

Abstract
Opa1 participates in inner mitochondrial membrane fusion and cristae morphogenesis. Here, we show that muscle‐specific Opa1 ablation causes reduced muscle fiber size, dysfunctional mitochondria, enhanced Fgf21, and muscle inflammation characterized by NF‐κB activation, and enhanced expression of pro‐inflammatory genes. Chronic sodium salicylate treatment ameliorated muscle alterations and reduced the muscle expression of Fgf21. Muscle inflammation was an early event during the progression of the disease and occurred before macrophage infiltration, indicating that it is a primary response to Opa1 deficiency. Moreover, Opa1 repression in muscle cells also resulted in NF‐κB activation and inflammation in the absence of necrosis and/or apoptosis, thereby revealing that the activation is a cell‐autonomous process and independent of cell death. The effects of Opa1 deficiency on the expression NF‐κB target genes and inflammation were absent upon mitochondrial DNA depletion. Under Opa1 deficiency, blockage or repression of TLR9 prevented NF‐κB activation and inflammation. Taken together, our results reveal that Opa1 deficiency in muscle causes initial mitochondrial alterations that lead to TLR9 activation, and inflammation, which contributes to enhanced Fgf21 expression and to growth impairment.
Keywords: endosome, mitochondrial dynamics, muscle disease, systemic inflammation
Subject Categories: Autophagy & Cell Death, Immunology, Metabolism
Introduction
Mitochondrial metabolism, quality control, response to apoptotic stimuli, and hormone activity are partly controlled through the balance between mitochondrial fusion and fission (Twig et al, 2008; Liesa et al, 2009; Sebastian et al, 2012; Liesa & Shirihai, 2013; Munoz et al, 2013). Mitochondrial fusion in mammalian cells is regulated by the proteins mitofusin 1 and mitofusin 2 (Mfn1 and Mfn2) and optic atrophy 1 (Opa1). Various Opa1 isoforms are located in the mitochondrial intermembrane space or inserted within the inner mitochondrial membrane (Olichon et al, 2003; Cipolat et al, 2004; Griparic et al, 2004; Ishihara et al, 2006). The existence of multiple Opa1 isoforms and cleavage mechanisms may explain the role of this protein beyond mitochondrial inner membrane fusion, such as in cristae remodeling, and supercomplex formation (Frezza et al, 2006; Cogliati et al, 2013).
In humans, OPA1 mutations have been reported in patients affected by autosomal dominant optic atrophy or ADOA (Alexander et al, 2000; Delettre et al, 2000). Overall, data support the view that the pathogenesis of ADOA occurs as a result of haploinsufficiency (Pesch et al, 2001). Human fibroblasts carrying OPA1 mutations show impaired oxidative phosphorylation and mitochondrial fusion (Amati‐Bonneau et al, 2008). Some of these OPA1 missense mutations are associated with altered mitophagy and parkinsonism (Carelli et al, 2015). In addition to dominant optic atrophy, OPA1 mutations also cause a multi‐systemic disorder called ADOA plus syndrome, which results in severe myopathy (Amati‐Bonneau et al, 2008; Hudson et al, 2008; Zeviani, 2008). Interestingly, these patients show multiple mitochondrial DNA (mtDNA) deletions in muscles, which suggests a role of OPA1 on mtDNA stability.
The appropriate homeostasis of mitochondria is essential in the maintenance of cellular health. Mitochondria are a source of damage‐associated molecular patterns (DAMPs), and among others, mtDNA has been shown to induce a pro‐inflammatory state (Zhang et al, 2010; Oka et al, 2012; Wenceslau et al, 2014). MtDNA has been reported to activate immunity through two distinct pathways, namely Toll‐like receptor 9 (TLR9; Zhang et al, 2010; Oka et al, 2012; Liu et al, 2015), and cGAS activation (White et al, 2014; West et al, 2015). Under basal conditions, TLR9 is located in the endoplasmic reticulum (ER), and upon stimulation by inducers, TLR9 translocates to the membrane of endosomes or to lysosomes, bind to ligands, and initiate cellular inflammation (Latz et al, 2004; Zhang et al, 2010; Wei et al, 2015; De Leo et al, 2016). Interaction of TLR9 with mtDNA activates the nuclear factor kappa B (NF‐κB) signaling and increases the expression of other pro‐inflammatory cytokines such as tumor necrosis factor‐α (TNF‐α), interleukin (IL)‐6, IL‐1β (Julian et al, 2013; Yu & Bennett, 2014; Zhang et al, 2014).
Recently, cytosolic mtDNA has been reported to engage cytosolic antiviral signaling and to enhance the expression of interferon‐stimulated genes (Rongvaux et al, 2014; White et al, 2014; West et al, 2015). Thus, cytosolic mtDNA activates the DNA sensor cGAS and promotes STING‐IRF3‐dependent signaling (Rongvaux et al, 2014; White et al, 2014; West et al, 2015). Furthermore, neutrophils extrude interferogenic mtDNA by a process dependent in part on lysosomal activity and that occurs in the presence of a constitutive defect in mitophagy (Caielli et al, 2016),
Based on the observations that OPA1 mutations cause mtDNA instability (Kim et al, 2005; Amati‐Bonneau et al, 2008; Hudson et al, 2008; Yu‐Wai‐Man et al, 2010), we reasoned that OPA1 deficiency in a non‐immune cells should not only alter mitochondrial morphology but also mitochondrial stability, and in consequence trigger immune responses. Based on this, we analyzed the impact of Opa1 loss‐of‐function in skeletal muscle. Our data indicate that Opa1 deletion in skeletal muscle causes mitochondrial inflammatory myopathy characterized by impaired mitochondrial function, pro‐inflammatory cytokine production, altered myofiber morphology, and muscle dysfunction. In addition, we found that the activation of NF‐κB in Opa1‐deficient muscle cells is mediated by TLR9 and by mtDNA.
Results
Skeletal muscle‐specific Opa1 ablation causes reduced body growth and premature death
In preliminary studies, we monitored skeletal muscle regeneration in a cardiotoxin (CTX) injury‐induced model in gastrocnemius muscles in wild‐type animals at a range of times (Fig EV1A). Two days post‐injury (dpi), when satellite cells become active, muscles were treated with adenoviruses encoding for engineered miRNAs against Opa1 (miR Opa1) or with control adenoviruses encoding for miRNA with no homology in the mouse genome (miR Ctrl; Fig EV1B). Opa1 deficiency (miR Opa1) not only caused impaired muscle regeneration but also increased the presence of immune cells and reduced muscle fiber size in regenerating myofibers (Figs 1A and B, and EV1C). Immunostaining of the developmental form of MHC (dMHC) revealed a reduction in controls between 9 and 12 days post‐CTX treatment, whereas Opa1‐deficient muscles showed sustained high expression of dMHC (Figs 1C and EV1D), indicating impaired muscle fiber maturation.
Figure EV1. Skeletal muscle‐specific Opa1 ablation causes reduced body growth and premature death.
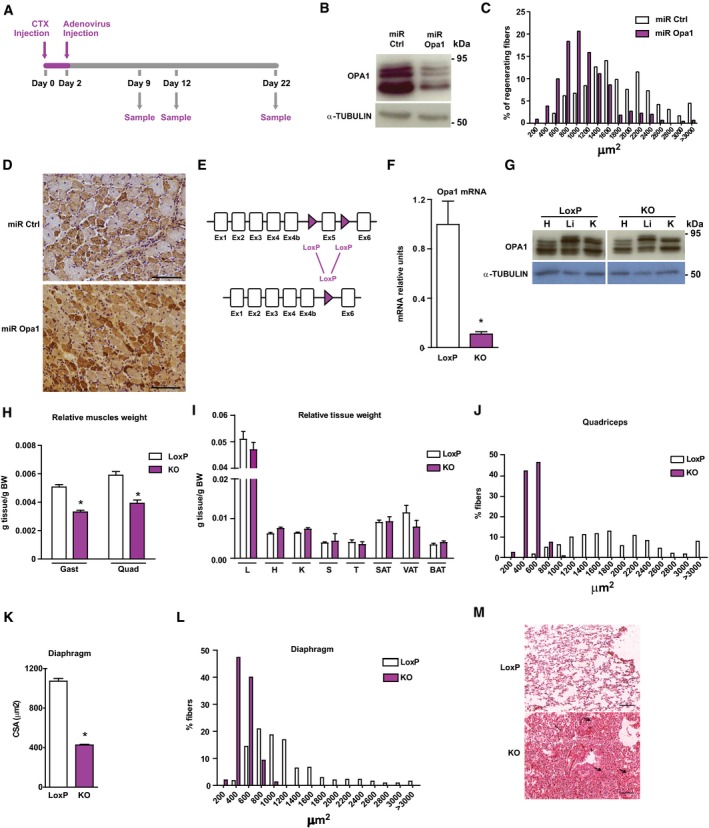
-
AScheme of the experimental design of CTX‐induced injury.
-
BOpa1 protein levels in control C2C12 myoblasts, which were transduced with control miRNA (miR Ctrl) or in Opa1 loss‐of‐function C2C12 myoblasts, which were transduced with adenoviruses encoding for miRNA against Opa1 (miR Opa1) (n = 5).
-
CDistribution of myofiber size in mice treated with miR Ctrl or miR Opa1 adenoviruses at dpi 12 (n = 5).
-
DRepresentative image of dMHC immunohistochemistry from gastrocnemius muscle treated with miR Ctrl or miR Opa1 adenoviruses (12 dpi). Scale bars, 100 μm.
-
EPartial genomic structure of the Opa1 gene showing the scission of exon 5, thus deleting all Opa1 protein isoforms.
-
FOpa1 mRNA levels in the gastrocnemius muscle of loxP (non‐expressing Cre Opa1loxP/loxP mice) and skeletal muscle‐specific KO mice (KO) (n = 10).
-
GOpa1 protein levels in tissue homogenates from control (loxP) and skeletal muscle‐specific KO mice (KO).
-
H, IRelative weight of muscles (gastrocnemius (Gast) and quadriceps (Quad)) (panel H) (n = 18) and organs (liver (L), heart (H), kidney (K), spleen (S), T thymus (T), subcutaneous adipose tissue (SAT), visceral adipose tissue (VAT), and brown adipose tissue (BAT)) (panel I) (n = 32) of 9‐week‐old loxP and KO mice. These data are expressed as g of tissue/g of body weight (BW).
-
JDistribution of myofiber size of 150 myofibers in quadriceps muscle.
-
K, LMean cross‐sectional area (CSA) (K) and distribution of myofiber size (L) of 150 myofibers in diaphragm muscle.
-
MRepresentative images of hematoxylin and eosin‐stained lung sections from loxP and KO mice. Thick arrows indicate congestion and atelectasis. Thin arrows indicate normal parenchyma. Asterisks show bronchioles. Scale bars, 100 μm.
Figure 1. Skeletal muscle‐specific Opa1 ablation causes reduced body growth and premature death.
