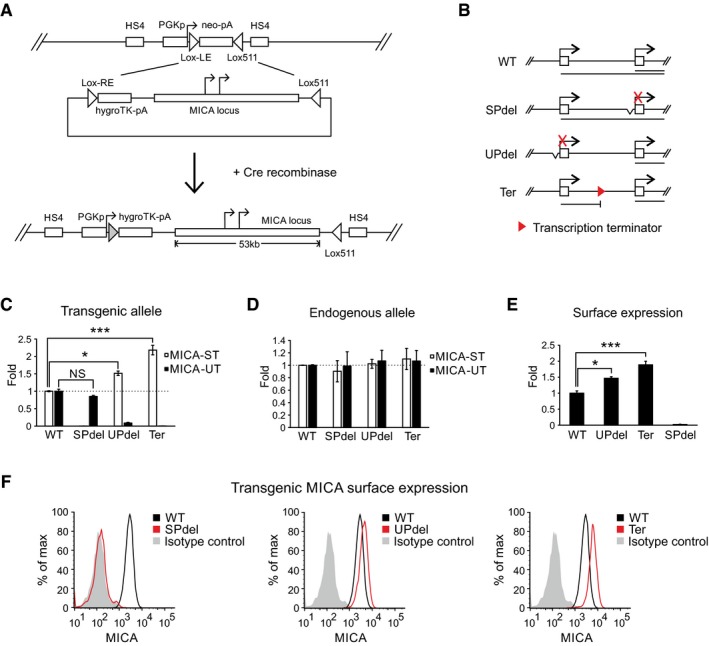
We hypothesized that the process of transcription from the upstream promoter inhibited transcription from the standard promoter. However, in plasmid‐based reporter assays we did not observe any inhibition of the standard promoter activity by the upstream promoter (Fig EV2F and G). Plasmid‐based reporter systems lack distal genetic elements and the concomitant chromatin structure and environment. Therefore, we created a set of isogenic cell lines using recombinase‐mediated cassette exchange to insert synthetic variants of the entire MICA locus into chromatin at the same genomic location (Fig 3A). These variants were constructed by BAC recombineering and included deletions of the core upstream or standard promoter or insertion of a transcription terminator between the two promoters (Fig 3B and Appendix Table S1). The endogenous MICA loci remained intact as controls for in trans effects. The host cell line was homozygous for the MICA*007 allele, which allows cell surface detection of the transgenic MICA*008 allele with an allele‐specific monoclonal antibody (Fig EV3A).
Figure 3. Transcriptional interference of the standard MICA promoter by the upstream promoter.
-
AGeneration of the site‐directed BAC reporter cell line. The acceptor cell line was generated such that it carries only a single copy of a landing site containing a neomycin‐resistance cassette under the control of a PGK promoter. The neomycin cassette was flanked by variants of LoxP sites (Lox‐LE and Lox511), and the entire inserted locus was insulated at both ends by HS4 insulators. The donor BAC constructs contain the MICA locus flanked on one side by a Lox‐RE site followed by a promoterless hygromycin‐resistance cassette, and on the other side by a Lox511 site compatible with the one in the landing site. Following co‐transfection of the donor BAC construct and a Cre recombinase expression plasmid into the acceptor cell line, the ensuing Cre‐Lox recombination results in irreversible exchange of the neomycin cassette in the cell with the insert containing the MICA locus from the BAC.
-
BDiagram of BAC constructs used to generate modified isogenic cell lines by recombinase‐mediated cassette exchange. Four different constructs were created: wild type (WT), deletion of the core standard promoter (SPdel), deletion of the core upstream promoter (UPdel) and insertion of a transcription terminator between the two promoters (Ter). The primary transcripts generated are shown below the constructs.
-
C, DTransgenic (C) or endogenous (D) MICA upstream and standard transcript expression measured by qPCR in modified isogenic cell lines carrying a transgenic 53‐kb MICA locus. Deletion of the upstream promoter or insertion of a transcription terminator between the promoters led to increased expression of the transgenic MICA standard transcript; no effect was seen on the endogenous transcripts. Error bars represent standard deviations of multiple independently generated clones (n = 2–4, Appendix Table S1). NS, not significant; *P < 0.05, ***P < 0.001, Student's t‐test.
-
E, FFlow cytometric analysis of transgenic MICA surface expression in isogenic cell lines carrying a transgenic 53‐kb MICA locus. Bar chart of mean fluorescent intensity is shown in (E), and histograms of representative clones in (F). Consistent with in cis transcriptional interference, expression of the transgenic MICA protein was upregulated by deletion of the upstream promoter or insertion of a transcription terminator between the two promoters. Error bars represent standard deviations of multiple independently generated clones (n = 2–4, Appendix Table S1). *P < 0.05, ***P < 0.001, Student's t‐test.
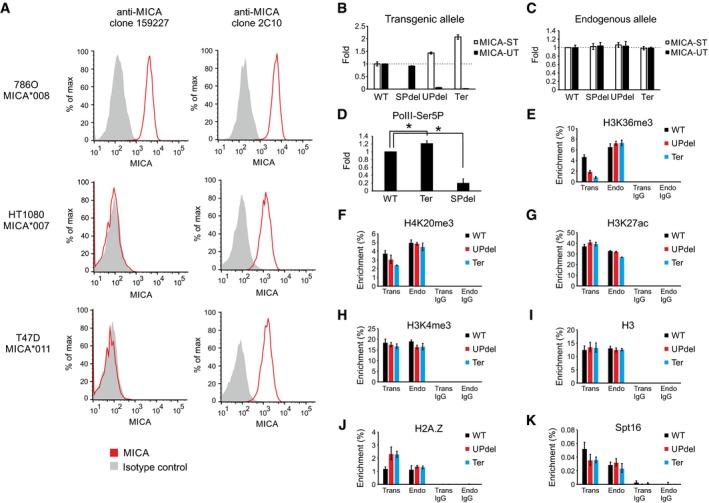
Figure EV3. Transcriptional interference of MICA .
-
AFlow cytometric analysis of MICA surface expression in cell lines homozygous for MICA alleles demonstrates allelic specificity of the clone 159227 anti‐MICA antibody for the MICA*008 allele. In contrast, the clone 2C10 anti‐MICA antibody does not display allele‐specific activity.
-
B, CqPCR analysis of transgenic (B) or endogenous (C) MICA upstream and standard transcript expression in isogenic cell lines carrying a transgenic 161‐kb MICA locus. Deletion of the upstream promoter or insertion of a transcription terminator between the upstream promoter and the standard promoter results in an increase in the level of the transgenic standard transcript. No effect was seen on the endogenous transcripts. Error bars represent standard deviations of multiple independently generated clones (n = 2–3, Appendix Table S1).
-
DChIP analysis of Pol II phospho‐Ser5 signal at the transgenic standard promoter region. Data are shown as fold change over the wild‐type construct and normalized to endogenous standard promoter Pol II phospho‐Ser5 signal. Error bars represent standard deviations of three independent experiments. *P < 0.05, Student's t‐test.
-
E–KChIP analysis of H3K36me3 (E), H4K20me3 (F), H3K27ac (G), H3K4me3 (H), H3 (I), H2A.Z (J) and Spt16 (K) at the transgenic (Trans) and endogenous (Endo) standard promoter regions. Error bars represent standard deviations of three replicates.
Deletion of the upstream promoter in a MICA transgene containing the native ~20‐kb flanking sequences downregulated expression of the transgenic upstream transcript and upregulated expression of the transgenic standard coding transcript as predicted (Fig 3C). Insertion of a transcription terminator between the upstream promoter and the standard promoter had a similar effect, which demonstrates that transcriptional elongation from the upstream promoter is required for repression of the standard promoter (Fig 3C). The slightly lesser effect of the upstream promoter deletion compared to the transcription terminator likely reflects residual low‐level transcription from around the deleted region of the gene. Deletion of the standard promoter had no effect on expression of the upstream transcript, which excludes promoter competition as a mechanism for the inhibitory effect of the upstream promoter (Fig 3C). These different manoeuvres had no effect on the endogenous MICA alleles, confirming the in cis nature of the inhibitory effect of the upstream promoter on transcription from the standard promoter (Fig 3D). Changes in expression of the transgenic standard transcript were reflected in MICA protein expression at the cell surface (Fig 3E and F). Similar results were obtained using larger genomic inserts, which included ~75‐kb flanking sequences on both sides of the MICA gene and incorporated the neighbouring HLA‐B and HCP5 loci (Fig EV3B and C). Further, we carried out ChIP for Ser5‐phoshorylated RNA Pol II, which is a marker of transcription initiation activity, but decreases substantially during transcription elongation (Harlen & Churchman, 2017). Deletion of the standard downstream promoter abolished most of the Pol II phospho‐Ser5 signal in this region, demonstrating that the majority of this signal arose from transcription initiation from the downstream promoter (Fig EV3D). Interposition of a transcription terminator between the two promoters caused a significant increase in the ChIP signal seen at the downstream promoter, indicating that run‐through transcription from the upstream promoter exerts an inhibitory effect on transcription initiation at the downstream promoter (Fig EV3D). Overall, these data together confirm that the upstream promoter represses MICA expression from the standard promoter in cis through transcriptional interference.
