

Abstract
Purpose
Selection bias in clinical trials has consequences for scientific validity and applicability of study results to the general population. There is concern that patients with clinically aggressive disease may not have enrolled in recent diffuse large B-cell lymphoma (DLBCL) trials due to the consent process and the inability to delay therapy for eligibility evaluation. We have examined the diagnosis-to-treatment interval (DTI) and its association with clinical factors and outcome in a clinic-based observational cohort of patients with DLBCL from the United States. Validation of results was performed in an independent, clinical trial-based cohort from Europe.
Patients and Methods
Patients were prospectively enrolled in the University of Iowa and Mayo Clinic Specialized Programs of Research Excellence Molecular Epidemiology Resource (MER; N = 986) or the Lymphoma Study Association (LYSA) LNH-2003 clinical trials program (N = 1,444). All patients received anthracycline-based immunochemotherapy at initial diagnosis. Associations of DTI with clinical factors and outcome were examined. Outcome was assessed using event-free survival at 24 months from diagnosis (EFS24).
Results
Median (range) DTI was 15 days (0 to 155 days in the MER and 23 days (0 to 215 days) in LYSA. Shorter DTI was strongly associated with adverse clinical factors, including elevated lactate dehydrogenase levels, poor performance status, B symptoms, and higher International Prognostic Index in both cohorts (all P < .001). Longer DTI was associated with improved EFS24 in both the MER (per-week odds ratio, 0.80; 95% CI, 0.74 to .0.87) and LYSA (per-week odds ratio, 0.90; 95% CI, 0.86 to 0.94); association with EFS24 remained significant after adjustment for International Prognostic Index.
Conclusion
DTI is strongly associated with prognostic clinical factors and outcome in newly diagnosed DLBCL. DTI should be reported in all clinical trials of newly diagnosed DLBCL and future trials should take steps to avoid selection bias due to treatment delay.
INTRODUCTION
The selection bias of patients participating in cancer clinical trials has been long postulated to limit the generalizability of clinical trials to the general population.1 Although selection bias is widely acknowledged as a product of trial inclusion and exclusion criteria,2 other factors, such as patient preference, provider preference, socioeconomic status, and access to medical care, may inadvertently play a role in patient selection. The latter factors typically do not reflect different disease biology. However, in an aggressive malignancy, real or perceived urgency for initiation of therapy has to be weighed against the time required for trial screening, the consent process, and any relevant pathology and biomarker assessment. The resulting exclusion of patients with rapidly progressive or symptomatic disease may lead to selection of patients with less aggressive disease to participate in clinical trials. This bias may not be captured by study eligibility criteria and standard prognostic factors.
The phenomenon of time-dependent selection bias in clinical trials may be particularly evident in the treatment of diffuse large B-cell lymphoma (DLBCL). DLBCL is an aggressive malignancy and most patients are cured using anthracycline-based immunochemotherapy regimens, typically given as a combination of rituximab, cyclophosphamide, doxorubicin hydrochloride (aka, hydroxydaunorubicin), vincristine sulfate, and prednisone (R-CHOP)3-6 or similar combinations. Gene expression profiling has identified molecular phenotypes of DLBCL, grouping most patients into one of two molecular classifications: germinal center B cell (GCB) and activated B cell,7,8 and recent trial designs in DLBCL have included molecular phenotyping as part of eligibility criteria or subset analyses. These studies have typically required central pathology review before enrollment or treatment, with some trials requiring additional research biopsy procedures before enrollment. Several therapy combinations showing promise in nonrandomized studies largely failed to demonstrate improvement over the control arm in randomized studies.9-12 Moreover, better-than-expected outcomes on the control (standard therapy) arm were observed despite attempts to limit inclusion to patients with high-risk disease as defined by available clinical and biologic prognostic factors.10,13
To evaluate the potential influence of the timing of initiation of therapy on outcome, we have examined the effect of diagnosis-to-treatment interval (DTI) in DLBCL in two large, independent, prospectively enrolled cohorts of patients: (1) the University of Iowa/Mayo Clinic Specialized Program of Research Excellence (SPORE) Molecular Epidemiology Resource (MER), and (2) patients enrolled in the LNH-2003 clinical trials program from the Lymphoma Study Association (LYSA) group.