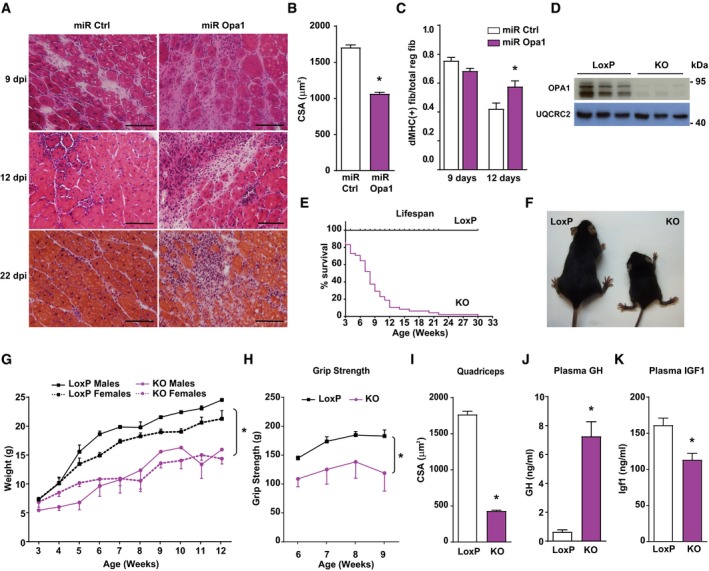
-
ATransversal sections of gastrocnemius muscles of 3‐month‐old mice injected with CTX as an injury‐induced model and, 2 days later, with adenoviruses encoding for non‐targeting miRNA (miR Ctrl) or miRNA targeting Opa1 (miR Opa1). Samples were taken on various days after the injury (dpi, n = 5). Scale bars, 100 μm.
-
BMean cross‐sectional area (CSA) of 150 myofibers per gastrocnemius muscle at dpi 12.
-
CQuantification of positive MHC myofibers vs. total regenerating myofibers of gastrocnemius muscle treated with miR Ctrl or miR Opa1 adenoviruses at dpi 9 and 12 (n = 5).
-
DOpa1 protein levels in tissue homogenates of control (loxP) and skeletal muscle‐specific KO mice (KO). The skeletal muscle used was gastrocnemius muscle (n = 6).
-
ESurvival curves for loxP and Opa1 KO mice (n = 25).
-
FPicture of loxP and KO mice at 9 weeks of age.
-
GBody weight of loxP and KO male and female mice (n = 25).
-
HGrip strength in loxP and KO mice. loxP (n = 7) and KO (n = 4).
-
IMean cross‐sectional area (CSA) of 150 myofibers in quadriceps muscle.
-
J–KPlasma concentration of growth hormone (GH) (J) and Igf1 (K) of loxP (n = 8) and KO mice (n = 10).
Based on these data, we analyzed the impact of Opa1 depletion on muscle homeostasis by the generation of skeletal muscle‐specific knockout mice. This was performed by crossing homozygous Opa1‐loxP/loxP mice with a mouse strain expressing Cre recombinase under the control of the myogenin promoter (Li et al, 2005; Figs 1D and EV1E–G). Opa1 ablation in this specific tissue induced a dramatic reduction in life span (Fig 1E) and impaired normal growth (Fig 1F and G). At 9 weeks of age, Opa1 KO mice showed a reduction in relative weight of skeletal muscle (Fig EV1H) and no change in various organs and tissues (Fig EV1I). Muscle force was also lower in Opa1 KO mice (Fig 1H). In keeping with this, histological analyses of KO mice indicated a marked decrease in fiber size and cross‐sectional area in quadriceps and diaphragm (Figs 1I and EV1J, K and L). Consistent with altered diaphragm morphology, we observed extensive congestion and atelectasis in the lungs of these mice (Fig EV1M). In keeping with the reduced growth, plasma levels of GH were high (Fig 1J) and IGF1 were low (Fig 1K). Characterization of the hepatic profile of Opa1 KO mice showed reduced phosphorylation of STAT5 (Fig 2A), and decreased expression of its target genes Igf1 and Fos (Fig 2B). Also, it revealed reduced expression of growth hormone receptor (Ghr) and increased expression of the STAT5 inhibitor Socs3 (Fig 2B).
Figure 2. Muscle‐specific Opa1 ablation induces growth hormone resistance.
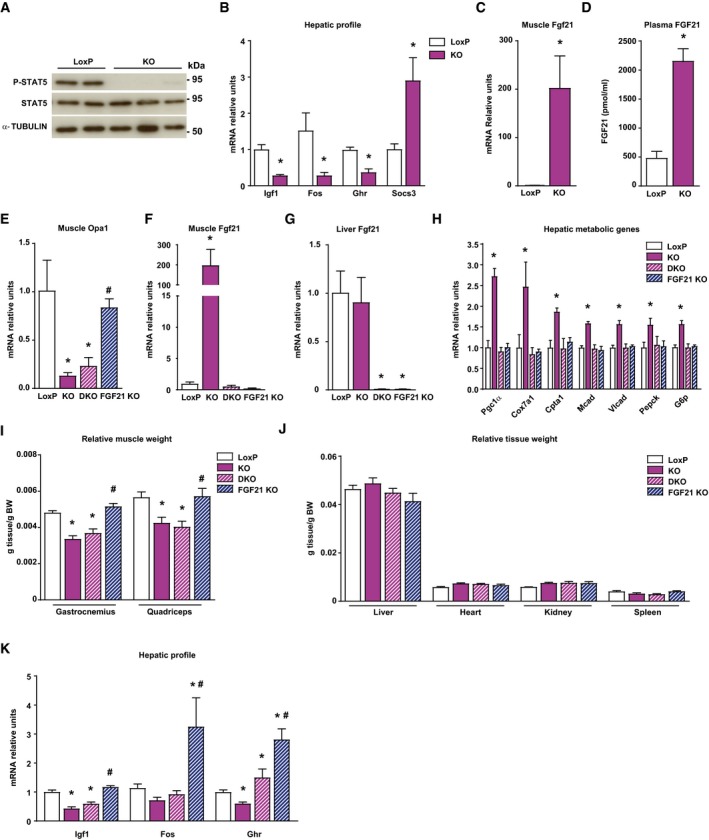
-
AStat5 phosphorylation in livers of loxP and KO mice (n = 10).
-
BmRNA levels of Igf1, Fos, growth hormone receptor (Ghr), and Socs3 in liver of loxP and KO mice (n = 5).
-
CFgf21 gene expression in muscle of loxP and KO mice (n = 8).
-
DFGF21 levels in plasma of loxP (n = 8) and KO mice (n = 10).
-
E–GOpa1 mRNA levels in muscle (E), and Fgf21 gene expression in muscle (F) and liver (G) of loxP, Opa1 KO, FGF21 KO, and double KO (Opa1 + FGF21 KO, DKO) (n = 8).
-
HHepatic expression of genes encoding Pgc1α, respiratory complex IV subunit Cox7a1, fatty acid oxidation components (Cpta1, Mcad, and Vlcad), and gluconeogenic enzymes (Pepck and G6p) in loxP, Opa1 KO, DKO, and Fgf21 KO mice (n = 7).
-
I, JRelative weight of gastrocnemius and quadriceps muscles, (I) and organs (J) of loxP, Opa1 KO, DKO, and Fgf21 KO mice (n = 10). These data are expressed as g of tissue/g of body weight (BW).
-
KIgf1, Fos, and Ghr mRNA levels in the livers of loxP, Opa1 KO, DKO, and Fgf21 KO mice (n = 10).
Gene set enrichment analysis of genomic profiling in muscles of control and Opa1 KO mice revealed the up‐regulation of ATF4 target genes in muscles of Opa1 KO mice (Dataset EV1; Kim et al, 2013) among the large number of genes with deregulated expression (Dataset EV2; 486 genes up‐regulated and 175 repressed). Fgf21, an ATF4 target gene, was greatly increased in the muscles of Opa1 KO mice (Fig 2C). Circulating Fgf21 levels were fourfold greater in these animals (Fig 2D). Given the observation that Fgf21 causes resistance to GH (Inagaki et al, 2008), we explored the possibility of a rescue by generating a double knockout (DKO) mouse for Fgf21 and Opa1 (Fig 2E–G). DKO mice normalized hepatic PGC‐1α expression consistent with the regulatory role of Fgf21 (Potthoff et al, 2009; Fig 2H). In keeping with these observations, Opa1 KO mice showed enhanced expression of hepatic genes relevant in gluconeogenesis, lipid metabolism, or mitochondrial respiration, and DKO mice showed a normalized expression (Fig 2H). In contrast, DKO mice showed similar muscle and tissue weights to those of Opa1 KO mice (Fig 2I and J). Furthermore, DKO failed to enhance the hepatic expression of Igf1 or Fos genes (Fig 2K). In all, these data indicate that Fgf21 mediates the metabolic alterations occurring in liver from Opa1 KO mice, but it is not involved in the reduced growth of Opa1 KO mice.
Opa1 ablation causes inflammatory myopathy
Histological inspection of Opa1‐deficient muscle sections revealed a substantial number of necrotic myofibers, as well as regenerating fibers (centrally located nuclei; Fig 3A). Given the preliminary data obtained from the CTX‐treated muscles (Fig 1A), we questioned whether Opa1‐deficiency promotes muscle inflammation, and whether that could be the reason for the growth impairment. Non‐specific esterase (NSE) and major histocompatibility complex (MHC) class I stainings revealed severe inflammation in muscles from Opa1 KO mice (Fig 3A). In addition, the expression of NF‐κB target genes was also induced (Fig 3B), as well as the plasma levels of pro‐inflammatory cytokines IL‐1β and IL‐6 (Fig 3C).
Figure 3. Opa1 ablation causes mitochondrial inflammatory myopathy.
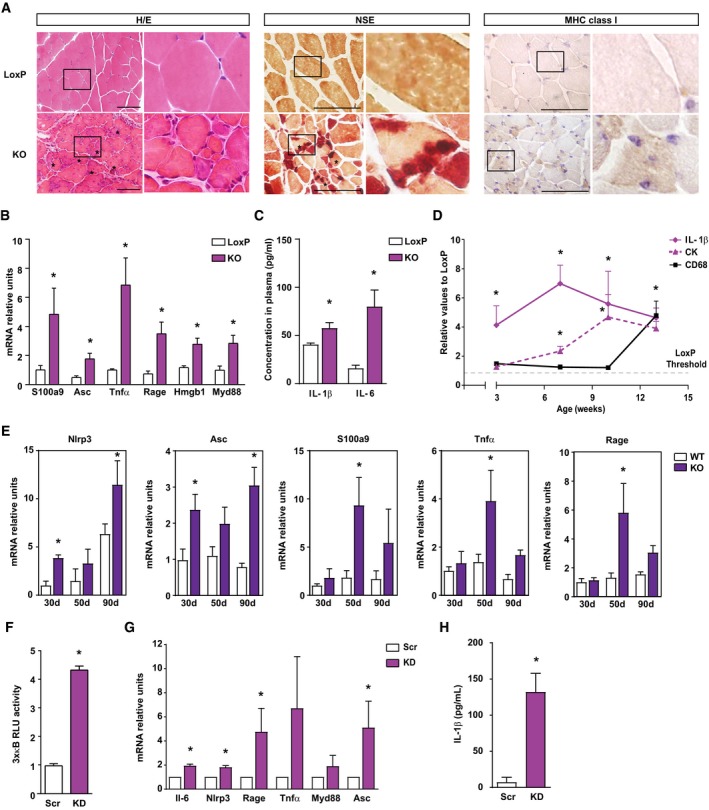
- Representative images of hematoxylin and eosin (H/E) staining, non‐specific esterase (NSE) staining and MHC class I expression in gastrocnemius muscle sections of 6‐week‐old loxP and skeletal muscle‐specific Opa1 KO mice. Inset panels are 4× magnification of the outlined areas. Asterisks in H/E indicate necrotic fibers. Asterisks in NSE indicate the presence of macrophages. Scale bars, 100 μm.