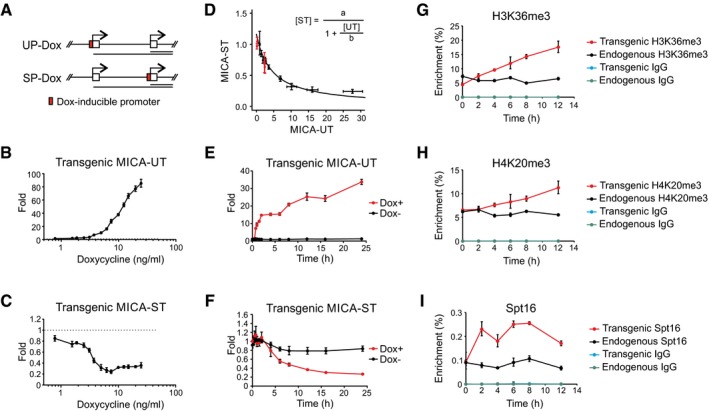
To define the input–output characteristics of the MICA dual‐promoter system, we created further sets of isogenic cell lines with the MICA gene engineered to be tuneable at the upstream or standard promoter (Fig 4A, Appendix Table S1). When the core upstream promoter was replaced with a doxycycline‐inducible promoter, doxycycline treatment induced dose‐dependent upregulation of the transgenic upstream transcript, downregulation of the standard transcript in cis and downregulation of the encoded MICA protein (Figs 4B and C, and EV4B). As predicted, no in trans effect was observed on expression of the endogenous upstream or standard MICA transcripts (Fig EV4C). The wild‐type upstream and standard promoters did not respond to doxycycline, confirming that repression of the transgenic standard promoter is caused directly by the induction of the upstream promoter with doxycycline (Fig EV4D). Mathematical analysis of the steady‐state response curve shows that the transcriptional activity arising from the standard promoter is in a simple reciprocal relationship with the transcriptional activity arising from the upstream promoter (Fig 4D). Expression of the transgenic upstream or standard MICA transcripts in cells with the native upstream promoter or with upstream promoter deletions maps onto this response curve (Fig 4D). This shows that different upstream promoters, which drive quantitatively similar levels of transcription, give rise to similar levels of transcriptional interference; it is the strength of transcription from the upstream promoter, rather than the identity of the promoter, that drives the observed transcriptional interference. Timecourse analysis of upstream and standard transcript expression following doxycycline treatment demonstrates that transcription from the upstream promoter is rapidly followed by a shutdown of transcription from the standard promoter (Fig 4E and F). The decline in the steady‐state level of the standard transcript is slower than the rise in the upstream transcript due to the longer half‐life of the standard transcript. Replacement of the standard MICA promoter with a doxycycline‐inducible promoter confers dose‐dependent induction of the standard MICA transcript and cell surface MICA protein expression, but has no effect on expression of the upstream transcript (Fig EV4A, E and F). This confirms that transcriptional interference between the two promoters is unidirectional, and further excludes promoter competition as the mechanism for transcriptional interference. This observation reflects the unidirectional nature of the underlying transcription.
Figure 4. Characterization of intragenic transcriptional interference in MICA .
-
ADiagram of BAC constructs used to generate modified isogenic cell lines in which a doxycycline‐inducible promoter was positioned to drive transcription of the upstream transcript (UP‐Dox) or standard transcript (SP‐Dox).
-
B, CDose‐dependent changes of transgenic upstream and standard transcript levels in isogenic cells in which expression of the upstream transcript are driven by doxycycline. Doxycycline induced expression of the transgenic upstream transcript (B) and caused a fourfold dose‐dependent reduction in expression of the transgenic standard transcript (C). Error bars represent standard deviations of three replicates.
-
DMathematical modelling of the dose‐dependent regulation of expression of the standard transcript by the upstream promoter (UP‐Dox). Data from doxycycline‐treated cells (black) up to the dose with maximum transcriptional interference (0–7.5 ng/ml) fitted to a reciprocal function as shown. Data from wild‐type clones and clones with deletion of the upstream promoter are represented in red. Error bars represent standard deviations of three replicates.
-
E, FTimecourse of change in expression of the transgenic upstream (E) or standard (F) transcript in an isogenic cell line in which expression of the upstream transcript is driven by doxycycline (7.5 ng/ml) or mock control. Error bars represent standard deviations of three replicates.
-
G–IChIP analysis of H3K36me3 (G), H4K20me3 (H) and Spt16 (I) modifications at the transgenic and endogenous standard promoter over time following induction of the transgenic upstream transcript by doxycycline (7.5 ng/ml). Error bars represent standard deviations of three replicates.
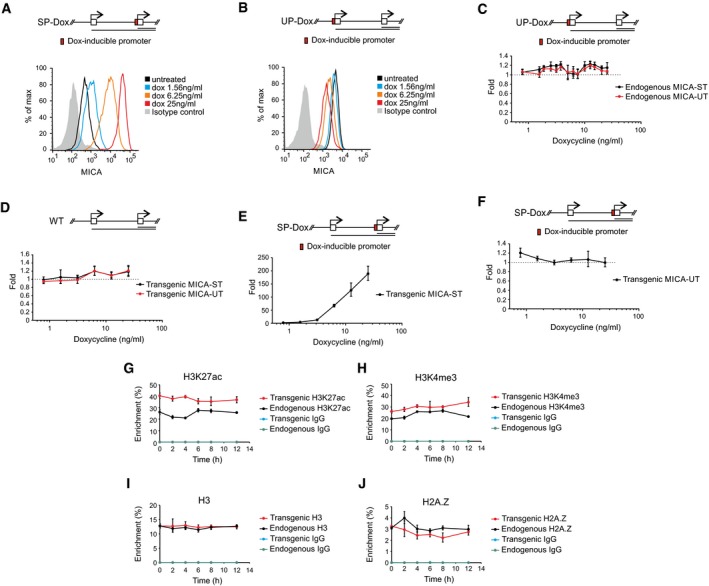
Figure EV4. Transcriptional interference demonstrated with doxycycline‐inducible promoters.
-
A, BRepresentative flow cytometry histograms showing dose‐dependent changes of opposite direction in transgenic MICA surface expression when expression of the standard (A) or upstream (B) transcript was under control of a doxycycline‐inducible promoter.
-
CqPCR analysis showing no change of endogenous upstream and standard transcript expression in modified isogenic cells in which the upstream transcript of the MICA transgene is under the direct control of a doxycycline‐inducible promoter.
-
DqPCR analysis showing no response of the transgenic upstream and standard transcript expression to doxycycline in isogenic cells with the transgenic wild‐type upstream and standard promoters.
-
E, FqPCR analysis of dose‐dependent changes in expression of the transgenic standard transcript (E) and upstream transcript (F) in modified isogenic cells in which the transgenic standard transcript is under the control of a doxycycline‐inducible promoter. The strong induction of the standard transcript is not associated with any change in the upstream transcript which confirms the unidirectional nature of the in cis transcriptional interference.
-
G–JChIP analysis of H3K27ac (G), H3K4me3 (H), H3 (I) or H2A.Z (J) at the transgenic and endogenous standard promoter over time following induction of the transgenic upstream transcript by doxycycline (7.5 ng/ml).
Transcriptional interference is synchronous with FACT recruitment
The process of transcription is associated with changes in histones along the path taken by the polymerase. We tested whether deposition of candidate histone modifications was involved in transcriptional interference in the MICA gene. By combining MICA allele‐specific restriction digestion and chromatin immunoprecipitation (ChIP), we were able to analyse independently a range of histone modifications at both the transgenic and endogenous MICA promoters in the sets of modified isogenic cell lines (Fig 3B). H3K36me3, a histone mark cotranscriptionally recruited by elongating polymerase and involved in silencing of cryptic transcription within gene bodies (Carrozza et al, 2005; Keogh et al, 2005), is decreased at the transgenic standard MICA promoter when transcriptional interference is inhibited by deletion of the upstream promoter or by interposition of a transcription terminator between the two promoters (Fig EV3E). The upstream promoter deletion and transcription terminator interposition both decrease the repressive mark H4K20me3 (Jorgensen et al, 2013; Fig EV3F) and increase the histone variant H2A.Z (Fig EV3J), but not pan‐histone H3, H3K27ac or the promoter mark H3K4me3 (Fig EV3G–I). No changes were seen at the endogenous loci, demonstrating the in cis nature of the underlying mechanisms.
To further clarify the role of these markers in transcriptional interference in MICA, we studied dynamic changes in histone modifications over time following induction of transcriptional interference (Fig 4A). When transcriptional interference was induced by expression of the upstream transcript from a doxycycline‐inducible promoter, there was progressive enrichment of H3K36me3 and H4K20me3 in cis at the transgenic standard MICA promoter (Fig 4G and H), but not of the other markers (Fig EV4G–J). However, the increases seen with H3K36me3 and H4K20me3 were gradual (Fig 4G and H), in contrast with the rapid induction of upstream transcript expression and the rapid onset of transcriptional interference (Fig 4E and F). This suggests that H3K36me3 and H4K20me3 deposition lags behind the transcriptional interference and, therefore, may not be acutely causative of the transcriptional interference.
The histone chaperone FACT plays a role in nucleosome remodelling during transcription (Belotserkovskaya et al, 2003), and ChIP analysis demonstrated that the signal for the Spt16 subunit of FACT at the transgenic standard downstream promoter was reduced by deletion of the upstream promoter or interposition of a transcription terminator between the two promoters (Fig EV3K). When expression of the upstream transcript was driven by the doxycycline promoter, the Spt16 signal rose rapidly at the downstream promoter (Fig 4I) with a timecourse that is synchronous with that of transcriptional interference itself and changes much faster than the changes in histone marks. This is consistent with a role in transcriptional interference for events involved in the nucleosome remodelling associated with elongation of the upstream transcript through the downstream standard promoter.
IFN‐γ and IL‐4 regulate MICA through transcriptional interference
Evolutionary conservation of the MICA tandem promoter arrangement suggests that transcriptional regulation of the upstream promoter and the associated transcriptional interference may have biological value. Bioinformatic analysis revealed a highly conserved binding site within the upstream promoter for the activating transcription factor interferon regulatory factor 1 (IRF1), and we analysed the possible regulatory function of this site (Fig 5A; Tanaka et al, 1993; Rettino & Clarke, 2013). ChIP assays demonstrated that IFN‐γ treatment of primary human arterial endothelial cells induces binding of IRF1 to the upstream promoter and not to the standard promoter (Fig 5B). Specific binding to the IRF1 site was confirmed using supershift and competition electrophoretic mobility shift assays (EMSAs) for IRF1 (Fig 5C and Appendix Fig S1). Reporter assays confirmed that the upstream promoter, but not the standard promoter, was inducible by IFN‐γ treatment of endothelial cells and that the induction was mediated through the IRF1 binding site (Fig 5D). Together, these data confirm that IFN‐γ activates the upstream promoter through inducible binding of IRF1 to the conserved IRF1‐binding site. Therefore, we hypothesized that activation of the upstream promoter through this site would act by in cis transcriptional interference to downregulate expression of the standard promoter and so of MICA protein expression. As predicted, IFN‐γ treatment activated the upstream promoter leading to expression of the upstream transcript and downregulation of the standard MICA transcript and of protein expression, consistent with transcriptional interference (Fig 5E and F).
Figure 5. Regulation of MICA by IFN‐γ through transcriptional interference.