PATIENTS AND METHODS
Discovery Cohort
This study was reviewed and approved by the human subjects institutional review boards at the Mayo Clinic and the University of Iowa, and written informed consent was obtained from all participants. Details on the cohort have been previously reported.6,14,15 Briefly, adult patients with newly diagnosed DLBCL without prior lymphoma history were prospectively enrolled in the MER from 2002 to 2013. All patients were within 9 months of initial diagnosis at the time of enrollment. All diagnoses were confirmed by a study hematopathologist. Cell-of-origin (COO) determination (GCB v non-GCB) was performed in accordance with Hans algorithm.16 All patients in this analysis received anthracycline-based immunochemotherapy (IC); patients with primary CNS lymphoma, posttransplant lymphoproliferative disorders, and primary mediastinal B-cell lymphoma were excluded. Baseline clinical, laboratory, and treatment data were abstracted from medical records by using a standard protocol. Patients were systematically contacted every 6 months for the first 3 years and annually after 3 years; treatments, progression, and deaths were validated by medical record review. The date of diagnosis was defined as the biopsy date from the first biopsy specimen containing lymphoma; this includes biopsy specimens in which the subtype was unable to be determined and later biopsy specimens were needed to determine the subtype, as well as biopsies performed at an outside institution.
Validation Cohort
A validation cohort was assembled from patients enrolled in the LYSA LNH03 clinical trial series.17-21 The LNH2003B program of the LYSA consisted of six prospective, multicenter studies of patients with DLBCL who were older than 18 years. Patients were stratified by age and age-adjusted International Prognosis Index (IPI) for treatment allocation in four randomized phase III and two phase II studies. All patients were included in initial intent-to-treat analysis; sensitivity analysis was performed on data from patients with central pathology–confirmed DLBCL. During the first 2 years after treatment, assessment consisted of physical examination and laboratory tests every 3 months and computed tomography scans of the chest, abdomen, and pelvis every 6 months. Thereafter, physical examination and laboratory tests were done every 6 months and computed tomography scans every year for 5 years.
DTI was defined as the number of days from date of diagnosis to the initiation of IC therapy. Event-free survival (EFS) was defined as time from the start of IC therapy to progression or relapse; initiation of new, unplanned lymphoma therapy for efficacy purposes; or death (any cause). The primary outcome analysis used the DLBCL-specific end point of event-free survival at 24 months (EFS24), which was defined using the EFS status at 24 months from initiation of treatment.6,22 Loess curves were used to identify the nature of the association between DTI and EFS24. Strength of associations between clinical outcomes and DTI were assessed using χ2 and Wilcoxon rank-sum tests. Associations with EFS24 were assessed using logistic regression, with case weights for patients censored for EFS before 24 months. Two-sided P values were used for all tests. Additional details on the statistical analysis approach can be found in the Appendix, Statistical Methods.
RESULTS
MER Cohort (Discovery)
A total of 986 patients with DLBCL treated with IC and available DTI were enrolled in the MER from 2002 to 2012. Median age at diagnosis was 63 years (range, 18 to 92 years) and 57% were men. Most patients (65%) had advanced-stage disease. Full patient characteristics are listed in Table 1. At a median follow-up of 84 months (by reverse Kaplan-Meier analysis), 451 patients (46%) had an event and 340 patients (34%) had died; the EFS24 failure rate by Kaplan-Meier analysis was 31%(95% CI, 28% to 34%). The median (range) time from diagnosis to treatment initiation was 15 days (range, 0 to 155 days; Fig 1A).
Table 1.
Patient Characteristics

Fig 1.

Distributions of DTI by cohort. (A) Data for three patients with DTI > 100 days not shown. (B) Data for 13 patients with DTI not shown. DTI, diagnosis-to-treatment interval.
Shorter DTI was strongly associated with adverse prognostic factors at initial diagnosis (Table 2; Appendix Table A1, online only). Compared with patients who initiated treatment ≥ 15 days after diagnosis, patients initiating treatment within 14 days had higher lactate dehydrogenase (LDH) levels (elevated LDH levels, 68% v 42%; P < .001), advanced-stage disease (stage III-IV, 74% v 56%; P < .001), worse Eastern Cooperative Oncology Group performance status (ECOG PS ≥ 2, 28% v 10%; P < .001), more frequent B symptoms(33% v 18%; P < .001), more bulky (ie, ≥ 10 cm) disease (14% v 8%; P = .0030), and worse age-adjusted IPI (aaIPI 2-3, 58% v 33%; P < .001). Associations were weaker between DTI and sex (Wilcoxon P = .026) and age (Wilcoxon P = .077; Table 2; Appendix Table A1).
Table 2.
Patient Characteristics by Diagnosis-to-Treatment Interval ≤ 14 days

Examining the functional form showed an approximately linear association of increasing DTI and achieving EFS24 (Fig 2), and DTI was thus analyzed as a linear variable in models for EFS24. DTI was strongly associated with EFS24 in unadjusted (per-week odds ratio [OR], 0.77 [95% CI, 0.71 to 0.84]; P < .001) and IPI-adjusted models (per-week OR, 0.83 [95% CI, 0.76 to 0.91; P < .001; Table 3). Kaplan-Meier EFS curves grouped by week of DTI are shown in Figure 3 and results from Cox models examining the association between continuous DTI and continuous EFS are shown in the Appendix Table A5.