- Expression of NF‐κB target genes in muscles of loxP and KO mice (n = 7).
- Circulating IL‐1β and IL‐6 measured in plasma of 8‐ to 10‐week‐old loxP and KO mice (n = 9).
- Quantification of IL‐1β and CD68 expression in gastrocnemius, and CK activity in plasma of loxP and Opa1 KO animals at 3, 7, 10, and 13 weeks of age. Values of Opa1 KO mice are represented relative to the loxP group. The staining quantification was performed using Trainable Weka Segmentation plugin from ImageJ (5 images per group).
- Expression of NF‐κB target genes in muscles of control and inducible skeletal muscle‐specific Opa1‐KO mice at 30, 50, and 90 days after tamoxifen treatment (n = 6).
- Transcriptional activity of NF‐κB in myoblast expressing scramble shRNA (Scr) or shRNA against Opa1 (KD) (n = 5).
- Expression of NF‐κB target genes in Scr and KD myoblasts (n = 6).
- Levels of IL‐1β in culture media from C2C12 myoblasts (n = 6).
In order to determine whether inflammation is a primary consequence of Opa1 depletion, we followed the presence of IL‐1β and CD68 in muscle and the activity of creatine kinase (CK) in plasma in 3‐, 7‐, 10‐, and 13‐week‐old mice. Highly positive labeling for IL‐1β was already detected at 3 weeks in Opa1‐deficient muscles and was maintained with age (Fig 3D and Appendix Fig S1A). Plasma CK activity was increased from week 7 (Fig 3D). However, pro‐inflammatory macrophages (stained for CD68) were not detected before 13 weeks of age (Fig 3D and Appendix Fig S1B) in these mice, thereby indicating that the inflammation in skeletal muscle caused by Opa1 deficiency precedes macrophage infiltration. No evidence of apoptosis was detected in muscles of control or KO mice (Fig EV2A).
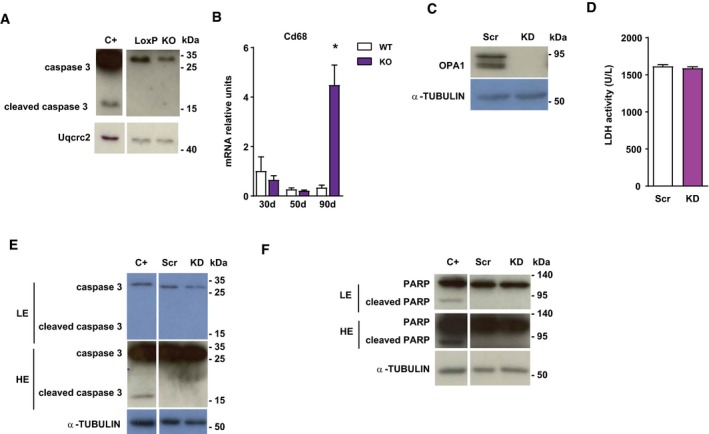
Figure EV2. Opa1 ablation causes mitochondrial inflammatory myopathy.
-
ACaspase 3 expression in loxP and skeletal muscle‐specific Opa1 KO mice.
-
BCD68 expression in muscles of control and inducible muscle‐specific Opa1 KO mice at 30, 50, and 90 days after tamoxifen treatment (n = 4).
-
COpa1 protein levels in C2C12 control myoblasts (Scr) and in stably depleted Opa1 myoblasts (KD).
-
DLDH activity in cultured media of Scr and Opa1 KD myoblasts (n = 8).
-
E, FCaspase 3 (E) and Parp (F) expression in Scr and Opa1 KD myoblasts. Apoptosis‐positive control was obtained by treating C2C12 myoblasts with 10 μm staurosporin for 4 h.
To further confirm inflammation being a primary event, inducible skeletal muscle‐specific Opa1 KO mice were treated with tamoxifen, in order to specifically ablate Opa1 in adult muscles, and the expression of NF‐κB target genes was measured at different times. Thirty days after the onset of tamoxifen treatment, enhanced NLRP3 and ASC expression was detected under conditions in which muscle mass is normal (Fig 3E). At 50 days of tamoxifen treatment, the expression of genes S100A9, TNFα, and RAGE was also enhanced in Opa1‐deficient muscles under conditions in which muscle loss was already apparent (Fig 3E). These changes in the expression of NF‐κB target genes were detectable before alterations in CD68, a marker of pro‐inflammatory macrophages (Fig EV2B). These results indicate that Opa1 deficiency triggers muscle inflammation even when depletion occurs in mature muscles from adult mice.
To determine whether cultured muscle cells show a cell‐autonomous response, we analyzed whether Opa1‐deficient C2C12 myoblasts exhibit an inflammatory state. Phenocopying the muscle profile, Opa1 KD myoblasts (Fig EV3C) showed a marked increase of NF‐κB promoter activity (Fig 3F) and the expression of NF‐κB target genes (Fig 3G). IL‐1β was also increased in culture media from Opa1‐deficient cells (Fig 3H). Opa1 deficiency did not cause necrosis (Fig EV2D) or apoptosis (Fig EV2E and F).
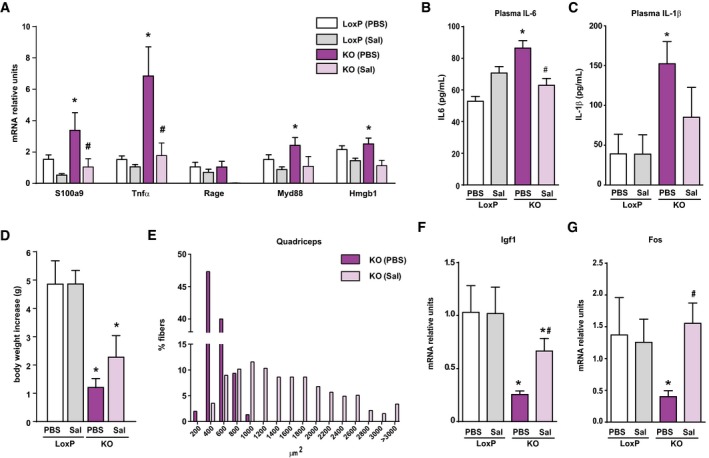
Figure EV3. Salicylate treatment reduces inflammation and improves growth in Opa1 KO mice.
-
AExpression of NF‐κB target genes in muscles of loxP and KO mice treated with sodium salicylate (Sal) or with PBS (PBS).
-
B, CCirculating IL‐6 (B) and IL‐1β (C) measured in plasma of loxP and KO mice treated or not with sodium salicylate.
-
DBody weight increase in loxP and KO mice treated or not with salicylate for 20 days.
-
EDistribution of myofiber size in quadriceps muscles from loxP and KO mice treated or not with salicylate.
-
F, GIgf1 (F) and Fos (G) mRNA levels in liver from salicylate‐treated and non‐treated mice.
In all, our data indicate that Opa1‐deficient muscle undergoes a primary inflammatory process characterized by NF‐κB activation, followed by necrosis and then macrophage infiltration. In addition, they reveal that Opa1 deficiency‐induced inflammation is a cells.
Salicylate treatment reduces inflammation and improves growth in Opa1 KO mice
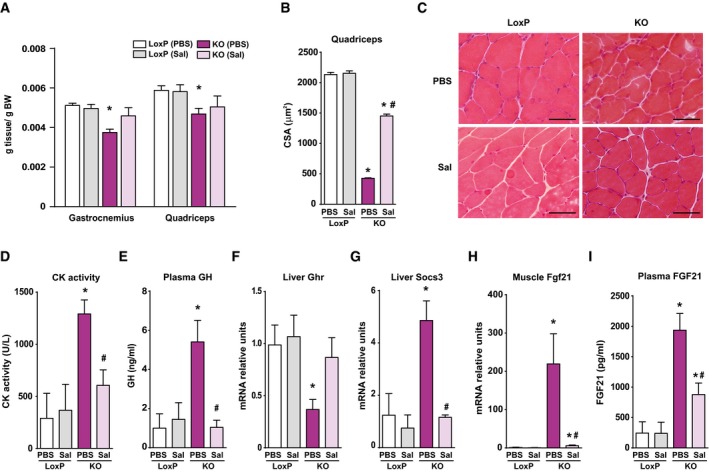
Based on the previous observations, we examined whether the pro‐inflammatory state was responsible for the reduced growth of Opa1 KO mice. To this end, mice were treated for 30 days with sodium salicylate, a known NF‐κB inhibitor (Kopp & Ghosh, 1994; Yin et al, 1998) or with PBS. Anti‐inflammatory efficiency was documented by low expression of NF‐κB target genes and by low circulating levels of IL‐6 and IL‐1β (Fig EV3A–C). Salicylate‐treated Opa1 KO mice showed a notable increase in body weight and relative muscle weight compared to PBS‐treated mice (Figs 4A and EV3D). Moreover, fiber size and distribution were markedly enhanced in KO mice treated with salicylate (Figs 4B and C, and EV3E), and also CK activity was normalized (Fig 4D), consistent with reduced muscle necrosis. To further confirm that growth reduction is due to inflammation, we analyzed circulating and hepatic markers. Salicylate treatment markedly reduced plasma GH (Fig 4E), increased the hepatic expression of Ghr, Igf1, and Fos (Figs 4F and EV3F and G), and reduced SOCS3 expression (Fig 4G). Furthermore, the muscle expression and circulating levels of Fgf21 were normalized in Opa1 KO mice treated with salicylate (Fig 4H and I). In all, our data indicate that inflammation is a key pathological event leading to many metabolic alterations detected under Opa1 deficiency.
Figure 4. Salicylate treatment reduces inflammation and improves growth in Opa1 KO mice.
-
ARelative weight of gastrocnemius and quadriceps muscles of loxP and KO mice treated with sodium salicylate (Sal) or PBS (PBS) for 30 days.
-
BMean CSA of 150 myofibers in quadriceps muscles.
-
CRepresentative H/E staining of quadriceps muscles of LoxP and KO mice treated with salicylate or PBS. Scale bars, 50 μm.
-
DCreatine kinase (CK) activity measured in plasma of salicylate‐treated and PBS‐treated mice.
-
ECirculating GH levels from salicylate‐treated and PBS‐treated animals.
-
F, GGrowth hormone receptor (GHR) (F) and SOCS3 (G) mRNA levels in livers of treated and PBS‐treated animals.
-
HFgf21 mRNA levels measured in tibialis muscle of salicylate‐treated and PBS‐treated mice.
-
IConcentration of Fgf21 in plasma of loxP and treated and PBS‐treated KO mice.