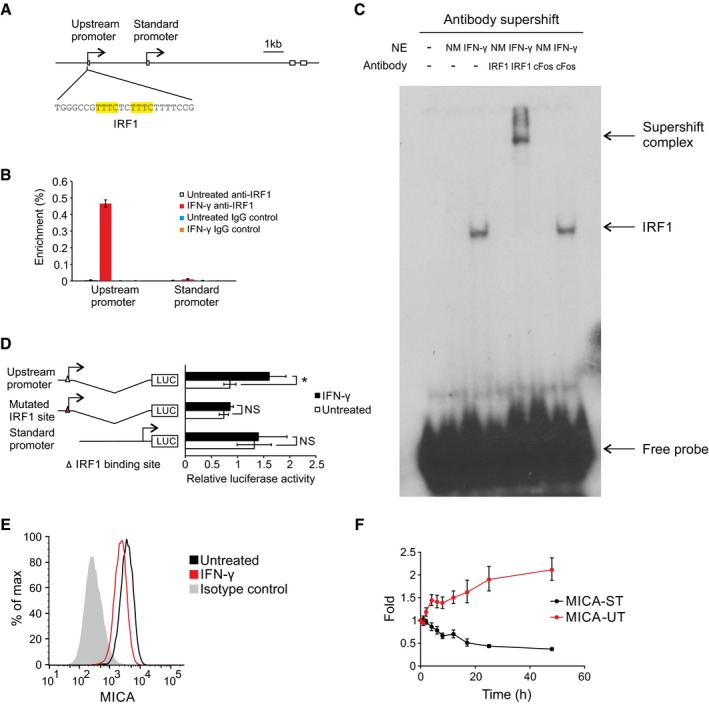
- Diagram of the upstream promoter with the IRF1 binding site highlighted in yellow.
- ChIP analysis of the upstream promoter and standard promoter regions of the endogenous MICA locus for binding of the transcription factor IRF1 in primary human arterial endothelial cells following interferon‐γ (IFN‐γ) treatment. Interferon‐γ causes substantial binding of IRF1 to the upstream promoter, but not to the standard promoter. Error bars represent standard deviations of three replicates.
- Electrophoretic mobility shift assay (EMSA) showing in vitro binding of IRF1 to the upstream promoter following interferon‐γ treatment of primary human arterial endothelial cells. Nuclear extracts (NE) of cells treated with interferon‐γ (IFN‐γ) or untreated (NM) were pre‐incubated with anti‐IRF1 antibody or control anti‐cFos antibody before the addition of 32P‐labelled probe containing the wild‐type IRF1 binding site of the MICA upstream promoter.
- Reporter assays in primary human arterial endothelial cells demonstrate that the upstream promoter activity is increased by interferon‐γ (IFN‐γ) treatment and that this effect is abolished by mutation of the IRF1 binding site. The standard promoter does not contain any predicted IRF1 binding site and did not respond to interferon‐γ. Error bars represent standard deviations of biological triplicates. NS, not significant; *P < 0.05, Student's t‐test.
- Flow cytometry of MICA surface expression in primary human arterial endothelial cells treated with interferon‐γ.
- Interferon‐γ treatment of primary human arterial endothelial cells increased expression of the upstream transcript (red) over time and reduced expression of the standard transcript (black). Fold change over mock‐treated control is shown. Error bars represent standard deviation of three replicates.
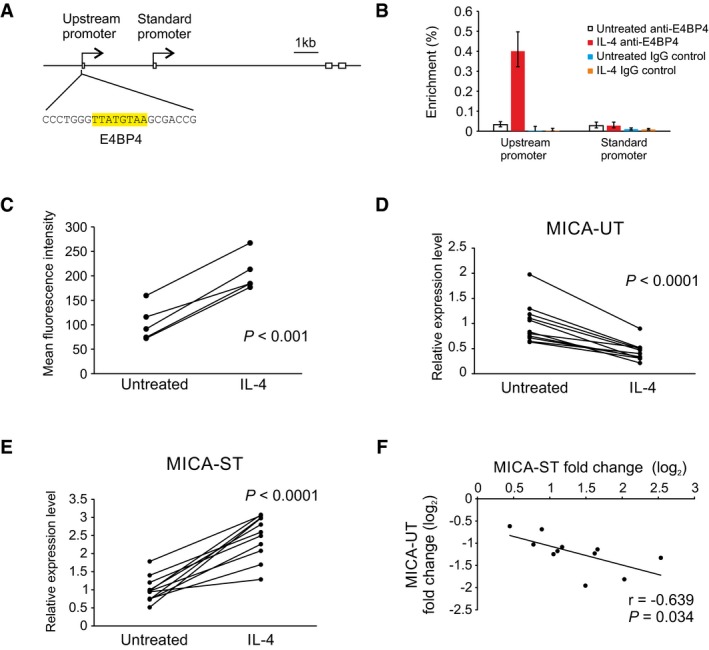
Conversely, further examination of the upstream promoter sequence revealed a conserved binding site for the transcription repressor E4BP4 (Fig 6A; Cowell et al, 1992). E4BP4 has been shown to play a role in the B‐cell response to interleukin‐4 (IL‐4), which promotes immunoglobulin class switching in B cells (Tangye et al, 2002; Kashiwada et al, 2010). We hypothesized that induction of E4BP4 by IL‐4 would downregulate activity of the upstream promoter and expression of the upstream transcript, so reducing transcriptional interference and increasing expression of the standard coding MICA transcript. Consistent with this, treatment of primary B cells with IL‐4 increased binding of E4BP4 to the upstream promoter as evidenced by ChIP (Fig 6B), downregulated expression of the upstream transcript and induced expression of the standard coding MICA transcript and of MICA protein expression at the cell surface (Fig 6C–E). Reporter assays and EMSA studies using cells transfected with E4BP4 confirmed that E4BP4 represses the upstream promoter activity through specific binding to the E4BP4 site (Appendix Fig S2A and B). Across multiple individual donors, the magnitude of downregulation of the upstream transcript correlated reciprocally with the upregulation of the standard transcript (Fig 6F).
Figure 6. Regulation of MICA by IL‐4 through transcriptional interference.
-
ADiagram of the upstream promoter with the E4BP4 binding site highlighted in yellow.
-
BChIP analysis of the upstream promoter and standard promoter regions of the endogenous MICA locus for binding of the transcription factor E4BP4 in primary human B cells following interleukin‐4 (IL‐4) treatment. Interleukin‐4 causes substantial binding of E4BP4 to the upstream promoter, but not to the standard promoter. Error bars represent standard deviations of three replicates.
-
CMICA surface expression on primary human CD19+ B cells in PBMC from five different donors treated with interleukin‐4 (IL‐4) for 3 days. Cells were gated on the CD3−CD19+ population. Data shown are mean fluorescence intensity with the isotype control subtracted. P‐value is from paired Student's t‐test.
-
D–FInterleukin‐4 (IL‐4) treatment of primary human B cells from 11 donors reduced expression of the upstream transcript (D) and increased expression of the standard transcript (E). P‐values are from paired Student's t‐test. Linear regression (F) demonstrates a reciprocal correlation between changes in expression of the upstream transcript and of the standard transcript in response to interleukin‐4 treatment. Pearson's correlation coefficient r and associated P‐value are shown.
CAGE‐seq data are consistent with transcriptional interference in other genes
The expression of transcripts from upstream promoters in many other human genes raises the possibility that transcriptional interference could be involved in transcriptional regulation in genes other than MICA. To survey this possibility, we analysed CAGE‐seq data sets because CAGE‐seq provides good clarity about which promoter a transcript arises from (Carninci et al, 2006). We identified promoters which had been experimentally validated (Dreos et al, 2013) and selected the subset of these promoters that formed tandem promoter systems. Data sets for analysis included CAGE‐seq data sets from the FANTOM5 project of transcript expression at multiple time points following a stimulus (Arner et al, 2015). We determined the trajectory of expression over time in all transcripts that were expressed. For tandem promoter systems, we identified patterns of monotonic transcript expression that were consistent with transcriptional interference—that is, an increase in the level of transcript arising from the upstream promoter associated with a fall in the level of transcript arising from the downstream promoter, or a fall in the level of transcript arising from the upstream promoter associated with an increase in the level of transcript arising from the downstream promoter (Table EV1). Across multiple human and mouse data sets, we found a substantial number of cases where there was a reciprocal pattern of transcript expression analogous to that seen with MICA. Examples of the transcript trajectories over time following a stimulus are illustrated in Fig 7. This analysis indicates that in higher eukaryotes there are multiple genes with tandem promoters displaying reciprocal patterns of transcript expression consistent with transcriptional interference.
Figure 7. Tandem promoter transcript expression patterns consistent with transcriptional interference.
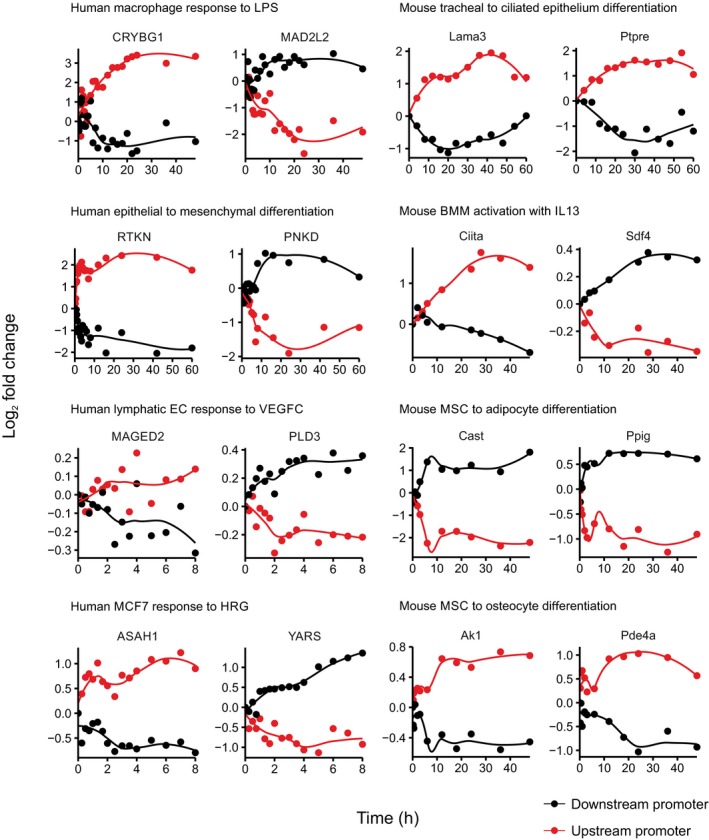
Transcript levels from tandem promoter pairs for genes from the FANTOM5 CAGE‐seq data sets over time following the indicated stimuli are shown as log2 fold change, with the upstream promoter transcript in red and downstream promoter transcript in black. Coordinates of promoter windows used for counting expression levels are shown in Appendix Table S5. Reciprocal changes in expression from tandem promoter pairs are consistent with transcriptional interference.
