

Abstract
Ivermectin (IVM) is an antiparasitic drug that is used worldwide and rescues hundreds of millions of people from onchocerciasis and lymphatic filariasis. It was discovered by Satoshi Ōmura and William C. Campbell, to whom the 2015 Nobel Prize in Physiology or Medicine was awarded. It kills parasites by activating glutamate‐gated Cl− channels, and it also targets several ligand‐gated ion channels and receptors, including Cys‐loop receptors, P2X4 receptors and fernesoid X receptors. Recently, we found that IVM also activates a novel target, the G‐protein‐gated inwardly rectifying K+ channel, and also identified the structural determinant for the activation. In this review, we aim to provide an update and summary of recent progress in the identification of IVM targets, as well as their modulation mechanisms, through molecular structures, chimeras and site‐directed mutagenesis, and molecular docking and modelling studies.
Keywords: ion channel, Receptor, Ivermectin
Introduction
Ivermectin (IVM) is a well‐known antiparasitic drug that rescues humans and domestic animals from parasitic infection such as onchocerciasis (river blindness) and lymphatic filariasis. In the 1970s, Satoshi Ōmura isolated a mixture that consists of eight components (A1a, A1b, A2a, A2b, B1a, B1b, B2a and B2b) from Streptomyces avermitilis. Then, he and his collaborator, William C. Campbell, identified several 16‐membered macrocyclic lactone derivatives (known as avermectins), which possess high anthelmintic activity that kills parasitic worms (Burg et al. 1979; Egerton et al. 1979). IVM is one of the avermectins and consists of a mixture of B1a and B1b components in an 80:20 ratio (Fig. 1). In the 1980s, IVM became a commercially available drug, since it was found to be safer and more potent than other avermectins (Chabala et al. 1980; Campbell et al. 1983; Campbell & Benz, 1984). To date, IVM has contributed to the improvement of the health of hundreds of millions of people in many developing communities and in consequence has been included on the Model List of Essential Medicines of the World Health Organization (Omura, 2016).
Figure 1. Chemical structure of IVM.
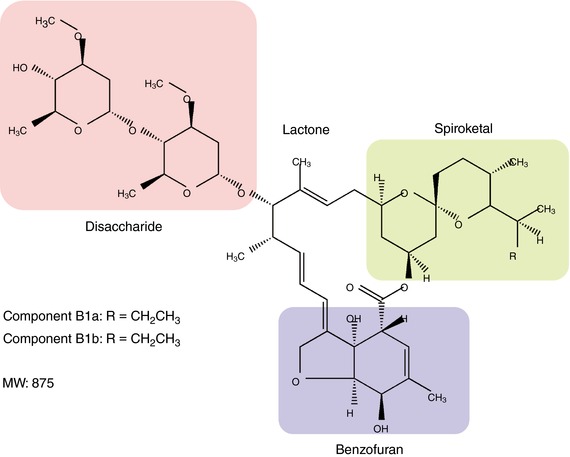
IVM is a 16‐membered macrocyclic lactone derivative with disaccharide, benzofuran and spiroketal moieties, and the molar mass is about 875 g mol−1. It is a complex of 80% B1a and 20% B1b components. Previous studies suggested that the benzofuran moiety plays the most critical role in its interaction with GluCl (Michael et al. 2001; Lynagh & Lynch, 2012).
IVM is highly hydrophobic and deeply inserts into the subunit interfaces of transmembrane domains (TMs) of Cys‐loop receptor family members (Lynagh & Lynch, 2012). When IVM binds to the glutamate‐gated Cl− channel (GluCl), a member of the Cys‐loop receptor family found in invertebrates, it potentiates the channel activity, resulting in hyperpolarization of parasite neurons and muscles, thereby killing the parasites (Cully et al. 1994; Arena et al. 1995). Mutations in GluCl reduce IVM sensitivity and thereby produce IVM resistance in parasites and insects (Kane et al. 2000; McCavera et al. 2009; Ghosh et al. 2012). IVM also interacts with the TMs of other Cys‐loop receptors, including the histamine‐gated Cl− channel (Zheng et al. 2002), the pH‐gated Cl− channel (Mounsey et al. 2007; Nakatani et al. 2016), the glycine receptor (GlyR) (Shan et al. 2001), the γ‐aminobutyric acid receptor (GABAAR) (Adelsberger et al. 2000) and the α7‐nicotinic acetylcholine receptor (nAChR) (Krause et al. 1998). IVM also modulates the P2X4 receptor and most likely binds to the TMs of this receptor, similarly to the Cys‐loop receptors described above (Silberberg et al. 2007).
The farnesoid X receptor (FXR), a nuclear receptor involved in metabolic regulation, is also a novel target of IVM (Jin et al. 2013). Also, it was reported that IVM acts as an inhibitor of the human ether‐à‐go‐go‐related gene (hERG) K+ channel with an IC50 of approximately 13 μm at 23°C and 24 μm at 37°C (Kauthale et al. 2015).
We recently revealed that IVM activates a novel target, the G‐protein‐gated inwardly rectifying K+ (GIRK) channel, especially subtype 2 (GIRK2) (Chen et al. 2017). In remarkable contrast to the critical site for IVM binding in Cys‐loop receptors and the P2X4 receptor, we found that the interface between the TMs and intracellular domains, rather than the TMs themselves, plays critical roles in IVM‐induced GIRK activation. Here we introduce novel information as to how IVM modulates ion channels and receptors, by summarizing the findings from structural and functional points of view.
Roles of IVM and the structural determinants for the modulation of targets
Cys‐loop receptors
IVM acts as an activator or modulator of Cys‐loop receptors, including invertebrate GluCl and human inhibitory anion‐permeable receptors (GlyR and GABAAR) and an excitatory cation‐permeable receptor (nAChR). Another human cation‐permeable Cys‐loop receptor, the type 3 5‐hydroxytryptamine receptor (5‐HT3R), is insensitive to IVM. These Cys‐loop receptors have a similar structure but are activated by different neurotransmitters, such as glycine, glutamate, acetylcholine and serotonin, while controlling excitatory or inhibitory synaptic transmission in the central nervous system.
In recent years, high resolution structures of several Cys‐loop receptors have been solved by X‐ray crystallography or cryo‐electron microscopy: GluCl (Hibbs & Gouaux, 2011), GABAAR (Miller & Aricescu, 2014), GlyR (Du et al. 2015; Huang et al. 2017) and nAChR (Unwin, 2005; Morales‐Perez et al. 2016). In general, Cys‐loop receptors are pentamers of the same or different subunits, in which each subunit contains an N‐terminal extracellular domain and four TMs, and the TM2 helices from each subunit line the channel pore. The Cys‐loop is one of the domains that connect the extracellular domains and the TMs. When a neurotransmitter binds to the pocket in the extracellular domain, conformational changes are transmitted to the TMs to open the channel pore. There are only two of the Cys‐loop receptor family members whose structures have been solved in an IVM‐bound complex: Caenorhabditis elegans GluCl (Hibbs & Gouaux, 2011) and the zebrafish and human GlyRs (Du et al. 2015; Huang et al. 2017) (Fig. 2 A). The IVM‐bound structures of other IVM‐targeted Cys‐loop receptors remain unknown.
Figure 2. IVM binding site and its structural determinants in GluCl and GlyR.
