

Abstract
Purpose
Terminal differentiation of erythropoietic progenitors requires the rapid accumulation of large amounts of iron, which is transported to the mitochondria, where it is incorporated into heme. Ferritin is the sole site of iron storage present in the cytosol. Yet the role of iron accumulation into ferritin in the context of red cell development had not been clearly defined. Early studies indicated that at the onset of terminal differentiation, iron initially accumulates in ferritin and precedes heme synthesis. Whether this accumulation is physiologically important for red cell development was unclear until recent studies defined an obligatory pathway of iron flux through ferritin.
Recent findings
The iron chaperone functions of poly rC-binding protein 1 (PCBP1) and the autophagic cargo receptor for ferritin, nuclear co-activator 4 (NCOA4) are required for the flux of iron through ferritin in developing red cells. In the absence of these functions, iron delivery to mitochondria for heme synthesis is impaired.
Summary
The regulated trafficking of iron through ferritin is important for maintaining a consistent flow of iron to mitochondria without releasing potentially damaging redox-active species in the cell. Other components of the iron trafficking machinery are likely to be important in red cell development.
Keywords: Erythropoiesis, iron chaperone, PCBP1, NCOA4, autophagy
Introduction
Terminal differentiation of erythroid precursors is a precisely orchestrated developmental process in which the contents of the cell are rapidly and progressively replaced with a highly concentrated solution of hemoglobin. This process of hemoglobinization requires the coordinated accumulation of iron, synthesis of heme, and translation of globin chains for assembly into hemoglobin [1]. Each of these components must be available in the proper amounts simultaneously. Deficiency or excess of any one of these components is toxic to the developing red cell. Excess cellular iron exerts its toxicity through mis-trafficking (competition with other divalent metals) and chemical reactivity (generation of reactive oxygen species) [2]. Because of the tremendous accumulation of iron, heme, and globin over a short period of development, erythroid cells exhibit multiple redundant systems to control this process and prevent toxicity. This review will focus on the role of ferritin in terminal erythroid differentiation.
Managing the flow of iron in terminal differentiation
Terminal differentiation begins with a dramatic increase in iron uptake [1]. This uptake is triggered by the transcriptional up-regulation of transferrin receptor 1(TfR1), which traffics to the surface of the cell and captures circulating diferric transferrin (Fe-Tf) from the extracellular milieu. TfR1 with bound Fe-Tf undergoes endocytosis and it is within the endosomal compartment that the Fe(III) is released from Tf, reduced to Fe(II), and transported into the cytosol. Divalent metal transporter 1 (DMT1) facilitates transport of Fe(II) across the endosomal membrane to the cytosol. Ultimately, the iron is delivered to the mitochondria, where it is transferred to the mitochondrial matrix for insertion into heme. The route and the machinery that intracellular iron takes to arrive in the mitochondria remained unclear until a series of studies confirmed the role of ferritin in the flux of iron through the developing erythroid cell. Unlike heme cofactors or globin protomers, iron can safely be accumulated and stored in a developing red cell through deposition into ferritin.
Ferritin is a ubiquitous iron storage protein that is composed of 24 subunits of H- and L-chains [3]. Ferritin H- and L-subunits assemble into a hollow sphere that can accumulate in its interior up to 4500 atoms of iron as ferric oxy-hydroxides. Fe(II) gains access to the interior through pores lined with iron-binding carboxylate side chains. H-ferritin has ferroxidase activity; the oxidation of Fe(II) to Fe(III) occurs at enzymatic active sites on the interior surface of the ferritin cage. Precipitates of Fe(III) within the ferritin interior are relatively inert and are not available for either iron-mediated redox reactions that contribute to iron toxicity or for ultilization as enzymatic cofactors. In order to mobilize the iron stored in ferritin, the protein cages must be degraded [4], the iron precipitates solubilized, and the Fe(III) reduced to Fe(II) [5]. These events occur within the acidified lumen of the lysosome [6]. Fe(II) can then be exported from the lysosome for use as an iron cofactor.