Discussion
We studied the function of a set of tandem intragenic promoters in the transcriptional regulation of MICA. We found that transcription from the upstream promoter represses MICA expression through transcriptional interference. Transcriptional interference of MICA occurs in cis through transcriptional elongation from the upstream promoter over the downstream standard MICA promoter. The transcriptional interference is independent of the transcript generated from the upstream promoter. Deletion of the upstream core promoter or insertion of a transcription terminator downstream of the upstream promoter removes the transcriptional interference, but only in cis, demonstrating the requirement for overlapping transcription. Quantitative analysis, using an experimental system in which the upstream promoter activity is tuneable under the control of doxycycline, showed that the level of transcriptional interference observed was similar with either the native upstream promoter or with a heterologous promoter of equivalent strength. Therefore, transcription from an upstream promoter is sufficient for transcriptional interference; the degree of transcriptional interference depends on the strength of transcription from the upstream promoter, rather than the identity of the upstream promoter. Variation in the extent of transcriptional interference between different cell types will be influenced by differing transcription factor landscapes and so the strength of the transcriptional drive from each promoter. Intragenic transcriptional interference thus constitutes a general mechanism available for regulation of human genes with tandem promoter configuration.
Current knowledge about the role of transcriptional interference in gene regulation in mammals is limited. The few known examples of mammalian transcriptional interference are developmentally or ontologically regulated genes studied in model organisms. These genes are all controlled by tissue‐specific promoters or enhancers that are subject to epigenetic modifications established during the cell differentiation programme, and some are special cases of genes that undergo developmental imprinting with irreversible epigenetic silencing due to antisense transcriptional interference (Abarrategui & Krangel, 2007; Racanelli et al, 2008; Latos et al, 2012; MacIsaac et al, 2012). In these examples, transcriptional interference has been demonstrated by genetic deletion of the interfering promoter or insertion of a transcription terminator. However, the long delay from genetic manipulation in stem cells to examining the effect of transcriptional interference in differentiated cells makes it difficult to establish the mechanism of transcriptional interference. In particular, it has been an open question whether overlapping transcription itself is sufficient for transcriptional interference without the involvement of the widespread epigenetic changes that occur during the cell differentiation process. It has also been unclear whether transcriptional interference can mediate real‐time regulatory changes in gene expression.
Given the shortcoming of the chronic steady‐state genetic approaches, we developed a cellular model with tuneable control of the interfering upstream promoter of MICA to study quantitative aspects of transcriptional interference in real time. Timecourse analysis of transcriptional activity from both upstream and standard downstream MICA promoters, as well as histone modification at the downstream promoter, show that transcriptional interference occurs rapidly without the involvement of histone modifications such as H3K36me3, previously known to be associated with transcriptional interference (Houseley et al, 2008). Therefore, overlapping transcription itself is sufficient to interfere with downstream promoter activity in real time for the MICA gene.
The timecourse of transcriptional interference is similar to that seen for changes in occupancy of the Spt16 subunit of the FACT histone chaperone at the downstream promoter. FACT plays a role in elongation of the polymerase through nucleosomes and, as the polymerase advances, is involved in both nucleosome destabilization and nucleosome reassembly (Belotserkovskaya et al, 2003). In yeast, FACT mutants are associated with cryptic transcription initiation, indicating a role for the FACT‐mediated nucleosomal recovery, which follows polymerase nucleosomal navigation, in the inhibition of transcription initiation (Kaplan et al, 2003). During transcriptional interference, elongation of Pol II through the downstream standard promoter will be associated with nucleosomal reassembly in the wake of the polymerase and this reassembly may render the promoter relatively unfavourable for transcription initiation compared to the situation when there is no run‐through transcription. This is consistent with a model of transcriptional interference in which transcription through the downstream promoter reduces the likelihood of initiation at that promoter (Fig 8).
Figure 8. Model of transcription interference in MICA .
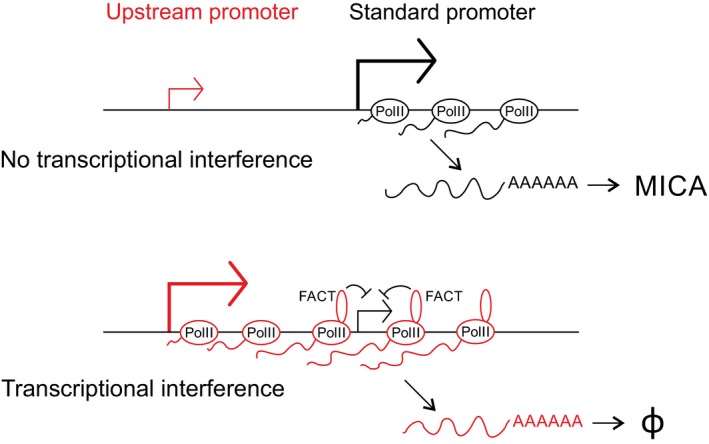
The upstream promoter of MICA encodes an unstable noncoding transcript which is subject to rapid degradation (φ), whereas the downstream promoter encodes the functional MICA protein. As illustrated in the lower panel, transcription from the upstream promoter inhibits transcription initiation at the downstream promoter and this transcriptional interference involves recruitment of the histone chaperone FACT.
We also used this tuneable system to study the stimulus–response relationship of transcriptional interference at equilibrium by varying the strength of the upstream promoter. The only previous attempt to quantitatively study eukaryotic transcriptional interference was in yeast and only assessed activity of the downstream promoter directly, leaving the quantitative relationship between downstream promoter activity and upstream promoter activity unclear (Buetti‐Dinh et al, 2009). Our quantitative analysis demonstrates a simple reciprocal relationship between the activities of the two promoters, analogous to the classical repressor model that describes the repression of gene expression through binding of a transcription repressor to a single‐copy binding site. This is consistent with transcriptional elongation over the downstream promoter as the physical basis for the transcriptional interference.
By virtue of its mechanism, the transcriptional interference that we have defined has distinct characteristics compared to other forms of gene regulation. Firstly, transcriptional interference is highly specific in its effect. Information about upstream promoter activity is transmitted locally in cis through the elongating transcription complex directly to the downstream promoter. The specificity of transcriptional interference is endowed by the physical linkage and linear proximity of the two promoters and is thus restricted locally in cis. This use of the local linear gene structure does not require the transport—by diffusion or other means—of soluble trans‐factors. In contrast, transcription factor or repressor or RNA‐based gene regulation requires the remote production of the soluble mediator, its physical movement to the target site in trans, and then specific molecular interaction between the regulator and target molecules.
Secondly, transcriptional interference allows a very fast response to a regulatory input: information from the upstream promoter is transmitted rapidly along the DNA molecule at the speed of transcription. With trans‐factors, there is the obligate time lag determined by the production or activation of the mediating factor and its transport to the site of action through either diffusion or active recruitment. Repressor‐based transcription silencing may trigger long‐term locuswide gene silencing through mechanisms such as heterochromatin formation with DNA methylation (Bintu et al, 2016). In contrast, the repressed state of transcriptional interference requires a strong and active upstream promoter within an active chromatin environment, which allows rapid upregulation of transcription from the downstream promoter when the transcriptional interference is reduced.
Thirdly, transcriptional interference allows the polarity of a regulatory input to be inverted, an input that increases transcription from the upstream promoter will reduce transcription from the downstream promoter. During evolution, a tandem promoter gene structure suitable for transcriptional interference may arise from a genetic duplication event or the insertion of transposable elements containing an upstream promoter. This can add sites for pre‐existing transcription factors as new regulatory inputs to the gene and if transcriptional interference occurs, then the effect of these transcription factors on the downstream promoter—and so on transcription of the coding transcript—will be opposite to the effect on the upstream promoter itself. In this way, for a given gene, activating regulatory pathways can be converted into repressive pathways or vice versa without the need for new molecules to evolve. Consequently, the binding of a cis‐acting transcription activator or repressor may have the opposite effect to that anticipated, if the binding affects the activity of an upstream promoter involved in transcriptional interference. This needs to be considered in bioinformatic and functional genomics studies of genetic variations associated with phenotypic traits or diseases in genomewide association studies (Hardy & Singleton, 2009; Price et al, 2015).
These characteristics of the transcriptional interference that we have identified are all pertinent to the evolution and function of MICA. Regulatory inputs which lower transcription from the upstream promoter can result in a rapid increase in transcription of the downstream coding transcript and so promote a prompt attack by immune cells expressing the NKG2D receptor (Bauer et al, 1999; Wu et al, 1999). A timely MICA response would be advantageous in defence against cancer or virus infection. The tandem promoter system in MICA allows for evolutionary selection within either promoter of binding sites for regulatory inputs, which allows expansion in the number and complexity of the physiological inputs that converge at the MICA promoter. Thus, a given regulatory input could drive transcription from either the upstream or the downstream promoter and so increase or decrease production of the downstream coding transcript. Such evolutionary selection at the MICA locus has no direct effects on the gene regulatory pathways themselves and so remains highly specific for MICA. Correct functional interpretation of the regulatory polymorphisms at the MICA locus is only possible if the polarity‐reversing effect of transcriptional interference on regulatory inputs is incorporated.
We demonstrated the conversion of an activator into a repressor and vice versa in the regulation of MICA by IFN‐γ and IL‐4 through the IRF1 and E4BP4 transcription factors, respectively. Downregulation of MICA mRNA expression by IFN‐γ has been observed previously (Zhang et al, 2008; Schwinn et al, 2009; Yadav et al, 2009), but the molecular mechanism was not clear. One report proposed that the miR‐520b miRNA might mediate this effect by direct action on both the MICA promoter and the MICA 3′UTR, but blocking miR‐520b failed to block IFN‐γ induced MICA expression (Yadav et al, 2009). Our results demonstrate that IFN‐γ acts via IRF1 to regulate MICA expression at the transcription level through transcriptional interference. Transcriptional interference alters the polarity of the IFN‐γ effect from activator to repressor. Conversely, IL‐4 acts via the transcriptional repressor E4BP4 to upregulate MICA. In this case, transcriptional interference inverts a negative gene regulation mechanism into one that positively upregulates expression of MICA.
The finding of intragenic transcriptional interference in MICA may lead to re‐evaluation of the function of additional upstream promoters in other human genes. Previous studies have focused largely on the protein coding potential of the transcripts from these promoters, some of which may encode proteins with altered N‐terminal sequence compared to the reference transcripts (Kimura et al, 2006). Our findings expand the scope of additional upstream promoter function. A tandem promoter gene structure results in promoter‐specific exon 1 usage with potentially shared downstream sequence and strictly implies overlapping transcription. Overlapping transcription is sufficient to cause transcriptional interference in MICA, so tandem promoter gene structures in other genes may also have undergone evolutionary selection for their capacity to shape gene expression through transcriptional interference. For some of these genes, the spliced transcripts from the additional upstream promoter may be merely by‐products of transcription. The production of these transcripts does impose an energetic cost, but their transcription is essential for transcriptional interference to occur. Early termination of the transcripts before they reach the downstream promoter would abolish the transcriptional interference. This requirement for their transcription will impose an evolutionary pressure to maintain them if the regulatory mechanisms that the upstream promoters confer have sufficient survival value. In MICA, the transcript arising from the upstream promoter is rapidly eliminated by the nonsense‐mediated decay pathway, which will minimize any further unnecessary energetic expenditure, such as nonproductive protein translation.