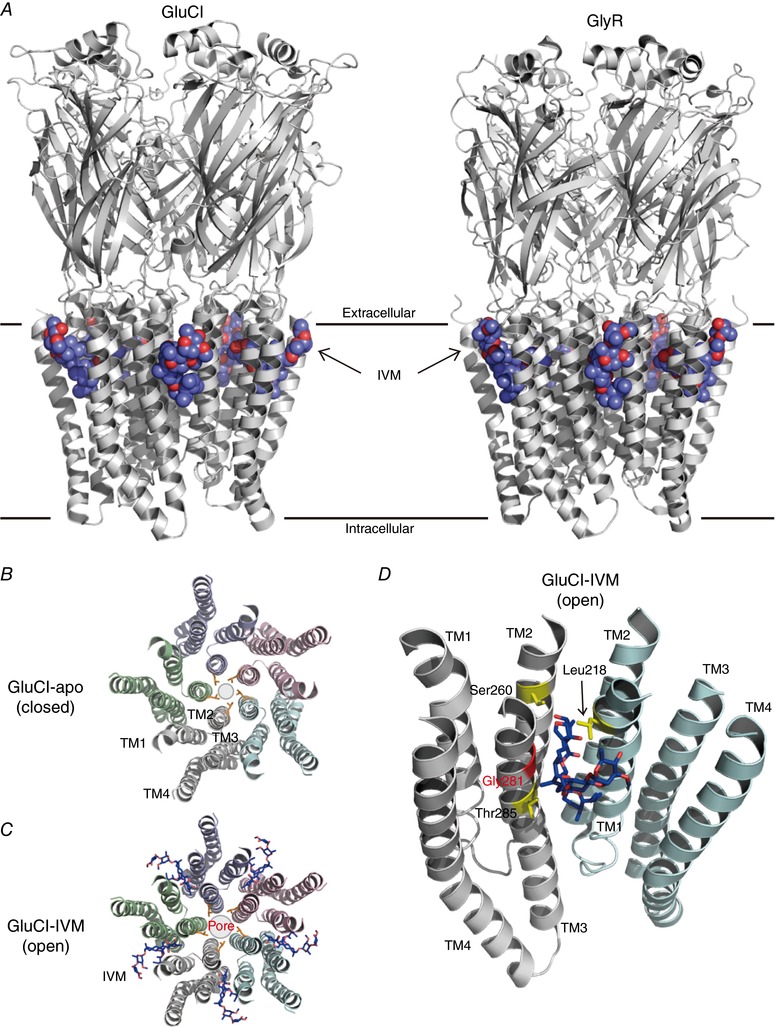
A, a lateral view (parallel to plasma membrane) of the structure of IVM‐bound C. elegans GluCl (left panel; PDB ID: 3RHW) (Hibbs & Gouaux, 2011) and zebrafish GlyR (right panel; PDB ID: 3JAF) (Du et al. 2015) pentamer. The IVM molecules are shown in sphere mode. B and C, a top view (perpendicular to plasma membrane) of TMs of GluCl from the extracellular side in an apo (closed) state (B; PDB ID: 4TNV) (Althoff et al. 2014) and an IVM‐bound (open) state (C; PDB ID: 3RHW). Each subunit is shown in a different colour and the shut gate of the channel pore, formed by the side chains of Leu254 in TM2 from each subunit, is shown in orange. The IVM molecules are shown in stick mode. The binding of IVM to GluCl enlarges the channel pore. D, IVM inserts into the subunit interface and interacts with Leu218 in TM1, Ser260 in TM2 and Thr285 in TM3 (shown in yellow) by forming hydrogen bonds. A key amino acid residue for IVM sensitivity in the Cys‐loop receptor family (Gly281 in GluCl) is shown in red.
In the case of GluCl, IVM acts as an activator with an EC50 of 140 nm or an allosteric modulator at a low concentration (5 nm) that potentiates glutamate binding (Cully et al. 1994). IVM binds to the TMs at the subunit interface of the GluCl pentamer, near the extracellular surface of the plasma membrane (Hibbs & Gouaux, 2011) (Fig. 2 A, left panel). By comparing the structures between an apo (closed) state and an IVM‐bound (open) state of GluCl, the insertion of IVM is shown to enlarge the channel pore of the TM region (Hibbs & Gouaux, 2011; Althoff et al. 2014) (Fig. 2 B and C). In the apo (closed) state, the side chain of Leu254, located in the narrowest position in the channel pore, acts as a closed gate. Althoff et al (2014) described that the activation of GluCl by IVM induces a rotation and a shift of the TMs toward the extracellular side (an upward movement of the TMs), thereby enlarging the pore gate formed by Leu254 and also expanding the space in the inter‐subunit interface between the TM1 and the TM3 helices (Althoff et al. 2014). Leu218 in TM1, Ser260 in TM2 and Thr285 in TM3 interact with IVM by forming hydrogen bonds (Fig. 2 D). Gly281 in TM3 also contributes to IVM sensitivity (Lynagh & Lynch, 2010a). IVM binding stabilizes the TMs in an open state through an interaction between Pro268 and Val45 located at the interface between the extracellular domains and TMs (Calimet et al. 2013; Althoff et al. 2014). Conformational changes in the TMs by IVM also induce a tilt of the extracellular domain, which may potentiate the binding of glutamate.
In the case of GlyR, IVM acts as an activator at high concentrations, with an EC50 between 1 and 5 μm toward different GlyR subunits (Lynagh & Lynch, 2010b). IVM also acts as an allosteric modulator that potentiates the glycine (saturating concentration, 250 μm)‐induced current at a low concentration (30 nm) (Shan et al. 2001). The IVM‐binding site in GlyR is similar to that in GluCl (Fig. 2 A, right panel). IVM also binds to the TMs at each subunit interface of the zebrafish GlyRα1 and human GlyRα3 pentamer, near the extracellular surface of the membrane (Du et al. 2015; Huang et al. 2017). In human GlyRα3, Pro230 in TM1 and Ala288 in TM3 (corresponding to Gly281 in GluCl) interact with IVM by forming hydrophobic interactions (Huang et al. 2017). The Ala288Gly mutation in GlyR increased IVM activity, suggesting that a smaller residue at this position gives a higher IVM sensitivity (Lynagh & Lynch, 2010b). Any residues larger than Gly at this corresponding position in Cys‐loop receptors disturbs IVM binding, and this may be one of the reasons why IVM activates GlyR less potently than GluCl (IVM activates GlyR at a micromolar concentration and activates GluCl at a nanomolar concentration) (Lynagh & Lynch, 2010a, 2012; Huang et al. 2017). Ile225 and Gln226 in TM1 and Ser267 and Arg287 in TM2 of human GlyRα3 interact with IVM by forming hydrogen bonds (Huang et al. 2017). All of these residues contribute to IVM binding and sensitivity. By comparing the structures of the strychnine‐bound (closed) state, glycine‐bound (open) state and glycine–IVM‐bound state of zebrafish GlyR, it was shown that there are two pore gates formed by Leu277 and Pro266 (Du et al. 2015). The insertion of IVM induces rotation of TMs and thus enlargement of the pore gate formed by Leu277 but constriction of the pore gate formed by Pro266 in TM2 (Du et al. 2015). Unlike in GluCl, the binding of IVM to glycine‐bound GlyR induces remarkable conformational changes in TMs but only limited changes in the extracellular domains.