Opa1 depletion causes mitochondrial dysfunction and mitochondrial DNA stress
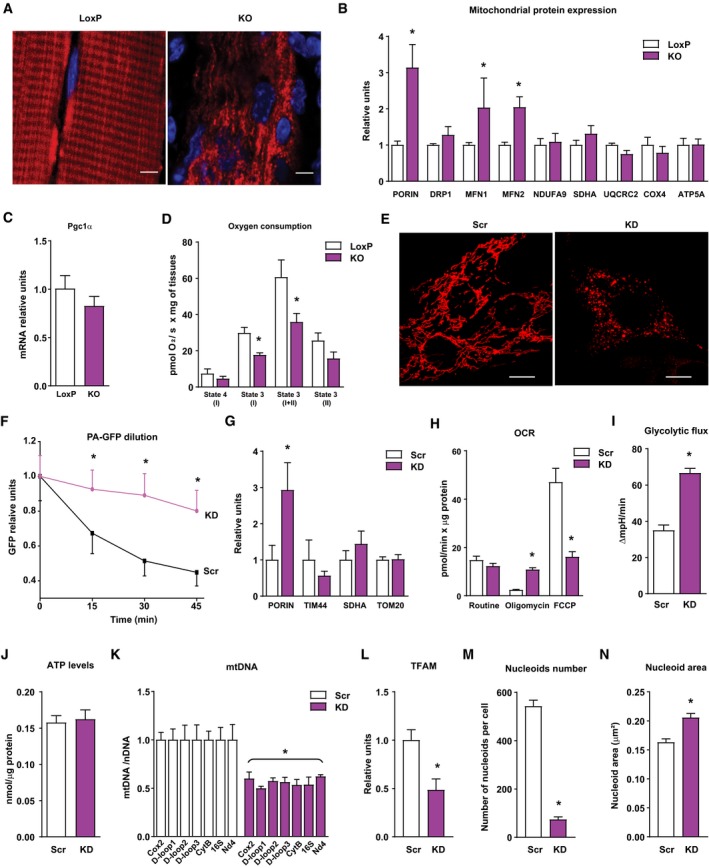
Next, we studied whether Opa1 ablation causes mitochondrial dysfunction in skeletal muscle. Analysis of mitochondrial morphology showed a markedly altered organization of the mitochondrial network and an enhanced mitochondrial density (Fig 5A). Opa1‐deficient muscles showed increased levels of various mitochondrial proteins, but neither of OXPHOS subunits nor of Pgc‐1α expression (Fig 5B and C and Appendix Fig S2A). Moreover, analysis of mitochondrial respiration in permeabilized muscles revealed a marked reduction in state 3 activity (Fig 5D). In parallel to muscles, MtDsRed labeling of Opa1 KD myoblasts revealed a complete fragmentation of the mitochondrial network (Fig 5E). In vivo cell imaging indicated that Opa1‐deficient cells showed movement of fragmented mitochondria and frequent mitochondria encounters but no fusion events (Movies EV1 and EV2). Mitochondrial fusion analysis by expression of a photo‐activatable mitochondrial GFP protein (PA‐GFPmt) revealed a time‐dependent dilution and diffusion of the fluorescence through the mitochondrial network in control cells, whereas Opa1‐deficient cells generated a static signal with time (Fig 5F and Appendix Fig S2B), compatible with lack of mitochondrial fusion. There were no changes in mitochondrial mass, as the expression of mitochondrial proteins PORIN, TIM44, SDHA, or TOM20 was not reduced in Opa1‐deficient cells (Fig 5G). Under these conditions, Opa1 deficiency also caused decreased mitochondrial respiration, which was characterized by enhanced respiratory leak and reduced maximal respiratory capacity (Fig 5H), increased glycolysis (Fig 5I), and unaltered ATP levels (Fig 5J).
Figure 5. Opa1 depletion causes mitochondrial dysfunction and mitochondrial DNA stress.
-
ARepresentative images of mitochondrial network of tibialis muscles of loxP and KO mice electroporated with cDNA encoding DsRed2‐Mito vector (n = 3). Scale bars, 5 μm.
-
BRelative mitochondrial protein expression of gastrocnemius muscles of loxP and KO mice. Values were corrected by α‐tubulin (10 observations per group).
-
CPGC‐1α expression in muscles of loxP and KO mice (n = 7).
-
DMitochondrial respiration assayed in isolated myofibers of tibialis muscles (2–4 mg) from loxP or KO mice (n = 6).
-
EMitochondrial morphology of control (Scr) and Opa1 KD (KD) C2C12 myoblasts. Scale bars, 10 μm.
-
FEstimation of mitochondrial fusion as PA‐GFP dilution in control (black line, n = 27) and Opa1‐silenced (purple line, n = 17) C2C12 myoblasts.
-
GExpression of mitochondrial proteins in Scr and Opa1 KD myoblasts (n = 4).
-
HMitochondrial oxygen consumption rates (OCR) measured in intact Scr or Opa1‐silenced C2C12 myoblasts (n = 5). Five mM glucose was used as a substrate. Proton leak (respiration independent of ATP synthesis) was induced by 1.25 μM oligomycin (complex V inhibitor). Respiratory electron transfer system (ETS) capacity was analyzed using 1 μM FCCP. Non‐mitochondrial electron transport OCR was determined by the addition of the complex III inhibitor antimycin A (0.1 μM) and subtracted from the total OCR in order to obtain mitochondrial OCR.
-
IGlycolytic flux assessed as extracellular acidification rates were measured in intact Scr or KD C2C12 myoblasts (n = 5). Five mM glucose was used as a substrate.
-
JATP content in Scr and Opa1 KD C2C12 myoblasts (n = 15).
-
KmtDNA levels relative to nuclear DNA (Tert) (n = 4).
-
LTFAM protein levels (n = 4).
-
M, NNucleoid composition in Scr and Opa1 KD myoblasts. Number of mtDNA nucleoids per cell (M) and area of each nucleoid (N). Data are from a representative experiment with 20 cells randomly quantified per condition.
Because some OPA1 mutations are linked to mtDNA instability (Amati‐Bonneau et al, 2008; Hudson et al, 2008), we analyzed the effect of Opa1 depletion on mtDNA. Indeed, Opa1 deficiency in muscle cells led to a near 50% reduction in mtDNA copy number measured by the amplification of seven different regions of the mtDNA (Fig 5K). Accordingly, TFAM protein levels were also reduced to a half (Fig 5L). Nucleoid composition analysis by confocal microscopy revealed a reduction in the number of nucleoids per cell (Fig 5M) and enhanced nucleoid size (Fig 5N). This pattern of changes suggests the existence of mitochondrial DNA stress similar to what detected upon TFAM repression (West et al, 2015).
Opa1 deficiency‐driven inflammation requires mtDNA and is independent of cGAS
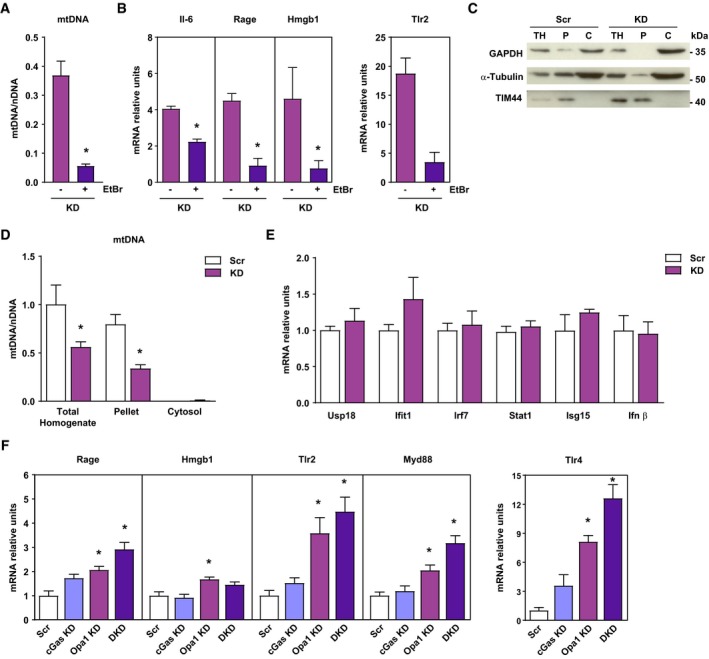
Based on the development of mtDNA stress in Opa1‐depleted cells and because mtDNA triggers inflammation (Arnoult et al, 2011), we analyzed whether Opa1‐deficiency causes inflammation through mtDNA. We depleted mtDNA in C2C12 muscle cells by chronic treatment with ethidium bromide (EtBr). EtBr‐treated cells showed a 95% reduction of mtDNA in Opa1‐deficient cells (Fig 6A and Appendix Fig S3A). EtBr‐induced mtDNA depletion abolished the expression of NF‐κB target genes Il‐6, Rage, Hmgb1, or Tlr2 in Opa1‐deficient cells (Fig 6B). These results indicate the necessity of mtDNA to trigger the inflammatory response.
Figure 6. Opa1 deficiency‐driven inflammation requires mtDNA and is independent of cGAS .
-
A, BmtDNA levels relative to nuclear DNA (D‐loop1 to Tert) and expression of NF‐κB target genes of Opa1 KD (KD) treated or not with EtBr. Relative values to untreated control myoblasts (n = 3).
-
CRepresentative experiment of cytosolic extraction by digitonin protocol. Total homogenate (TH), pellet fraction after cytosolic extraction (P), and cytosolic extract (C).
-
DmtDNA levels relative to nuclear DNA (D‐loop1 to Tert) in total homogenate, pellet and cytosolic fraction of Scr and Opa1 KD (KD) cells (n = 3).
-
EScr and KD mRNA expression of interferon‐stimulated genes (ISG) and Ifnβ (n = 6).
-
FExpression of NF‐κB target genes in control (Scr), cGas depleted (cGas KD), Opa1 depleted (Opa1 KD), and double cGas‐Opa1 ablated (DKD) cells (n = 6).
MtDNA stress has been reported to cause inflammation when it is found in the cytosol, through its recognition by cGAS resulting preferentially in a type I interferon (IFN) response (Rongvaux et al, 2014; White et al, 2014). Thus, we next analyzed the presence of mtDNA in the cytosol by using permeabilization with digitonin as reported (West et al, 2015). The method permits to isolate cytosol from the rest of the cellular material (Fig 6C). Under these conditions, mtDNA was retained in organelles and it was not detected in the cytosolic fraction either in Opa1‐depleted or in control cells (Fig 6D and Appendix Fig S3B). In this connection, we did not detect induction of interferon‐stimulated genes neither Ifnβ in Opa1‐deficient cells (Fig 6E), which lie downstream of cGAS. Furthermore, cGAS loss‐of‐function (Appendix Fig S3C and D) did not rescue NF‐κB activation in Opa1‐deficient cells (Fig 6F).
Opa1 deficiency disrupts mitophagy completion in muscle cells
Because Opa1 depletion does not lead to the release of mtDNA to the cytosol, we examined whether mtDNA is maintained to mitochondria. Analysis of the distribution of mtDNA and TOM20 (an outer mitochondrial membrane protein) revealed a complete co‐localization of both markers in control muscle cells (Appendix Fig S4A and B). The two markers also showed close apposition in Opa1‐deficient muscle cells (Appendix Fig S4A and B) suggesting that mtDNA and TOM20 were in the same compartment and that mtDNA is unlikely to be located in the cytosol.