Overall, these findings demonstrate that intragenic transcriptional interference plays a role in the transcriptional regulation of the MICA gene and is mediated by read‐through transcription. These findings provide a roadmap for manipulating MICA expression for therapeutic benefit, especially in cancer immunotherapy. Intragenic transcriptional interference in an endogenous human gene represents a hitherto unrecognized and potentially important modality of transcriptional regulation. A large number of human genes express transcripts from upstream promoters, so intragenic transcriptional interference may be widespread. The CAGE‐seq analysis indicates that multiple human and mouse genes with tandem promoter systems display a pattern of reciprocal changes in transcript expression similar to that seen with transcriptional interference in MICA. This analysis demonstrates that transcriptional interference may be involved in the regulation of many genes in higher eukaryotes, which has implications for the study of transcriptional regulation in such genes. A tandem promoter system allows evolutionary variation in the MICA gene to capture a wide range of activating transcription factor pathways to upregulate standard MICA expression through the standard promoter or to downregulate it through the upstream promoter. Intragenic transcriptional interference actuates an integration of two opposing sets of inputs, one set from each promoter. The evolutionary conservation of the tandem promoter configuration in this gene and many other human genes attests to its likely survival value.
Materials and Methods
Reagents
Chemicals were obtained from Sigma, and enzymes for molecular biology from NEB, unless otherwise stated. Interferon‐γ and interleukin‐4 were from eBioscience. Primers were from Invitrogen or Integrated DNA Technologies. Primer sequences are listed in Appendix Table S2.
Cell lines and primary cells
HeLa, HT1080, 293T, T47D cell lines and human skin fibroblasts were maintained in Dulbecco's Modified Eagle Medium (Sigma) supplemented with 10% foetal calf serum (First Link) (D10). Human aortic arterial endothelial cells (Invitrogen) were maintained in Medium 200 with Low Serum Growth Supplement (Invitrogen). Human iPSC cells SFC840‐03‐03 (Fernandes et al, 2016) were cultured in mTeSR1 (StemCell Technologies) on plates coated with hESC‐qualified Matrigel Matrix (BD Biosciences). Cell lines were confirmed to be free of mycoplasma contamination using MycoAlert Mycoplasma Detection Kit (Lonza). Peripheral blood mononuclear cells (PBMCs) were enriched from fresh human blood by Ficoll density centrifugation. Prior written consent was obtained from healthy blood donors (Ethical approval: South Central‐Hampshire B, reference 13/SC/0392). Primary B cells were isolated from PBMC by MACS using CD19 MicroBeads (Miltenyi Biotec). The purity of B cells was consistently over 95% as determined by flow cytometry using anti‐CD19‐APC (eBioscience 17‐0199‐73) and anti‐CD20‐FITC (AbD Serotec MCA1822F) antibodies. Purified PBMC or B cells were cultured in RPMI1640 medium with 10% foetal calf serum. Primary monocytes and T cells were isolated from PBMC by MACS using CD14 or CD3 MicroBeads (Miltenyi Biotec), respectively.
Plasmid construction
BAC modification/retrofitting plasmids were constructed by standard cloning techniques using conditional replicating pSG80A plasmid in the pir116 strain. Plasmid pBS‐Landing carrying the landing site for site‐directed recombinase‐mediated cassette exchange (RMCE) was constructed by sequential insertion of functional cassettes into the pBluescript plasmid. Luciferase reporter plasmids were constructed by cloning the MICA standard or upstream promoter into the pGL3 Basic or pGL4.10 plasmid. pHR‐SIN‐rtTA3 the lentiviral plasmid for expression of the rtTA3 doxycycline‐responsive transactivator was constructed by cloning rtTA3 into pHR‐SIN‐BX‐IRES‐Emerald. CRISPR plasmids were constructed based on the PX458 plasmid (Ran et al, 2013). dCas9‐based targeted transcription activation plasmids were constructed based on the UniSam plasmid with mCherry replaced with EGFP (Fidanza et al, 2017). The source of plasmids and detailed steps of plasmid construction are listed in Appendix Table S3. The inserts for all of the plasmids were verified by sequencing.
BAC modification
BAC modification was carried out by lambda red‐based recombineering. Briefly, pRed/ET plasmid was first transformed into DH10B cells harbouring the target BAC to make the cells recombineering‐proficient. Markerless mutations were then introduced into the BAC using an rpsL‐neo‐based positive/negative selection system, by first tagging the mutation site with an rpsL‐neo cassette using a PCR product with 50‐bp homologous arms and selection with kanamycin. The rpsL‐neo cassette was then replaced using linear DNA fragments cut from plasmids that carry the intended mutations and 500‐ to 1,000‐bp homology arms with streptomycin counterselection. BAC truncation, concatenation and retrofitting steps were carried out by recombineering using linearized plasmid or BAC fragments containing bacterial selection markers and over 50‐bp homology arms. Following recombineering, temperature‐sensitive pRed/ET plasmid was removed by plating cells at 37°C.
In total, reporters with six different 53‐kb BAC inserts and four different 161‐kb BAC inserts were constructed. The 53‐kb BAC reporters were generated from BAC CH501‐248L24, and the 161‐kb BAC reporters from BAC CH501‐248L24 and CH501‐181B23 (BACPAC Resources Center, CHORI). Both CH501‐248L24 and CH501‐181B23 are derived from the PGF cell line, which carries a MICA*00804 haplotype. All the BAC constructs were engineered to contain synonymous mutations in the MICA exon 2 coding region to facilitate discrimination of transgenic and endogenous MICA by allele‐specific PCR. Appendix Table S4 lists the detailed steps of the BAC modifications. Each step was verified by restriction digest and pulsed‐field gel electrophoresis using the CHEF II system (Bio‐Rad). Markerless mutations introduced into the BAC were verified by sequencing. Endotoxin‐free BAC DNA was purified using the Phaseprep BAC DNA extraction kit (Sigma).
Generation of acceptor cell line for BAC RMCE
The acceptor cell line carrying a single‐copy landing site was generated by transient transfection of ScaI‐linearized pBS‐Landing plasmid into HT1080 cells by nucleofection using Cell Line Nucleofector Kit V and Nucleofector II Device (Lonza), followed by selection with 500 μg/ml G418 (Calbiochem). Single clones were picked using cloning cylinders, expanded and screened first by PCR to check the integrity of the landing site, then by Southern blot using three different restriction enzymes to check the copy number of the insert. One clone designated HT1080‐L3N9 was confirmed to carry a single‐copy landing site and was used for the subsequent generation of BAC RMCE clones. The stable HT1080‐L3N9 acceptor cell line was maintained in D10 medium with 250 μg/ml G418.
Generation of site‐directed BAC RMCE clones
BAC RMCE clones were generated by co‐transfection of BAC and pCre‐Pac plasmid into the HT1080‐L3N9 acceptor cell line using GeneJuice transfection reagent (Merck) followed by selection with 250 μg/ml hygromycin (Calbiochem). Single clones were picked using cloning cylinders (Sigma), expanded, and genomic DNA was extracted using the ZR‐96 Genomic DNA Kit (Zymo Research) for verification by PCR. PCR‐verified clones were then screened by flow cytometry to exclude hyperploid clones based on forward and side‐scattering properties and propidium iodide staining for DNA content. On average, 8% of picked clones successfully passed the whole screening process (Appendix Table S1). At least two independent BAC RMCE clones were generated for each BAC construct. Stable BAC RMCE clones were maintained in D10 medium with 125 μg/ml hygromycin. For functional assays, clones were cultured in hygromycin‐free medium for at least 48 h before the start of the experiments. Transgenic MICA expression in BAC RMCE clones was stable and homogeneous without hygromycin selection for at least 2 months. BAC clones were confirmed to be free of mycoplasma contamination using MycoAlert Mycoplasma Detection Kit.
Lentivirus production and infection
Lentivirus was generated by co‐transfection of the lentiviral expression plasmid with pMD2.G and psPax2 packaging plasmids into 293T cells. The supernatant was harvested, filtered through a 0.4‐μm filter (Millipore) and titrated in HT1080 cells. For the generation of doxycycline‐inducible cells, BAC clones were infected with pHR‐SIN‐rtTA3 lentivirus at an MOI of 10 and expanded and used between 5 and 7 days post‐infection. The rtTA3 transactivator expression is stable with near 100% unimodal expression as indicated by GFP expression from the bicistronic construct.
RNA ligase‐mediated rapid amplification of cDNA ends (RLM‐RACE)
RLM‐RACE was carried out using HeLa and HT1080 total RNA and the ExactStart Eukaryotic mRNA 5′‐ & 3′‐RACE Kit (Epicentre) following the manufacture's protocol with the following modifications. A longer RNA ligation oligo GCUGAUGGCGAUGAAUGAACACUGCGUUUGCUGGCUUUGAUGAAA was used in the ligation reaction, and reverse transcription was carried out using random hexamers (Qiagen) and BioScript reverse transcriptase (Bioline). The RACE product was amplified by nesting PCR using primers CO4210/606 followed by CO4211/1109, and cloned using the Strataclone PCR cloning kit (Stratagene) for sequencing.
RNA analysis
Total RNA was prepared using the Trizol plus kit (Invitrogen) with the on‐column DNase digestion step to remove genomic DNA. Total bovine and porcine adult kidney RNA (Zyagen), and total human adult kidney RNA (Clontech) were cleaned up using the Purelink RNA Mini kit (Invitrogen) with DNase digestion. Total RNA was reverse transcribed into cDNA using BioScript reverse transcriptase and random hexamers. Where indicated, oligo‐dT was used instead of random hexamers. Human adult and foetal tissue cDNA panels were from Clontech. qPCR was carried out using the Fast SYBR Green Master Mix and StepOne real‐time PCR system (Applied Biosystems). qPCR results were analysed using the ΔΔC t method and normalized to GAPDH expression for gene expression studies. Semi‐quantitative RT–PCR was carried out using Biotaq polymerase (Bioline).