In the case of GABAAR, the X‐ray structure of the human GABAAR β3 homopentamer revealed the agonist (benzamidine)‐binding pocket in the extracellular domains (Miller & Aricescu, 2014), whereas the IVM‐bound structure still remains unsolved. GABAARs are usually composed of multiple kinds of subunits, and the major isoform in brain is composed of α1, β2 and γ2 subunits (Sigel & Steinmann, 2012). IVM activates the α1β2γ2 GABAAR with an EC50 of approximately 2−7 μm (Adelsberger et al. 2000; Westergard et al. 2015) and potentiates the native GABA‐induced Cl− current in mouse hippocampal neurons at a low concentration (0.1 μm) (Zemkova et al. 2014). Based on functional studies and docking analyses, it is most likely that IVM binds to a similar position in the TM region as in GluCl or GlyR (Estrada‐Mondragon & Lynch, 2015; Westergard et al. 2015). According to the results of mutagenesis at Ala291 in the α1 subunit, Met286 in the β2 subunit and Ser301 in the γ2 subunit (corresponding to Gly281 in GluCl or Ala288 in GlyR), IVM induces different effects when it binds to different subunit interfaces: (1) the binding of IVM to the α1–β2 interface (in an anticlockwise orientation of subunit stoichiometry in a top view from the extracellular side) potentiates the GABA‐induced current; (2) the binding of IVM to the γ2–β2 interface induces irreversible activation of the channel; (3) IVM cannot bind to the β2–α1 interface because of the large side chain of Met286 in the β2 subunit (Estrada‐Mondragon & Lynch, 2015). Further investigations are needed to obtain more detailed information on the modulation mechanisms of GABAAR by IVM.
In the case of nAChR, the IVM‐bound structure also remains to be determined. Application of IVM alone does not activate the nAChR, but it potentiates the ACh‐induced current at a concentration of 30 μm (Krause et al. 1998). Based on docking analyses and functional studies of rat nAChR α7–mouse 5‐HT3A chimeras and mutagenesis experiments (Collins & Millar, 2010), IVM also interacts with the amino acid residues located in the upper region of the TMs. However, the predicted IVM‐binding site in the nAChR is most likely to be different from those of other Cys‐loop receptors (Lynagh & Lynch, 2012). IVM binds to the intra‐subunit cavity between TM1 and TM4 (in the same subunit) (Sattelle et al. 2009; Collins & Millar, 2010), and this site is distinct from the IVM‐binding site in GluCl, GlyR and GABAAR, which is located in the subunit interface, as described above. Ala225Asp, Gln272Val, Thr456Tyr and Cys459Tyr mutations reduced the IVM potency (Collins & Millar, 2010). Surprisingly, Ser222Met (in TM1), Met253Leu (in TM2) and Ser276Val (in TM3) mutations induced a change of the IVM effect from potentiation to inhibition.
P2X receptors
IVM acts as an allosteric positive modulator of purinergic ATP‐gated P2X receptors (seven P2X subunits, P2X1–7), especially P2X4 but not P2X2 or P2X3 receptors (Khakh et al. 1999). IVM alone does not directly activate the P2X4 receptor but potentiates the amplitude of the ATP‐induced current with an EC50 of approximately 0.25 μm (Priel & Silberberg, 2004; Gao et al. 2015). IVM also potentiates the ATP‐induced current of the human P2X7 receptor, but has only a limited effect on the mouse and rat P2X7 receptors (Norenberg et al. 2012). Recent findings suggest that IVM mediates alcohol intake, sensorimotor gating and dopamine‐induced motor behaviour through modulation of P2X4 receptors (Bortolato et al. 2013; Franklin et al. 2014, 2015; Khoja et al. 2016). IVM also possesses a potential anti‐cancer effect: it kills breast cancer cells through potentiating P2X4/P2X7 signalling (Draganov et al. 2015).
The crystal structures of an apo (closed) state and an ATP‐bound (open) state of the zebrafish P2X4 receptor have been solved (Kawate et al. 2009; Hattori & Gouaux, 2012) (Fig. 3), while the IVM‐bound state still remains unknown. A previous study showed that extracellular application of IVM modulates human P2X4 receptors better than when applied from the intracellular side, suggesting that IVM does not interact with the intracellular domains (Priel & Silberberg, 2004). The authors also proposed that there are two separate IVM‐binding sites in the extracellular domains of the P2X4 receptor: (1) IVM binds to the high affinity site to increase ATP‐induced current by reducing channel desensitization; and (2) IVM binds to the low affinity site to decelerate current deactivation by inducing stabilization of the open state. Based on their subsequent functional analyses of the rat P2X4–rat P2X2 chimeras and mutants, it was shown that IVM most likely interacts with the TMs of open P2X4 receptors, near the extracellular surface of the plasma membrane (Silberberg et al. 2007). This predicted IVM binding site, which is located in the upper region of the TMs (Fig. 3), is similar to those of Cys‐loop receptors. By scanning mutagenesis of all amino acid residues of TM1 and TM2 to Ala or Trp, it was observed that mutations of Val28, Ile39, Tyr42, Val43 and Val47 in TM1 and Gly340, Gly342, Leu345 and Val348 in TM2 lowered IVM occupancy (Silberberg et al. 2007). Other studies also indicated that Gln36, Leu40, Val43, Trp46, Val47 and Trp50 in TM1 and Asn338, Gly342, Leu346, Ala349 and Ile356 in TM2 contribute to the IVM effect (Jelinkova et al. 2008; Popova et al. 2013). Taken together, the residues in TMs near the extracellular surface of the plasma membrane are critical for IVM action.
Figure 3. Critical amino acid residues for the IVM effect and the predicted binding site in the P2X4 receptor.
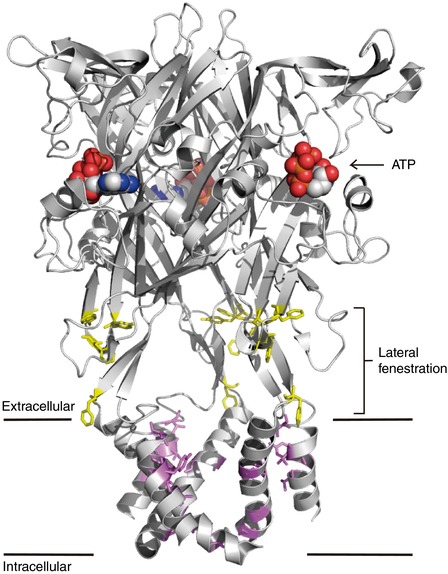
A lateral view of the structure of the ATP‐bound (open) state zebrafish P2X4 receptor (PDB ID: 4DW1) (Hattori & Gouaux, 2012) trimer. The structure of the IVM‐bound P2X4 receptor is still unsolved. ATPs that bind to the extracellular domains are shown in sphere mode. The amino acid residues of the rat P2X4 receptor, which contribute to the IVM response, located in the TMs (violet) and lateral fenestration (yellow), are shown in their corresponding positions in the zebrafish P2X4 structure. The violet residues (TM regions) are the predicted IVM‐binding sites, and the yellow residues (lateral fenestration regions) also contribute to the IVM response.