Based on data indicating that autophagy malfunction caused mtDNA‐mediated inflammation (Nakahira et al, 2011; Oka et al, 2012), we next assessed the functionality of the mitochondrial quality control system. Mitochondrial autophagy was analyzed upon treatment of control of Opa1‐deficient muscle cells with the depolarizing agent CCCP. A rapid recruitment of LC3‐II to mitochondrial enriched fractions was detected upon incubation with CCCP for 30 min in both control and Opa1‐deficient cells, suggesting a normal mitophagy induction in Opa1‐deficient cells (Fig 7A and Appendix Fig S4C). Alterations in mitophagy resolution were assessed by inducing mitochondrial damage for longer times and analyzing the rate of mitochondrial nucleoid disappearance (Lazarou et al, 2015). While control cells responded with a marked reduction in mitochondrial nucleoids upon 9‐h incubation with CCCP, Opa1‐deficient cells showed no mitophagic response (Fig 7B and C). Mitochondrial autophagy was analyzed in skeletal muscle by measuring the abundance of LC3‐II in mitochondrial enriched fractions after treatment for 5 days with the lysosomal inhibitor chloroquine. Control muscles responded to chloroquine by enhanced LC3‐II abundance (Fig 7D, and Appendix Fig S4D) in keeping with prior data (Sebastian et al, 2016). In contrast, Opa1 ablated muscles only showed a weak non‐significant increase in LC3‐II (Fig 7D and Appendix Fig S4D). In keeping with these data, ultrastructural analysis of Opa1‐ablated myofibers of 3‐week‐old mice showed a dramatic accumulation of abnormal mitochondria (Fig 7E).
Figure 7. Opa1 deficiency affects mitophagy.
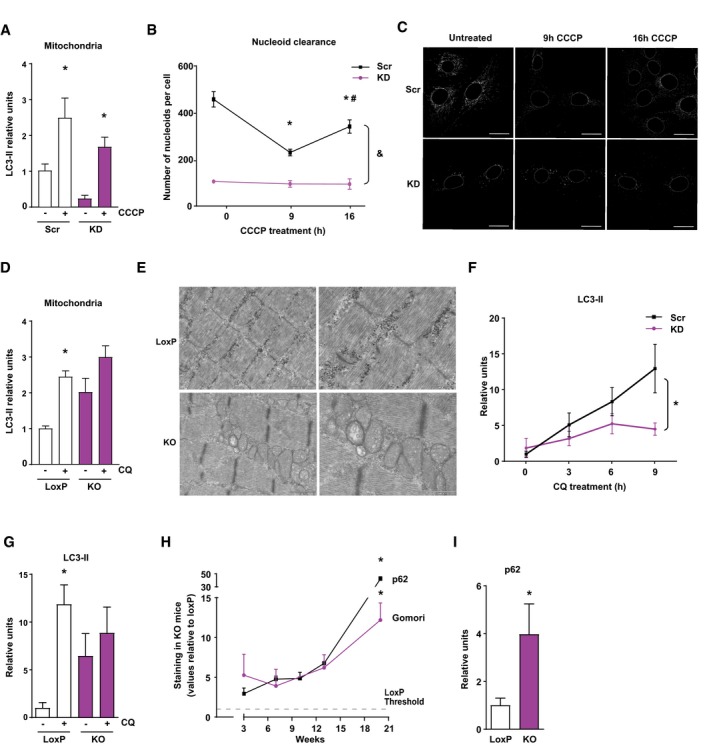
- LC3‐II recruitment to mitochondrial enriched fractions in Scr and KD myoblasts after 30 min of CCCP treatment (n = 4).
- Study of mitophagy completion by mtDNA nucleoid clearance upon long CCCP treatment in Scr and KD cells. Data represent mean ± SEM of three independent experiment with 20 cells randomly quantified per condition. *P < 0.05 vs. Scr untreated, # P < 0.05 vs. 9 h of treatment, and & P < 0.05 KD vs. Scr.
- Representative images of the nucleoid clearance upon CCCP treatment. Scale bars 20 μm.
- Relative LC3‐II protein levels in mitochondrial enriched fractions of quadriceps muscle after 5 days of intraperitoneal treatment with chloroquine (CQ) (10 observations per group).
- Electron micrographs of gastrocnemius muscles from 3‐week‐old loxP and KO mice. Scale bars, 1 μm in left panels, and 500 nm in right panels.
- Time‐course study of LC3‐II protein levels in Scr and KD C2C12 myoblasts under CQ treatment (n = 6).
- Relative LC3‐II protein levels in total homogenates of gastrocnemius muscle after 5 days of intraperitoneal treatment with CQ (six observations per group).
- Quantification of p62 expression and Gomori's Trichrome staining in gastrocnemius muscle of Opa1 KO animals at 3, 7, 10, 13, and 20 weeks of age. Values of Opa1 KO mice are represented relative to the loxP group. This quantification was performed using Trainable Weka Segmentation plugin from ImageJ (five images per group).
- Relative expression of 3‐week‐old mice of p62 (seven and four observations in control and KO mice, respectively).
Studies in Opa1‐deficient muscle cells also revealed a defective autophagy assessed by the reduced buildup of LC3‐II upon incubation in the presence of chloroquine (Fig 7F and Appendix Fig S4E). Opa1‐depleted muscle also showed an impaired autophagy flux and treatment for 5 days with chloroquine caused a marked buildup of the autophagic protein LC3‐II in muscles from control mice, whereas only a weak induction was detectable in Opa1 KO mice (Fig 7G and Appendix Fig S4F). Time‐course studies revealed significantly increased levels of p62 and presence of ragged red fibers in the muscles of 3‐week‐old mice, which further accumulated with age (Fig 7H, Appendix Fig S4H and I). In addition, 3‐week‐old Opa1 KO mice showed enhanced p62 abundance compared to controls by Western blot (Fig 7I and Appendix Fig S4G). In all, our data are coherent with the view that Opa1 deficiency results in a defective mitophagy characterized by normal induction followed by defective resolution. However, measurements of mitophagy are difficult in skeletal muscle and therefore further studies are necessary to document the impact of Opa1 ablation on muscle mitophagy.
Localization of mitochondrial DNA in Opa1‐deficient muscle cells
Recent data have established a relationship between autophagy malfunction and presence of undegraded mtDNA, which is recognized by TLR9 and results in inflammation (Nakahira et al, 2011; Oka et al, 2012). Based on the observation indicating that Opa1 deficiency causes an impairment in the resolution of mitophagy in muscle cells, next we explored the localization of mitochondrial DNA, in order to provide preliminary evidence on a re‐routing of mitochondria‐containing autophagosomes to the endosomal pathway, which would enable the recognition of mtDNA by TLR9. To this end, we analyzed the cellular localization of mtDNA regarding the different endo‐lysosomal compartments. In some studies, we carried out the co‐staining of mtDNA and VPS4 (a subunit of ESCRT‐III complex, involved in the formation of multivesicular bodies). We detected the presence of aggregation of VPS4 in Opa1 KD cells compared to a diffuse cytosolic, barely noticeable in control cells (Appendix Fig S5A). In addition, we detected a close co‐apposition of the signals corresponding to VPS4 and mtDNA in Opa1 KD cells (60% of mtDNA was close to VPS4; Appendix Fig S5A and B). There was no co‐localization between LAMP1 and VPS4; thus, we suggest the structures labeled by these markers are different (Appendix Fig S5C). The lack of triple staining with a mitochondrial protein or with a late endosomal marker does not permit us to conclude at this stage whether mitochondrial DNA or mitochondria are located in late endosomes.
Analysis of the co‐staining of mtDNA and early endosomes (EEA1 protein marker) showed a complete exclusion of the markers under control or Opa1‐deficient conditions (Appendix Fig S6A). Given the reported presence of TLR9 in the lysosomes (De Leo et al, 2016), we examined whether this could be a relevant compartment. To this end, we performed double and triple staining using LAMP1 as a lysosomal marker. These studies showed no co‐localization between LAMP1 and TOM20 (containing 100% of mtDNA nucleoids; Appendix Fig S6B). Triple labeling of mtDNA, LAMP1, and VPS4 in control and Opa1‐deficient cells showed exclusion localization between mtDNA and LAMP1, and apposition of mtDNA and VPS4 signals (Appendix Fig S6C). Based on the close apposition of the mtDNA and VPS4 signals but lack of triple staining, a co‐localization in the same structure may be still possible, but is not sufficiently supported by data at this stage.
TLR9 mediates inflammation upon Opa1 deficiency
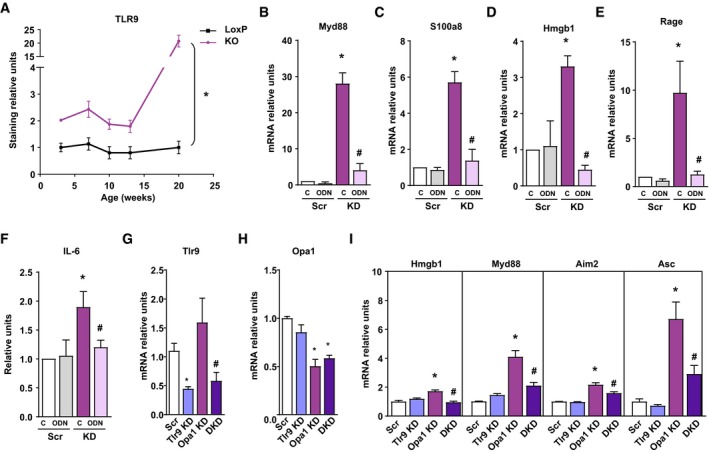
We next analyzed whether mtDNA co‐localizes with TLR9 in Opa1‐deficient muscle cells. In control cells, TLR9 was barely detectable, which was markedly enhanced in Opa1‐deficient cells (Appendix Fig S7A). In addition, Opa1 KD cells showed co‐localization of the mtDNA in TLR9 signals (Appendix Fig S7A). More than 50% of mtDNA signal co‐localized with TLR9 (Appendix Fig S7B). In addition, TLR9 did not co‐localize with lysosomes, when using LAMP1 as a marker, in control or in Opa1‐deficient cells (Appendix Fig S7C). Furthermore, TLR9 expression was increased in Opa1‐deficient muscles at various postnatal ages compared to controls (Fig 8A and Appendix Fig S7D). These data support the concept that TLR9 is activated upon Opa1 deficiency. Based on this, cells were treated with the TLR9 antagonist ligand ODN2088 or with the corresponding negative control. Incubation of Opa1 KD muscle cells with ODN2088 attenuated the expression of NF‐κB target genes, Myd88, S100a8, Hmgb1, or Rage (Fig 8B–E). In addition, the enhanced release of IL‐6 of Opa1‐deficient cells was inhibited by incubation with ODN2088 compared to the negative control (Fig 8F). Lastly, we analyzed whether similar effects were detected upon genetic depletion of TLR9. To this end, TLR9 was depleted in scramble or Opa1‐depleted muscle cells (Fig 8G and H). Depletion of TLR9 markedly reduced the induction of NF‐κB genes in Opa1‐deficient cells (Fig 8I), in agreement with the data obtained by the treatment with TLR9 antagonists. In all, our data are coherent with a TLR9 activation mediated by Opa1 deficiency.
Figure 8. TLR9 mediates inflammation upon Opa1 deficiency.