Current models suggest that in most cell types, ferritin serves exclusively to store iron not needed to meet immediate metabolic requirements [3]. However, previous studies have indicated that the role of ferritin in erythroid development extends beyond the storage of excess iron when uptake exceeds a threshold for utilization. A study tracking the flow of radiolabeled Fe-Tf through isolated reticulocytes indicated that iron first accumulated in ferritin then appeared in hemoglobin [7]. Even when Fe-Tf uptake was restricted to very low levels, iron was trafficked to ferritin, indicating that ferritin iron did not represent the overflow from excess accumulation, but instead suggested that the flux was an obligatory part of iron trafficking through the cell. A later study using early erythroid precursors isolated from peripheral blood confirmed that radioiron introduced as Fe-Tf initially appeared in ferritin before appearing in hemoglobin [8]. Ferritin levels rapidly rise early in terminal differentiation, then fall at later points, suggesting that ferritin degradation and the release of ferritin iron occurs as erythrocytes develop [9,10]. Inhibitors of lysosomal (and endosomal) function inhibit both the degradation of ferritin and heme synthesis in this model of erythrocyte development.
A recent report examined the genetic role for H-ferritin in the hemoglobinization of the human K562 erythroleukemia cell line. In both stable and transient models of RNA-interference-mediated H-ferritin depletion, K562 cells lacking ferritin exhibited markedly reduced hemoglobinization and reduced expression of mRNAs characteristic of erythroid differentiation [11]. Although the authors attributed the block in hemoglobinization to expression of the microRNA mir-150, other recent studies suggest that additional mechanisms are also likely involved.
Some experimental evidence supports a ferritin-independent path for iron trafficking in terminal erythroid differentiation. Circulating reticulocytes contain relatively low levels of ferritin and, although they have largely completed hemoglobinization, can incorporate newly acquired iron into heme. A direct transfer of iron from endosomes to mitochondria has been proposed to account for this ferritin-independent mechanism and microscopic analyses suggest that contacts between endosomes and mitochondria are detectable [12-14]. A recent study also indicates that transferrin receptor 2 (Tfr2) selectively traffics Fe-Tf to the lysosome, where the iron is released and ultimately used for heme synthesis [15]. Furthermore, a mouse model of inducible ferritin deletion exhibits relatively normal red blood cell development, although other changes in iron parameters may compensate for the loss of ferritin [16]. A mouse model of erythrocyte-specific ferritin deletion has not been reported; thus, the relative contribution of these compensatory changes is not known.
Iron trafficking through ferritin in erythroid cells: The role of PCBP1
Cytosolic/nuclear iron chaperones of the poly rC-binding protein (PCBP) family assist in the delivery of iron to ferritin [17]. PCBP1 (also called hnRNP E1 or α-CP1) was identified as an iron chaperone after a genetic screen determined that it could facilitate the transfer of iron into ferritin when both were expressed exogenously in baker’s yeast [18]. PCBP1 can bind iron directly and deliver it to ferritin via a metal-mediated protein-protein interaction. Subsequent studies indicate that other members of the PCBP family can also function as iron chaperones to ferritin [19]. In Huh7 cells, siRNA-mediated depletion of either PCBP1 or its close paralog, PCBP2, impairs the loading of iron into ferritin and in HEK293 cells, PCBP1 and PCBP2 can be detected in complex with ferritin. PCBP1 and PCBP2 are also involved in the delivery of iron cofactors to cytosolic mono- and di-nuclear iron enzymes [20,21].
Recent studies regarding the role of PCBP1 and 2 in terminal erythroid differentiation confirm the importance of ferritin iron flux in red cell development [22]. PCBP1 and PCBP2 are highly expressed in G1E-ER4 cells, a murine cell line that recapitulates the terminal differentiation of red cells from the proerythroblast to the orthochromatic erythroblast stage. In G1E-ER4 cells, PCBP1 is readily detected in a complex with ferritin. Cells depleted of PCBP1 exhibit markedly impaired accumulation of iron into ferritin, as well as defective heme and hemoglobin synthesis. These studies confirmed that PCBP1 functions as an iron chaperone for ferritin in developing erythroid cells and further indicate that delivery of iron to ferritin is required for efficient heme and hemoglobin synthesis.