Recently, functional studies identified a novel structural determinant of the rat P2X4 receptor for IVM sensitivity, the lateral fenestration (Kawate et al. 2009) (Fig. 3), which acts as a linker to connect the extracellular domains and the pore‐forming TMs (Rokic et al. 2013; Gao et al. 2015). Several aromatic residues (Tyr195, Phe198, Phe200 and Phe330) located in the lateral fenestration contribute to IVM sensitivity and thus modulate current deactivation (Gao et al. 2015). A specific negatively charged residue in the human P2X4 receptor, Glu51, located on the top of the extracellular entrance of the channel pore, is also critical for the IVM effect (Samways et al. 2012).
G‐protein‐gated inwardly rectifying K+ channels
IVM acts as a novel activator of GIRK channels (Su et al. 2016; Chen et al. 2017). GIRK channels (four GIRK subunits, Kir3.1–4) control various physiological functions: Kir3.1–Kir3.2 heterotetramers in the brain regulate the excitability of neurons and Kir3.1–Kir3.4 heterotetramers in the heart regulate heart rate (Kubo et al. 1993; Krapivinsky et al. 1995; Hibino et al. 2010). GIRK channels are known to be directly activated by Gβγ and also directly modulated by phosphatidylinositol 4,5‐bisphosphate (PIP2) and Na+ (Logothetis et al. 1987; Reuveny et al. 1994; Huang et al. 1998; Ho & Murrell‐Lagnado, 1999). We recently observed that IVM activates Kir3.1–Kir3.2 remarkably (EC50 of 3.5 μm) and Kir3.1–Kir3.4 weakly (EC50 of 7.5 μm) in a PIP2‐dependent, Gβγ‐independent manner, and identified the structural determinants for IVM‐mediated activation (Chen et al. 2017).
The crystal structures of the mouse Kir3.2 in an apo (Arg201Ala, closed) state, a PIP2‐bound (closed) state (Fig. 4 A) and a Gβγ‐bound (pre‐open) state have been solved (Whorton & MacKinnon, 2011, 2013), while the full open state is still unknown, and no complexes with IVM have been solved. Based on our observation that IVM activates Kir3.2 more efficiently than Kir3.4 (Fig. 4 B and C), we identified the structural determinants by constructing Kir3.2–Kir3.4 chimeras and single‐point mutants (Chen et al. 2017). We found that the TMs are not responsible for IVM‐mediated GIRK activation, in remarkable contrast to IVM activation in Cys‐loop receptors and the P2X4 receptor. A single amino acid residue in Kir3.2, Ile82, located in the slide helix at the end of the N‐terminus cytoplasmic region, is critical for the IVM response (Fig. 4 A). Mutation of Kir3.2 Ile82 (corresponding to Kir3.4 Leu77) to Leu reduced IVM‐induced current, and the reverse mutation of Kir3.4 Leu77 (corresponding to Kir3.2 Ile82) to Ile increased the IVM effect. Ile82 is oriented with a methyl group of the branched side chain toward the inner transmembrane helix (TM2), and this methyl group acts as a switch for IVM‐mediated activation. Trp91Ala and Ile195Ala mutations also reduced GIRK current induced by IVM, suggesting that Ile82, Trp91 and Ile195, located in the interface between the TMs and the cytoplasmic tail domains (CTDs), determine IVM‐mediated GIRK activation, presumably by forming an IVM‐binding pocket (Fig. 4 D and E). We speculated that, when IVM binds to the pocket, hydrophobic interactions are induced between IVM and Ile82/Trp91/Ile195, which reinforce the hydrophobic core around the TM–CTD interface, stabilizing the open state of GIRK channel. Since the PIP2‐binding site is close to the pocket formed by Ile82/Trp91/Ile195, the association of PIP2 may support IVM binding by inducing stabilization of the TM–CTD interface (Chen et al. 2017).
Figure 4. Critical amino acid residues for the IVM effect and the predicted binding site in GIRK channels.
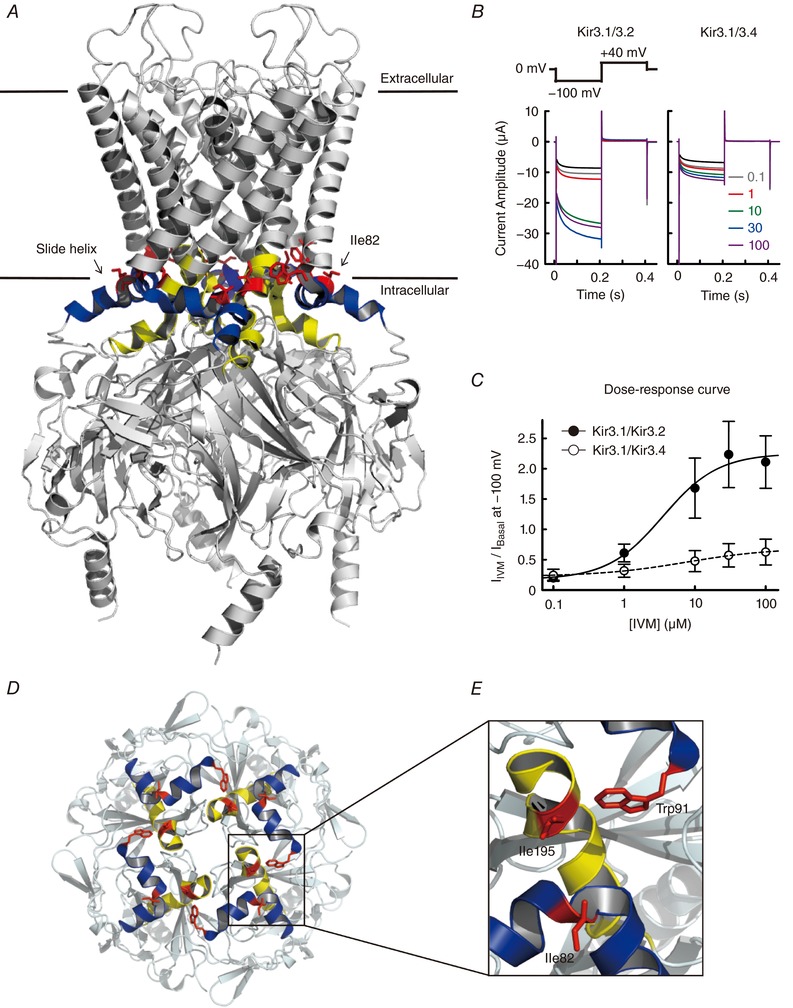
A, a lateral view of the structure of the PIP2‐bound (closed) state mouse Kir3.2 (PDB ID: 3SYA) (Whorton & MacKinnon, 2011) tetramer. The structure of the IVM‐bound GIRK channel is still unsolved. The slide helices are shown in blue; TM‐CTD linkers are shown in yellow; Ile82, Trp91 and Ile195 (the amino acid residues contributing to the IVM response) are shown in red. B, currents in the absence (black trace) and presence of IVM (0.1−100 μm, coloured traces) in oocytes expressing Kir3.1−Kir3.2 (left) and Kir3.1−Kir3.4 (right) were recorded using the protocol shown above (modified from Fig. 1 A of Chen et al. 2017). C, dose–response curves for Kir3.1−Kir3.2 (filled circles) and Kir3.1−Kir3.4 (open circles) were constructed by the ratio of I Basal and I IVM, where I Basal refers to the basal current before the application of IVM, I IVM refers to the maximum current in the presence of IVM at a given concentration at −100 mV, and the basal current was subtracted (modified from Fig. 1 D of Chen et al. 2017). D, the top view of the TM–CTD interfaces and intracellular domains of Kir3.2 from the extracellular side. E, the expanded view of Ile82, Trp91 and Ile195 located in the TM–CTD interfaces. We speculated that Ile82/Trp91/Ile195 contribute to IVM sensitivity by forming a binding pocket in the TM–CTD interface (Chen et al. 2017).
