

Abstract
Key points
Recovery from the potentially devastating consequences of stroke depends largely upon plastic changes occurring in the lesion periphery and its inputs.
In a focal model of stroke in mouse somatosensory cortex, we found that the recovery of sensory responsiveness occurs at the level of synaptic inputs, without gross changes of the intrinsic electrical excitability of neurons, and also that recovered responses had longer than normal latencies.
Under normal conditions, one somatosensory cortex inhibits the responsiveness of the other located in the opposite hemisphere (interhemispheric inhibition) via activation of GABAB receptors.
In stroke‐recovered animals, the powerful interhemispheric inhibition normally present in controls is lost in the lesion periphery.
By contrast, contralateral hemisphere activation selective contributes to the recovery of sensory responsiveness after stroke.
Abstract
Recovery after stroke is mediated by plastic changes largely occurring in the lesion periphery. However, little is known about the microcircuit changes underlying recovery, the extent to which perilesional plasticity occurs at synaptic input vs. spike output level, and the connectivity behind such synaptic plasticity. We combined intrinsic imaging with extracellular and intracellular recordings and pharmacological inactivation in a focal stroke in mouse somatosensory cortex (S1). In vivo whole‐cell recordings in hindlimb S1 (hS1) showed synaptic responses also to forelimb stimulation in controls, and such responses were abolished by stroke in the neighbouring forelimb area (fS1), suggesting that, under normal conditions, they originate via horizontal connections from the neighbouring fS1. Synaptic and spike responses to forelimb stimulation in hS1 recovered to quasi‐normal levels 2 weeks after stroke, without changes in intrinsic excitability and hindlimb‐evoked spike responses. Recovered synaptic responses had longer latencies, suggesting a long‐range origin of the recovery, prompting us to investigate the role of callosal inputs in the recovery process. Contralesional S1 silencing unmasked significantly larger responses to both limbs in controls, a phenomenon that was not observed when GABAB receptors were antagonized in the recorded area. Conversely, such GABAB‐mediated interhemispheric inhibition was not detectable after stroke: callosal input silencing failed to change hindlimb responses, whereas it robustly reduced recovered forelimb responses. Thus, recovery of subthreshold responsiveness in the stroke periphery is accompanied by a loss of interhemispheric inhibition and this is a result of pathway‐specific facilitatory action on the affected sensory response from the contralateral cortex.
Keywords: stroke‐driven plasticity, interhemispheric inhibition, cortical physiology
Key points
Recovery from the potentially devastating consequences of stroke depends largely upon plastic changes occurring in the lesion periphery and its inputs.
In a focal model of stroke in mouse somatosensory cortex, we found that the recovery of sensory responsiveness occurs at the level of synaptic inputs, without gross changes of the intrinsic electrical excitability of neurons, and also that recovered responses had longer than normal latencies.
Under normal conditions, one somatosensory cortex inhibits the responsiveness of the other located in the opposite hemisphere (interhemispheric inhibition) via activation of GABAB receptors.
In stroke‐recovered animals, the powerful interhemispheric inhibition normally present in controls is lost in the lesion periphery.
By contrast, contralateral hemisphere activation selective contributes to the recovery of sensory responsiveness after stroke.
Introduction
Stroke is a leading cause of mortality and disability worldwide not only as a result of the loss of the affected neurons, but also because of functional deficits in their target areas. Neuroprotection strategies such as modulating NDMA receptors, anti‐adhesion antibodies and membrane stabilization, as reviewed for example by Onwuekwe & Ezeala‐Adikaibe (2012), aim to limit the halo of neuronal death around the core of the ischaemic area and are of some help in the (sub)acute time window after stroke. However, a significant degree of the recovery of the lost neurological function occurs over a longer time scale and is based on plastic changes of the brain connectivity that has the potential to take over the function at least partially (Silasi & Murphy, 2014). By contrast, stroke‐associated plasticity can also contribute to additional clinical problems (e.g. hyperexcitability that might cause to post‐stroke epilepsy and spasticity). Because opto‐molecular tools that modify the activity of specific cell types are now available, it is important to fulfill our gap of knowledge about the precise microcircuit changes underlying stroke‐induced plasticity. In that respect, a focal model of stroke is particularly useful because it allows the recovery process to be studied in the stroke vicinity (i.e. in an identified functional area), rendering more controlled and reproducible results. Indeed, the stroke outcome, as well as the vicariant mechanisms, are not only time‐dependent (Biernaskie et al. 2004), but also dependent on lesion size, even within the same species (e.g. rodents).
One example of a reliable mouse focal stroke model uses both a voltage‐sensitive dye (Brown et al. 2009) and calcium imaging (Winship & Murphy, 2008). This previous model shows that lesioning fS1 leads to a receptive field expansion of neurons in the neighbouring hS1 and to a subsequent cortical remapping. The experimental model also gives rises to detectable behavioural deficits (Brown et al. 2009) that go in parallel with the recovery of neuronal responses.
A largely debated issue on the circuit changes behind recovery is the degree to which any recovery is attributable to changes occurring in the lesion periphery vs. those occurring in distant cortical areas. In that respect, the role of the contralateral, healthy hemisphere remains controversial in part because it is both time‐ (Marshall et al. 2000) and size‐dependent (Silasi & Murphy, 2014). Furthermore, direct electrophysiological measurements of the changes of synaptic strength of this connection after lesion are not available in vivo. Indeed, cortical map plasticity can be attributable to changes in either synaptic inputs (subthreshold plasticity) or in the action potential (AP) generating mechanisms (suprathreshold plasticity), as well as to a combination of these two types of plasticity. To address these issues, we used a combination of intracellular recordings from identified cells and extracellular recordings at different times after stoke, combined with acute callosal silencing experiments.
We found that the process of recovery of sensory responses in the stroke periphery occurs largely at the level of synaptic inputs without major changes in intrinsic cell excitability (AP generating mechanisms). Our data indicate that recovery from stroke is a result of the combined effect of the loss of the synaptic GABAB‐dependent inhibition between the two hemispheres observed in controls in the stroke periphery, as well as a pathway‐specific facilitation of the recovered sensory response of callosal origin.
Methods
Ethical approval and animal procedures
Young male adult (postnatal day 30, weighing between 16 and 20 g) C57BL/6J mice (Charles River, Erkrath, Germany) were used. The animals were housed in the Umeå Centre for Comparative Biology's premises under a 12:12 h light/dark cycle and were provided with ad libitum access to food and water. All procedures were conducted in accordance with the Ethical Committee of the Italian Institute of Technology, Genova, Italy, and the Swedish Ethical Committee for Northern Sweden (permit number A 44‐13) and they conformed to the principles and regulations as described by Grundy (2015). All animal procedures and study design were performed in agreement with the ARRIVE guidelines for terminal experiments. Mice were anaesthetized with 20% urethane (0.8–1 g kg−1) (i.p.). Anaesthesia depth was maintained on the surgical level during experiments with additional doses (5–10% of the initial dose). Anaesthesia depth was monitored by pinch and corneal reflexes, electrocardiogram and breathing rate. Before surgery, dexamethasone was injected (0.01 mg kg−1 i.m.) to prevent brain and mucosal oedemas and local anaesthetic (bupivacaine 2.5 mg mL−1) was topically applied in the incision skin area. Humidified oxygen was administrated through a nose cannula, and the body temperature was held at 37°C. A metal recording chamber was placed onto the skull and fixed with acrylic glue and dental cement to allow intrinsic optical imaging (IOI) followed by electrophysiology [in vivo whole‐cell recordings, local field potentials (LFPs)]. The region of the skull of interest was thinned by a dental drill until the vasculature underneath became visible. At the end of electrophysiological recordings, animals were killed with an i.p. bolus of urethane.
When IOI was used to induce stroke, mice were anaesthetized with ketamine 100 mg mL−1 + xylazine 20 mg mL−1; 0.1 mL per 10 g of weight (i.p.). The animal head was secured to a stereotaxic apparatus and the skull was exposed without thinning to minimize invasiveness. Next, the small plastic imaging chamber was placed onto the exposed skull.
After surgery, the antibiotic steroid cream (Fucidin 2%; Leo Laboratories Limited, Maidenhead, UK) was applied along the surgical suture and pain killer was given (ketoprofen 5 mg kg−1, i.m.). Then, the animal was placed on a heated pad and surveyed until it gained back full mobility and then left to recover in individual cages. Food softened with water was placed at the bottom of the cage after surgery. We carefully controlled physical (hydration, weight) and behavioural recovery from anaesthesia and from the procedure (Swedish Ethical Committee for Northern Sweden, permit number A 44‐13).
IOI
Mapping the cortical representation of the forelimb and hindlimb area was performed by IOI as described previously (Iurilli et al. 2013). A vasculature image was acquired under 540 nm illumination before starting imaging. During IOI, the cortex was illuminated with monochromatic light of 630 nm. Images were acquired using a cooled 50 Hz CCD camera connected to a frame grabber (Imager 3001; Optical Imaging Ltd, Rabin Science Park, Israel) and defocused 500–600 μm below the pia. Data frame duration was 200 ms and a spatial binning of 3 × 3 was applied over the images, which were 4 × 4 mm. Stimulations of the limbs were achieved by touching the limbs with a sponge glued to a piezoelectric wafer (Physik Instrumente GmbH, Karlsruhe, Germany). Stimulation frequency was 100 Hz for 10 s, with a linear displacement of 2 mm. All image frames obtained during stimulus presentation were divided by the average image of the first 10 frames acquired just before stimulus presentation (Schuett et al. 2002). The relative decrease of reflectance, averaged over the stimulus presentation period, was then outlined. The spot area was taken as the area where the decrease in reflectance was 50% of the peak. This region was overlaid with the vasculature image.