The delivery of iron to ferritin by PCBP1 is a regulated process in G1E-ER4 cells [23]. PCBP1 binding to ferritin is maximal in the early hours of differentiation, when ferritin iron accumulation is high but hemoglobin synthesis has not yet begun. Later in differentiation PCBP1 binding activity lessens, despite the increasing levels of ferritin and sustained levels of PCBP1. These changes in binding activity are directly mediated by iron and can be reproduced by iron chelation or supplementation, both in cells and in vitro. These observations suggest that that early in differentiation, when cellular iron is low, PCBP1 efficiently transfers iron to ferritin, which serves to keep cytosolic iron levels low. Later in differentiation, iron levels rise in ferritin and the cytosol, and the amount of iron directed by PCBP1 into ferritin falls. Thus, later stages of terminal differentiation may feature a ferritin-independent pathway of iron trafficking to mitochondria.
Although PCBP1 and PCBP2 appear to function similarly as iron chaperones in general cell culture models, this similarity is not evident in the G1E-ER4 cell model, where PCBP2 has the opposite effect on iron flux [22]. PCBP2 is relatively abundant in developing G1E-ER4 cells, but it is not detectable in a complex with ferritin. Loss-of-function experiments indicate that cells depleted of PCBP2 exhibit enhanced trafficking of iron to ferritin and increased levels of heme and hemoglobin synthesis. More PCBP1 is found in complex with ferritin when PCBP2 is absent and the enhanced iron loading observed with PCBP2 depletion is dependent on expression of PCBP1. PCBP2 has iron chaperone activity that differs from PCBP1 in general cultured cell models. PCBP2, but not PCBP1, is reported to bind to and enhance the activity of membrane transporters involved in iron import and efflux [24,25], although G1E-ER4 cells depleted of PCBP2 did not exhibit defects in Fe-Tf accumulation. PCBP1 and PCBP2 can form heterodimeric complexes in cells [19,21]. Thus, PCBP2 could serve to divert PCBP1 away from ferritin to other potential sites of iron delivery.
The role of PCBP1 in red cell development is further supported by studies in mice. In a murine model of inducible PCBP1 gene deletion, mice with PCBP1 deficiency exhibited lower levels of heme and hemoglobin in cells isolated from bone marrow as well as a mild microcytic anemia, indicating iron-limited erythropoiesis [22]. These animals also exhibit compensatory responses to the anemia, with elevated erythropoietin mRNA detected in kidney, elevated erythroferrone mRNA in the bone marrow, and suppressed hepcidin mRNA in the liver. These changes in regulatory hormones serve to enhance iron uptake and delivery to the erythron as well as to stimulate erythropoiesis. Ex vivo differentiation of primary erythroid progenitors isolated from PCBP1-deficient mice also exhibit impaired iron accumulation in ferritin and impaired heme and hemoglobin synthesis. In sum, PCBP1-mediated iron delivery to ferritin appears to serve a critical step in the flow of iron through developing red cells.
Iron trafficking through ferritin in erythroid cells: The role of NCOA4
While iron chaperone activity accounts for the delivery of iron to ferritin, the release of iron from ferritin requires a wholly distinct process. Ferritin iron release occurs through degradation of the protein nanocage, a process that occurs mostly within the lumen of the lysosome [4,6]. Ferritin is introduced to the lysosome via the autophagosome [26] and recent studies have identified machinery that accounts for the selective recruitment of ferritin to the autophagosome. Nuclear coactivator 4 (NCOA4) was initially described as a coactivator for a variety of nuclear hormone receptors [27]. Subsequent proteomic studies identified NCOA4 as a component of autophagosomes [28,29]. Further analysis revealed that it functions as a receptor for autophagic cargo, with the primary target being cytosolic ferritin. The carboxyl terminal of NCOA4 specifically binds to H-ferritin subunits, recruiting them to nascent autophagosomes [30]. Cells lacking NCOA4 exhibit delayed degradation of ferritin in response to iron starvation. In K562 cells lacking NCOA4, hemoglobin synthesis is inhibited. Fish embryos injected with morpholinos against NCOA4 exhibit defects in red cell development and hemoglobin formation.
Studies in G1E-ER4 cells confirm that the defects in red cell development associated with loss of NCOA4 are due to a block in iron flux through ferritin [22]. Similar to cells lacking PCBP1, developing G1E-ER4 cells depleted of NCOA4 have greatly reduced levels of heme synthesis and hemoglobin. However, in contrast to cells without PCBP1, the amount of iron detected in ferritin is greatly increased, indicating that NCOA4 is required for the mobilization of iron from ferritin in developing erythroid progenitors. The ferritin iron pool dramatically expands in these cells, yet the stored iron is unavailable for heme synthesis.