Fig 2.

Functional form of DTI versus EFS24. DTI, diagnosis-to-treatment interval; EFS24, event-free survival at 24 months; LYSA, Lymphoma Study Association; MER, Molecular Epidemiology Resource.
Table 3.
Logistic Regression Models of Continuous Diagnosis-to-Treatment Interval for Event-Free Survival at 24 Months

Fig 3.

(A and B) Kaplan-Meier Curves of event-free survival by diagnosis-to-treatment interval grouped by week.
We closely examined the MER data to assess if referral bias may explain MER results. There was no evidence of a strong association between the distance from a patient’s residence to their enrolling MER location with either DTI (Spearman correlation = 0.053; P = .093) or EFS24 (P = .61). Primary results were also consistent when stratified by patients who enrolled in MER before initiating therapy (56%) versus patients who enrolled after initiation of therapy (44%; results not shown).
LYSA Cohort (Validation)
A total of 1,446 patients were enrolled in the LYSA 2003 program from 2003 to 2009; DTI data were available for 1,444. Median age at diagnosis was 61 years (range, 17 to 95 years) and 54% were men. Most patients (73%) had advanced-stage disease. Full patient characteristics are listed in Table 1. At a median follow-up of 41 months (by reverse Kaplan-Meier analysis), 551 patients (38%) had an event and 352 patients (24%) had died; the EFS24 failure rate by Kaplan-Meier analysis was 34% (95% CI, 31% to 36%). The median (range) time from diagnosis to treatment was 23 days (range, 0 to 215 days), and 418 patients (29%) initiated treatment within 14 days of diagnosis (Fig 1B).
Similar to the MER, shorter DTI was associated with poor prognostic factors (Table 2; Appendix Table A1). Patients initiating treatment within 14 days had higher LDH levels (elevated LDH levels, 79% v 53%; P < .001), worse ECOG PS (ECOG PS≥ 2, 27% v 13%; P < .001), more frequent B symptoms(47% v 30%; P < .001), more bulky disease (30% v 17%; P < .001), and worse aaIPI (aaIPI 2-3, 63% v 47%; P < .001). The association was weaker between DTI and advanced-stage disease (stage III-IV, 76% v 71%; P = .050) and between DTI and sex (Wilcoxon P = .089); there was no evidence of significant association between DTI and age (Wilcoxon P = .14). Results were consistent in the subset of patients with central pathology confirmed DLBCL (Appendix Table A4).
DTI was also strongly associated with outcome in the LYSA cohort. The functional form analysis confirmed a linear association of improving EFS24 with longer DTI (Fig 2). Longer DTI was associated with improved EFS24 in unadjusted (per-week OR, 0.88 [95% CI, 0.84 to 0.92]; P < .001) and IPI-adjusted models (per-week OR, 0.93 [95% CI, 0.88 to 0.98]; P = .0050). The association was slightly stronger in the subset of patients (n = 1,222) with central pathology–confirmed DLBCL (IPI-adjusted OR, 0.92 [95% CI, 0.86 to 0.987], P = .0023; Appendix Table A2). Kaplan-Meier EFS curves grouped by per-week DTI are shown in Figure 3 and results from Cox models examining association between continuous DTI and continuous EFS are reported in Appendix Table A5.
Subset and Sensitivity Analyses in Combined Cohort
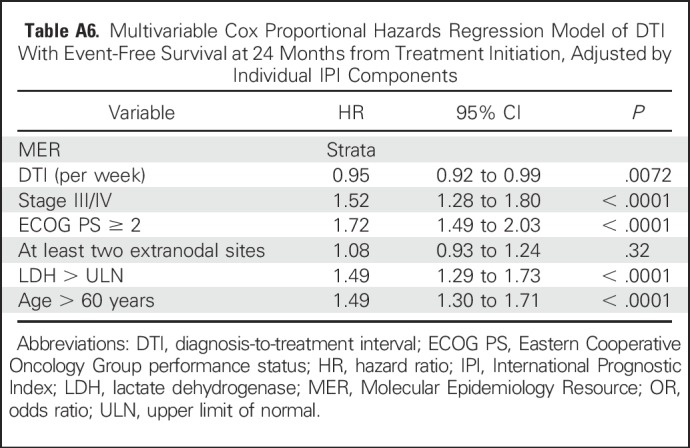
To increase the sample size to assess DTI in specific patient subsets, we combined the MER and LYSA cohorts with analysis adjusted by cohort (MER v LYSA). The association between longer DTI and improved EFS24 was consistent in GCB (OR, 079 [95% CI, 0.71 to 0.86]; P < .001) and non-GCB (OR, 0.82 [95% CI, 0.73 to 0.92]; P < .001) COO subsets and remained significant when examined in low-risk (IPI 0-2: OR,0.88 [95% CI, 0.82 to 0.93]; P < .001), and high-risk (IPI 3-5: OR, 0.89 [95% CI, 0.84 to 0.95]; P < .001) patient subsets (Table 3), as well as when adjusting for individual IPI components in a multivariable model (OR, 0.93 [95% CI, 0.88 to 0.97], P = .0014; Appendix Tables A3 and A6).