Phototrombosis
To induce a focal stroke in fS1, we used the photothrombotic model (Brown et al. 2009; Takatsuru et al. 2009). Guided by the IOI functional cortical map, an optic fibre (NA 0.63, diameter 500 μm) was positioned on top of fS1. The area was illuminated with white light (11 mW, 15 min), 5 min after an i.p. injection of 1% Rose Bengal in physiological solution (100 mg kg−1).
Electrophysiology
All the recordings were performed in hS1 as defined by IOI and the responses to both limbs were recorded. In the case of stroked animals, we took care to compare the hindlimb cortical representations as defined by IOI right before the stroke and at the time of recording and we also carefully checked to perform our recordings within the area of overlap between the pre‐stroke and post‐stroke hindlimb representation (Fig. 2 D). This ensured that our electrophysiological recordings were performed in the same anatomo‐functional region (i.e. compared to pre‐stroke condition). We failed to observe major shifts of the hS1, in line with previous report showing that it is the representation of the stroked forelimb area that undergoes a major spatial shift (Brown et al. 2009).
Figure 2. Anatomical and functional controls of the focal stroke model.
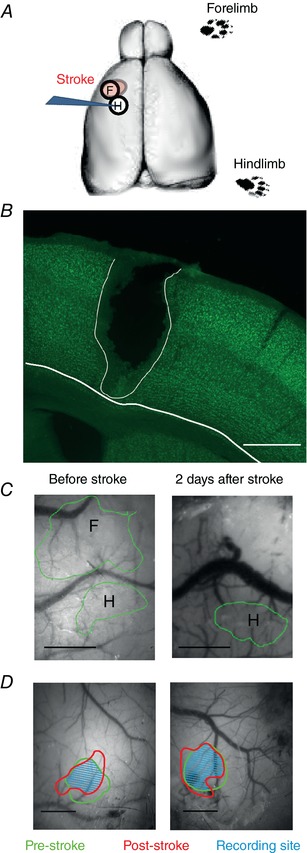
A, sketch of the experimental stroke protocol, showing an IOI‐guided phototrombotic stroke in the forelimb representation (F) in S1. Note that all recordings were performed in the neighboring hindlimb representation (H). The contralateral limbs were stimulated. B, anatomical control of the size of the phototrombotic stroke. The lesioned area matched the size of the forelimb area as defined by IOI (600–700 μm in diameter) and the lesion was reaching the white matter. Section thickness = 60 μm. Scale bar = 500 μm. C, IOI was used for cortical mapping of fS1 and hS1 in controls, as well as to position the fibre to induce focal stroke in fS1 (left image). IOI was also used to confirm the spatial selectivity of the lesion after 2 days (right image). Scale bars = 500 μm. D, comparison of the hindlimb cortical representations as defined by IOI right before the stroke and at the time of recording. All the recordings were perform within the area of overlap between the pre‐stroke and post‐stroke hindlimb representation. The IOI spots are drawn on the pre‐stroked ‘green’ vasculature image. Scale bars = 500 μm. [Color figure can be viewed at http://wileyonlinelibrary.com]
In vivo whole cell recording
Patch pipettes (5–8 MΩ) were filled with intracellular solution (in mm: 135 K‐gluconate, 10 Hepes, 10 phosphocreatine‐Na, 4 KCl, 4 ATP‐Mg salt and 0,3 GTP, pH 7.2, osmolarity 291 mOsm) and lowered until layer 2/3 was reached (Margrie et al. 2002). Positive pressure (300–400 mbar) was applied before and also when lowering the pipette into the brain. Once the layer of interest was reached, the pressure was diminished to 30 mmHg to search for cells. Cells were searched in voltage clamp when advancing in 2 μm steps. When cells were approached, the pressure was relieved, the pipette potential was hyperpolarized and light suction was applied to facilitate gigaseal formation. After compensation for electrode capacitance, usually a ramp of negative pressure led to whole‐cell configuration. Recordings were performed with an EPC10 double plus amplifier (HEKA, Lambrecht, Germany) in voltage follower mode (current clamp) and the signal was digitized (20 kHz) and acquired with Patchmaster software (HEKA, Lambrecht, Germany).
Limbs were stimulated by computer‐controlled movement of a piezoelectric wafer and by a single touch delivered 30 times every 5–7 s (on–off interval: 2000 ms; linear displacement: 2 mm).
Extracellular recordings
Glass pipettes (0.8–1.2 M Ω) filled with saline solution (0.9% NaCl) were inserted in layer 2/3 of mouse hS1. For LFP recordings, the signal was band‐filtered (0.1–100 Hz) and amplified (1000×) with a DAM 50 differential amplifier (World Precision Instruments, Hitchin, UK). After reaching the area of interest, the two limbs were stimulated separately following the same stimulation protocol used for in vivo whole‐cell recordings.
Analysis of cell excitability parameters and of the sensory responses
V m values were calculated as the average membrane potential in the 200 ms time window before stimulus onset. The AP threshold was measured at the peak of the second derivative of the V m trace (Wilent & Contreras, 2005). To analyse subthreshold responses, the sweeps were averaged after removal of the APs by linear interpolation. The time window for peak amplitude and latency calculations was 300 ms after stimulus onset. For analysis of the AP responses, spike counts were computed with 50 ms bins. An AP response was considered significant if, in the peristimulus time histogram, there was at least one bin that exceeded the mean + 2.5 SD spontaneous (prestimulus) firing frequency in a time window of 300 ms after stimulus onset. The peak AP response was taken as the highest firing frequency within that time window. For LFP response analysis, the peak amplitudes were relative to the average value of the first 200 ms pre‐stimulus onset and the time window for peak search was also 0–300 ms poststimulus. For a grand average calculation of LFPs, the single averaged sweeps obtained from different animals were aligned so that the average voltage value of the prestimulus time was the same.
In vivo pharmacology
Callosal silencing
We silenced the contralateral representation of both forelimb and hindlimb representations guided by IOI and by placing a borosilicate pipette (0.7–1.2 MΩ resistance) in layer 2/3 to inject the GABAA agonist muscimol (0.9 μL of 10 mm muscimol solution in saline) in between the IOI representations of the two limbs. The recording pipette inserted in the stroked hemisphere was not moved during and after the muscimol injection.
GABAB blockade
We blocked GABAB receptors by topical application of CGP52432 on recording site (hS1) as in (Palmer et al. 2012), at concentrations known not to cause epileptiform activity (1 μm in physiological solution) (Iurilli et al. 2012). We made sure that the craniotomy was covered by the drug solution for 40 min before starting the recordings.
Anatomy
In all animals tested subacutely (2 days) after stroking fS1, the forelimb‐IOI signal disappeared (Fig. 2 C). In this animal group, we checked that the anatomical lateral extent of the stroke was matching the IOI representation of the forelimb before stroke, as well as that the lesion reached the white matter (Fig. 2 B). Accordingly, mice were deeply anaesthetized by means of i.p. urethane overdose. Next, after confirming the complete absence of any responses to tail/toe pinches and corneal reflexes, animals were transcardially perfused with 50 mL of phosphate buffer 0.1 m, followed by 100 ml of 4% paraformaldehyde (PFA) in 0.1 m phosphate buffer. Brains were post fixed in PFA overnight and afterwards 60 μm thick coronal sections were cut with a sliding microtome (SM2010 R; Leica Biosystems, Nußloch, Germany). Sections were then stained for the neuronal nuclear marker (NeuN). Briefly, sections were incubated in PBS‐based blocking solution containing 0.3% Triton and 5% normal goat serum for 2 h on room temperature. Afterwards, the sections were incubated with mouse anti‐NeuN (cat. no. ab190195; 1:200; Abcam), 0.2% Triton and 5% normal goat serum at 4°C overnight. Slices were then washed with PBS, mounted with Vectashield and analysed under a fluorescence microscope (Zeiss Axio; Carl Zeiss, Carl Zeiss, Oberkochen, Germany) To analyse lesion size and depth, we used a 5× (Zeiss N‐Achroplan, NA 0.15). As shown in Fig. 2 B, in all tested cases (n = 5), both the IOI signal for fS1 and the stroke size covered a cortical surface area having a diameter of ∼600 μm and reached the white matter, in line with the results of a previous study (Brown et al. 2009).
Statistical analysis
Prism, version 6.0. (GraphPad Software Inc., San Diego, CA, USA) was used. Normality was checked with the D'Agostino & Pearson omnibus test. Normally distributed data were compared using one‐way ANOVA. Non‐normally distributed data were compared using the Kruskal–Wallis test and, in the case of no multiple comparison, the Mann–Whitney rank test was used. For comparison of paired data, the Wilcoxon paired test was used. Data are presented as the mean ± SD when data were normally distributed and as medians when the data were not normally distributed. In box plots, the line inside the box is the median, whereas the upper and lower segment of the box represent the 75th and 25th percentile, respectively, and the outer lines represent the whole data range.