The G1E-ER4 cell model also indicates that the activity of NCOA4 in erythroid precursors is regulated both developmentally and by cellular iron levels [23]. NCOA4 protein and RNA levels rise as erythroid precursors develop and reach maximal levels at stages where iron incorporation into heme is highest. The accumulation of NCOA4 is blocked, however, in cells treated with supplemental iron. Iron induces the degradation of NCOA4 in G1E-ER4 cells by directing it to the lysosome. This process requires the E3 ubiquitin ligase HERC2; cells depleted of HERC2 fail to exhibit iron-induced degradation of NCOA4. HERC2 protein and mRNA levels increase during erythroid terminal differentiation, but expression levels are not regulated by changes in iron. NCOA4 stoichiometrically binds iron in vitro [30]; it is likely that directly binding of cytosolic iron by NCOA4 mediates its interaction with HERC2 and subsequent ubiquitin-mediated trafficking to the lysosome. Thus, in developing erythroid cells with low levels of cytosolic iron, NCOA4 is stabilized and promotes the turnover of ferritin, freeing iron to be delivered to mitochondria for heme synthesis. In cells with excess iron, NCOA4 is degraded and ferritin is not turned over, instead retaining its stored iron (Figure 1).
Figure 1. Regulation of iron flux through ferritin: Cytosolic iron levels can affect activity of PCBP1 and NCOA4.
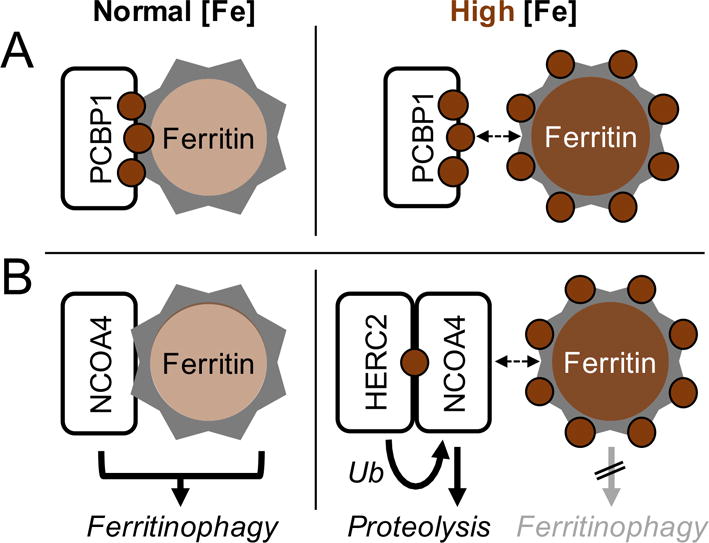
A. Under normal [Fe] conditions, labile iron in the cytosol is low. PCBP1-bound iron is efficiently delivered to ferritin. Under high iron conditions, both PCBP1 and ferritin iron-binding sites are saturated, and iron is not delivered to ferritin. B. Under normal [Fe] conditions, NCOA4 progressively accumulates and binds ferritin, directing it to the lysosome via ferritinophagy. Under high [Fe] conditions, NCOA4 binds iron, which is recognized and ubiquitinated by HERC2. Ubiquitination targets NCOA4 for degradation in the lysosome. Reproduced from [23]. Ryu MS, Duck KA, Philpott CC: Ferritin iron regulators, PCBP1 and NCOA4, respond to cellular iron status in developing red cells. Blood Cells Mol Dis 2017.
Mice deleted for NCOA4 confirm the importance of ferritin iron flux in iron homeostasis and erythroid development [31]. These mice exhibit high levels of ferritin and iron in the spleen and liver and have a mild microcytic anemia that is very similar to that of PCBP1-deficient mice. NCOA4-deleted mice are extremely sensitive to nutritional iron deprivation and rapidly develop severe anemia when fed an iron-deficient diet. These defects may largely be due to a failure to adequately mobilize iron stored in splenic and liver ferritin, but the observations in G1E-ER4 and K562 cells clearly indicate an intrinsic defect in red cell iron trafficking and utilization. The animal studies also indicate that erythroid cells must have a mechanism to bypass NCOA4-mediated ferritin turnover and deliver iron to mitochondria, as the anemia is relatively mild and hemoglobin synthesis clearly occurs.