DISCUSSION
In this study, we identified that the simple measure of the number of days between diagnosis and treatment initiation identifies patients with vastly different outcomes in newly diagnosed DLBCL. Patients who initiate therapy sooner have significantly worse outcomes compared with patients with longer time from diagnosis to initiation of treatment. This effect appears to be a function of disease aggressiveness: Shorter DTI was strongly associated with adverse disease–related prognostic factors, including elevated LDH level, poor performance status, symptomatic disease, and bulky disease. Importantly, however, the prognostic value of DTI was independent of the IPI. The effect between DTI and outcome appears to be continuous, which is important for translation to other datasets, because a typical DTI will likely vary based on regional clinical and pathology practices. This observation has important implications in any research efforts that may restrict patient participation based on time from diagnosis to treatment.
Strengths of the study include demonstration of these findings in two large, prospective cohorts, including a US clinic–based cohort and a European clinical trials cohort. Limitations include the lack of COO classification on the complete cohorts, though results were consistent in the subset with available data. Clinical parameters in the MER were captured as performed clinically under routine care; when multiple assessments were performed before the beginning treatment, the assessment closest to diagnosis was used. Thus, there may be changes in these clinical variables before the initiation of therapy; however, assessment of the effect of this variation is beyond the scope of the current study.
Patients enrolled in the MER are from two tertiary care centers in the upper midwestern United States.15 All MER patients were seen at either the Mayo Clinic or University of Iowa within 9 months of initial diagnosis and some may have initiated treatment locally in the community. We did not see any association between the distance from a patient’s residence to their enrolling MER center with either DTI or outcome; other demographic factors such as age and sex were not strongly associated with DTI. Results were also consistent in patients with pretherapy versus posttherapy MER consent. Two recent Canadian studies examined the timing of treatment in DLBCL, focusing on delay in therapy and potential detrimental effect.23,24 Neither study found an association between socioeconomic status, income, or distance to treatment center with timing of therapy, but both identified variables related to more aggressive disease such as IPI, bone marrow involvement, and inpatient chemotherapy initiation, to be related to prevention of a treatment delay. In addition, patients with none to 1 week of treatment delay had the worst overall survival in the Toronto series,23 whereas outcome was inferior for patients with DTI of no longer than 4 weeks in a series from the British Columbia Cancer Agency.24 These findings further support DTI being a function of disease aggressiveness and not because of referral bias.
The association of DTI with clinical factors and outcomes were validated in an independent cohort of European patients. Patients in the LYSA cohort initiated therapy (median DTI, 23 days) later than the MER cohort (median DTI, 15 days), with only 29% of the LYSA cohort initiating therapy within 14 days. Associations between EFS24 and DTI in the LYSA cohort were also slightly weaker than in the MER, though they remained statistically significant and independent of the IPI. These differences are not unexpected, because all the LYSA patients were enrolled in the trial (v 9% in MER), so presumably some patient selection was present and the LYSA cohort may underrepresent patients with clinically aggressive disease who needed urgent therapy. Validation of the association of DTI with outcome in such a cohort provides strong evidence that the observed associations in the MER are not a spurious finding. Furthermore, similar associations among DTI and prognostic factors and outcome have recently been reported25 from the GOYA (GA101 for diffuse large B-cell lymphoma) trial.9
These findings have important consequences for design and interpretation of clinical trials in DLBCL. DTI must be taken into account when analyzing results of clinical trials or other efforts to compare otherwise similar cohorts. This is especially of concern in single-arm studies where success rules are predicated on the basis of historical data; these data suggest the improvement in outcomes may be strictly due to a lengthier process to enroll the patient in the trial. DTI should be used to evaluate single-arm clinical trials to gauge potential selection bias and help inform validity of results when compared with historical data or other trials in the setting of newly diagnosed DLBCL. DTI can similarly be used to evaluate the control arm for randomized trials and provide inference when outcomes are better than expected. The observation that DTI has an independent association with outcome beyond established clinical and biologic prognostic factors suggests that current trial inclusion criteria are not sufficient for nonbiased patient selection. Efforts to include patients with clinically aggressive disease in need of urgent therapy are critically important to ensure a representative group of patients enrolled in the trial. Strategies for this may include streamlined enrollment procedures, rapid review of pathological material, or allowing bridging therapy before starting the trial.26,27 Single-arm trials can consider determination of biomarker-based eligibility while patients are undergoing the first cycle of experimental treatment, with ineligible patients receiving subsequent therapy off trial if determined ineligible.28 Multiarm trials could consider beginning all patients on standard-of-care therapy (eg, R-CHOP) while phenotyping is ongoing, and assigning study therapies after cycle 1. Researchers may also want to consider an upper limit on time from diagnosis as part of entry criteria.