Results
Subthreshold‐only forelimb inputs in the hindlimb representation of S1 in control mice
In most sensory cortices, the receptive fields assessed by synaptic inputs (subthreshold receptive field) are larger compared to those obtained with spike output measurements (suprathreshold receptive field) (Brecht et al. 2003; Medini, 2011). This was true also in the limb representations of S1. We performed in vivo whole‐cell recordings from layer 2/3 pyramids in hS1 as identified by IOI (see Methods), in normal mice to begin with. As shown in the example of Fig. 1 A, stimulations of both hind‐ and fore‐limbs gave rise to clearly detectable postsynaptic potentials (PSPs) in hS1 in control mice, although the synaptic responses were significantly smaller upon forelimb stimulation (median: 11.2 vs. 4.9 mV, N = 13 animals, n = 25 cells; Wilcoxon paired test, P < 0.0001) (Fig. 1 B, top). All recorded neurons were receiving synaptic inputs from both the forelimb and the hindlimb, whereas only a minority generated a significant AP response to forelimb stimulation (see example of peristimulus time histogram in Fig. 1 A, bottom; at the population level, 18 out of 25 neurons gave a significant response to hindlimb stimulation, whereas only three responded to the forelimb stimulation). The quantitative analysis of AP responses was restricted to those cells where there was a significant spike response to at least one limb (see Methods). AP responses to hindlimb stimulation in hS1 were significantly larger compared to those obtained after forelimb stimulation (median: 0.21 and 0.10 spikes s−1) (see example in Fig. 1 A, bottom peristimulus time histograms, as well as the population values in Fig. 1 B, bottom). Thus, our data indicate that forelimb‐driven synaptic inputs reach all neurons also in hS1, although this synaptic input was able to drive neurons to fire APs only in a minority of cells. This sharpening of receptive fields at suprathreshold level is consistent with the rectifying, non‐linear action of the AP threshold (Lu et al. 2012).
Figure 1. Hindlimb S1 neurons receive subthreshold‐only forelimb inputs in control mice.
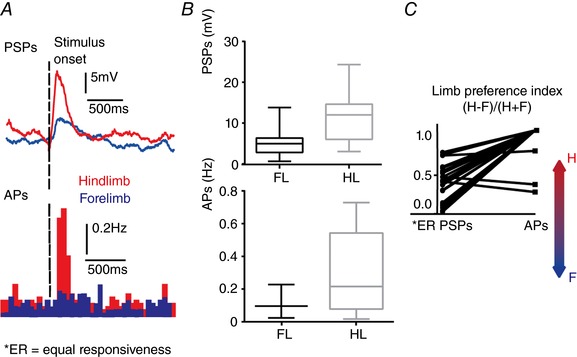
A, top: example of averaged PSPs driven by hindlimb (red) and by forelimb (blue) stimulation, recorded from a layer 2/3 pyramidal neuron in hS1. Sweeps were averaged over 30 presentations. Bottom: peristimulus time histograms for AP counts of the same neuron (bin 50 ms). Note that forelimb stimulation evoked a subthreshold depolarization that doesn't drive the neuron to fire APs. B, top: PSP responses upon forelimb (FL) and hindlimb (HL) stimulation; bottom: significant AP responses upon forelimb (FL) and hindlimb (HL) stimulation. C, plot showing the relative preference index for the hindlimb and forelimb for each hS1 neuron, calculated for PSP and AP responses, separately. The index was calculated as (H – F)/(H + F), where H and F are the peak responses to hindlimb and forelimb stimulation. The limb preference of AP responses was more skewed towards the hindlimb compared to PSPs (Wilcoxon paired test, P < 0.0001). [Color figure can be viewed at http://wileyonlinelibrary.com]
To express this difference quantitatively, we compared a ‘limb preference index’ for PSP and AP responses for each recorded neuron. The index is computed as (H – F)/(H + F), where H and F are the peak responses upon independent stimulation of the hindlimb and forelimb, respectively. Such an index varies from −1 to +1 for neurons responding only to the forelimb or to the hindlimb, respectively, and is 0 for neurons that gave equal responses to stimulation of the two limbs. As expected, in hS1, all neurons had index values higher than 0 or equal to 1 (in the case of AP responses), indicating the dominance of hindlimb responses. A paired statistical analysis showed that responses were more skewed in favour of the hindlimb when APs were compared (Fig. 1 C, medians of limb preference indexes are 0.38 for PSPs vs. 1 for APs, Wilcoxon paired test, P < 0.0001).
Recovery from stroke occurs at subthreshold (synaptic input) level
We used IOI to phototrombotically lesion fS1 (Fig. 2 A) because this method generates reproducible focal lesions (Maxwell & Dyck, 2005). Histological analysis showed that the stroked area size matched the size of the forelimb area as defined by IOI (600–700 μm in diameter) and that lesions were reaching the white matter (Fig. 2 B). To control for the focality of the lesion, we analysed the IOI maps in the subacute phase, 2 days after stroke (Fig. 2 C). The functional mapping showed that hS1 was spared by the stroke, whereas the representation of the forelimb was no longer detectable (see Methods).
Because hS1 and fS1 are neighbouring areas, we took the changes of synaptic responses to forelimb stimulation in hS1 over time as a measure of the plasticity occurring in the stroke periphery. We compared the subacute effects of the stroke (2 days) with those occurring after a longer recovery (2 weeks) with IOI‐targeted in vivo whole‐cell recordings in hS1 after stroke in fS1. The amplitudes of the PSPs induced by hindlimb stimulation in hS1 did not change neither after 2 days, nor by 2 weeks post‐stroke (controls: 11.2 ± 5.8 mV vs. 2 days post‐stroke: 12.4 ± 4.9 mV, N = 9 animals, n = 20 cells vs. 2 weeks post‐stroke recovery group: 10.5 ± 4.8 mV, N = 11 animals, n = 28 cells; F 2,70 = 0.77, P = 0.47, one‐way ANOVA test; r 2 = 0.02) (Fig. 3 A, top), confirming the spatial selectivity of the stroke at functional level. The same was true for hindlimb‐evoked AP responses (controls: 0.21 spikes s–1 vs. 2 days post‐stroke 0.26 spikes s–1, N = 9 animals, n = 9 responsive cells vs. 2 weeks post‐stroke 0.17 spikes s−1, N = 11 animals, n = 16 responsive cells; Kruskal–Wallis test, P = 0.94) (Fig. 3 A, bottom;). On other hand, forelimb‐evoked responses in the same cells decreased dramatically after 2 days from stroke, whereas a significant recovery was observed thereafter (2 weeks after stroke). PSP responses to forelimb in hS1 dropped from 4.9 mV to 2.4 mV after 2 days and recovered to 3.6 mV after 2 weeks (median values are given; Kruskal–Wallis test followed by post hoc tests, P = 0.001) (Fig. 3 B, top). The fact that forelimb responses in hS1 dropped right after stroke of fS1 indicates that they originate from fS1‐to‐hS1 intracortical connections that are destroyed as a consequence of stroke. After 2 weeks from stroke, responses recovered also at suprathreshold level: no neuron was responsive with detectable AP responses 2 days after stroke, whereas we observed that, after 2 weeks, a minority of cells gave rise to detectable AP responses, to a similar extent observed in controls (four out of 28 neurons in 2 weeks recovered animals compared to three out of 25 neurons in controls). Also, the absolute values of these responses attained statistically similar amplitudes, with identical medians (0.1 spikes s–1; Mann–Whitney rank test, P = 0.74).
Figure 3. Recovery of sensory synaptic responses 2 weeks after stroke.
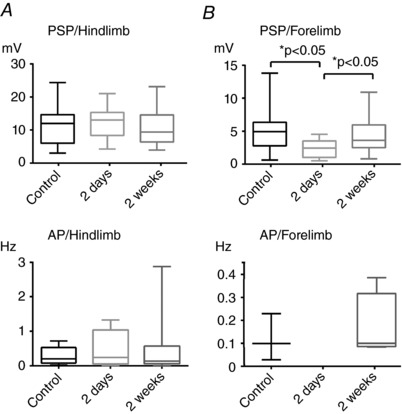
A, no effect of stroke on the hindlimb responses in hS1. Top: PSP responses upon hindlimb stimulation in hS1 in controls, 2 days and 2 weeks after stroke were statistically comparable (F 2,70 = 0.77, P = 0.47, one‐way ANOVA test; r 2 = 0.02), confirming the focal nature of the lesion protocol. Bottom: AP responses upon hindlimb stimulation in hS1 in controls, 2 days and 2 weeks after stroke were also statistically comparable (Kruskal–Wallis test, P = 0.94). B, recovery of sensory responsiveness to forelimb stimulation in hS1. Top: PSP responses upon forelimb stimulation in hS1 in controls, 2 days and 2 weeks after stroke. Note the recovery of spike responses after 2 weeks (Kruskal–Wallis test followed by post hoc tests, P = 0.001). Bottom: APs upon forelimb stimulation in hS1 in controls, 2 days and 2 weeks after stroke (median: 0.1 spikes s−1; Mann–Whitney rank test P = 0.74).
Lack of changes in intrinsic excitability and longer latencies of recovered synaptic responses
Excitability of neurons in the periinfarct area has been reported to change after stroke. For example, downregulation of phasic inhibition (Schiene et al. 1996; Imbrosci et al. 2013) accompanied by upregulation of NMDA receptors (Lee et al. 1999) has been reported and, simultaneously, an increase in tonic inhibition with behavioural relevance for recovery has been documented (Clarkson et al. 2010). However, the net effect of such changes in the intact network in vivo remains elusive. Surprisingly, we failed to observe significant changes in the main parameters that measure intrinsic excitability of layer 2/3 pyramidal neurons, such as V m values (mean: −67.1 ± 12.8 mV in controls, −68.0 ± 13.5 mV 2 days after stroke, −66.0 ± 15.7 mV 2 weeks after stroke; F 2,70 = 0.1206, P = 0.89, one‐way ANOVA test; r 2 = 0.003) (Fig. 4 A) and spontaneous AP rates (median: 0.003 Hz in controls, 0.005 Hz 2 days after stroke, 0.020 Hz 2 weeks after stroke; Kruskal–Wallis test, P = 0.76) (Fig. 4 B). Another important parameter describing the intrinsic AP generating mechanism is the AP threshold measured as the V m value at the time point of V m maximal acceleration right before AP peak (Wilent & Contreras, 2005). Also in this case, we did not find significant differences among experimental groups, albeit a non‐significant trend for increased AP threshold values (suggesting hypoexcitability) was observed (mean: −41.3 ± 4.1 mV in controls, −37.0 ± 6.1 mV in 2 days after stroke, −37.5 ± 5.8 mV in 2 weeks after stroke; F 2,34 = 2.49, P = 0.1, one‐way ANOVA test; r 2 = 0.13) (Fig. 4 C).