Farnesoid X receptors
IVM acts as a novel ligand of FXR (Jin et al. 2013), a receptor in the cytoplasm belonging to the nuclear hormone receptor superfamily. One of the physiological ligands of FXR is bile acid. It is highly expressed in liver, small intestine, kidney and adrenals (Wang et al. 2008). When a ligand binds to FXR, it targets to the DNA and regulates the expression of genes that are involved in the metabolism of bile acids, lipid and glucose (Wang et al. 2008). Therefore, the novel FXR ligand, IVM, has a potential to treat metabolic syndromes, such as non‐alcoholic fatty liver disease (Wang et al. 2008; Jin et al. 2013, 2015; Zheng et al. 2017). IVM shows a higher FXR selectivity and affinity (EC50 of approximately 0.2 μm) than the physiological FXR ligand, bile acid (EC50 of approximately 10 μm) (Jin et al. 2013; Ding et al. 2015).
The crystal structure of the ligand‐binding domain of IVM‐bound human FXR has been solved (Akwabi‐Ameyaw et al. 2009; Jin et al. 2013) (Fig. 5 A). The conformational change of the ligand‐binding pocket induced by IVM was different from that induced by a widely used FXR ligand, GW4064. The analyses of the transcriptional activity of FXR mutants revealed several important residues for IVM binding (Jin et al. 2013, 2015) (Fig. 5 B). The Ala291Trp mutation reduces the size of the ligand‐binding pocket, thereby preventing the binding of FXR ligands (IVM, IVM analogues or GW4064), as well as the transcriptional activity induced by these ligands. The Asn283Leu and Phe284His mutations of FXR abolished the transcriptional activity induced by IVM by disrupting a hydrogen bond between IVM and Asn283, and a hydrophobic interaction between IVM and the hydrophobic side chain of Phe284. Surprisingly, these mutants were still able to be activated by GW4064. On the other hand, Leu287Thr, Arg331Met and His447Phe mutations increased IVM (or its analogues)‐activated transcriptional activity, while abolishing the GW4064 effect. It is most likely that these amino acid residues play important roles in the ligand selectivity of FXR.
Figure 5. IVM‐binding site and structural determinants for binding in FXR.
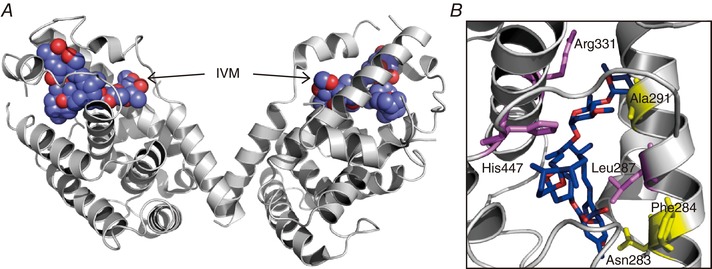
A, the structure of the IVM‐bound human FXR ligand‐binding domain (PDB ID: 4WVD) (Jin et al. 2013) as a dimer. The IVM molecules are shown in sphere mode. The binding of IVM activates FXR and thus regulates the expression of FXR‐targeted genes. B, the expanded view of the IVM binding site in FXR. Mutations in Asn283, Phe284 and Ala291 (yellow) abolish the IVM response, whereas mutations in Leu287, Arg331 and His447 (violet) increase the IVM response.
Comparison of the effects of IVM analogues on target proteins
There are several IVM analogues known to modulate ion channels and receptors like IVM does. These compounds, including abamectin (ABM), doramectin (DOM), eprinomectin (EPM) and emamectin (EMM), share a backbone of a 16‐membered macrocyclic lactone with different functional groups at the benzofuran, spiroketal and disaccharide moieties (Figs 1 and 6 A). Based on the IVM‐bound Cys‐loop receptor complexes (Fig. 2), the benzofuran moiety is inserted deeply toward the pore of the channel, the spiroketal moiety interacts with TM1 and the disaccharide moiety orients toward the outside of the channel (Hibbs & Gouaux, 2011; Du et al. 2015). In a study that compared the binding of IVM in GluCl and GlyR, it was shown that IVM is inserted deeper into the subunit interfaces in GluCl than in GlyR, supporting the notion that Gly281 in GluCl provides a larger space for IVM binding than Ala288 in GlyR (Huang et al. 2017). Therefore, the size of IVM analogues may also influence their binding to target proteins, due to the limited space of the binding pocket.
Figure 6. Comparison of chemical structures and effects of IVM analogues on target proteins.
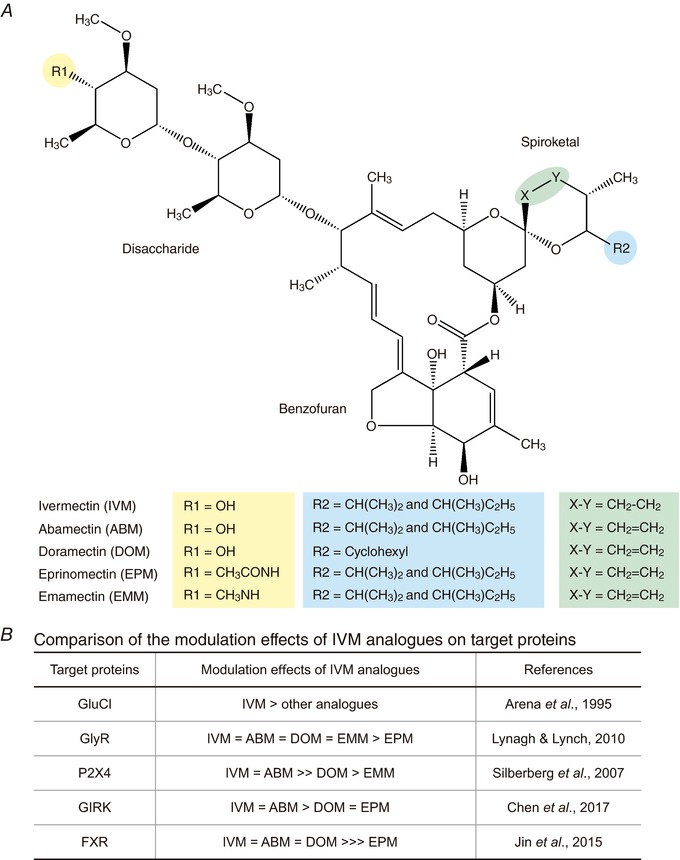
A, chemical structures of ivermectin (IVM) and its analogues: abamectin (ABM), doramectin (DOM), eprinomectin (EPM) and emamectin (EMM). B, a comparison of the effects of IVM analogues on modulation of the function of target proteins: GluCl (Arena et al. 1995), GlyR (Lynagh & Lynch, 2010b), P2X4 receptor (Silberberg et al. 2007), GIRK (Kir3.1−Kir3.2) channel (Chen et al. 2017) and FXR (Jin et al. 2015). IVM is the most potent among the analogues to activate GluCl; ABM possesses a similar efficacy to IVM in the activation of GlyR, the P2X4 receptor, GIRK channels and FXR; DOM shows a lesser activation or potentiation effect on the P2X4 receptor and GIRK channels than IVM; EPM does not modulate FXR‐mediated signalling.