Prognostic scores such as the IPI are designed to help stratify patients, with the goal of lessening bias in clinical trials and assisting clinicians in identifying the most appropriate treatment of a given patient based on results of prior studies. Our observation that DTI is a prognostic factor independent of IPI suggests clinicians can identify patient factors that lead them to accelerate initiation of therapy that are, indeed, associated with a worse prognosis yet are not accounted for in standard prognostic tools. This supports ongoing efforts to continue to refine prognostic scales such as the IPI and demonstrates clinical judgement remains a vital factor in determining the right treatment of the right patient even in this era of personalized molecular medicine.
Our observations regarding importance of DTI in the newly diagnosed setting may also have importance in relapsed and refractory DLBCL, where the same principles may apply. The effect of the ability to delay therapy on response rate and outcome is unknown in the relapsed setting, but recent data from the MER29 and the SCHOLAR-1 study30 suggest significant diversity in clinical outcomes in the relapsed setting. The observed associations among DTI, clinically aggressive disease, and outcomes at diagnosis are especially concerning for patient selection bias and, subsequently, optimistic response rates and outcomes in single-arm studies of novel agents that require therapy delays because of specimen testing and/or preparation of individualized therapy, as well as potential delays while patients await slots in the trial to open at their treatment location.
Our results for DLBCL are, in fact, broadly relevant in all malignancies as the regulatory, pathologic, and/or genetic requirements to enroll in a trial have become stricter, and more laborious and time consuming. The danger lies in patients who enroll in the trial who do not accurately represent the target patient population for the therapy. Many precision medicine trials are not randomized and there is concern of being too optimistic about single-arm trial results, because of patient selection for known and unknown factors. There is concern that exceptional outcomes in small, uncontrolled studies of targeted agents with biomarker-based testing are influenced by selection of patients healthy enough to remain in a holding pattern while waiting for trial testing; our results suggest that this selection may bias toward a more favorable clinical disease as well.
In conclusion we have identified that the time from diagnosis to treatment is an important factor in newly diagnosed DLBCL and is strongly associated with adverse clinical factors and poor outcome. DTI should be reported in all clinical trials of newly diagnosed DLBCL. Steps must be taken in clinical trials in newly diagnosed DLBCL to avoid selection bias due to treatment delay (Appendix Tables A4-A6.
Appendix
Statistical Methods
Diagnosis-to-treatment interval (DTI) was defined as the number of days from date of first lymphoma-containing biopsy specimen to the initiation of immunochemotherapy. Patient’s residence distance from Molecular Epidemiology Resource location was defined using the zip code of patient residence and the enrolling center (Rochester, MN, or Iowa City, IA). Event-free survival (EFS) was defined as time from initiation of therapy to progression or relapse; initiation of new, unplanned lymphoma therapy; or death (any cause). The primary outcome analysis used the diffuse large B-cell lymphoma–specific end point of event-free survival at 24 months (EFS24), which was defined using the EFS status at 24 months from initiation of therapy.6,22 Patients with < 24 months of follow-up were included, using case weights based on probability of achieving EFS24, as previously described (Maurer MJ, et al: Am J Hematol 91:179-184, 2016). Association of continuous DTI with clinical factors was assessed using Wilcoxon rank-sum and Spearman correlation; clinical factors were also assessed using the median split (0 to 14 days) of DTI from the discovery dataset via χ2 and Wilcoxon rank-sum tests. Associations between DTI and EFS24 were assessed using logistic regression models with case weights for patients censored for EFS before 24 months. The functional form of association between continuous DTI with EFS24 was assessed using loess curves (Cleveland: J Am Stat Assoc 74:829-836) and a linear fit through 60 days from diagnosis was used for modeling DTI with EFS24. DTI was grouped by week from enrollment for Kaplan-Meier analyses of continuous EFS and Cox proportional hazards models were used to assess associations between DTI and continuous EFS. Validation of Molecular Epidemiology Resource results was performed in the Lymphoma Study Association (LYSA) cohort using all patients enrolled on the LYSA trials as an intent-to-treat approach. Sensitivity analyses were performed on the data from the subset of patients in the LYSA cohort who had diffuse large B-cell lymphoma on central pathology review. Two-sided P values were used for all tests. All analyses were performed using SAS, version 9.4 (SAS Institute, Cary, NC) and R, version 3.3.1 (https://www.r-project.org/).
Table A1.
Association of Patient Characteristics With Continuous DTI

Table A2.