Figure 4. Lack of changes in intrinsic neuronal excitability in stroke periphery and longer latencies of recovered synaptic responses.
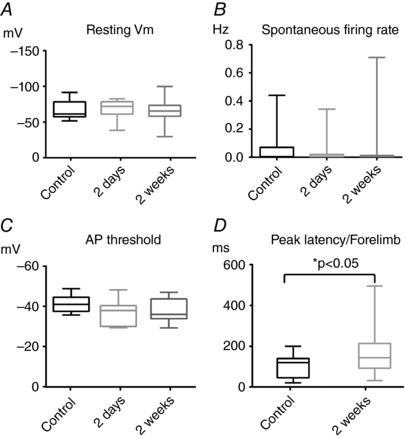
A, resting V m values showed no significant difference among groups (controls, 2 days and 2 weeks after stroke; F 2,70 = 0.1206, P = 0.89, one‐way ANOVA test; r 2 = 0.003). B, median values of spontaneous firing rates were statistically comparable among groups (Kruskal–Wallis test, P = 0.76). C, AP thresholds values did not show statistically difference among groups (F 2,34 = 2.49, P = 0.1, one‐way ANOVA test; r 2 = 0.13). D, comparison of the peak latencies of the synaptic responses 2 weeks after stroke upon forelimb stimulation with those of control animals. Note the longer latencies of the recovered synaptic responses (Mann–Whitney test, P = 0.02).
Comparing the temporal kinetics of the recovered synaptic responses with those obtained from controls can give hints on their possible synaptic origin. Accordingly, we compared peak latencies of the synaptic responses upon forelimb stimulation in the controls and 2 weeks stroked mice. Recovered synaptic responses had longer peak latencies compared to controls (median: 143 vs. 120 ms, Mann–Whitney rank test, P = 0.02) (Fig. 4 D), suggesting the possibility that recovery might be due to plasticity of long‐range connections projecting to hS1.
Similar assessment of stroke‐induced plasticity with whole‐cell and local field potential recordings
Extracellular LFPs integrate synaptic responses within a radius of ∼250 μm (Katzner et al. 2009). To confirm whether LFPs can be used to assess stroke‐induced synaptic plasticity in the stroke periphery, we analysed the changes in LFPs recorded from layer 2/3 in hS1 in response to different limb stimulation before and after stroke (2 days and 2 weeks, as we did for our in vivo whole‐cell recording series). Stroke‐induced plasticity can be quantified by the change in the relative strength of hindlimb‐ and forelimb‐responses, as quantified by the H‐F index. Thus, we compared the distributions of the H‐F indexes for in vivo whole‐cell recordings and for LFP recordings within each experimental groups (N = 7 for controls, N = 6 for 2 days after stroke, N = 9 for 2 weeks after stroke). The distributions of H‐F indexes obtained with LFPs and in vivo whole‐cell recordings, respectively, were statistically comparable within each experimental group (median: for PSPs and LFPs are reported; left: 0.38 vs. 0.29 for controls; middle: 0.64 vs. 0.78 for 2 days after stroke; right: 0.44 vs. 0.29 for 2 weeks after stroke; Mann–Whitney rank tests, P = 0.73, 0.39 and 0.10, respectively) (Fig. 5). Thus, in vivo whole‐cell recordings and LFP recordings provide a comparable assessment of stroke‐induced plasticity in layer 2/3 in our experimental model.
Figure 5. Similar assessment of stroke‐induced synaptic plasticity with whole‐cell recordings and local field potentials.
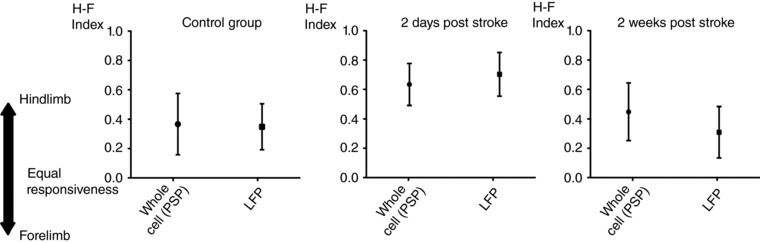
The distributions of the H‐F indexes, which express the relative strength of the synaptic inputs driven stimulation of the two limbs, were comparable within each experimental group, when calculated after in vivo whole‐cell recordings and LFPs (controls: left; 2 days after stroke: middle; 2 weeks after stroke: right; Mann–Whitney rank tests, P = 0.73, 0.39 and 0.10, respectively). Data are the mean ± SD.
GABAB‐dependent interhemispheric inhibition (IHI) in controls
Increased activation of the contralesional hemisphere as a consequence of stroke has been observed previously, although its extent depends on many factors, such as timing, size of the lesion and the technical approach employed (Caleo, 2015). Most importantly, the functional role of callosal connectivity in the process of recovery of function remains uncertain because controversial results have been obtained by different groups (Di Pino et al. 2014). Having shown that the recovered synaptic responses had longer than normal latencies, we wanted to examine how silencing of contralateral S1 influenced the recovered sensory responses in the stroke periphery in our lesion model.
We started with control mice by recording LFP responses of layer 2/3 hS1 to independent stimulation forelimb and hindlimb, before and after acute pharmacological silencing of both limb representation in contralateral S1 (Fig. 6 A). In the control group, silencing contralateral S1 caused a dramatic increase in sensory responses to separate stimulation of both limbs (median LFP peak responses for hindlimb: 452 μV vs. 2918 μV before and after contralateral S1 silencing, respectively; for forelimb: 287 μV before vs. 1156 μV after silencing; N = 7 animals; Wilcoxon paired tests, P = 0.01) (Fig. 6 B, representative examples on the left, grand averages in the middle, and a plot showing the paired statistics on the right; hindlimb and forelimb responses are shown on the top and bottom rows, respectively). These data indicate that, in controls, one hemisphere powerfully inhibits contralateral S1, in line with previous studies (Ferbert et al. 1992; Gerloff et al. 1998; Di Lazzaro et al. 1999; Grefkes et al. 2008).
Figure 6. GABAB‐dependent interhemispheric inhibition in controls.
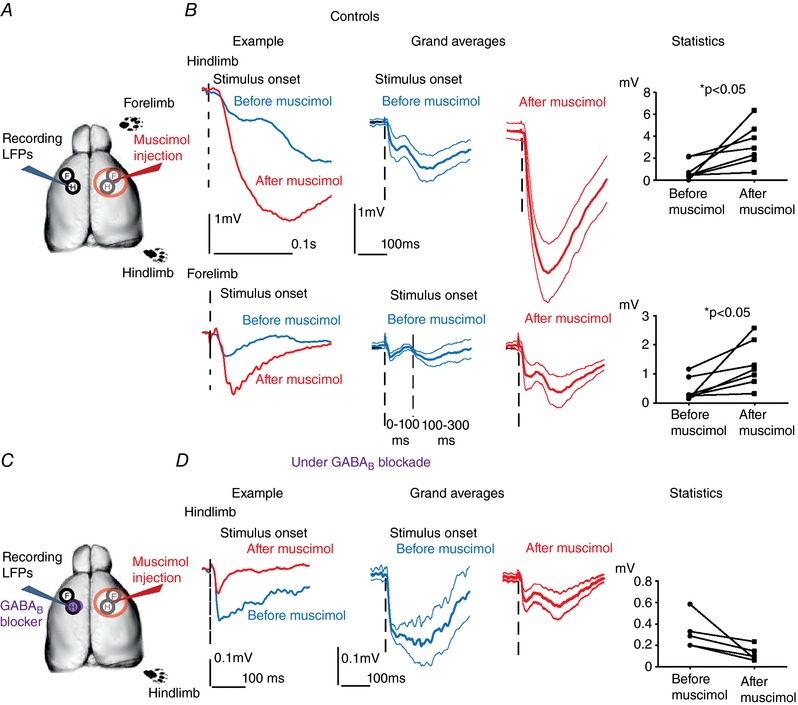
A, experimental sketch for LFPs from hS1 before and after acute silencing of contralateral S1 with muscimol in controls. B, interhemispheric inhibition in controls. Left: examples of LFP responses to separate stimulation of the hindlimb (top) and of the forelimb (bottom) before (in blue) and after (in red) silencing contralateral S1. Note the dramatic increase of sensory responses. Middle: comparison of the grand averages of the LFP responses to hindlimb (top) and forelimb (bottom) before and after contralater S1 silencing (blue and red, respectively). Mean ± SEM values are shown by the thick and thinner lines, respectively. Sensory responses significantly increase after inactivation of contralateral hemisphere. Right: paired statistical comparisons of the amplitudes of the sensory responses to separate hindlimb (top) and forelimb (bottom) stimulation before and after contralateral S1 silencing show a significant increase (Wilcoxon paired tests, P = 0.01). C, experimental sketch for LFPs from hS1 before and after acute silencing of contralateral S1 with muscimol under GABAB blockade in the recording site, done in controls. D, interhemispheric inhibition was not observed under GABAB blockade in controls. Left: example of LFP responses to hindlimb stimulation in hS1 before (in blue) and after (in red) silencing contralateral S1. Note that there is non‐significant trend for response reduction upon contralateral S1 silencing under GABAB blockade. Middle: comparison of the grand averages of the LFP responses to hindlimb before and after contralater S1 silencing (blue and red, respectively). Mean ± SEM values are shown by the thick and thinner lines, respectively. Sensory responses non‐significantly decrease after inactivation of contralateral hemisphere. Right: paired statistical comparisons of the amplitudes of the sensory responses to hindlimb stimulation before and after contralateral S1 silencing show non‐significant decrease under GABAB blockade (Wilcoxon paired test, P = 0.06). [Color figure can be viewed at http://wileyonlinelibrary.com]
The time windows for our analysis was in the interval 0–300 ms after stimulus onset (see Methods). Because the FP responses were often biphasic, with a first peak occurring before 100 ms and a second one occurring between 100 and 300 ms (see example with dashed line in the grand average shown in Fig. 6 B), we repeated the analysis to control whether the IHI was differentially occurring in these two time windows. Our results showed that this was not the case. Indeed, forelimb responses increased upon muscimol blockade from a median value of 255 μV to 1156 μV (Wilcoxon paired test, P = 0.01) when the peak was searched in the 0–100 ms time window and from 286 μV to 967 μV (Wilcoxon paired test, P = 0.01 time window 100–300 ms). The same was true for hindlimb responses (0–100 ms; median: 452 μV vs. 2918 μV, Wilcoxon paired test P = 0.01, 100–300 ms: 660 μV vs. 2498 μV, Wilcoxon paired test, P = 0.03).