-
AQuantification of TLR9 expression immunostaining in gastrocnemius muscle of Opa1 KO animals at 3, 7, 10, 13, and 20 weeks of age. This quantification was performed using Trainable Weka Segmentation plugin from ImageJ (five images per group). Represented values are relative to loxP group.
-
B–EMyD88, S100A8, HMGB1, and RAGE expression in Scr and KD myoblasts treated with ODN2088 (ODN) or ODN2088 negative control (C) 1 μM (n = 9).
-
FIL‐6 levels detected in cultured medium of myoblasts (Scr and KD) treated with ODN2088 (ODN) or ODN2088 negative control (C) (n = 9).
-
G, HExpression of genes of TLR9 (G) and Opa1 (H) upon single or double knock‐down. n = 8 (G); n = 3 (H).
-
IExpression of NF‐κB target genes in control (Scr), Tlr9 depleted (Tlr9 KD), Opa1 depleted (Opa1 KD), and double Tlr9‐Opa1 ablated (DKD) cells (n = 3).
Discussion
In this study, we show that specific ablation of the mitochondrial fusion protein Opa1 in skeletal muscle results in whole body growth impairment, systemic inflammation, and premature death. The characterization of the Opa1 KO mice growth defects revealed low levels of circulating Igf1, high plasma growth hormone, and high Fgf21 expression in muscle as well as in circulation. We also found hepatic alterations in Opa1 KO mice such as enhanced Socs3 expression, reduced STAT5 phosphorylation, reduced gene expression of growth hormone receptor, and lower expression of growth hormone target genes. These data are consistent with the view that Opa1 deficiency in muscle causes global growth defects in conjunction with an altered Igf1 pathway (Wong et al, 2016). Given that high plasma concentrations of FGF21 have been observed to reduce growth hormone signaling, resulting in a marked growth defect (Inagaki et al, 2008), we examined whether the phenotype could be rescued by knocking out FGF21 expression globally. Analysis of double knockout mice proved that, although the hepatic expression of metabolic genes was restored, the growth defects due to Opa1 deficiency were independent of FGF21. Our observations also support the notion that muscle Opa1 exerts non‐cell‐autonomous actions on growth.
Inflammation has been reported to regulate growth (O'Connor et al, 2008). Growth inhibition is commonly observed in children with chronic inflammatory diseases such as juvenile idiopathic arthritis, chronic kidney disease, or chronic inflammatory bowel disease (Wong et al, 2016). In these patients, circulating concentrations of pro‐inflammatory cytokines such as IL‐1β and IL‐6 are elevated (Kutukculer et al, 1998; Ji et al, 2002). The use of mouse models has documented that overexpression of IL‐6 causes reduced circulating levels of Igf1 and reduced growth which is rescued by blocking anti‐IL‐6 antibodies (De Benedetti et al, 1997). In addition, TNF‐α, IL‐1β, and IL‐6 induce hepatic growth hormone resistance in mice (Zhao et al, 2014). In agreement with these data, here we report that Opa1 KO mice show systemic inflammation characterized by increased levels of IL‐1β and IL‐6. Furthermore, chronic treatment with salicylate resulted in substantial amelioration of muscle mass, body weight, muscle necrosis, and hepatic growth hormone resistance. Interestingly, anti‐inflammatory treatment also normalized FGF21 expression and its circulation levels, suggesting that the enhancement of the mitokine is downstream of inflammation. For now, we support the view that reduced growth in Opa1 KO mice is, at least in part, secondary to systemic inflammation.
In this study, we document the existence of a connection between Opa1 deficiency and muscle inflammation. Thus, we have demonstrated that skeletal muscle‐specific Opa1 ablation causes an inflammatory myopathy in mice, which is characterized by enhanced expression of NF‐κB target genes, and increased circulating levels of pro‐inflammatory cytokines IL‐1β and IL‐6. Inflammation was initially detected in 3‐week‐old Opa1 KO mice by enhanced IL‐1β abundance and normal CD68 labeling (a marker of macrophage infiltration), whereas at greater ages (13 weeks of age), CD68 labeling was enhanced, thereby indicating macrophage infiltration. In parallel to these data, tamoxifen‐induced Opa1 ablation revealed that the expression of some NF‐κB target genes such as NLRP3 and ASC was induced early under conditions in which muscle mass was normal. On the basis of these data, we conclude that Opa1 deficiency causes muscle inflammation which is independent of muscle mass loss, that occurs before macrophage infiltration, and that it is mediated via activation of NF‐κB. Cell culture studies in myoblasts also support the view that Opa1 deficiency causes a primary autonomous cellular response leading to inflammation. Opa1 deficiency in C2C12 muscle cells caused increased NF‐κB transcriptional activity, expression of NF‐κB target genes, and production of IL‐1β. This pattern of changes was not associated with cell death activation.
During the revision of this manuscript, two studies were published reporting the phenotype of muscle‐specific Opa1 depleted mice (Pereira et al, 2017; Tezze et al, 2017). In agreement with our data, they reported that ablation of Opa1 in skeletal muscle reduces muscle mass, and premature death. In addition, they also detected an enhanced muscle expression of Fgf21 in Opa1‐deficient muscles (Pereira et al, 2017; Tezze et al, 2017). We propose that the muscle inflammation driven by Opa1 deficiency is a primary event leading to the many alterations detected in this condition.
Dysregulation of mitochondrial dynamics has a major impact on mitochondrial function leading to a number of diseases (Liesa et al, 2009). OPA1 mutations causes an autosomal dominant optic atrophy (ADOA), and some OPA1 mutations lead to a multi‐systemic disorder called ADOA plus syndrome, which results in severe myopathy (Alexander et al, 2000; Delettre et al, 2000; Amati‐Bonneau et al, 2008; Hudson et al, 2008). Here, we report that Opa1 deficiency causes mitochondrial dysfunction both in skeletal muscle and in cultured muscle cells. This is characterized by mitochondrial fragmentation, reduced mitochondrial respiration, normal or enhanced expression of mitochondrial proteins, and unaltered expression of the mitochondrial biogenesis factor PGC1α.
Some OPA1 mutations are linked to mtDNA deletions, which has permit to propose a role of OPA1 on mtDNA stability (Amati‐Bonneau et al, 2008; Hudson et al, 2008). Here, we document that Opa1 deficiency was characterized by reduced mtDNA copy number, parallel to diminished TFAM protein abundance, reduced number of nucleoids, and increased nucleoid size. Opa1 deficiency has been reported to induce low mtDNA copy number and nucleoids abundance in human fibroblasts, which suggests that those alterations are not just restricted to muscle cells (Liao et al, 2017). Our data support the existence of mtDNA stress induced by Opa1 deficiency, which includes mtDNA loss and reduced nucleoids number.
Damaged mitochondria have been implicated in the induction of inflammation through the production of reactive oxygen species and the release of damage‐associated molecular patterns (DAMPs), namely formyl peptides and mtDNA (Zhang et al, 2010; Oka et al, 2012; Wenceslau et al, 2014). In this connection, we document that the inflammation detected in Opa1‐deficient muscle cells requires the presence of mtDNA so ethidium bromide‐treated Opa1‐deficient cells with more than 95% depletion of mtDNA, no longer show induction of NF‐κB target genes.
MtDNA stress triggered by TFAM deficiency has been reported to release mtDNA to the cytosol causing the activation of the DNA sensor cGAS, and to promote STING‐IRF3‐dependent signaling to induce interferon‐stimulated genes (West et al, 2015). However, here we report that Opa1 depletion causes mtDNA stress, but this occurs in the absence of leakage of mtDNA to the cytosol, and with no induction of interferon‐stimulated genes, and instead it causes induction of NF‐κB dependent on TLR9 activity.
A potential explanation for these different cellular responses under condition of mtDNA instability may lie on mitochondrial morphology. West et al (2015) showed a hyperfused phenotype of the mitochondrial network, and it has been reported the need for mitochondria to be fragmented prior to undergo mitophagy (Twig et al, 2008). Therefore, TFAM‐deficient mitochondria may not be included into autophagosomes, thus enabling mtDNA leakage to the cytosol. On the contrary, Opa1‐deficient mitochondria are completely fragmented which favors their entry into the mitophagic pathway, thus restraining the potential leakage to the inside of the vesicular system. Further studies regarding when and how mtDNA is released from mitochondria should be performed not only to understand whether there is an active form of transport of this mtDNA, but also to understand whether mtDNA has any control on the destination of the mito‐autophagosome. In this regard, it remains unclear the nature of the mechanisms by which Opa1 deficiency causes alterations in late stages of mitophagy, as well as in autophagy in muscle cells and whether this alteration also occurs in skeletal muscle. Studies need to be performed to understand whether this blockage of mitophagy completion is a consequence of general autophagy alterations, meaning that there are common players directly affected by Opa1 deficiency, or whether there are distinguishable defects for each pathway.
Based on the pattern of alterations detected, we speculate that in Opa1‐deficient cells, mitochondria‐containing autophagosomes are mainly diverted into the formation of amphisomes or amphisome‐like organelles, which is linked to the activation of TLR9. In this connection, further studies are required to determine whether Opa1 deficiency is linked to re‐routing of the mito‐autophagosome to the endosomal pathway, and if so, to address whether such a re‐routing may be an active programmed cause‐response mechanism or rather a consequence of the saturation of the lysosomal capacity due to the large amount of material to be degraded in an Opa1‐deficient context. The existence of communication mechanisms linking mitochondria and lysosomes may be responsible for the potential alterations in Opa1‐deficient cells. Thus, mitochondrial dysfunction has been reported to promote enhanced lysosomal biogenesis in some conditions, and reduced lysosomal function (Baixauli et al, 2015; Demers‐Lamarche et al, 2016; Fernandez‐Mosquera et al, 2017). Alternatively, Opa1 deficiency may regulate mTOR, which has been reported to regulate mitochondrial activity through translational control (Morita et al, 2013).
The existence of TLR9 activation is mainly suggested by the normalization of NF‐κB target genes expression upon TLR9 antagonist treatment or genetic depletion. The precise mechanism by how mtDNA may interact with TLR9 remains elusive, and our preliminary data indicate that mtDNA and TLR9 may be present in an intracellular compartment. However, a robust demonstration is lacking. We cannot exclude the possibility that Opa1 deficiency causes the activation of TLR9 by mtDNA through the formation of mitochondria‐derived vesicles, in spite of being reported under conditions of mitochondrial elongation (Neuspiel et al, 2008; McLelland et al, 2014, 2016).
In all, we document that Opa1 is required for mtDNA stability and mitochondrial quality control. As a result, Opa1 deficiency leads to the engagement of TLR9 resulting in the activation of NF‐κB inflammatory program. This is the mechanism by which Opa1 deficiency triggers muscle inflammation, which eventually becomes systemic, and leads to altered growth hormone/Igf1 axis, enhanced Fgf21 expression, and reduced growth. We propose that a deficient Opa1 activity may participate in the development of human inflammatory myopathies.
Materials and Methods
Refer to Appendix Supplementary Methods for a detailed description of all methods used.
Animal care
All animal experiments were done in compliance with the guidelines established by the Committee on Animal Care of the University of Barcelona. Mice were kept under a 12‐h dark‐light period and provided with a standard chow diet and water ad libitum. For detailed information of mouse strain generation, see Appendix Supplementary Methods.