The following primers were used for SYBR green‐based qPCR analysis: MICA‐ST, CO3706/3707; MICA‐UT, CO3705/3708; GAPDH, CO3744/3745; 18S rRNA, CO3746/3747; ITGB5, CO3740/3741. For qPCR analysis of endogenous and transgenic MICA transcripts in BAC RMCE clones, the following primers were used: transgenic MICA‐ST, CO3526/3525; transgenic MICA‐UT, CO3705/3525; endogenous MICA‐ST, CO3526/4080; endogenous MICA‐UT, CO3705/4080. For semi‐quantitative RT–PCR analysis of tissue samples, the following primers were used: MICA‐ST, CO1111/1109; MICA‐UT, CO1099/1109; GAPDH, CO631/632; POLR2F, CO1831/1832. For semi‐quantitative RT–PCR analysis of MIC homologs in human, pig and cow kidney tissues, the following primers were used: human MICA‐ST, CO1111/1109; human MICA‐UT, CO1099/1109; pig MIC2‐ST, CO5217/5221; pig MIC2‐UT, CO5219/5221; cow MIC1‐ST, CO5218/5222; cow MIC1‐UT, CO5220/5222; human, pig or cow GAPDH, CO5223/5224. Primer sequences are listed in Appendix Table S2.
mRNA stability measurement
mRNA stability was measured by either global transcription inhibition using actinomycin D, or metabolic labelling with 4‐thiouridine (4sU, Carbosynth). For measurements based on actinomycin D treatment, cells were treated with 5 μM actinomycin D, and MICA‐ST or MICA‐UT mRNA was quantified independently by qPCR using cDNA prepared from the same amount of total RNA.
Measurement of mRNA half‐life by 4sU metabolic labelling was based on the protocol described previously (Dolken et al, 2008). Briefly, total and 4sU‐labelled RNA were isolated from proliferating cells pulsed with 500 μM 4sU for 2 h, and reverse transcribed into cDNA using BioScript reverse transcriptase and random hexamers. Enrichment of 4sU‐labelled RNA was measured by qPCR. The half‐life of mRNA measured by 4sU labelling is influenced by the combined effects of mRNA decay and dilution due to cell proliferation. The half‐life was calculated using 18S rRNA as an internal control, assuming that mRNA decay follows first‐order kinetics and that the half‐life of stable 18S rRNA is dominated by cell proliferation.
Polysome fractionation
Polysome fractionation was carried out based on protocols described previously (Powley et al, 2009) with the following modifications: heparin in the sucrose gradient and cell lysis buffer was replaced with RNasin RNase inhibitor (Promega) at 40 or 100 U/ml, respectively. RNA was isolated using Trizol‐LS (Invitrogen) and Purelink RNA Mini kit for RNA analysis.
Flow cytometry
Flow cytometry was carried out as described previously (Lin et al, 2012). The pan‐allelic anti‐MICA antibody clone 2C10 (Santa Cruz sc‐23870) was used for measuring MICA surface expression in general. The allele‐specific anti‐MICA clone 159227 (R&D Systems MAB1300) was used to detect transgenic MICA surface expression in RMCE BAC clones. This antibody is an allele‐specific antibody that recognizes the transgenic MICA*008 allele carried by the BAC, but not the endogenous MICA*007 allele of HT1080 cells. For flow cytometry of B cells, cells were blocked with Fc receptor‐blocking reagent (Miltenyi Biotec) and stained with anti‐CD19‐APC (eBioscience 17‐0199‐73), anti‐CD3‐PE‐Cy7 (BD Biosciences 557851), anti‐MICA‐PE (Santa Cruz sc‐23870 PE) and the viability dye LIVE/DEAD® Fixable Near‐IR stain (Invitrogen); the viable CD3−CD19+ B‐cell population was gated on for analysis of MICA expression. Flow cytometry was performed using a BD FACSCanto system (Becton Dickinson), and data were analysed using FlowJo software (FlowJo. LLC).
Western blot
Cells were lysed in lysis buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1% CHAPS) with 1× protease inhibitor cocktail (Roche), and proteins were separated by SDS–PAGE and transferred to Immobilon‐P PVDF membrane (Millipore). Myc‐tagged proteins were detected using anti‐c‐myc (clone 9E10) antibody and goat anti‐mouse IgG‐HRP secondary antibody (Abcam ab20043). HRP‐conjugated anti‐beta actin antibody (Abcam ab49900) was used as control. HRP signals were detected using ECL Prime reagent (GE Healthcare) and a ChemiDoc MP imaging system (Bio‐Rad).
siRNA knockdown
Cells were transfected with Silencer Select siRNA targeting MICA‐UT (target sequence GCAGUGGCGCCUAAAGUCU) or Silencer Select Negative Control siRNA #1 (Ambion) using Oligofectamine Reagent (Invitrogen).
Electrophoretic mobility shift assay (EMSA)
EMSAs were carried out as described previously (Lin et al, 2012). For EMSA of IRF1, nuclear extracts were prepared from HAECs treated with 20 ng/ml interferon‐γ for 24 h. Wild‐type probe containing the IRF‐1 binding site of the MICA upstream promoter was prepared by annealing of oligonucleotides CO3611/3612 and mutant probe CO3615/3616. Cold competition assays were carried out by pre‐incubation of nuclear extract with 100× excess of unlabelled cold probe. For supershift assays, nuclear extract was pre‐incubated with 2 μg anti‐IRF1 antibody (Santa Cruz sc‐497X) or anti‐c‐Fos control antibody (Santa Cruz sc‐52X) for 30 min on ice before adding 32P‐labelled probe. EMSA for E4BP4 was carried out using nuclear extracts from 293T cells transfected with plasmids expressing N‐ or C‐ terminal myc‐tagged E4BP4 or control ATF2 transcription factor, and 32P‐labelled probes prepared by annealing oligonucleotides CO2717/2718.
Reporter assay
Reporter assays were carried out by co‐transfection of firefly luciferase reporter plasmid carrying MICA promoter fragments and control pRL‐SV Renilla luciferase plasmid, followed by cell lysis and luciferase assay using the Dual‐Luciferase Reporter Assay System (Promega) and a TD‐2020 luminometer (Turner Designs). 293T, HeLa and HT1080 cells were transfected using GeneJuice and primary human arterial endothelial cells were transfected using HCAEC Nucleofector Kit (Lonza), and cells were lysed 48 h post‐transfection. Firefly luciferase reporters were constructed in pGL3‐based plasmids, with the exception of the E4BP4 experiment, in which pGL4‐based plasmids were used. This was due to concern about the presence of an E4BP4 site within the luciferase gene of the pGL3 plasmid. In the interferon‐γ experiment, the upstream promoter reporters carry −78 bp upstream promoter (MICA‐UT‐P‐78 bp and MICA‐UT‐P‐78 bp‐ISREmut) and the standard promoter reporter carries −2,779 bp standard promoter. In the E4BP4 experiment, the upstream promoter reporters carry −702 bp upstream promoter (MICA‐UT‐P‐702 bp and MICA‐UT‐P‐702 bp‐E4BP4mut). Results represented as relative luciferase activity have been normalized to Renilla luciferase activity and pGL3P promoter control plasmid luciferase activity for pGL3‐based reporters, or pGL4.23 control plasmid activity for pGL4‐based reporters.
Determination of MICA promoter haplotypes
The haplotypes of the MICA promoter in selected cell lines were determined by PCR sequencing to facilitate identification of suitable restriction enzymes for allele‐specific PCR analysis of the standard or upstream MICA promoters in ChIP assays and fluorescent PCR assay. For HT1080 cells homozygous for the MICA*007 allele, the promoter haplotype was assembled directly (deposited as GenBank KF724603). For the primary human fibroblasts heterozygous for MICA*004/010 alleles, the promoter haplotype linked to each allele was determined by sequencing of the −6‐kb MICA promoter region of the fibroblasts as well as selected EBV‐transformed B cells homozygous for MICA*004 or MICA*010 alleles (IHWG Cell and DNA Bank). The promoter haplotype linked to the MICA*004 allele in the fibroblasts was deposited as GenBank KF724624, and MICA*010 as KF724587.
Chromatin immunoprecipitation
Allele‐specific ChIPs were carried out based on the Q2ChIP protocol (Dahl & Collas, 2007). The following antibodies were used: H3K36me3 (Abcam ab9050), H3K27ac (Abcam ab4729), H3K4me3 (Abcam ab8580), H4K20me3 (Abcam ab9053), H2A.Z (Abcam ab4174), H3 (Abcam ab1791), Spt16 (clone 8D2, BioLegend 607002), PolII phospho‐Ser5 (clone 1H4B6, Millipore MABE954), and normal rabbit IgG control (Santa Cruz sc‐2027X), mouse IgG2a isotype control (eBioscience 14‐4724‐85), and rat IgG2b isotype control (clone RTK4530, BioLegend 400601). Phosphatase inhibitor cocktail PhosSTOP (Roche) was included in the buffers for Pol II phospho‐Ser5 ChIP. ChIP DNA was purified using the PCR purification kit (Qiagen). For allele‐specific amplification of the MICA standard promoter region from the transgenic MICA allele in RMCE clones, ChIP and input control samples were digested with BfaI to disrupt the endogenous MICA*007 allele promoter region that differs from the transgenic MICA at SNP rs116135464T, which is sensitive to BfaI. For allele‐specific amplification of the same region from the endogenous allele, samples were treated with TspRI to disrupt the transgenic MICA*008 allele that differs from the endogenous MICA at the same SNP. The digestion efficiencies of BfaI and TspRI exceed 99% as assessed by qPCR analysis of ChIP input samples prepared from HT1080 cells that carry only the endogenous MICA*007 allele or 293T cells that carry only the transgenic MICA*008 allele, respectively. BfaI‐ or TspRI‐treated samples were analysed by qPCR using primer pair CO6351/6358 for the MICA standard promoter.
ChIP for IRF1 was carried out similarly using anti‐IRF1 antibody (Santa Cruz sc‐497X) or rabbit IgG control (Santa Cruz sc‐2027X) on human arterial endothelial cells treated with 20 ng/ml interferon‐γ for 24 h. ChIP for E4BP4 was carried out using anti‐E4BP4 antibody (Santa Cruz sc‐9550X) or goat IgG control (Santa Cruz sc‐2028) on primary human B cells treated with 20 ng/ml IL‐4 for 24 h. ChIP and input control samples were digested with BseRI to disrupt homologous sequences in the genome similar to the MICA upstream promoter. BseRI‐treated samples were then analysed by qPCR using primer pair CO6696/6699 for the MICA upstream promoter or CO6351/6358 for the standard promoter.