IHI has been proven to be mediated by GABAB receptors in rat hS1 (Palmer et al. 2012). We aimed to control whether the IHI that we documented in mouse S1 is also GABAB‐dependent. Accordingly, we topically applied the GABAB blocker CGP52432 (at a concentration of 1 μm; Iurilli et al. 2012; Palmer et al. 2012) before starting LFP recordings (Fig. 6 C). Under GABAB blockade, responses did not increase after contralateral silencing. In other words, we failed to observe any sign of IHI (as an example, see the grand averages and paired data plot in Fig. 6 D). By contrast, we observed a non‐significant trend for response reduction upon contralateral S1 silencing under GABAB blockade (median: 288 μV vs. 99 μV before and after contralateral S1 silencing, respectively; N = 5 animals; Wilcoxon paired test, P = 0.06). The observed trend for a decreased response amplitude after callosal silencing under GABAB blockade can be explained by the fact that, under GABAB blockade, the callosal pathway has a ‘pure’ excitatory influence on the recording site that is not masked by IHI (see callosal pathway depicted in orange in Fig. 8 A). As a result, callosal silencing in these conditions reduces the sensory response measured before silencing because the latter is the summed response of the callosal and of the direct activation by the contralateral limb (depicted in orange and green, respectively in Fig. 8 A). Overall, these results indicate that the IHI we documented is GABAB‐mediated, in line with the previous report in rat hS1 (Palmer et al. 2012).
Figure 8. Proposed model of the synaptic changes occurring in callosal connectivity in the periphery of a focal stroke.
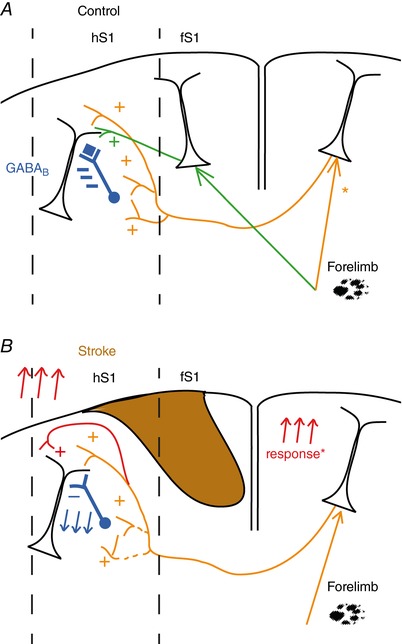
A, normal condition: callosal inputs (orange pathway) inhibits sensory responses mediated by the direct activation of the contralateral limb (green pathway) via activation of locally residing interneurons that act via GABAB (blue). *Under normal conditions limb tactile stimulation activates the ipsilateral cortex (e.g. (Takatsuru et al. 2009); Kokinovic and Medini, unpublished observation). B, after a focal stroke, recovery of forelimb responses cannot occur via horizontal connections from the stroked area. Instead, recovery happens via the combined effect of reduced strength of IHI (blue) and by potentiation of the direct callosal excitatory input (red). The latter can be accounted for increased strength of the local excitatory synapses present in the stroke periphery and/or by an increased activation of the contralesional cortex (Reinecke et al. 2003; Takatsuru et al. 2009). [Color figure can be viewed at http://wileyonlinelibrary.com]
Loss of IHI and facilitation of recovered responses of callosal origin in stroke periphery
The same approach was then used in animals after 2 weeks of recovery from the phototrombotic stroke (Fig. 7 A). Unexpectedly, we did not find any evidence of the powerful IHI normally observed in controls in this case. Responses to stimulation of hindlimb were statistically comparable before and after muscimol silencing of contralateral hemisphere, showing no sign of IHI. Not only we did not observe any sign of response increase but a trend to a decrease actually was observed for hindlimb responses in hS1 that was far from being significant (median value of LFP before 631 μV vs. 246 μV after contralateral silencing; N = 9 animals; Wilcoxon paired test, P = 0.3) (Fig. 7 B, top row). Of relevance, the recovered responses to stimulation of forelimb were instead significantly (two‐fold) reduced after inactivation of contralateral S1 (median value of LFP before and after contralateral silencing 471.8 μV vs. 70 μV, respectively; N = 9 animals, Wilcoxon paired test, P = 0.008) (Fig. 7 B, bottom row).
Figure 7. Loss of interhemispheric inhibition and selective facilitation of recovered responses of callosal origin in the stroke periphery.
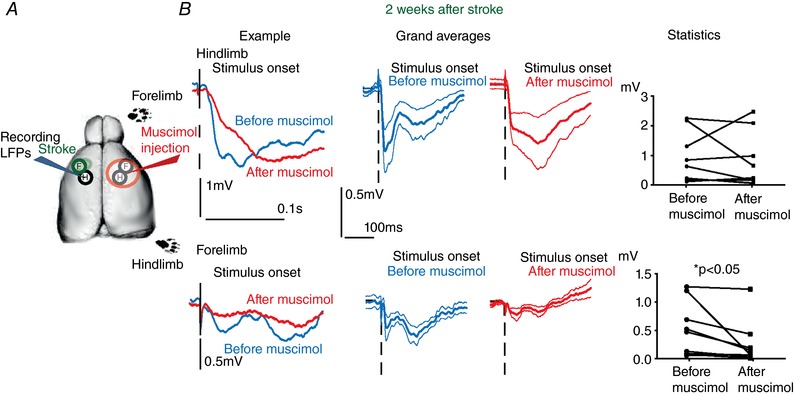
A, experimental sketch for LFPs from hS1 before and after acute silencing of contralateral (contralesional) S1 with muscimol in the animals that displayed full recovery (2 weeks after stroke). B, loss of interhemispheric inhibition after recovery from stroke. Left: example of LFP responses to separate stimulation of the hindlimb (top) and of the forelimb (bottom) before (in blue) and after (in red) silencing contralateral S1. Note that the response to hindlimb did not show the dramatic increase of sensory responses observed in controls, whereas a reduction of the response to forelimb stimulation was observed. Middle: comparison of the grand averages of the LFP responses to hindlimb (top) and forelimb (bottom) before and after contralateral S1 silencing (blue and red, respectively). Mean ± SEM values are given. In this case, sensory responses to hindlimb stimulation were not significantly affected by contralateral S1 silencing (top), whereas the recovered forelimb responses were almost suppressed by the same manipulation. Right: paired statistical comparison of the amplitudes of the sensory responses to separate hindlimb (top) and forelimb (bottom) before and after contralateral S1 silencing. Note the lack of effect of contralateral S1 silencing for hindlimb responses after callosal silencing (top: Wilcoxon paired test, P = 0.3), whereas forelimb responses showed a significant decrease after the very same manipulation (Wilcoxon paired test, P = 0.01). [Color figure can be viewed at http://wileyonlinelibrary.com]
In summary, our data indicate a dramatic loss of IHI of synaptic sensory responses on the stroke periphery compared to controls, accompanied by a pathway‐specific facilitation of recovered responses of callosal origin.
Discussion
We investigated the sub‐ and suprathreshold effects of stroke‐driven plasticity in the excitatory network of a functionally identified region in mouse S1 using a focal stroke model. Recovery from stroke in the lesion periphery occurred primarily at subthreshold (synaptic input) level, without gross changes in neuronal intrinsic excitability. We found that the source of synaptic recovery was the homotopic region of the contralateral hemisphere. In particular, the usually strong GABAB‐mediated IHI present in controls was absent in the stroke periphery and was replaced instead by a pathway‐specific facilitation of the recovered sensory response of callosal origin.