Mice treatment
Sodium salicylate (200 mg/Kg, Sigma‐Aldrich) was administered daily by intraperitoneal injection for 30 days. Chloroquine (Sigma) was administered via i.p twice a day during 5 days at a concentration of 50 mg/kg into 3‐week‐old mice.
In vivo gene transfer and fluorescence microscopy of muscle sections
Left tibialis anterior was injected with the empty vector, while pDsRed2‐Mito (Clontech) was injected in the right muscle, and then muscles were electroporated. After 8 days, muscles were removed and fixed. 10‐μm cryosections were analyzed using a Leica TCS SP2 AOBS Systems confocal scanning microscope.
Histological analysis
For light microscopy, muscles were removed and embedded in OCT solution (TissueTek). Cryosections of 10 μm were stained with hematoxylin and eosin, Gomori's modified trichrome stain, or non‐specific esterase (Sigma) following standard protocols. Immunohistochemistry was performed with the indicated antibodies and labeling with the Vectastain ABC kit (Vector Laboratories), following the manufacturer's instructions.
Muscle regeneration studies
Regeneration of skeletal muscle was induced by intramuscular injection of 75 μl of 1 μM Cardiotoxin (CTX, Latoxan) in the gastrocnemius muscle (Kherif et al, 1999). Two days later, adenoviruses (microRNA Ctrl or microRNA Opa1) were injected in the region of the previous injury. Muscles were removed at 9, 12, and 22 days post‐injury. CSA was analyzed as described.
Respiration measurements in muscle
The respiration of permeabilized muscle was measured at 37°C by high‐resolution respirometry with an Oxygraph‐2k (Oroboros Instruments), as described (Kuznetsov et al, 2008).
Transmission electron microscopy
Gastrocnemius muscles were dissected, fixed, embedded, and then semi‐ and ultrathin cut and mounted, as described in the Appendix Supplementary Methods. JEM‐1010 electron microscope (Jeol, Japan) equipped with a CCD camera SIS Megaview III and the AnalySIS software were used for image acquisition.
Cell culture and stable knock‐down generation
Cells were grown in DMEM (Invitrogen) with 10% FBS and 100 U/ml of penicillin/streptomycin (Invitrogen) at 37°C in a humidified atmosphere of 5% CO2/95% O2. Lentiviral infection was used to deliver shRNA to C2C12 myoblasts using a procedure described in (Munoz & Zorzano, 2015).
Oxygen consumption measurements in C2C12 myoblasts
C2C12 myoblasts (Scr or Opa1 KD) were plated on SeaHorse Bioscience XF24 plates. After 48 h, oxygen consumption was measured using a Seahorse Bioscience XF24 extracellular flux analyzer. Extracellular acidification rate of C2C12 cells (ECAR) was measured in parallel. For detailed methods, see Appendix Supplementary Methods.
ATP content
ATP cellular content was measured using the ATP Determination Kit (Molecular Probes), following the manufacturer's instructions.
Immunofluorescence
Cells were fixed, permeabilized, and immunostained for the indicated components. For a detailed description of the protocol followed, see Appendix Supplementary Methods.
Mitochondrial fusion assays
One micro gram of mtPA‐GFP was transfected into C2C12 myoblasts (Scr or Opa1 KD) using Lipofectamine® 2000 (Lovy et al, 2012). Experiments were performed 48 h after transfection, and the protocol was followed as described previously.
Mitophagy assessment
To analyze mitophagy initiation, Scr and Opa1 KD C2C12 cells were treated with 30 μM CCCP (Sigma) for 30 min. Mitochondrial fractions were examined for recruited LC3‐II protein. To analyze mitophagy resolution, cells were treated with 30 μM CCCP for 9 and 16 h, and then, together with control untreated cells, fixed and immunostained for dsDNA. MtDNA nucleoid number was quantified using ImageJ software.
Protein extraction and Western blotting
Mitochondrial enriched fractions, cytosolic fractions, total cellular homogenates, and tissue homogenates were used for Western blotting. For detailed methods and antibodies used, see Appendix Supplementary Methods.
Plasma and cell culture media measurements
Plasma concentrations of creatine kinase (Thermo Scientific), lactate dehydrogenase (LDH) (BioVision), IGF‐1, IL‐6, and IL‐1β (Abnova), and GH and FGF21 (Millipore) were measured following the manufacturer's instructions. Cell culture media was concentrated, and cytokine concentration was assessed in a similar way.
Statistics
The data presented here were analyzed using the Student's t‐test or analysis of variance (ANOVA) with an appropriate post hoc test. Data are presented as mean ± SEM unless stated otherwise. Significance was established at P < 0.05.
Author contributions
AR‐N and AD‐R performed cellular and animal experiments and wrote the manuscript, and contributed equally to this work; EN, FD‐S, XD, JPM, MR, NP, DS, JS, and JV performed some cellular or animal experiments; CT, VR, and MS performed some animal studies; ML and OS participated in some cell studies; EP performed statistical analyses; JG and MB performed histological analysis; FR, SRD, and FV contributed with materials and analysis tools; MP analyzed the experimental data; AZ directed the research, revised the experimental data, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Movie EV1
Movie EV2
Review Process File
Acknowledgements
We thank Steve Forrow, Herbert Auer, the Unit of Electron Cryo‐Microscopy (UB), the Functional Genomics core facility (IRB Barcelona), and the Advanced Digital Microscopy core facility (IRB Barcelona) for technological assistance. A.R‐N. was the recipient of a FPI fellowship from the “Ministerio de Educación y Cultura,” Spain. E.N. was the recipient of a FPI fellowship from the “Ministerio de Educación y Cultura,” Spain. This study was supported by research grants from the MINECO (SAF2013‐40987R and SAF2016‐75246R), Grant 2014SGR48 from the Generalitat de Catalunya, CIBERDEM, and INFLAMES (PIE14/00045) grants from the “Instituto de Salud Carlos III”, grant “Todos somos raros, todos somos únicos” from Fundación Isabel Gemio, and INTERREG IV‐B‐SUDOE‐FEDER (DIOMED, SOE1/P1/E178). A.Z. is a recipient of an ICREA “Academia” (Generalitat de Catalunya). IRB Barcelona is the recipient of a Severo Ochoa Award of Excellence from MINECO (Government of Spain).
The EMBO Journal (2018) 37: e96553 29632021
References
- Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, Rodriguez M, Kellner U, Leo‐Kottler B, Auburger G, Bhattacharya SS, Wissinger B (2000) OPA1, encoding a dynamin‐related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet 26: 211–215 [DOI] [PubMed] [Google Scholar]
- Amati‐Bonneau P, Valentino ML, Reynier P, Gallardo ME, Bornstein B, Boissiere A, Campos Y, Rivera H, de la Aleja JG, Carroccia R, Iommarini L, Labauge P, Figarella‐Branger D, Marcorelles P, Furby A, Beauvais K, Letournel F, Liguori R, La Morgia C, Montagna P et al (2008) OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain 131: 338–351 [DOI] [PubMed] [Google Scholar]
- Arnoult D, Soares F, Tattoli I, Girardin SE (2011) Mitochondria in innate immunity. EMBO Rep 12: 901–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baixauli F, Acin‐Perez R, Villarroya‐Beltri C, Mazzeo C, Nunez‐Andrade N, Gabande‐Rodriguez E, Ledesma MD, Blazquez A, Martin MA, Falcon‐Perez JM, Redondo JM, Enriquez JA, Mittelbrunn M (2015) Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell Metab 22: 485–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caielli S, Athale S, Domic B, Murat E, Chandra M, Banchereau R, Baisch J, Phelps K, Clayton S, Gong M, Wright T, Punaro M, Palucka K, Guiducci C, Banchereau J, Pascual V (2016) Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med 213: 697–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carelli V, Musumeci O, Caporali L, Zanna C, La Morgia C, Del Dotto V, Porcelli AM, Rugolo M, Valentino ML, Iommarini L, Maresca A, Barboni P, Carbonelli M, Trombetta C, Valente EM, Patergnani S, Giorgi C, Pinton P, Rizzo G, Tonon C et al (2015) Syndromic parkinsonism and dementia associated with OPA1 missense mutations. Ann Neurol 78: 21–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L (2004) OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci USA 101: 15927–15932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana‐Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC, Perales‐Clemente E, Salviati L, Fernandez‐Silva P, Enriquez JA, Scorrano L (2013) Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155: 160–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Benedetti F, Alonzi T, Moretta A, Lazzaro D, Costa P, Poli V, Martini A, Ciliberto G, Fattori E (1997) Interleukin 6 causes growth impairment in transgenic mice through a decrease in insulin‐like growth factor‐I. A model for stunted growth in children with chronic inflammation. J Clin Investig 99: 643–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Leo MG, Staiano L, Vicinanza M, Luciani A, Carissimo A, Mutarelli M, Di Campli A, Polishchuk E, Di Tullio G, Morra V, Levtchenko E, Oltrabella F, Starborg T, Santoro M, Di Bernardo D, Devuyst O, Lowe M, Medina DL, Ballabio A, De Matteis MA (2016) Autophagosome‐lysosome fusion triggers a lysosomal response mediated by TLR9 and controlled by OCRL. Nat Cell Biol 18: 839–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc‐Carel C, Perret E, Astarie‐Dequeker C, Lasquellec L, Arnaud B, Ducommun B, Kaplan J, Hamel CP (2000) Nuclear gene OPA1, encoding a mitochondrial dynamin‐related protein, is mutated in dominant optic atrophy. Nat Genet 26: 207–210 [DOI] [PubMed] [Google Scholar]
- Demers‐Lamarche J, Guillebaud G, Tlili M, Todkar K, Belanger N, Grondin M, Nguyen AP, Michel J, Germain M (2016) Loss of mitochondrial function impairs lysosomes. J Biol Chem 291: 10263–10276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Mosquera L, Diogo CV, Yambire KF, Santos GL, Luna Sanchez M, Benit P, Rustin P, Lopez LC, Milosevic I, Raimundo N (2017) Acute and chronic mitochondrial respiratory chain deficiency differentially regulate lysosomal biogenesis. Sci Rep 7: 45076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, Scorrano L (2006) OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126: 177–189 [DOI] [PubMed] [Google Scholar]
- Griparic L, van der Wel NN, Orozco IJ, Peters PJ, van der Bliek AM (2004) Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J Biol Chem 279: 18792–18798 [DOI] [PubMed] [Google Scholar]
- Hudson G, Amati‐Bonneau P, Blakely EL, Stewart JD, He L, Schaefer AM, Griffiths PG, Ahlqvist K, Suomalainen A, Reynier P, McFarland R, Turnbull DM, Chinnery PF, Taylor RW (2008) Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain 131: 329–337 [DOI] [PubMed] [Google Scholar]