ABM shares the most similar structure to IVM among these analogues, and their structure differs only at a position between C22 and C23 in the spiroketal moiety (indicated as X–Y in Fig. 6 A): double bonds in ABM reduced to a single bond in IVM (Omura, 2016). Therefore, ABM induces modulation of most of the target proteins with a similar efficacy to that of IVM. They show equal effects on the activation of GlyR (Lynagh & Lynch, 2010b) and GIRK current (Chen et al. 2017), the potentiation of ATP‐gated P2X4 current (Silberberg et al. 2007) and the regulation of metabolism by modulation of FXR signalling (Jin et al. 2015). In the case of the invertebrate GluCl, IVM is most potent and ABM and others are less effective (Arena et al. 1995) (Fig. 6 B).
DOM contains a larger functional group in the spiroketal moiety (indicated as R2 in Fig. 6 A: cyclohexyl). This analogue has less effect on the modulation of P2X4 receptor and GIRK than IVM (Silberberg et al. 2007; Chen et al. 2017), while it has similar effects on that of Cys‐loop receptors and FXR (Arena et al. 1995; Lynagh & Lynch, 2010b; Jin et al. 2015) (Fig. 6 B). Therefore, we speculate that the IVM‐binding space in P2X4 and GIRK may be narrower than that in Cys‐loop receptors and FXR.
EPM and EMM contain a larger functional group in the disaccharide moiety (indicated as R1 in Fig. 6 A). These two analogues have weaker effects on most of the target proteins than IVM (see detail in Fig. 6 B). For example, EPM did not rescue the metabolic syndrome by targeting FXR (Jin et al. 2015). These results suggest that the structural difference in disaccharide moiety also influences its binding.
Summary
Since IVM is such a big and hydrophobic compound, the size of the binding cavity between the α‐helices of the TMs or the binding domains generally governs IVM binding. Some of the amino acid residues, whose side chains are oriented to the binding cavity, stabilize IVM binding by forming a hydrophobic interaction and hydrogen bond network.
IVM modulates various ion channels and receptors by different mechanisms: (1) IVM binds to the TMs of Cys‐loop receptors near the extracellular surface of the plasma membrane and induces the rotation of TMs, thereby facilitating opening of the channel pore. A small amino acid residue located in TM3 (such as Gly281 in GluCl and Ala288 in GlyR) plays a key role in IVM sensitivity (Fig. 2 D). (2) IVM most likely interacts with the TMs of the P2X4 receptor by binding to the subunit interfaces, and this is similar to the IVM binding site in most of the Cys‐loop receptors (Fig. 3). (3) Unlike in Cys‐loop receptors and the P2X4 receptor, the TMs of GIRK channels are not critical for the IVM response. We found that the hydrophobic side chain of Ile82 (Kir3.2) at the slide helix, which is located in the interface between the TMs and intracellular domains, is critical for IVM sensitivity (Fig. 4 A, D and E). (4) IVM not only embeds into lipid bilayers and then influences the functions of membrane proteins, but also presumably permeates through the membrane and then interacts with the nuclear receptor FXR (Fig. 5).
IVM shows higher affinity to GluCl, P2X4 and FXR (EC50 ≤ 0.25 μm) than to other targeted channels (EC50 1−10 μm). Therefore, the application of IVM to the treatment of parasitic infections, cancers and metabolic syndrome, through modulation of GluCl‐, P2X4‐ and FXR‐mediated signalling, is considered to be saved from side effects due to the modulation of other targets by IVM. On the other hand, since IVM analogues possess a relatively low efficiency compared to IVM toward a specific receptor (Fig. 6 B), it will be possible to reduce an undesirable influence due to the activation of a specific receptor by replacing IVM with a suitable analogue. Further research is awaited to advance our knowledge of this multifaceted drug, IVM, and its analogues.
Additional information
Competing interests
The authors declare no competing financial interests.
Author contributions
I‐S.C. and Y.K. conceived and wrote the review. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This study was supported by JSPS KAKENHI Grant Number JP16K18999 (to I‐S.C.), JP26293044 and JP17H04021 (to Y.K.).
Acknowledgements
We thank Dr Anthony Collins (Saba University School of Medicine, Dutch Caribbean) for text editing and correction. We also thank Dr Takushi Shimomura (National Institute for Physiological Sciences, Okazaki) for valuable comments on the manuscript.
Biographies
I‐Shan Chen received her PhD from Osaka University (where her supervisor was Prof. Yoshihisa Kurachi) where she started the GPCR‐GIRK studies. After performing post‐doctoral research in the same laboratory, she moved to the National Institute for Physiological Sciences as an assistant professor (where her principal investigator (PI) is Prof. Yoshihiro Kubo) in 2015.

Yoshihiro Kubo received his PhD from The University of Tokyo (where his supervisor was the late Prof. Kunitaro Takahashi) and performed post‐doctoral research in University of California San Francisco (where his PI was Prof. Lily Jan). He engaged in structure–function studies of membrane proteins in Tokyo Metropolitan Institute for Neurosciences and then in Tokyo Medical and Dental University. He has been a professor of the National Institute for Physiological Sciences since 2003.
This review was presented at the symposium ‘Shared and unique aspects of the gating mechanisms of ligand‐ and voltage‐gated ion channels’ which took place at IUPS 38th World Congress, Rio de Janeiro, Brazil, 1–5 August 2017.
Edited by: Ole Petersen & David Adams
This is an Editor's Choice article from the 15 May 2018 issue.