Plasticity of local microcircuits after stroke: sub‐ vs. suprathreshold effects
Around brain lesions, hypoexcitability is followed by hyperexcitability and then by sensory input remapping (Carmichael, 2012). Changes in both excitatory and inhibitory transmissions occur. Schiene et al. (1996) and Imbrosci et al. (2013) reported decreased phasic inhibition coupled with hyperexcitability, whereas (Clarkson et al. 2010) showed an increase of tonic inhibition that was behaviourally relevant. Furthermore, excitatory NMDA currents increase in stroke periphery (Huemmeke et al. 2004) and this is accompanied by a time‐dependent AMPA current modulation (Clarkson et al. 2011). However, the net effect on local microcircuits in vivo remains unclear. Our data suggest that such changes might balance each other to some extent. Indeed, V m, spontaneous AP rates and AP threshold were normal in lesion periphery. Because in vivo whole‐cell recordings are taken mostly from somatas (Margrie et al. 2002), we cannot exclude changes in dendritic excitability (e.g. as a result of modifications of ion channel composition). Moreover, because we intracellularly recorded layer 2/3 pyramids, we cannot exclude changes in the intrinsic excitability of deeper (e.g layer 5) neurons in that callosal influences are layer‐specific (Palmer et al. 2013). A major change of the excitability of GABAergic cells appears improbable because V m, spontaneous AP rates and AP threshold are sensitive to modifications of GABAergic transmission (Caillard, 2011). Taken together, our data indicate that post‐stroke recovery of sensory responsiveness is largely a result of changes in the synaptic inputs received by neurons in the stroke periphery rather than changes in their AP generating mechanism.
Plasticity of interarea connections after stroke: effects of callosal inputs on stroke periphery
hS1 neurons display subthreshold‐only responses also to forelimb stimulation in controls. Such responses presumably originate from excitatory fS1‐to‐hS1 connections because they disappear subacutely after fS1 lesioning. Moreover, we show that contralateral S1 inhibits sensory responses in controls. Thus, our data suggest that in hS1 interhemispheric, inhibitory connections, antagonize horizontal, excitatory connections from fS1 (Fig. 8 A). This antagonism is in line with callosal fibres sharpening tactile fields in rat S1 (Palmer et al. 2012).
In the complex and still controversial literature on the role of callosum on stroke recovery there is, at least to some degree, consensus that: (i) contralesional activation is size‐dependent, being larger for larger lesions (Grefkes et al. 2008; Di Pino et al. 2014); (ii) this activation appears to be beneficial for larger lesions and detrimental for smaller ones (Biernaskie et al. 2005); and (iii) in case of small lesions, an initial contralesional activation is followed by re‐activation of perilesional areas, with this re‐activation correlating with functional recovery (Dijkhuizen et al. 2001).
However, clinical studies show that the role of the contralateral activation in stroke recovery remains controversial. Indeed, transcranial magnetic stimulation (TMS) inhibiting the contralesional hemisphere interfered with neurological recovery (Dancause et al. 2015), whereas other studies showed no benefit (Talelli et al. 2007, 2012) or even a facilitatory action (Butefisch et al. 2003; Song et al. 2005; Bradnam et al. 2012).
Importantly, attempts to promote stroke recovery by inhibiting the contralesional hemisphere rest on the so called ‘interhemispheric competition model’, which is based on the idea that inhibiting the healthy hemisphere will facilitate recovery by reducing IHI on the stroked hemisphere (Silasi & Murphy, 2014; Caleo, 2015). Clinical data already showed the necessity of revising such a model (Di Pino et al. 2014).
In that respect, our data show that the assumption that IHI persists in the stroke periphery needs to be revisited. Indeed, we show that, in a reproducible model of focal stroke, the normal interhemispheric suppression of sensory responsiveness observed in controls is simply not detectable in the stroke periphery. Instead, we found a switch of the action of callosal input from inhibitory to facilitatory because contralateral S1 silencing selectively suppresses the recovered (forelimb) responses, without affecting the hindlimb responses, whereas, in controls, the same manipulation caused dramatic increases of limb responses.
Second, our data, taken together with the existing literature, indicate a postsynaptic origin of this switch within the network of the stroke periphery. Indeed, forelimb responses increase in the contralesional hemisphere in a similar mouse model of S1 stroke (Takatsuru et al. 2009) and increased contralesional activation and synaptic remodelling after stroke in S1 was also shown by (Biernaskie & Corbett, 2001; Reinecke et al. 2003). Thus, if IHI from the healthy side would have still been present, we should have observed an even larger percentage increase of responsiveness in stroke periphery after contralateral silencing as a result of the hyperactivation of the contralesional hemisphere. The fact that, instead, we failed to see any increase of responsiveness after contralateral silencing in stroked animals argues in favour of the idea that the synaptic change(s) responsible for lack of IHI must have occurred locally in the microcircuits of the stroke periphery. Furthermore, the fact that contralateral silencing caused a response reduction both in controls under GABAB blockade and on stroke‐recovered responses is in line with the view that GABAB‐dependent IHI is lost in the stroke periphery (Fig. 8).
In our stroke model, contralateral S1 silencing had no effect on hindlimb but suppressed recovered forelimb‐responses in stroke periphery, suggesting the co‐existence of: (i) a generalized reduction of IHI and (ii) a selective facilitation of recovered responses of callosal origin. A plausible mechanism underlying the latter observation could be a higher activation of the contralesional fS1 (compared to the contralesional hS1), as a result of interhemispheric innervation and the accompanying inhibition presumably being denser for somatotopically corresponding regions in mouse S1 (Wise & Jones, 1976; Zhou et al. 2013).
Possible synaptic and molecular mechanisms of interhemispheric plasticity
IHI is present both in human and rodents S1. Callosal connections activate layer 1 interneurons that in turn inhibit layer 5 neurons via GABAB receptors (Palmer et al. 2012). Contralateral silencing did not increase sensory responses in controls under GABAB blockade, indicating that the IHI we have studied in the present study is GAABAB‐mediated, presumably relying on the same cellular mechanism described by Palmer et al. (2012). Interestingly, GABAB receptors play also a role in IHI in humans (Irlbacher et al. 2007). What could be the cellular mediator of IHI and how could this subcircuit be affected by stroke? One candidate are neurogliaform cells, which can generate long‐lasting inhibition in many neighbouring pyramids (Rudy et al. 2011).
One economical explanation of our results could be that layer 1 interneurons in hS1 mediating IHI are more sensitive to ischaemia compared to pyramids. However, this does not appear to plausible because global ischaemia not only does not cause such a loss, but instead triggers neurogenesis of GABAergic cells selectively in layer 1 (Ohira et al. 2010).
An alternative explanation could instead be pruning or retraction of callosal synapses from the layer 1 interneurons responsible for IHI, possibly accompanied by a potentiation of the direct effect of glutamateric callosal fibres onto local pyramids (see the model proposed in Fig. 8 B).
We observe callosal‐dependent recovery of responses 2 weeks but not 2 days after stroke, suggesting that slow, structural rearrangements might have occurred. This is plausible as two‐photon imaging showed dendritic remodelling after stroke, with increased spine formation in stroke periphery peaking when functional plasticity is also maximal (Brown et al. 2007, 2010). Such processes are accompanied by upregulation of axonal growth markers such as GAP‐43 (Caleo, 2015) and CAP 23 (Caleo, 2015), as well as growth inhibiting factors such as ephrin‐A5 (Overman et al. 2012) and chondroitin sulphate proteoglycans (Soleman et al. 2012; Quattromani et al. 2018), which are known to play a role in functional recovery.
Behavioural relevance
The role of callosal input in recovery after human stroke remains controversial and a matter of debate, as effectively summarized by (Buetefisch, 2015): ‘Meta‐analyses on the effectiveness of repetitive transcranial magnetic stimulation or transcranial direct current stimulation in stroke rehabilitation therapy do not agree on the available evidence to either support or reject it’.
Our results indicate that IHI is not detectable in the stroke periphery of mouse S1 at a time point where recovery of responsiveness was complete. It is not straightforward to compare the result of our direct pharmacological inactivation with the clinical results using TMS because the cellular mechanisms and the net effects of TMS protocols on microcircuits remain largely unknown. Even a simple TMS pulse recruits the very same dendritic inhibitory mechanisms involved in IHI (Murphy et al. 2016), implying that even a single TMS pulse is certainly not purely excitatory. Species‐specific differences, as well as the possible effect of lesion size on interhemispheric interactions after stroke, could also explain some discrepancies with the clinical literature.
Our view on the relevance of our results for functional recovery after stroke is that the loss of IHI in the stroke periphery could play a permissive role for the plasticity of those excitatory connections (that we have shown to be also of callosal origin) underlying recovery of neuronal responsiveness. Indeed, a reduction of inhibitory neurotransmission facilitates experience‐dependent plasticity in vivo because the strength of inhibitory connections is one of the major determinants for the opening and closure of the critical period for ocular dominance plasticity in the primary visual cortex, a classical paradigm of experience‐dependent plasticity (Hensch et al. 1998; Huang et al. 1999; Harauzov et al. 2010). Inhibition is indeed a determinant of the temporal integration properties of synapses (Pouille & Scanziani, 2001), which is the core mechanism underlying input‐specific plasticity. More strictly related to our work, diminution of inhibitory neurotransmission in the stroke periphery (Mittmann et al. 1994) is indeed accompanied by a facilitation of classical forms of synaptic plasticity, such as long‐term potentiation (Mittmann & Eysel, 2001).
There are behavioural observations conducted out in preclinical models very close to ours (focal strokes in rodent S1) that are in line with our observations. It was reported previously that lidocaine contralesional silencing indeed counteracted the behavioural recovery of the affected limb in a way that was dependent on the lesion size (Biernaskie et al. 2005). In addition, a significantly decreased function of the recovered forelimb was found after acute lesioning of the previously healthy hemisphere (Shanina et al. 2006), after one full week from the initial stroke, and a trend for a decreased function was observable but not significant at later times.