- Inagaki T, Lin VY, Goetz R, Mohammadi M, Mangelsdorf DJ, Kliewer SA (2008) Inhibition of growth hormone signaling by the fasting‐induced hormone FGF21. Cell Metab 8: 77–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N, Fujita Y, Oka T, Mihara K (2006) Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J 25: 2966–2977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Pettit A, Ohmura K, Ortiz‐Lopez A, Duchatelle V, Degott C, Gravallese E, Mathis D, Benoist C (2002) Critical roles for interleukin 1 and tumor necrosis factor alpha in antibody‐induced arthritis. J Exp Med 196: 77–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julian MW, Shao G, Vangundy ZC, Papenfuss TL, Crouser ED (2013) Mitochondrial transcription factor A, an endogenous danger signal, promotes TNFalpha release via RAGE‐ and TLR9‐responsive plasmacytoid dendritic cells. PLoS One 8: e72354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kherif S, Lafuma C, Dehaupas M, Lachkar S, Fournier JG, Verdiere‐Sahuque M, Fardeau M, Alameddine HS (1999) Expression of matrix metalloproteinases 2 and 9 in regenerating skeletal muscle: a study in experimentally injured and mdx muscles. Dev Biol 205: 158–170 [DOI] [PubMed] [Google Scholar]
- Kim JY, Hwang JM, Ko HS, Seong MW, Park BJ, Park SS (2005) Mitochondrial DNA content is decreased in autosomal dominant optic atrophy. Neurology 64: 966–972 [DOI] [PubMed] [Google Scholar]
- Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, Kim SS, Kim do H, Hur KY, Kim HK, Ko T, Han J, Kim HL, Kim J, Back SH, Komatsu M, Chen H, Chan DC, Konishi M, Itoh N et al (2013) Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med 19: 83–92 [DOI] [PubMed] [Google Scholar]
- Kopp E, Ghosh S (1994) Inhibition of NF‐kappa B by sodium salicylate and aspirin. Science 265: 956–959 [DOI] [PubMed] [Google Scholar]
- Kutukculer N, Caglayan S, Aydogdu F (1998) Study of pro‐inflammatory (TNF‐alpha, IL‐1alpha, IL‐6) and T‐cell‐derived (IL‐2, IL‐4) cytokines in plasma and synovial fluid of patients with juvenile chronic arthritis: correlations with clinical and laboratory parameters. Clin Rheumatol 17: 288–292 [DOI] [PubMed] [Google Scholar]
- Kuznetsov AV, Veksler V, Gellerich FN, Saks V, Margreiter R, Kunz WS (2008) Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat Protoc 3: 965–976 [DOI] [PubMed] [Google Scholar]
- Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, Lien E, Nilsen NJ, Espevik T, Golenbock DT (2004) TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol 5: 190–198 [DOI] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524: 309‐314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Czubryt MP, McAnally J, Bassel‐Duby R, Richardson JA, Wiebel FF, Nordheim A, Olson EN (2005) Requirement for serum response factor for skeletal muscle growth and maturation revealed by tissue‐specific gene deletion in mice. Proc Natl Acad Sci USA 102: 1082–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao C, Ashley N, Diot A, Morten K, Phadwal K, Williams A, Fearnley I, Rosser L, Lowndes J, Fratter C, Ferguson DJ, Vay L, Quaghebeur G, Moroni I, Bianchi S, Lamperti C, Downes SM, Sitarz KS, Flannery PJ, Carver J et al (2017) Dysregulated mitophagy and mitochondrial organization in optic atrophy due to OPA1 mutations. Neurology 88: 131–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesa M, Palacin M, Zorzano A (2009) Mitochondrial dynamics in mammalian health and disease. Physiol Rev 89: 799–845 [DOI] [PubMed] [Google Scholar]
- Liesa M, Shirihai OS (2013) Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab 17: 491–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Yan W, Tohme S, Chen M, Fu Y, Tian D, Lotze M, Tang D, Tsung A (2015) Hypoxia induced HMGB1 and mitochondrial DNA interactions mediate tumor growth in hepatocellular carcinoma through Toll‐like receptor 9. J Hepatol 63: 114–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovy A, Molina AJ, Cerqueira FM, Trudeau K, Shirihai OS (2012) A faster, high resolution, mtPA‐GFP‐based mitochondrial fusion assay acquiring kinetic data of multiple cells in parallel using confocal microscopy. J Vis Exp 65: e3991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLelland GL, Soubannier V, Chen CX, McBride HM, Fon EA (2014) Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J 33: 282–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLelland GL, Lee SA, McBride HM, Fon EA (2016) Syntaxin‐17 delivers PINK1/parkin‐dependent mitochondrial vesicles to the endolysosomal system. J Cell Biol 214: 275–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M, Gravel SP, Chenard V, Sikstrom K, Zheng L, Alain T, Gandin V, Avizonis D, Arguello M, Zakaria C, McLaughlan S, Nouet Y, Pause A, Pollak M, Gottlieb E, Larsson O, St‐Pierre J, Topisirovic I, Sonenberg N (2013) mTORC1 controls mitochondrial activity and biogenesis through 4E‐BP‐dependent translational regulation. Cell Metab 18: 698–711 [DOI] [PubMed] [Google Scholar]
- Munoz JP, Ivanova S, Sanchez‐Wandelmer J, Martinez‐Cristobal P, Noguera E, Sancho A, Diaz‐Ramos A, Hernandez‐Alvarez MI, Sebastian D, Mauvezin C, Palacin M, Zorzano A (2013) Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J 32: 2348–2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz JP, Zorzano A (2015) Analysis of mitochondrial morphology and function under conditions of mitofusin 2 deficiency. Methods Mol Biol 1265: 307–320 [DOI] [PubMed] [Google Scholar]
- Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12: 222–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuspiel M, Schauss AC, Braschi E, Zunino R, Rippstein P, Rachubinski RA, Andrade‐Navarro MA, McBride HM (2008) Cargo‐selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr Biol 18: 102–108 [DOI] [PubMed] [Google Scholar]
- O'Connor JC, McCusker RH, Strle K, Johnson RW, Dantzer R, Kelley KW (2008) Regulation of IGF‐I function by proinflammatory cytokines: at the interface of immunology and endocrinology. Cell Immunol 252: 91–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, Akira S, Yamamoto A, Komuro I, Otsu K (2012) Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485: 251–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, Lenaers G (2003) Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem 278: 7743–7746 [DOI] [PubMed] [Google Scholar]
- Pereira RO, Tadinada SM, Zasadny FM, Oliveira KJ, Pires KMP, Olvera A, Jeffers J, Souvenir R, McGlauflin R, Seei A, Funari T, Sesaki H, Potthoff MJ, Adams CM, Anderson EJ, Abel ED (2017) OPA1 deficiency promotes secretion of FGF21 from muscle that prevents obesity and insulin resistance. EMBO J 36: 2126–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesch UE, Leo‐Kottler B, Mayer S, Jurklies B, Kellner U, Apfelstedt‐Sylla E, Zrenner E, Alexander C, Wissinger B (2001) OPA1 mutations in patients with autosomal dominant optic atrophy and evidence for semi‐dominant inheritance. Hum Mol Genet 10: 1359–1368 [DOI] [PubMed] [Google Scholar]
- Potthoff MJ, Inagaki T, Satapati S, Ding X, He T, Goetz R, Mohammadi M, Finck BN, Mangelsdorf DJ, Kliewer SA, Burgess SC (2009) FGF21 induces PGC‐1alpha and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc Natl Acad Sci USA 106: 10853–10858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rongvaux A, Jackson R, Harman CC, Li T, West AP, de Zoete MR, Wu Y, Yordy B, Lakhani SA, Kuan CY, Taniguchi T, Shadel GS, Chen ZJ, Iwasaki A, Flavell RA (2014) Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 159: 1563–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastian D, Hernandez‐Alvarez MI, Segales J, Sorianello E, Munoz JP, Sala D, Waget A, Liesa M, Paz JC, Gopalacharyulu P, Oresic M, Pich S, Burcelin R, Palacin M, Zorzano A (2012) Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci USA 109: 5523–5528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastian D, Sorianello E, Segales J, Irazoki A, Ruiz‐Bonilla V, Sala D, Planet E, Berenguer‐Llergo A, Munoz JP, Sanchez‐Feutrie M, Plana N, Hernandez‐Alvarez MI, Serrano AL, Palacin M, Zorzano A (2016) Mfn2 deficiency links age‐related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J 35: 1677–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tezze C, Romanello V, Desbats MA, Fadini GP, Albiero M, Favaro G, Ciciliot S, Soriano ME, Morbidoni V, Cerqua C, Loefler S, Kern H, Franceschi C, Salvioli S, Conte M, Blaauw B, Zampieri S, Salviati L, Scorrano L, Sandri M (2017) Age‐associated loss of OPA1 in muscle impacts muscle mass, metabolic homeostasis, systemic inflammation, and epithelial senescence. Cell Metab 25: 1374–1389. e1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS (2008) Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 27: 433–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Shao B, He Z, Ye T, Luo M, Sang Y, Liang X, Wang W, Luo S, Yang S, Zhang S, Gong C, Gou M, Deng H, Zhao Y, Yang H, Deng S, Zhao C, Yang L, Qian Z et al (2015) Cationic nanocarriers induce cell necrosis through impairment of Na(+)/K(+)‐ATPase and cause subsequent inflammatory response. Cell Res 25: 237–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenceslau CF, McCarthy CG, Szasz T, Spitler K, Goulopoulou S, Webb RC, Working Group on DiCD (2014) Mitochondrial damage‐associated molecular patterns and vascular function. Eur Heart J 35: 1172–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Khoury‐Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, Kaech SM, Smiley JR, Means RE, Iwasaki A, Shadel GS (2015) Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520: 553–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, van Delft MF, Bedoui S, Lessene G, Ritchie ME, Huang DC, Kile BT (2014) Apoptotic caspases suppress mtDNA‐induced STING‐mediated type I IFN production. Cell 159: 1549–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SC, Dobie R, Altowati MA, Werther GA, Farquharson C, Ahmed SF (2016) Growth and the growth hormone‐insulin like growth factor 1 axis in children with chronic inflammation: current evidence, gaps in knowledge, and future directions. Endocr Rev 37: 62–110 [DOI] [PubMed] [Google Scholar]
- Yin MJ, Yamamoto Y, Gaynor RB (1998) The anti‐inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase‐beta. Nature 396: 77–80 [DOI] [PubMed] [Google Scholar]
- Yu EP, Bennett MR (2014) Mitochondrial DNA damage and atherosclerosis. Trends Endocrinol Metab 25: 481–487 [DOI] [PubMed] [Google Scholar]
- Yu‐Wai‐Man P, Sitarz KS, Samuels DC, Griffiths PG, Reeve AK, Bindoff LA, Horvath R, Chinnery PF (2010) OPA1 mutations cause cytochrome c oxidase deficiency due to loss of wild‐type mtDNA molecules. Hum Mol Genet 19: 3043–3052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeviani M (2008) OPA1 mutations and mitochondrial DNA damage: keeping the magic circle in shape. Brain 131: 314–317 [DOI] [PubMed] [Google Scholar]
- Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ (2010) Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464: 104–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JZ, Liu Z, Liu J, Ren JX, Sun TS (2014) Mitochondrial DNA induces inflammation and increases TLR9/NF‐kappaB expression in lung tissue. Int J Mol Med 33: 817–824 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhao Y, Xiao X, Frank SJ, Lin HY, Xia Y (2014) Distinct mechanisms of induction of hepatic growth hormone resistance by endogenous IL‐6, TNF‐alpha, and IL‐1beta. Am J Physiol Endocrinol Metab 307: E186–E198 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Movie EV1
Movie EV2
Review Process File