A final common path for iron: From endo-lysosomes to mitochondria
Studies in cell and animal models of erythropoiesis demonstrate the importance of iron flux through ferritin to the lysosome. The final step in heme synthesis, insertion of iron into protoporphyrin IX by ferrochelatase, occurs within the mitochondrial matrix. The mechanisms that account for the transfer of iron from the endosome or lysosome to mitochondria are not completely clear, but some likely components have been identified. DMT1 has been localized to the lysosomal membrane, where it could potentially function in the translocation of iron from the lumen of the lysosome to the cytosol [32]. But quantitative transfer to the cytosol of lysosomal iron released from ferritin seems unlikely. Measurement of the kinetically active, labile iron pool in the cytosol of K562 cells suggests that labile iron levels are low even when iron transport and heme synthesis is high [33,34]. Furthermore, iron regulatory proteins in developing erythroid precursors respond to iron supplementation and deprivation, which suggests that the cytosol is neither iron-saturated or -depleted during normal development [22]. Ferroportin, an iron efflux pump, is highly expressed in developing erythroid precursors [35,36]. But ferroportin transport activity would tend to direct “free” cytosolic iron out of the cell, rather than to the mitochondria. For these reasons, a more direct path from the endolysosomal compartment to the mitochondria is likely. In addition to DMT1, mucolipin-1 (MCOLN1, TRPML1) is a lysosomal ion channel that can mediate iron transfer out of the lysosome [37]. A recent study of mice deleted for MCOLN1 indicates that they exhibit anemia, delays in erythroid cell differentiation, and defects in mitochondrial morphology [15]. Direct contacts between lysosomes and mitochondria were initially detected in melanocytes and more recently in CD34+ human erythroid progenitors [15,38]. Contact sites in both systems require Mitofusin 2, a mitochondrial outer membrane protein that is also required for mitochondrial-endosomal contacts and for the fusion of autophagosomes with lysosomes. Depletion of Mitofusin 2 in human erythroid progenitors resulted in loss of contact sites between lysosomes and mitochondria and reduced heme synthesis in these cells. Thus, lysosomal iron may be transferred to mitochondria via a direct mechanism in developing erythrocytes.
Conclusion
Why do developing red cells direct iron through ferritin and lysosomes on its way to mitochondria for heme synthesis in terminal differentiation (Figure 2)? Why is this process regulated on so many levels? First, sequestering ferrous iron as ferric oxyhydroxides protects the cell from the damaging reactive oxygen species that are readily produced by reactive iron in respiring cells. Second, the regulated flux of iron through ferritin allows cells to buffer red cell iron in a readily available reservoir and thereby ensure a continuous flow of iron to mitochondria. Serum iron and transferrin saturation levels can fluctuate by 100% over the course of a day in normal, healthy individuals. The fluctuations in transferrin-iron uptake and the flow of iron to mitochondria would be greatly affected without the buffering effects of ferritin, which ensures a consistent flux of iron for heme synthesis.
Figure 2. Iron flux through ferritin in the developing erythrocyte.
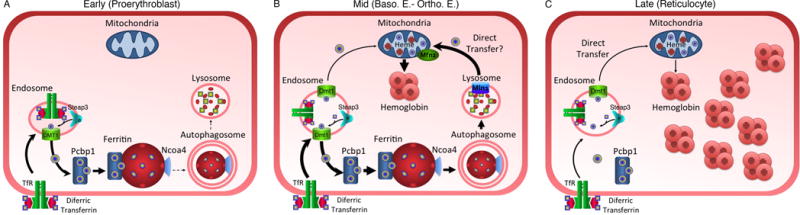
A. Early stages of terminal differentiation (Proerythroblast) are characterized by PCBP1-mediated, iron accumulation in ferritin. NCOA4 levels are low and ferritin turnover is slow. B. Mid stages of development (basophilic to orthochromatic erythroblast) are characterized by high levels of iron flux through ferritin, high rates of ferritin turnover, and high rates of iron transfer to the mitochondria. A direct mechanism involving Mucolipin1 (Mln1) and Mitofusin 2 (Mfn2) may be operating. C. Late stage development (Reticulocyte) is characterized by low levels of iron uptake and heme synthesis. Ferritin levels are low and intracellular organelles are rapidly lost. Iron may be directly transferred to mitochondria. Modified from [22]. Ryu MS, Zhang D, Protchenko O, Shakoury-Elizeh M, Philpott CC: PCBP1 and NCOA4 regulate erythroid iron storage and heme biosynthesis. J Clin Invest 2017, 127:1786-1797.