References
- Adelsberger H, Lepier A & Dudel J (2000). Activation of rat recombinant α1β2γ2S GABAA receptor by the insecticide ivermectin. Eur J Pharmacol 394, 163–170. [DOI] [PubMed] [Google Scholar]
- Akwabi‐Ameyaw A, Bass JY, Caldwell RD, Caravella JA, Chen L, Creech KL, Deaton DN, Madauss KP, Marr HB, McFadyen RB, Miller AB, Navas F 3rd, Parks DJ, Spearing PK, Todd D, Williams SP & Bruce Wisely G (2009). FXR agonist activity of conformationally constrained analogs of GW 4064. Bioorg Med Chem Lett 19, 4733–4739. [DOI] [PubMed] [Google Scholar]
- Althoff T, Hibbs RE, Banerjee S & Gouaux E (2014). X‐ray structures of GluCl in apo states reveal a gating mechanism of Cys‐loop receptors. Nature 512, 333–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arena JP, Liu KK, Paress PS, Frazier EG, Cully DF, Mrozik H & Schaeffer JM (1995). The mechanism of action of avermectins in Caenorhabditis elegans: correlation between activation of glutamate‐sensitive chloride current, membrane binding, and biological activity. J Parasitol 81, 286–294. [PubMed] [Google Scholar]
- Bortolato M, Yardley MM, Khoja S, Godar SC, Asatryan L, Finn DA, Alkana RL, Louie SG & Davies DL (2013). Pharmacological insights into the role of P2X4 receptors in behavioural regulation: lessons from ivermectin. Int J Neuropsychopharmacol 16, 1059–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burg RW, Miller BM, Baker EE, Birnbaum J, Currie SA, Hartman R, Kong YL, Monaghan RL, Olson G, Putter I, Tunac JB, Wallick H, Stapley EO, Oiwa R & Omura S (1979). Avermectins, new family of potent anthelmintic agents: producing organism and fermentation. Antimicrob Agents Chemother 15, 361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calimet N, Simoes M, Changeux JP, Karplus M, Taly A & Cecchini M (2013). A gating mechanism of pentameric ligand‐gated ion channels. Proc Natl Acad Sci USA 110, E3987–E3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell WC & Benz GW (1984). Ivermectin: a review of efficacy and safety. J Vet Pharmacol Ther 7, 1–16. [DOI] [PubMed] [Google Scholar]
- Campbell WC, Fisher MH, Stapley EO, Albers‐Schonberg G & Jacob TA (1983). Ivermectin: a potent new antiparasitic agent. Science 221, 823–828. [DOI] [PubMed] [Google Scholar]
- Chabala JC, Mrozik H, Tolman RL, Eskola P, Lusi A, Peterson LH, Woods MF, Fisher MH, Campbell WC, Egerton JR & Ostlind DA (1980). Ivermectin, a new broad‐spectrum antiparasitic agent. J Med Chem 23, 1134–1136. [DOI] [PubMed] [Google Scholar]
- Chen IS, Tateyama M, Fukata Y, Uesugi M & Kubo Y (2017). Ivermectin activates GIRK channels in a PIP2‐dependent, Gβγ‐independent manner and an amino acid residue at the slide helix governs the activation. J Physiol 595, 5895–5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins T & Millar NS (2010). Nicotinic acetylcholine receptor transmembrane mutations convert ivermectin from a positive to a negative allosteric modulator. Mol Pharmacol 78, 198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cully DF, Vassilatis DK, Liu KK, Paress PS, Van der Ploeg LH, Schaeffer JM & Arena JP (1994). Cloning of an avermectin‐sensitive glutamate‐gated chloride channel from Caenorhabditis elegans . Nature 371, 707–711. [DOI] [PubMed] [Google Scholar]
- Ding L, Yang L, Wang Z & Huang W (2015). Bile acid nuclear receptor FXR and digestive system diseases. Acta Pharmaceutica Sinica B 5, 135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draganov D, Gopalakrishna‐Pillai S, Chen YR, Zuckerman N, Moeller S, Wang C, Ann D & Lee PP (2015). Modulation of P2X4/P2X7/Pannexin‐1 sensitivity to extracellular ATP via Ivermectin induces a non‐apoptotic and inflammatory form of cancer cell death. Sci Rep 5, 16222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Lu W, Wu S, Cheng Y & Gouaux E (2015). Glycine receptor mechanism elucidated by electron cryo‐microscopy. Nature 526, 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egerton JR, Ostlind DA, Blair LS, Eary CH, Suhayda D, Cifelli S, Riek RF & Campbell WC (1979). Avermectins, new family of potent anthelmintic agents: efficacy of the B1a component. Antimicrob Agents Chemother 15, 372–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada‐Mondragon A & Lynch JW (2015). Functional characterization of ivermectin binding sites in α1β2γ2L GABAA receptors. Front Mol Neurosci 8, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KM, Asatryan L, Jakowec MW, Trudell JR, Bell RL & Davies DL (2014). P2X4 receptors (P2X4Rs) represent a novel target for the development of drugs to prevent and/or treat alcohol use disorders. Front Neurosci 8, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KM, Hauser SR, Lasek AW, Bell RL & McBride WJ (2015). Involvement of purinergic P2X4 receptors in alcohol intake of high‐alcohol‐drinking (HAD) rats. Alcohol Clin Exp Res 39, 2022–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C, Yu Q, Xu H, Zhang L, Liu J, Jie Y, Ma W, Samways DS & Li Z (2015). Roles of the lateral fenestration residues of the P2X4 receptor that contribute to the channel function and the deactivation effect of ivermectin. Purinergic Signal 11, 229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh R, Andersen EC, Shapiro JA, Gerke JP & Kruglyak L (2012). Natural variation in a chloride channel subunit confers avermectin resistance in C. elegans . Science 335, 574–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori M & Gouaux E (2012). Molecular mechanism of ATP binding and ion channel activation in P2X receptors. Nature 485, 207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbs RE & Gouaux E (2011). Principles of activation and permeation in an anion‐selective Cys‐loop receptor. Nature 474, 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I & Kurachi Y (2010). Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90, 291–366. [DOI] [PubMed] [Google Scholar]
- Ho IH & Murrell‐Lagnado RD (1999). Molecular mechanism for sodium‐dependent activation of G protein‐gated K+ channels. J Physiol 520, 645–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CL, Feng S & Hilgemann DW (1998). Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gβγ. Nature 391, 803–806. [DOI] [PubMed] [Google Scholar]
- Huang X, Chen H & Shaffer PL (2017). Crystal structures of human GlyRα3 bound to ivermectin. Structure 25, 945–950.e2. [DOI] [PubMed] [Google Scholar]
- Jelinkova I, Vavra V, Jindrichova M, Obsil T, Zemkova HW, Zemkova H & Stojilkovic SS (2008). Identification of P2X4 receptor transmembrane residues contributing to channel gating and interaction with ivermectin. Pflugers Arch 456, 939–950. [DOI] [PubMed] [Google Scholar]
- Jin L, Feng X, Rong H, Pan Z, Inaba Y, Qiu L, Zheng W, Lin S, Wang R, Wang Z, Wang S, Liu H, Li S, Xie W & Li Y (2013). The antiparasitic drug ivermectin is a novel FXR ligand that regulates metabolism. Nat Commun 4, 1937. [DOI] [PubMed] [Google Scholar]
- Jin L, Wang R, Zhu Y, Zheng W, Han Y, Guo F, Ye FB & Li Y (2015). Selective targeting of nuclear receptor FXR by avermectin analogues with therapeutic effects on nonalcoholic fatty liver disease. Sci Rep 5, 17288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane NS, Hirschberg B, Qian S, Hunt D, Thomas B, Brochu R, Ludmerer SW, Zheng Y, Smith M, Arena JP, Cohen CJ, Schmatz D, Warmke J & Cully DF (2000). Drug‐resistant Drosophila indicate glutamate‐gated chloride channels are targets for the antiparasitics nodulisporic acid and ivermectin. Proc Natl Acad Sci USA 97, 13949–13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauthale RR, Dadarkar SS, Husain R, Karande VV & Gatne MM (2015). Assessment of temperature‐induced hERG channel blockade variation by drugs. J Appl Toxicol 35, 799–805. [DOI] [PubMed] [Google Scholar]