LYSA Sensitivity Analysis of DTI With Event-Free Survival at 24 Months in Patients With Central Pathology–Confirmed DLBCL
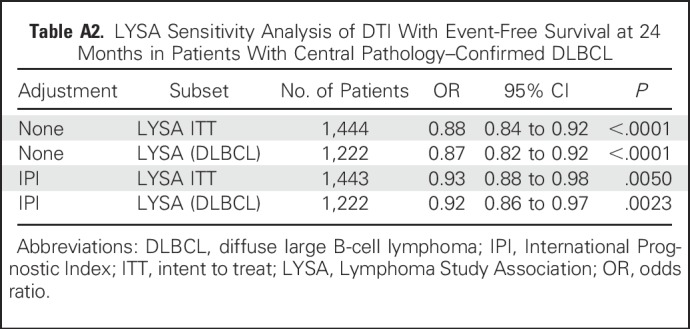
Table A3.
Multivariable Logistic Regression Model of DTI With Event-Free Survival at 24 Months Adjusted by Individual IPI Components
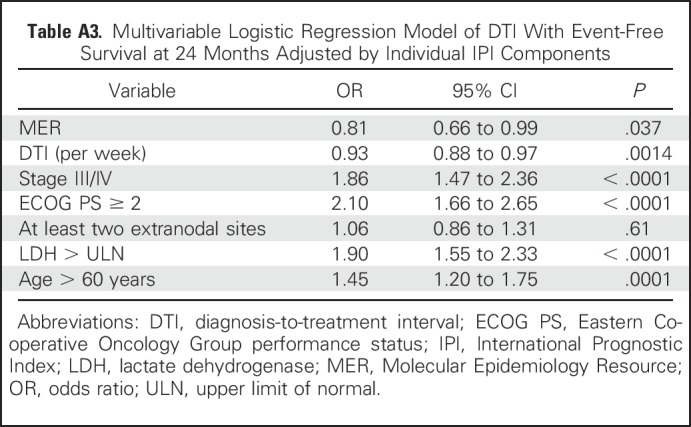
Table A4.
LYSA Sensitivity Analysis of DTI With Clinical Characteristics in Patients With Central Pathology–Confirmed DLBCL

Table A5.
Cox Models of Continuous DTI with Event-Free Survival

Table A6.
Multivariable Cox Proportional Hazards Regression Model of DTI With Event-Free Survival at 24 Months from Treatment Initiation, Adjusted by Individual IPI Components
Footnotes
Supported in part by National Cancer Institute Specialized Programs of Research Excellence in Human Cancer (Grants No. P50 CA97274 and U01 CA195568), and the Henry J. Predolin Foundation.
Presented in part at the 58th American Society of Hematology Annual Meeting, San Diego, CA, December 4, 2016; 59th American Society of Hematology Annual Meeting, Atlanta, GA, December 11, 2017.
AUTHOR CONTRIBUTIONS
Conception and design: Matthew J. Maurer, Hervé Ghesquières, Thomas M. Habermann, Andrew L. Feldman, James R. Cerhan, Gilles A. Salles, Thomas E. Witzig, Grzegorz S. Nowakowski
Collection and assembly of data: All authors
Data analysis and interpretation: Matthew J. Maurer, Hervé Ghesquières, Brian K. Link, Jean-Philippe Jais, Thomas M. Habermann, Carrie A. Thompson, Corinne Haioun, Cristine Allmer, Patrick B. Johnston, Richard Delarue, Ivana N. Micallef, Frederic Peyrade, David J. Inwards, Nicolas Ketterer, Umar Farooq, William R. Macon, Thierry J. Molina, Sergei Syrbu, Andrew L. Feldman, Susan L. Slager, George J. Weiner, Stephen M. Ansell, James R. Cerhan, Gilles A. Salles, Thomas E. Witzig, Hervé Tilly, and Grzegorz S. Nowakowski
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Diagnosis-to-Treatment Interval Is an Important Clinical Factor in Newly Diagnosed Diffuse Large B-cell Lymphoma and Has Implication for Bias in Clinical Trials
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Matthew J. Maurer
Research Funding: Celgene (Inst)
Hervé Ghesquières
No relationship to disclose
Brian K. Link
Consulting or Advisory Role: Roche, AbbVie, Gilead Sciences, Celgene
Research Funding: Roche (Inst)
Travel, Accommodations, Expenses: Roche, Celgene
Jean-Philippe Jais
No relationship to disclose
Thomas M. Habermann
No relationship to disclose
Carrie A. Thompson
Research Funding: Kite Pharma
Corinne Haioun
Honoraria: Amgen, Roche, Celgene, Janssen-Cilag, Gilead Sciences, Takeda
Consulting or Advisory Role: Roche, Celgene
Cristine Allmer
Employment: Roche (I)
Patrick B. Johnston
No relationship to disclose
Richard Delarue
No relationship to disclose
Ivana N. Micallef
No relationship to disclose
Frederic Peyrade
Consulting or Advisory Role: Merck
Research Funding: MSD Oncology
David J. Inwards
No relationship to disclose
Nicolas Ketterer
No relationship to disclose
Umar Farooq
Honoraria: Celgene
Olivier Fitoussi
Consulting or Advisory Role: Roche, Janssen-Cilag
William R. Macon
No relationship to disclose
Thierry J. Molina
Consulting or Advisory Role: Merck
Travel, Accommodations, Expenses: Takeda
Sergei Syrbu
No relationship to disclose
Andrew L. Feldman
Consulting or Advisory Role: Infinity Pharmaceuticals
Patents, Royalties, Other Intellectual Property: Inventor of technology for which Mayo Clinic holds an unlicensed patent or has submitted a patent application (Inst)
Susan L. Slager
No relationship to disclose
George J. Weiner
Consulting or Advisory Role: Sandoz, Checkmate Pharmaceuticals
Research Funding: Checkmate Pharmaceuticals
Stephen M. Ansell
Honoraria: WebMD, Research to Practice