Key Points.
The iron chaperone PCBP1 and the autophagic cargo receptor NCOA4 are responsible for moving iron into ferritn and directing ferritin to autophagosomes, respectively, in developing erythroid cells.
Genetic evidence in both cell and animal models indicate that the flux of iron through ferritin is an obligatory step in the early-to middle-stages of terminal erythroid differentiation.
The flux of iron through ferritin is regulated by the levels of iron in the cytosol and adds a layer of rapid, fine control to the flow of iron to mitochondria.
Evidence supports a direct transfer of iron from endolysosomes to mitochondria during red cell development.
Acknowledgments
The author thanks Dr. Moon-Suhn Ryu and members of the Genetics and Metabolism Section for helpful discussions of these studies.
Financial Support and Sponsorship – This work was supported by the Intramural Research Program of the National Institutes of Diabetes and Digestive and Kidney Diseases, National Institutes of Health.
Footnotes
Conflicts of Interest – The author declares no conflicts of interest.
References
- 1.Ganz T, Nemeth E. Iron metabolism: interactions with normal and disordered erythropoiesis. Cold Spring Harb Perspect Med. 2012;2:a011668. doi: 10.1101/cshperspect.a011668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Philpott CC, Ryu MS. Special delivery: distributing iron in the cytosol of mammalian cells. Front Pharmacol. 2014;5:173. doi: 10.3389/fphar.2014.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arosio P, Elia L, Poli M. Ferritin, cellular iron storage and regulation. IUBMB Life. 2017;69:414–422. doi: 10.1002/iub.1621. [DOI] [PubMed] [Google Scholar]
- 4.Kidane TZ, Sauble E, Linder MC. Release of iron from ferritin requires lysosomal activity. Am J Physiol Cell Physiol. 2006;291:C445–455. doi: 10.1152/ajpcell.00505.2005. [DOI] [PubMed] [Google Scholar]
- 5.Ohgami RS, Campagna DR, McDonald A, Fleming MD. The Steap proteins are metalloreductases. Blood. 2006;108:1388–1394. doi: 10.1182/blood-2006-02-003681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Y, Mikhael M, Xu D, Li Y, Soe-Lin S, Ning B, Li W, Nie G, Zhao Y, Ponka P. Lysosomal proteolysis is the primary degradation pathway for cytosolic ferritin and cytosolic ferritin degradation is necessary for iron exit. Antioxid Redox Signal. 2010;13:999–1009. doi: 10.1089/ars.2010.3129. [DOI] [PubMed] [Google Scholar]
- 7.Speyer BE, Fielding J. Ferritin as a cytosol iron transport intermediate in human reticulocytes. Br J Haematol. 1979;42:255–267. doi: 10.1111/j.1365-2141.1979.tb01130.x. [DOI] [PubMed] [Google Scholar]
- 8.Vaisman B, Fibach E, Konijn AM. Utilization of intracellular ferritin iron for hemoglobin synthesis in developing human erythroid precursors. Blood. 1997;90:831–838. [PubMed] [Google Scholar]
- 9.Beaumont C, Dugast I, Renaudie F, Souroujon M, Grandchamp B. Transcriptional regulation of ferritin H and L subunits in adult erythroid and liver cells from the mouse. Unambiguous identification of mouse ferritin subunits and in vitro formation of the ferritin shells. J Biol Chem. 1989;264:7498–7504. [PubMed] [Google Scholar]
- 10.Konijn AM, Hershko C, Izak G. Ferritin synthesis and iron uptake in developing erythroid cells. Am J Hematol. 1979;6:373–379. doi: 10.1002/ajh.2830060409. [DOI] [PubMed] [Google Scholar]
- 11.Zolea F, Battaglia AM, Chiarella E, Malanga D, De Marco C, Bond HM, Morrone G, Costanzo F, Biamonte F. Ferritin Heavy Subunit Silencing Blocks the Erythroid Commitment of K562 Cells via miR-150 up-Regulation and GATA-1 Repression. Int J Mol Sci. 2017;18 doi: 10.3390/ijms18102167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Das A, Nag S, Mason AB, Barroso MM. Endosome-mitochondria interactions are modulated by iron release from transferrin. J Cell Biol. 2016;214:831–845. doi: 10.1083/jcb.201602069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sheftel AD, Zhang AS, Brown C, Shirihai OS, Ponka P. Direct interorganellar transfer of iron from endosome to mitochondrion. Blood. 2007;110:125–132. doi: 10.1182/blood-2007-01-068148. [DOI] [PubMed] [Google Scholar]