Transient CRISPR
Primary human arterial endothelial cells or fibroblasts were transfected with CRISPR plasmids targeting the MICA upstream promoter (pair 1: PX458‐C40/C42 or pair 2: PX458‐C41/C43), or HLA‐B exon1 (PX458‐C50/C52) or PDPN exon2 (PX458‐C14) as negative controls by nucleofection using the HCAEC Nucleofector Kit or NHDF Nucleofector Kit, respectively (Lonza). Cells were cultured for 3 days before being lifted for flow cytometric analysis of surface MICA expression, or for cell sorting of GFP‐positive cells directly into Trizol‐LS using a MoFlo MLS sorter (Beckman Coulter) for RNA analysis.
Stable CRISPR and allele‐specific DNA analysis
Primary fibroblasts were transfected by nucleofection with CRISPR pairs PX458‐C41/43 targeting deletion of the MICA upstream promoter. Cells were expanded, stained with anti‐MICA antibody (clone 2C10) followed by goat anti‐mouse IgG Alexa Fluor 647 secondary antibody (Invitrogen A‐21236), and sorted into MICA‐high and MICA‐low populations based on MICA surface expression using a MoFlo MLS sorter. Sorted cells were further expanded, and genomic DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen). For analysis of allele‐specific MICA upstream promoter deletion, genomic DNA was treated with BsrDI, specific for rs2596539T within the upstream promoter linked to the MICA*010 allele, or BsgI, specific for rs2596539C linked to the MICA*004 allele, for analysis of the MICA*004 and MICA*010 alleles, respectively. BsrDI‐ or BsgI‐treated DNA samples were used for PCR amplification using CO6392 and 5′ 6‐FAM‐labelled CO6395, and 6‐FAM‐labelled PCR samples were mixed with GeneScan 500 LIZ size standard and analysed using a 3730xl DNA Analyzer and Peak Scanner software (Applied Biosystems).
dCas9‐based transcriptional activation
293T cells were transfected with EGFP‐based UniSam plasmids targeting the MICA upstream promoter (UniSamG‐MICAUT), MICA standard promoter (UniSamG‐MICAST), or CD43 (UniSamG‐CD43) or CD36 (UniSamG‐CD36) promoter as negative controls using GeneJuice. Cells were harvested 2 days post‐transfection for flow cytometric analysis of surface MICA expression.
Genomewide analysis of CAGE‐seq data
Sets of transcriptional start sites for the hg19 and mm9 reference genomes were downloaded from the Eukaryotic Promoter Database (Dreos et al, 2013) and extended to create 200‐bp promoter windows centred on each transcriptional start site. Each promoter window was associated with the nearest UCSC knownGene annotated gene. Overlapping and bookended windows were merged using BEDTools (Quinlan & Hall, 2010), giving a total of 25,718 and 21,119 promoter windows covering 17,842 and 17,564 genes in the human and mouse genomes, respectively. FANTOM5 CAGE‐seq timecourse data sets were downloaded from the DNA Data Bank of Japan (accession numbers DRA000991, DRA002711, DRA002747, and DRA002748) and analysed for promoter window tag counts using featureCounts (Liao et al, 2014; Arner et al, 2015). Raw counts were converted to relative log expression‐normalized counts per million (CPM) using edgeR (Anders & Huber, 2010; Robinson et al, 2010), and libraries with a median normalized log2CPM < −1 were excluded from the analysis. Mean expression at each time point was calculated from three biological replicates in R, and only promoters with a minimum expression of 1 CPM were retained. The Kendall rank correlation coefficient was calculated for each promoter's expression over time. Promoters were paired by gene associations and then filtered to include only pairs with a positive correlation coefficient for one promoter and a negative coefficient for the other. Expression values were converted to log2 (fold change) compared to time 0, and diverging expression changes over time were visualized for each promoter pair passing the filter. All plots were generated in R (R Core Team, 2015).
Data availability
Sequences for MICA promoter haplotypes have been submitted to Genbank with accession numbers KF724603, KF724624 and KF724587.
Author contributions
DL and CAO designed the experiments. DL performed the experiments and TKH carried out CAGE‐seq analysis. DL and CAO wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Review Process File
Acknowledgements
We are grateful to Dr Ben Davies for advice and assistance with BAC modification, to Dr Drew Worth for assistance with cell sorting, to Dr Matthew Cockman for advice and assistance with polysome fractionation, and to Professor Nicholas Proudfoot for providing the βΔ5‐7 plasmid carrying the minimal beta‐globin transcription terminator. This work was supported by the Medical Research Council (G116/165) and the Novo Nordisk Foundation (Grant Number NNF15CC0018346) and National Institute for Health Research Oxford Comprehensive Biomedical Research Centre Program.
The EMBO Journal (2018) 37: e97138 29643123
References
- Abarrategui I, Krangel MS (2007) Noncoding transcription controls downstream promoters to regulate T‐cell receptor alpha recombination. EMBO J 26: 4380–4390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11: R106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ard R, Tong P, Allshire RC (2014) Long non‐coding RNA‐mediated transcriptional interference of a permease gene confers drug tolerance in fission yeast. Nat Commun 5: 5576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ard R, Allshire RC (2016) Transcription‐coupled changes to chromatin underpin gene silencing by transcriptional interference. Nucleic Acids Res 44: 10619–10630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arner E, Daub CO, Vitting‐Seerup K, Andersson R, Lilje B, Drablos F, Lennartsson A, Ronnerblad M, Hrydziuszko O, Vitezic M, Freeman TC, Alhendi AM, Arner P, Axton R, Baillie JK, Beckhouse A, Bodega B, Briggs J, Brombacher F, Davis M et al (2015) Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science 347: 1010–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahram S, Bresnahan M, Geraghty DE, Spies T (1994) A second lineage of mammalian major histocompatibility complex class I genes. Proc Natl Acad Sci USA 91: 6259–6263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, Spies T (1999) Activation of NK cells and T cells by NKG2D, a receptor for stress‐inducible MICA. Science 285: 727–729 [DOI] [PubMed] [Google Scholar]
- Belotserkovskaya R, Oh S, Bondarenko VA, Orphanides G, Studitsky VM, Reinberg D (2003) FACT facilitates transcription‐dependent nucleosome alteration. Science 301: 1090–1093 [DOI] [PubMed] [Google Scholar]
- Bintu L, Yong J, Antebi YE, McCue K, Kazuki Y, Uno N, Oshimura M, Elowitz MB (2016) Dynamics of epigenetic regulation at the single‐cell level. Science 351: 720–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buetti‐Dinh A, Ungricht R, Kelemen JZ, Shetty C, Ratna P, Becskei A (2009) Control and signal processing by transcriptional interference. Mol Syst Biol 5: 300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, Ponjavic J, Semple CA, Taylor MS, Engstrom PG, Frith MC, Forrest AR, Alkema WB, Tan SL, Plessy C, Kodzius R, Ravasi T, Kasukawa T, Fukuda S, Kanamori‐Katayama M, Kitazume Y et al (2006) Genome‐wide analysis of mammalian promoter architecture and evolution. Nat Genet 38: 626–635 [DOI] [PubMed] [Google Scholar]
- Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, Shia WJ, Anderson S, Yates J, Washburn MP, Workman JL (2005) Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123: 581–592 [DOI] [PubMed] [Google Scholar]
- Cerboni C, Zingoni A, Cippitelli M, Piccoli M, Frati L, Santoni A (2007) Antigen‐activated human T lymphocytes express cell‐surface NKG2D ligands via an ATM/ATR‐dependent mechanism and become susceptible to autologous NK‐ cell lysis. Blood 110: 606–615 [DOI] [PubMed] [Google Scholar]
- Cowell IG, Skinner A, Hurst HC (1992) Transcriptional repression by a novel member of the bZIP family of transcription factors. Mol Cell Biol 12: 3070–3077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl JA, Collas P (2007) Q2ChIP, a quick and quantitative chromatin immunoprecipitation assay, unravels epigenetic dynamics of developmentally regulated genes in human carcinoma cells. Stem Cells 25: 1037–1046 [DOI] [PubMed] [Google Scholar]
- Davuluri RV, Suzuki Y, Sugano S, Plass C, Huang TH (2008) The functional consequences of alternative promoter use in mammalian genomes. Trends Genet 24: 167–177 [DOI] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, Xue C, Marinov GK, Khatun J, Williams BA, Zaleski C, Rozowsky J, Roder M, Kokocinski F, Abdelhamid RF, Alioto T et al (2012) Landscape of transcription in human cells. Nature 489: 101–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolken L, Ruzsics Z, Radle B, Friedel CC, Zimmer R, Mages J, Hoffmann R, Dickinson P, Forster T, Ghazal P, Koszinowski UH (2008) High‐resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA 14: 1959–1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreos R, Ambrosini G, Cavin Perier R, Bucher P (2013) EPD and EPDnew, high‐quality promoter resources in the next‐generation sequencing era. Nucleic Acids Res 41: D157–D164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantom Consortium and the RIKEN PMI and CLST (2014) A promoter‐level mammalian expression atlas. Nature 507: 462–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes HJ, Hartfield EM, Christian HC, Emmanoulidou E, Zheng Y, Booth H, Bogetofte H, Lang C, Ryan BJ, Sardi SP, Badger J, Vowles J, Evetts S, Tofaris GK, Vekrellis K, Talbot K, Hu MT, James W, Cowley SA, Wade‐Martins R (2016) ER stress and autophagic perturbations lead to elevated extracellular alpha‐synuclein in GBA‐N370S Parkinson's iPSC‐derived dopamine neurons. Stem Cell Reports 6: 342–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidanza A, Lopez‐Yrigoyen M, Romano N, Jones R, Taylor AH, Forrester LM (2017) An all‐in‐one UniSam vector system for efficient gene activation. Sci Rep 7: 6394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greger IH, Aranda A, Proudfoot N (2000) Balancing transcriptional interference and initiation on the GAL7 promoter of Saccharomyces cerevisiae . Proc Natl Acad Sci USA 97: 8415–8420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh V, Bahram S, Bauer S, Herman A, Beauchamp M, Spies T (1996) Cell stress‐regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc Natl Acad Sci USA 93: 12445–12450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gummalla M, Maeda RK, Castro Alvarez JJ, Gyurkovics H, Singari S, Edwards KA, Karch F, Bender W (2012) abd‐A regulation by the iab‐8 noncoding RNA. PLoS Genet 8: e1002720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hainer SJ, Pruneski JA, Mitchell RD, Monteverde RM, Martens JA (2011) Intergenic transcription causes repression by directing nucleosome assembly. Genes Dev 25: 29–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Singleton A (2009) Genomewide association studies and human disease. N Engl J Med 360: 1759–1768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlen KM, Churchman LS (2017) The code and beyond: transcription regulation by the RNA polymerase II carboxy‐terminal domain. Nat Rev Mol Cell Biol 18: 263–273 [DOI] [PubMed] [Google Scholar]