For the sake of completeness, the results of studies where chronic silencing (Mansoori et al. 2014) or callosotomy (Jin et al. 2011) were performed simultaneously with the stroke induction could also be discussed. However, it is difficult to compare possible behavioural extrapolations of our results with these studies because the effects of acute silencing experiments are testing more directly the actual role of the corpus callosum on the already recovered functional outcome. In addition, such studies also report conclusions that, to a significant extent, are not consistent with each other because chronic silencing is worsening the behavioural outcome, even though callosotomy did not affect the behavioural recovery.
Accordingly, any interpretation of the results of contralesional silencing experiments should take into account possible deficits caused by the acute silencing on the function on the initially unaffected limb. Indeed, the interpretation of the data of tests that measure the relative use of the two forelimbs (such as the glass cylinder test and the foot‐fault test) could be affected by this type of ipsilesional deficit. Even when the performance of a motor act performed by a single limb is measured (such as in reaching and grasping tests), the performance of a certain limb during ipsilateral silencing can be affected by postural deficits imposed to the limb contralateral to the acutely silenced hemisphere (a phenomenon clearly present also in controls) (Biernaskie et al. 2005). Thus, our results, taken together with other studies that are beginning to clarify the circuits mediating IHI in vivo, prompt the need to perform such experiments during selective (e.g. optogenetic) silencing of callosally projecting neurons and circuits (that are now at least partially known) (Palmer et al. 2012). The latter is indeed a manipulation that should minimize ipsilesional forelimb deficits on one side as a result of the lower number of neurons affected and also because callosally projecting cells usually do not project subcortically(Molnar & Cheung, 2006). By contrast, if such deficits are anyhow measurable, they should be taken into account in the data interpretation.
Generalized relevance for other cortical areas
Could these findings in S1 be extended to other cortical areas, where the synaptic circuits mediating interhemispheric communication could differ? Indeed, interhemispheric inputs are primarily excitatory in visual areas, as demonstrated by the reduced binocularity of visual cortical neurons after callosal inactivation in both cats (Yinon et al. 1992) and rodents (Pietrasanta et al. 2012). These data raise the question of whether IHI is area‐specific, and additionally, how interhemispheric communication is affected by stroke in non‐sensorimotor cortices, such as in visual areas.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
BK contributed to acquisition, analysis and interpretation of data for the work, as well as drafting the work and revising it critically for important intellectual content. PM contributed to conception and design of the work and drafting the work and revising it critically for important intellectual content. We confirm that all authors approved the final version of the manuscript submitted for publication, and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship all listed.
Funding
This work was supported by the Swedish Brain Foundation (Hjärnfonden) – (grant number FO2014‐0103); Åke‐Wiber Foundation (grant number 218577683); Kempe Foundation (grant number: SMK‐1245); and Umeå University.
Acknowledgements
We thank Dr Umberto Olcese for initial help with the experiments, Per Utsi for mechanical assistance, Roger Widmark for technical help, and Dr Umberto Olcese and Kamil Antos for programming the analysis routines.
Biography
Bojana Kokinovic finished her PhD studies in Neuroscience and Brain Technology at Italian Institute of Technology in Genova, Italy, studying lesion‐driven plasticity and cellular and synaptic mechanisms of it in cortical circuits in vivo. She is currently working as postdoctoral fellow in Neurophysiology at the Department of Integrative Medical Biology at Umeå University, Sweden in the laboratory of Professor Paolo Medini, applying her knowledge in different advanced electrophysiological and imaging techniques to further investigate plasticity of the brain after stroke and explore interhemispheric communication and its role in post‐stroke recovery.

Linked articles This article is highlighted by a Perspective by Caleo. To read this Perspective, visit https://doi.org/10.1113/JP276081.
Edited by: Ole Paulsen & Diego Contreras
Contributor Information
Bojana Kokinovic, Email: bojana.kokinovic@umu.se.
Paolo Medini, Email: paolo.medini@umu.se.
References
- Biernaskie J, Chernenko G & Corbett D (2004). Efficacy of rehabilitative experience declines with time after focal ischemic brain injury. J Neurosci 24, 1245–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biernaskie J & Corbett D (2001). Enriched rehabilitative training promotes improved forelimb motor function and enhanced dendritic growth after focal ischemic injury. J Neurosci 21, 5272–5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biernaskie J, Szymanska A, Windle V & Corbett D (2005). Bi‐hemispheric contribution to functional motor recovery of the affected forelimb following focal ischemic brain injury in rats. Eur J Neurosci 21, 989–999. [DOI] [PubMed] [Google Scholar]
- Bradnam LV, Stinear CM, Barber PA & Byblow WD (2012). Contralesional hemisphere control of the proximal paretic upper limb following stroke. Cereb Cortex 22, 2662–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brecht M, Roth A & Sakmann B (2003). Dynamic receptive fields of reconstructed pyramidal cells in layers 3 and 2 of rat somatosensory barrel cortex. J Physiol 553, 243–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CE, Aminoltejari K, Erb H, Winship IR & Murphy TH (2009). In vivo voltage‐sensitive dye imaging in adult mice reveals that somatosensory maps lost to stroke are replaced over weeks by new structural and functional circuits with prolonged modes of activation within both the peri‐infarct zone and distant sites. J Neurosci 29, 1719–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CE, Boyd JD & Murphy TH (2010). Longitudinal in vivo imaging reveals balanced and branch‐specific remodeling of mature cortical pyramidal dendritic arbors after stroke. J Cereb Blood Flow Metab 30, 783–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CE, Li P, Boyd JD, Delaney KR & Murphy TH (2007). Extensive turnover of dendritic spines and vascular remodeling in cortical tissues recovering from stroke. J Neurosci 27, 4101–4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butefisch CM, Netz J, Wessling M, Seitz RJ & Homberg V (2003). Remote changes in cortical excitability after stroke. Brain 126, 470–481. [DOI] [PubMed] [Google Scholar]
- Buetefisch CM (2015). Role of the Contralesional Hemisphere in Post-Stroke Recovery of Upper Extremity Motor Function. Front Neurol 6, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caillard O (2011). Pre & postsynaptic tuning of action potential timing by spontaneous GABAergic activity. PLoS ONE 6, e22322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caleo M (2015). Rehabilitation and plasticity following stroke: insights from rodent models. Neuroscience 311, 180–194. [DOI] [PubMed] [Google Scholar]
- Carmichael ST (2012). Brain excitability in stroke: the yin and yang of stroke progression. Arch Neurol 69, 161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson AN, Huang BS, Macisaac SE, Mody I & Carmichael ST (2010). Reducing excessive GABA‐mediated tonic inhibition promotes functional recovery after stroke. Nature 468, 305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson AN, Overman JJ, Zhong S, Mueller R, Lynch G & Carmichael ST (2011). AMPA receptor‐induced local brain‐derived neurotrophic factor signaling mediates motor recovery after stroke. J Neurosci 31, 3766–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dancause N, Touvykine B & Mansoori BK (2015). Inhibition of the contralesional hemisphere after stroke: reviewing a few of the building blocks with a focus on animal models. Prog Brain Res 218, 361–387. [DOI] [PubMed] [Google Scholar]
- Di Lazzaro V, Oliviero A, Profice P, Insola A, Mazzone P, Tonali P & Rothwell JC (1999). Direct demonstration of interhemispheric inhibition of the human motor cortex produced by transcranial magnetic stimulation. Exp Brain Res 124, 520–524. [DOI] [PubMed] [Google Scholar]
- Di Pino G, Pellegrino G, Assenza G, Capone F, Ferreri F, Formica D, Ranieri F, Tombini M, Ziemann U, Rothwell JC & Di Lazzaro V (2014). Modulation of brain plasticity in stroke: a novel model for neurorehabilitation. Nat Rev Neurol 10, 597–608. [DOI] [PubMed] [Google Scholar]
- Dijkhuizen RM, Ren J, Mandeville JB, Wu O, Ozdag FM, Moskowitz MA, Rosen BR & Finklestein SP (2001). Functional magnetic resonance imaging of reorganization in rat brain after stroke. Proc Natl Acad Sci USA 98, 12766–12771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferbert A, Priori A, Rothwell JC, Day BL, Colebatch JG & Marsden CD (1992). Interhemispheric inhibition of the human motor cortex. J Physiol 453, 525–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerloff C, Cohen LG, Floeter MK, Chen R, Corwell B & Hallett M (1998). Inhibitory influence of the ipsilateral motor cortex on responses to stimulation of the human cortex and pyramidal tract. J Physiol 510, 249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grefkes C, Eickhoff SB, Nowak DA, Dafotakis M & Fink GR (2008). Dynamic intra‐ and interhemispheric interactions during unilateral and bilateral hand movements assessed with fMRI and DCM. Neuroimage 41, 1382–1394. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harauzov A, Spolidoro M, DiCristo G, De Pasquale R, Cancedda L , Pizzorusso T, Viegi A, Berardi N & Maffei L (2010). Reducing intracortical inhibition in the adult visual cortex promotes ocular dominance plasticity. J Neurosci 30, 361–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensch TK, Fagiolini M, Mataga N, Stryker MP, Baekkeskov S & Kash SF (1998). Local GABA circuit control of experience‐dependent plasticity in developing visual cortex. Science 282, 1504–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF, Maffei L & Tonegawa S (1999). BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell 98, 739–755. [DOI] [PubMed] [Google Scholar]