Research Funding: Bristol-Myers Squibb (Inst), Celldex (Inst), Seattle Genetics (Inst), Merck (Inst), Affimed Therapeutics (Inst), Trillium Therapeutics (Inst)
James R. Cerhan
No relationship to disclose
Gilles A. Salles
Honoraria: Roche, Amgen, Janssen, Bristol-Myers Squibb, Celgene, Servier, Gilead Sciences, Novartis
Consulting or Advisory Role: Roche, Gilead Sciences, Janssen, Celgene, Novartis, NOvimmune, Merck
Research Funding: Roche (Inst)
Travel, Accommodations, Expenses: Roche, Sanofi
Thomas E. Witzig
No relationship to disclose
Hervé Tilly
No relationship to disclose
Grzegorz S. Nowakowski
Consulting or Advisory Role: Celgene (Inst), MorphoSys (Inst), Genetech (Inst)
Research Funding: Celgene (Inst)
REFERENCES
- 1.Antman K, Amato D, Wood W, et al. : Selection bias in clinical trials. J Clin Oncol 3:1142-1147, 1985 [DOI] [PubMed] [Google Scholar]
- 2.Unger JM, Barlow WE, Martin DP, et al. : Comparison of survival outcomes among cancer patients treated in and out of clinical trials. J Natl Cancer Inst 106:dju002, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coiffier B, Lepage E, Brière J, et al. : CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 346:235-242, 2002 [DOI] [PubMed] [Google Scholar]
- 4.Habermann TM, Weller EA, Morrison VA, et al. : Rituximab-CHOP versus CHOP alone or with maintenance rituximab in older patients with diffuse large B-cell lymphoma. J Clin Oncol 24:3121-3127, 2006 [DOI] [PubMed] [Google Scholar]
- 5.Pfreundschuh M, Trümper L, Österborg A, et al. : CHOP-like chemotherapy plus rituximab versus CHOP-like chemotherapy alone in young patients with good-prognosis diffuse large-B-cell lymphoma: A randomised controlled trial by the MabThera International Trial (MInT) Group. Lancet Oncol 7:379-391, 2006 [DOI] [PubMed] [Google Scholar]
- 6.Maurer MJ, Ghesquières H, Jais J-P, et al. : Event-free survival at 24 months is a robust end point for disease-related outcome in diffuse large B-cell lymphoma treated with immunochemotherapy. J Clin Oncol 32:1066-1073, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alizadeh AA, Eisen MB, Davis RE, et al. : Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403:503-511, 2000 [DOI] [PubMed] [Google Scholar]
- 8.Rosenwald A, Wright G, Chan WC, et al. : The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 346:1937-1947, 2002 [DOI] [PubMed] [Google Scholar]
- 9.Vitolo U, Trněný M, Belada D, et al. : Obinutuzumab or rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated diffuse large B-cell lymphoma. J Clin Oncol 35:3529-3537, 2017 [DOI] [PubMed] [Google Scholar]
- 10.Leonard JP, Kolibaba KS, Reeves JA, et al. : Randomized phase II study of R-CHOP with or without bortezomib in previously untreated patients with non–germinal center B-cell–like diffuse large B-cell lymphoma. J Clin Oncol 35:3538-3546, 2017 [DOI] [PubMed] [Google Scholar]
- 11.Wilson WH, sin-Ho J, Pitcher BN, et al. : Phase III randomized study of R-CHOP versus DA-EPOCH-R and molecular analysis of untreated diffuse large B-cell lymphoma: CALGB/Alliance 50303. Blood 128:469, 2016 [Google Scholar]
- 12.Offner F, Samoilova O, Osmanov E, et al. : Frontline rituximab, cyclophosphamide, doxorubicin, and prednisone with bortezomib (VR-CAP) or vincristine (R-CHOP) for non-GCB DLBCL. Blood 126:1893-1901, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goy A: Succeeding in breaking the R-CHOP ceiling in DLBCL: Learning from negative trials. J Clin Oncol 35:3519-3522, 2017 [DOI] [PubMed] [Google Scholar]
- 14.Thompson CA, Ghesquieres H, Maurer MJ, et al. : Utility of routine post-therapy surveillance imaging in diffuse large B-cell lymphoma. J Clin Oncol 32:3506-3512, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cerhan JR, Link BK, Habermann TM, et al. : Cohort profile: The Lymphoma Specialized Program of Research Excellence (SPORE) Molecular Epidemiology Resource (MER) Cohort Study. Int J Epidemiol 46:1753-1754i, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hans CP, Weisenburger DD, Greiner TC, et al. : Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 103:275-282, 2004 [DOI] [PubMed] [Google Scholar]