- Hongay CF, Grisafi PL, Galitski T, Fink GR (2006) Antisense transcription controls cell fate in Saccharomyces cerevisiae . Cell 127: 735–745 [DOI] [PubMed] [Google Scholar]
- Houseley J, Rubbi L, Grunstein M, Tollervey D, Vogelauer M (2008) A ncRNA modulates histone modification and mRNA induction in the yeast GAL gene cluster. Mol Cell 32: 685–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hug N, Longman D, Caceres JF (2016) Mechanism and regulation of the nonsense‐mediated decay pathway. Nucleic Acids Res 44: 1483–1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen S, Schotta G, Sorensen CS (2013) Histone H4 lysine 20 methylation: key player in epigenetic regulation of genomic integrity. Nucleic Acids Res 41: 2797–2806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan CD, Laprade L, Winston F (2003) Transcription elongation factors repress transcription initiation from cryptic sites. Science 301: 1096–1099 [DOI] [PubMed] [Google Scholar]
- Kasahara M, Sutoh Y (2015) Comparative genomics of the NKG2D ligand gene family. Immunol Rev 267: 72–87 [DOI] [PubMed] [Google Scholar]
- Kashiwada M, Levy DM, McKeag L, Murray K, Schroder AJ, Canfield SM, Traver G , Rothman PB (2010) IL‐4‐induced transcription factor NFIL3/E4BP4 controls IgE class switching. Proc Natl Acad Sci USA 107: 821–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keogh MC, Kurdistani SK, Morris SA, Ahn SH, Podolny V, Collins SR, Schuldiner M, Chin K, Punna T, Thompson NJ, Boone C, Emili A, Weissman JS, Hughes TR, Strahl BD, Grunstein M, Greenblatt JF, Buratowski S, Krogan NJ (2005) Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell 123: 593–605 [DOI] [PubMed] [Google Scholar]
- Kimura K, Wakamatsu A, Suzuki Y, Ota T, Nishikawa T, Yamashita R, Yamamoto J, Sekine M, Tsuritani K, Wakaguri H, Ishii S, Sugiyama T, Saito K, Isono Y, Irie R, Kushida N, Yoneyama T, Otsuka R, Kanda K, Yokoi T et al (2006) Diversification of transcriptional modulation: large‐scale identification and characterization of putative alternative promoters of human genes. Genome Res 16: 55–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, Nureki O, Zhang F (2015) Genome‐scale transcriptional activation by an engineered CRISPR‐Cas9 complex. Nature 517: 583–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Kato N, Urabe Y, Takahashi A, Muroyama R, Hosono N, Otsuka M, Tateishi R, Omata M, Nakagawa H, Koike K, Kamatani N, Kubo M, Nakamura Y, Matsuda K (2011) Genome‐wide association study identifies a susceptibility locus for HCV‐induced hepatocellular carcinoma. Nat Genet 43: 455–458 [DOI] [PubMed] [Google Scholar]
- Lanier LL (2015) NKG2D receptor and its ligands in host defense. Cancer Immunol Res 3: 575–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latos PA, Pauler FM, Koerner MV, Senergin HB, Hudson QJ, Stocsits RR, Allhoff W, Stricker SH, Klement RM, Warczok KE, Aumayr K, Pasierbek P, Barlow DP (2012) Airn transcriptional overlap, but not its lncRNA products, induces imprinted Igf2r silencing. Science 338: 1469–1472 [DOI] [PubMed] [Google Scholar]
- Le Clerc S, Delaneau O, Coulonges C, Spadoni JL, Labib T, Laville V, Ulveling D, Noirel J, Montes M, Schachter F, Caillat‐Zucman S, Zagury JF (2014) Evidence after imputation for a role of MICA variants in nonprogression and elite control of HIV type 1 infection. J Infect Dis 210: 1946–1950 [DOI] [PubMed] [Google Scholar]
- Li Z, Groh V, Strong RK, Spies T (2000) A single amino acid substitution causes loss of expression of a MICA allele. Immunogenetics 51: 246–248 [DOI] [PubMed] [Google Scholar]
- Li P, Morris DL, Willcox BE, Steinle A, Spies T, Strong RK (2001) Complex structure of the activating immunoreceptor NKG2D and its MHC class I‐like ligand MICA. Nat Immunol 2: 443–451 [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930 [DOI] [PubMed] [Google Scholar]
- Lin D, Lavender H, Soilleux EJ, O'Callaghan CA (2012) NF‐kappaB regulates MICA gene transcription in endothelial cell through a genetically inhibitable control site. J Biol Chem 287: 4299–4310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIsaac JL, Bogutz AB, Morrissy AS, Lefebvre L (2012) Tissue‐specific alternative polyadenylation at the imprinted gene Mest regulates allelic usage at Copg2. Nucleic Acids Res 40: 1523–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens JA, Wu PY, Winston F (2005) Regulation of an intergenic transcript controls adjacent gene transcription in Saccharomyces cerevisiae . Genes Dev 19: 2695–2704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MT, Moncayo G, Hiron TK, Jakobsen NA, Valli A, Soga T, Adam J, O'Callaghan CA (2017) Purine nucleotide metabolism regulates expression of the human immune ligand MICA. J Biol Chem 293: 3913–3924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Han B, Tsoi LC, Stuart PE, Ellinghaus E, Tejasvi T, Chandran V, Pellett F, Pollock R, Bowcock AM, Krueger GG, Weichenthal M, Voorhees JJ, Rahman P, Gregersen PK, Franke A, Nair RP, Abecasis GR, Gladman DD, Elder JT et al (2014) Fine mapping major histocompatibility complex associations in psoriasis and its clinical subtypes. Am J Hum Genet 95: 162–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AC, Egan JB, Shearwin KE (2011) Transcriptional interference by RNA polymerase pausing and dislodgement of transcription factors. Transcription 2: 9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petruk S, Sedkov Y, Riley KM, Hodgson J, Schweisguth F, Hirose S, Jaynes JB, Brock HW, Mazo A (2006) Transcription of bxd noncoding RNAs promoted by trithorax represses Ubx in cis by transcriptional interference. Cell 127: 1209–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powley IR, Kondrashov A, Young LA, Dobbyn HC, Hill K, Cannell IG, Stoneley M, Kong YW, Cotes JA, Smith GC, Wek R, Hayes C, Gant TW, Spriggs KA, Bushell M, Willis AE (2009) Translational reprogramming following UVB irradiation is mediated by DNA‐PKcs and allows selective recruitment to the polysomes of mRNAs encoding DNA repair enzymes. Genes Dev 23: 1207–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Spencer CC, Donnelly P (2015) Progress and promise in understanding the genetic basis of common diseases. Proc Biol Sci 282: 20151684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2015) R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; [Google Scholar]
- Racanelli AC, Turner FB, Xie LY, Taylor SM, Moran RG (2008) A mouse gene that coordinates epigenetic controls and transcriptional interference to achieve tissue‐specific expression. Mol Cell Biol 28: 836–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8: 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettino A, Clarke NM (2013) Genome‐wide identification of IRF1 binding sites reveals extensive occupancy at cell death associated genes. J Carcinog Mutagen (Spec Iss Apoptosis) pii: S6‐009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J, Halliwell JA, Hayhurst JD, Flicek P, Parham P, Marsh SG (2015) The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res 43: D423–D431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwinn N, Vokhminova D, Sucker A, Textor S, Striegel S, Moll I, Nausch N, Tuettenberg J, Steinle A, Cerwenka A, Schadendorf D, Paschen A (2009) Interferon‐gamma down‐regulates NKG2D ligand expression and impairs the NKG2D‐mediated cytolysis of MHC class I‐deficient melanoma by natural killer cells. Int J Cancer 124: 1594–1604 [DOI] [PubMed] [Google Scholar]
- Shearwin KE, Callen BP, Egan JB (2005) Transcriptional interference–a crash course. Trends Genet 21: 339–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N, Kawakami T, Taniguchi T (1993) Recognition DNA sequences of interferon regulatory factor 1 (IRF‐1) and IRF‐2, regulators of cell growth and the interferon system. Mol Cell Biol 13: 4531–4538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tangye SG, Ferguson A, Avery DT, Ma CS, Hodgkin PD (2002) Isotype switching by human B cells is division‐associated and regulated by cytokines. J Immunol 169: 4298–4306 [DOI] [PubMed] [Google Scholar]
- Ullrich E, Koch J, Cerwenka A, Steinle A (2013) New prospects on the NKG2D/NKG2DL system for oncology. Oncoimmunology 2: e26097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataraman GM, Suciu D, Groh V, Boss JM, Spies T (2007) Promoter region architecture and transcriptional regulation of the genes for the MHC class I‐related chain A and B ligands of NKG2D. J Immunol 178: 961–969 [DOI] [PubMed] [Google Scholar]
- Wang X, Hou J, Quedenau C, Chen W (2016) Pervasive isoform‐specific translational regulation via alternative transcription start sites in mammals. Mol Syst Biol 12: 875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Song Y, Bakker AB, Bauer S, Spies T, Lanier LL, Phillips JH (1999) An activating immunoreceptor complex formed by NKG2D and DAP10. Science 285: 730–732 [DOI] [PubMed] [Google Scholar]
- Yadav D, Ngolab J, Lim RS, Krishnamurthy S, Bui JD (2009) Cutting edge: down‐regulation of MHC class I‐related chain A on tumor cells by IFN‐gamma‐induced microRNA. J Immunol 182: 39–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Niu J, Zhang J, Wang Y, Zhou Z, Zhang J, Tian Z (2008) Opposing effects of interferon‐alpha and interferon‐gamma on the expression of major histocompatibility complex class I chain‐related A in tumors. Cancer Sci 99: 1279–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Liao D, Yang L, Hou S (2016) Association between functional MICA‐TM and Behcet's disease: a systematic review and meta‐analysis. Sci Rep 6: 21033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Wang J, Zou H, Ward MM, Weisman MH, Espitia MG, Xiao X, Petersdorf E, Mignot E, Martin J, Gensler LS, Scheet P, Reveille JD (2014) MICA, a gene contributing strong susceptibility to ankylosing spondylitis. Ann Rheum Dis 73: 1552–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Bresnahan W, Taylor RT, Stastny P (2005) Effect of human cytomegalovirus on expression of MHC class I‐related chains A. J Immunol 174: 3098–3104 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Review Process File
Data Availability Statement
Sequences for MICA promoter haplotypes have been submitted to Genbank with accession numbers KF724603, KF724624 and KF724587.