- Kawate T, Michel JC, Birdsong WT & Gouaux E (2009). Crystal structure of the ATP‐gated P2X4 ion channel in the closed state. Nature 460, 592–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh BS, Proctor WR, Dunwiddie TV, Labarca C & Lester HA (1999). Allosteric control of gating and kinetics at P2X4 receptor channels. J Neurosci 19, 7289–7299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoja S, Shah V, Garcia D, Asatryan L, Jakowec MW & Davies DL (2016). Role of purinergic P2X4 receptors in regulating striatal dopamine homeostasis and dependent behaviors. J Neurochem 139, 134–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapivinsky G, Gordon EA, Wickman K, Velimirovic B, Krapivinsky L & Clapham DE (1995). The G‐protein‐gated atrial K+ channel IKACh is a heteromultimer of two inwardly rectifying K+‐channel proteins. Nature 374, 135–141. [DOI] [PubMed] [Google Scholar]
- Krause RM, Buisson B, Bertrand S, Corringer PJ, Galzi JL, Changeux JP & Bertrand D (1998). Ivermectin: a positive allosteric effector of the α7 neuronal nicotinic acetylcholine receptor. Mol Pharmacol 53, 283–294. [DOI] [PubMed] [Google Scholar]
- Kubo Y, Reuveny E, Slesinger PA, Jan YN & Jan LY (1993). Primary structure and functional expression of a rat G‐protein‐coupled muscarinic potassium channel. Nature 364, 802–806. [DOI] [PubMed] [Google Scholar]
- Logothetis DE, Kurachi Y, Galper J, Neer EJ & Clapham DE (1987). The βγ subunits of GTP‐binding proteins activate the muscarinic K+ channel in heart. Nature 325, 321–326. [DOI] [PubMed] [Google Scholar]
- Lynagh T & Lynch JW (2010a). A glycine residue essential for high ivermectin sensitivity in Cys‐loop ion channel receptors. Int J Parasitol 40, 1477–1481. [DOI] [PubMed] [Google Scholar]
- Lynagh T & Lynch JW (2010b). An improved ivermectin‐activated chloride channel receptor for inhibiting electrical activity in defined neuronal populations. J Biol Chem 285, 14890–14897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynagh T & Lynch JW (2012). Ivermectin binding sites in human and invertebrate Cys‐loop receptors. Trends Pharmacol Sci 33, 432–441. [DOI] [PubMed] [Google Scholar]
- McCavera S, Rogers AT, Yates DM, Woods DJ & Wolstenholme AJ (2009). An ivermectin‐sensitive glutamate‐gated chloride channel from the parasitic nematode Haemonchus contortus . Mol Pharmacol 75, 1347–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael B, Meinke PT & Shoop W (2001). Comparison of ivermectin, doramectin, selamectin, and eleven intermediates in a nematode larval development assay. J Parasitol 87, 692–696. [DOI] [PubMed] [Google Scholar]
- Miller PS & Aricescu AR (2014). Crystal structure of a human GABAA receptor. Nature 512, 270–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales‐Perez CL, Noviello CM & Hibbs RE (2016). X‐ray structure of the human α4β2 nicotinic receptor. Nature 538, 411–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mounsey KE, Dent JA, Holt DC, McCarthy J, Currie BJ & Walton SF (2007). Molecular characterisation of a pH‐gated chloride channel from Sarcoptes scabiei . Invert Neurosci 7, 149–156. [DOI] [PubMed] [Google Scholar]
- Nakatani Y, Furutani S, Ihara M & Matsuda K (2016). Ivermectin modulation of pH‐sensitive chloride channels in the silkworm larvae of Bombyx mori . Pestic Biochem Physiol 126, 1–5. [DOI] [PubMed] [Google Scholar]
- Norenberg W, Sobottka H, Hempel C, Plotz T, Fischer W, Schmalzing G & Schaefer M (2012). Positive allosteric modulation by ivermectin of human but not murine P2X7 receptors. Br J Pharmacol 167, 48–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omura S (2016). A splendid gift from the earth: The origins and impact of the avermectins (Nobel Lecture). Angew Chem 55, 10190–10209. [DOI] [PubMed] [Google Scholar]
- Popova M, Trudell J, Li K, Alkana R, Davies D & Asatryan L (2013). Tryptophan 46 is a site for ethanol and ivermectin action in P2X4 receptors. Purinergic Signal 9, 621–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priel A & Silberberg SD (2004). Mechanism of ivermectin facilitation of human P2X4 receptor channels. J Gen Physiol 123, 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuveny E, Slesinger PA, Inglese J, Morales JM, Iniguez‐Lluhi JA, Lefkowitz RJ, Bourne HR, Jan YN & Jan LY (1994). Activation of the cloned muscarinic potassium channel by G protein βγ subunits. Nature 370, 143–146. [DOI] [PubMed] [Google Scholar]
- Rokic MB, Stojilkovic SS, Vavra V, Kuzyk P, Tvrdonova V & Zemkova H (2013). Multiple roles of the extracellular vestibule amino acid residues in the function of the rat P2X4 receptor. PLoS One 8, e59411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samways DS, Khakh BS & Egan TM (2012). Allosteric modulation of Ca2+ flux in ligand‐gated cation channel (P2X4) by actions on lateral portals. J Biol Chem 287, 7594–7602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattelle DB, Buckingham SD, Akamatsu M, Matsuda K, Pienaar IS, Jones AK, Sattelle BM, Almond A & Blundell CD (2009). Comparative pharmacology and computational modelling yield insights into allosteric modulation of human α7 nicotinic acetylcholine receptors. Biochem Pharmacol 78, 836–843. [DOI] [PubMed] [Google Scholar]
- Shan Q, Haddrill JL & Lynch JW (2001). Ivermectin, an unconventional agonist of the glycine receptor chloride channel. J Biol Chem 276, 12556–12564. [DOI] [PubMed] [Google Scholar]
- Sigel E & Steinmann ME (2012). Structure, function, and modulation of GABAA receptors. J Biol Chem 287, 40224–40231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberberg SD, Li M & Swartz KJ (2007). Ivermectin interaction with transmembrane helices reveals widespread rearrangements during opening of P2X receptor channels. Neuron 54, 263–274. [DOI] [PubMed] [Google Scholar]
- Su Z, Brown EC, Wang W & MacKinnon R (2016). Novel cell‐free high‐throughput screening method for pharmacological tools targeting K+ channels. Proc Natl Acad Sci USA 113, 5748–5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unwin N (2005). Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol 346, 967–989. [DOI] [PubMed] [Google Scholar]
- Wang YD, Chen WD, Moore DD & Huang W (2008). FXR: a metabolic regulator and cell protector. Cell Res 18, 1087–1095. [DOI] [PubMed] [Google Scholar]
- Westergard T, Salari R, Martin JV & Brannigan G (2015). Interactions of L‐3,5,3′‐triiodothyronine [corrected], allopregnanolone, and ivermectin with the GABAA receptor: evidence for overlapping intersubunit binding modes. PLoS One 10, e0139072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorton MR & MacKinnon R (2011). Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell 147, 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorton MR & MacKinnon R (2013). X‐ray structure of the mammalian GIRK2–βγ G‐protein complex. Nature 498, 190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemkova H, Tvrdonova V, Bhattacharya A & Jindrichova M (2014). Allosteric modulation of ligand gated ion channels by ivermectin. Physiol Res 63(Suppl 1), S215–S224. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Hirschberg B, Yuan J, Wang AP, Hunt DC, Ludmerer SW, Schmatz DM & Cully DF (2002). Identification of two novel Drosophila melanogaster histamine‐gated chloride channel subunits expressed in the eye. J Biol Chem 277, 2000–2005. [DOI] [PubMed] [Google Scholar]
- Zheng Z, Zhao Z, Li S, Lu X, Jiang M, Lin J, An Y, Xie Y, Xu M, Shen W, Guo G, Huang Y, Li S, Zhang X & Xie W (2017). Altenusin, a non‐steroidal microbial metabolite, attenuates non‐alcoholic fatty liver disease by activating the farnesoid X receptor. Mol Pharmacol 92, 425–436. [DOI] [PMC free article] [PubMed] [Google Scholar]