- 17.Récher C, Coiffier B, Haioun C, et al. : Intensified chemotherapy with ACVBP plus rituximab versus standard CHOP plus rituximab for the treatment of diffuse large B-cell lymphoma (LNH03-2B): An open-label randomised phase 3 trial. Lancet 378:1858-1867, 2011 [DOI] [PubMed] [Google Scholar]
- 18.Fitoussi O, Belhadj K, Mounier N, et al. : Survival impact of rituximab combined with ACVBP and upfront consolidation autotransplantation in high-risk diffuse large B-cell lymphoma for GELA. Haematologica 96:1136-1143, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peyrade F, Jardin F, Thieblemont C, et al. : Attenuated immunochemotherapy regimen (R-miniCHOP) in elderly patients older than 80 years with diffuse large B-cell lymphoma: A multicentre, single-arm, phase 2 trial. Lancet Oncol 12:460-468, 2011 [DOI] [PubMed] [Google Scholar]
- 20.Ketterer N, Coiffier B, Thieblemont C, et al. : Phase III study of ACVBP versus ACVBP plus rituximab for patients with localized low-risk diffuse large B-cell lymphoma (LNH03-1B). Ann Oncol 24:1032-1037, 2013 [DOI] [PubMed] [Google Scholar]
- 21.Delarue R, Tilly H, Mounier N, et al. : Dose-dense rituximab-CHOP compared with standard rituximab-CHOP in elderly patients with diffuse large B-cell lymphoma (the LNH03-6B study): A randomised phase 3 trial. Lancet Oncol 14:525-533, 2013 [DOI] [PubMed] [Google Scholar]
- 22.Jakobsen LH, Bøgsted M, Brown PN, et al. : Minimal loss of lifetime for patients with diffuse large B-cell lymphoma in remission and event free 24 months after treatment: A Danish population-based study. J Clin Oncol 35:778-784, 2017 [DOI] [PubMed] [Google Scholar]
- 23.Nikonova A, Guirguis HR, Buckstein R, et al. : Predictors of delay in diagnosis and treatment in diffuse large B-cell lymphoma and impact on survival. Br J Haematol 168:492-500, 2015 [DOI] [PubMed] [Google Scholar]
- 24.Hay K, Lee B, Goktepe O, et al. : Impact of time from diagnosis to initiation of curative chemotherapy on survival of patients with diffuse large B-cell lymphoma. Leuk Lymphoma 57:1-7, 2015 [DOI] [PubMed] [Google Scholar]
- 25.Szafer-Glusman E, Liu J, Peale FV, et al. : A simulation analysis to evaluate the effect of prospective biomarker testing on progression-free survival (PFS) in DLBCL. Blood 130:419, 2017 [Google Scholar]
- 26.Nowakowski GS, Vitolo U: Recent advances in clinical studies and the evolving role of subtyping for patients with diffuse large B-cell lymphoma. Future Oncol 13:859-862, 2017 [DOI] [PubMed] [Google Scholar]
- 27.King RL, Nowakowski GS, Witzig TE, et al. : Rapid, real-time central pathology review for E1412: A novel and successful paradigm for future National Clinical Trials Network diffuse large B cell lymphoma studies. J Clin Oncol 35:7547, 2017. (suppl 15) [Google Scholar]
- 28.Micallef INM, Maurer MJ, Wiseman GA, et al. : Epratuzumab with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone chemotherapy in patients with previously untreated diffuse large B-cell lymphoma. Blood 118:4053-4061, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Farooq U, Maurer MJ, Thompson CA, et al. : Clinical heterogeneity of diffuse large B cell lymphoma following failure of front-line immunochemotherapy. Br J Haematol 179:50-60, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crump M, Neelapu SS, Farooq U, et al. : Outcomes in refractory diffuse large B-cell lymphoma: Results from the international SCHOLAR-1 study. Blood 130:1800-1808, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]