- Huemmeke M, Eysel UT & Mittmann T (2004). Lesion‐induced enhancement of LTP in rat visual cortex is mediated by NMDA receptors containing the NR2B subunit. J Physiol 559, 875–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbrosci B, Neubacher U, White R, Eysel UT & Mittmann T (2013). Shift from phasic to tonic GABAergic transmission following laser‐lesions in the rat visual cortex. Pflugers Arch 465, 879–893. [DOI] [PubMed] [Google Scholar]
- Irlbacher K, Brocke J, Mechow JV & Brandt SA (2007). Effects of GABA(A) and GABA(B) agonists on interhemispheric inhibition in man. Clin Neurophysiol 118, 308–316. [DOI] [PubMed] [Google Scholar]
- Iurilli G, Ghezzi D, Olcese U, Lassi G, Nazzaro C, Tonini R, Tucci V, Benfenati F & Medini P (2012). Sound‐driven synaptic inhibition in primary visual cortex. Neuron 73, 814–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iurilli G, Olcese U & Medini P (2013). Preserved excitatory‐inhibitory balance of cortical synaptic inputs following deprived eye stimulation after a saturating period of monocular deprivation in rats. PLoS One 8, e82044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Xie L, Sun F, Mao X & Greenberg DA (2011). Corpus callosum and experimental stroke: studies in callosotomized rats and acallosal mice. Stroke 42, 2584–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzner S, Nauhaus I, Benucci A, Bonin V, Ringach DL & Carandini M (2009). Local origin of field potentials in visual cortex. Neuron 61, 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Zipfel GJ & Choi DW (1999). The changing landscape of ischaemic brain injury mechanisms. Nature 399, A7–14. [DOI] [PubMed] [Google Scholar]
- Lu U, Roach SM, Song D & Berger TW (2012). Nonlinear dynamic modeling of neuron action potential threshold during synaptically driven broadband intracellular activity. IEEE Trans Biomed Eng 59, 706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansoori BK, Jean‐Charles L, Touvykine B, Liu A, Quessy S & Dancause N (2014). Acute inactivation of the contralesional hemisphere for longer durations improves recovery after cortical injury. Exp Neurol 254, 18–28. [DOI] [PubMed] [Google Scholar]
- Margrie TW, Brecht M & Sakmann B (2002). In vivo, low‐resistance, whole‐cell recordings from neurons in the anaesthetized and awake mammalian brain. Pflugers Arch 444, 491–498. [DOI] [PubMed] [Google Scholar]
- Marshall RS, Perera GM, Lazar RM, Krakauer JW, Constantine RC & DeLaPaz RL (2000). Evolution of cortical activation during recovery from corticospinal tract infarction. Stroke 31, 656–661. [DOI] [PubMed] [Google Scholar]
- Maxwell KA & Dyck RH (2005). Induction of reproducible focal ischemic lesions in neonatal mice by photothrombosis. Dev Neurosci 27, 121–126. [DOI] [PubMed] [Google Scholar]
- Medini P ( 2011). Cell‐type‐specific sub‐ and suprathreshold receptive fields of layer 4 and layer 2/3 pyramids in rat primary visual cortex. Neuroscience 190, 112–126. [DOI] [PubMed] [Google Scholar]
- Mittmann T & Eysel UT (2001). Increased synaptic plasticity in the surround of visual cortex lesions in rats. Neuroreport 12, 3341–3347. [DOI] [PubMed] [Google Scholar]
- Mittmann T, Luhmann HJ, Schmidt‐Kastner R, Eysel UT, Weigel H & Heinemann U (1994). Lesion‐induced transient suppression of inhibitory function in rat neocortex in vitro. Neuroscience 60, 891–906. [DOI] [PubMed] [Google Scholar]
- Molnar Z & Cheung AF (2006). Towards the classification of subpopulations of layer V pyramidal projection neurons. Neurosci Res 55, 105–115. [DOI] [PubMed] [Google Scholar]
- Murphy SC, Palmer LM, Nyffeler T, Muri RM & Larkum ME (2016). Transcranial magnetic stimulation (TMS) inhibits cortical dendrites. Elife 5, e13598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohira K, Furuta T, Hioki H, Nakamura KC, Kuramoto E, Tanaka Y, Funatsu N, Shimizu K, Oishi T, Hayashi M, Miyakawa T, Kaneko T & Nakamura S (2010). Ischemia‐induced neurogenesis of neocortical layer 1 progenitor cells. Nat Neurosci 13, 173–179. [DOI] [PubMed] [Google Scholar]
- Onwuekwe I & Ezeala‐Adikaibe B (2012). Ischemic stroke and neuroprotection. Ann Med Health Sci Res 2, 186–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overman JJ, Clarkson AN, Wanner IB, Overman WT, Eckstein I, Maguire JL, Dinov ID, Toga AW & Carmichael ST (2012). A role for ephrin‐A5 in axonal sprouting, recovery, and activity‐dependent plasticity after stroke. Proc Natl Acad Sci USA 109, E2230–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer LM, Schulz JM & Larkum ME (2013). Layer‐specific regulation of cortical neurons by interhemispheric inhibition. Commun Integr Biol 6, e23545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer LM, Schulz JM, Murphy SC, Ledergerber D, Murayama M & Larkum ME (2012). The cellular basis of GABA(B)‐mediated interhemispheric inhibition. Science 335, 989–993. [DOI] [PubMed] [Google Scholar]
- Pietrasanta M, Restani L & Caleo M (2012). The corpus callosum and the visual cortex: plasticity is a game for two. Neural Plast 2012, 838672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouille F & Scanziani M (2001). Enforcement of temporal fidelity in pyramidal cells by somatic feed‐forward inhibition. Science 293, 1159–1163. [DOI] [PubMed] [Google Scholar]
- Quattromani MJ, Pruvost M, Guerreiro C, Backlund F, Englund E, Aspberg A, Jaworski T, Hakon J, Ruscher K, Kaczmarek L, Vivien D & Wieloch T (2018). Extracellular matrix modulation is driven by experience‐dependent plasticity during stroke recovery. Mol Neurobiol 55, 2196–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinecke S, Dinse HR, Reinke H & Witte OW (2003). Induction of bilateral plasticity in sensory cortical maps by small unilateral cortical infarcts in rats. Eur J Neurosci 17, 623–627. [DOI] [PubMed] [Google Scholar]
- Rudy B, Fishell G, Lee S & Hjerling‐Leffler J (2011). Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev Neurobiol 71, 45–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiene K, Bruehl C, Zilles K, Qu M, Hagemann G, Kraemer M & Witte OW (1996). Neuronal hyperexcitability and reduction of GABAA‐receptor expression in the surround of cerebral photothrombosis. J Cereb Blood Flow Metab 16, 906–914. [DOI] [PubMed] [Google Scholar]
- Schuett S, Bonhoeffer T & Hubener M (2002). Mapping retinotopic structure in mouse visual cortex with optical imaging. J Neurosci 22, 6549–6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanina EV, Schallert T, Witte OW & Redecker C (2006). Behavioral recovery from unilateral photothrombotic infarcts of the forelimb sensorimotor cortex in rats: role of the contralateral cortex. Neuroscience 139, 1495–1506. [DOI] [PubMed] [Google Scholar]
- Silasi G & Murphy TH (2014). Stroke and the connectome: how connectivity guides therapeutic intervention. Neuron 83, 1354–1368. [DOI] [PubMed] [Google Scholar]
- Soleman S, Yip PK, Duricki DA & Moon LD (2012). Delayed treatment with chondroitinase ABC promotes sensorimotor recovery and plasticity after stroke in aged rats. Brain 135, 1210–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song YM, Lee JY, Park JM, Yoon BW & Roh JK (2005). Ipsilateral hemiparesis caused by a corona radiata infarct after a previous stroke on the opposite side. Arch Neurol 62, 809–811. [DOI] [PubMed] [Google Scholar]
- Takatsuru Y, Fukumoto D, Yoshitomo M, Nemoto T, Tsukada H & Nabekura J (2009). Neuronal circuit remodeling in the contralateral cortical hemisphere during functional recovery from cerebral infarction. J Neurosci 29, 10081–10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talelli P, Greenwood RJ & Rothwell JC (2007). Exploring Theta Burst Stimulation as an intervention to improve motor recovery in chronic stroke. Clin Neurophysiol 118, 333–342. [DOI] [PubMed] [Google Scholar]
- Talelli P, Wallace A, Dileone M, Hoad D, Cheeran B, Oliver R, VandenBos M, Hammerbeck U, Barratt K, Gillini C, Musumeci G, Boudrias MH, Cloud GC, Ball J, Marsden JF, Ward NS, Di Lazzaro V, Greenwood RG & Rothwell JC (2012). Theta burst stimulation in the rehabilitation of the upper limb: a semirandomized, placebo‐controlled trial in chronic stroke patients. Neurorehabil Neural Repair 26, 976–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilent WB & Contreras D (2005). Stimulus‐dependent changes in spike threshold enhance feature selectivity in rat barrel cortex neurons. J Neurosci 25, 2983–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winship IR & Murphy TH (2008). In vivo calcium imaging reveals functional rewiring of single somatosensory neurons after stroke. J Neurosci 28, 6592–6606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise SP & Jones EG (1976). The organization and postnatal development of the commissural projection of the rat somatic sensory cortex. J Comp Neurol 168, 313–343. [DOI] [PubMed] [Google Scholar]
- Yinon U, Chen M & Gelerstein S (1992). Binocularity and excitability loss in visual cortex cells of corpus callosum transected kittens and cats. Brain Res Bull 29, 541–552. [DOI] [PubMed] [Google Scholar]
- Zhou J, Wen Y, She L, Sui YN, Liu L, Richards LJ & Poo MM (2013). Axon position within the corpus callosum determines contralateral cortical projection. Proc Natl Acad Sci USA 110, E2714–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]