- 14.Zhang AS, Sheftel AD, Ponka P. Intracellular kinetics of iron in reticulocytes: evidence for endosome involvement in iron targeting to mitochondria. Blood. 2005;105:368–375. doi: 10.1182/blood-2004-06-2226. [DOI] [PubMed] [Google Scholar]
- 15*.Khalil S, Holy M, Grado S, Fleming R, Kurita R, Nakamura Y, Goldfarb A. A specialized pathway for erythroid iron delivery through lysosomal trafficking of transferrin receptor 2. Blood Advances. 2017;1:1181–1194. doi: 10.1182/bloodadvances.2016003772. The authors identify a role for MCOLN1 and Mfn2 in the trafficking of iron from the lysosome to the mitochondria in erythroid precursors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Darshan D, Vanoaica L, Richman L, Beermann F, Kuhn LC. Conditional deletion of ferritin H in mice induces loss of iron storage and liver damage. Hepatology. 2009;50:852–860. doi: 10.1002/hep.23058. [DOI] [PubMed] [Google Scholar]
- 17.Philpott CC, Ryu MS, Frey A, Patel S. Cytosolic iron chaperones: Proteins delivering iron cofactors in the cytosol of mammalian cells. J Biol Chem. 2017;292:12764–12771. doi: 10.1074/jbc.R117.791962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi H, Bencze KZ, Stemmler TL, Philpott CC. A cytosolic iron chaperone that delivers iron to ferritin. Science. 2008;320:1207–1210. doi: 10.1126/science.1157643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leidgens S, Bullough KZ, Shi H, Li F, Shakoury-Elizeh M, Yabe T, Subramanian P, Hsu E, Natarajan N, Nandal A, et al. Each member of the poly-r(C)-binding protein 1 (PCBP) family exhibits iron chaperone activity toward ferritin. J Biol Chem. 2013;288:17791–17802. doi: 10.1074/jbc.M113.460253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frey AG, Nandal A, Park JH, Smith PM, Yabe T, Ryu MS, Ghosh MC, Lee J, Rouault TA, Park MH, et al. Iron chaperones PCBP1 and PCBP2 mediate the metallation of the dinuclear iron enzyme deoxyhypusine hydroxylase. Proc Natl Acad Sci U S A. 2014;111:8031–8036. doi: 10.1073/pnas.1402732111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nandal A, Ruiz JC, Subramanian P, Ghimire-Rijal S, Sinnamon RA, Stemmler TL, Bruick RK, Philpott CC. Activation of the HIF Prolyl Hydroxylase by the Iron Chaperones PCBP1 and PCBP2. Cell Metab. 2011;14:647–657. doi: 10.1016/j.cmet.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22**.Ryu MS, Zhang D, Protchenko O, Shakoury-Elizeh M, Philpott CC. PCBP1 and NCOA4 regulate erythroid iron storage and heme biosynthesis. J Clin Invest. 2017;127:1786–1797. doi: 10.1172/JCI90519. The authors characterize the roles of PCBP1 and NCOA4 in controlling the necessary flux of iron through ferritin in developing erythrocytes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23*.Ryu MS, Duck KA, Philpott CC. Ferritin iron regulators, PCBP1 and NCOA4, respond to cellular iron status in developing red cells. Blood Cells Mol Dis. 2017 doi: 10.1016/j.bcmd.2017.09.009. The authors describe the iron-dependent regulation of NCOA4 and PCBP1 activity in erythrocytes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yanatori I, Richardson DR, Imada K, Kishi F. Iron Export through the Transporter Ferroportin 1 Is Modulated by the Iron Chaperone PCBP2. J Biol Chem. 2016;291:17303–17318. doi: 10.1074/jbc.M116.721936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yanatori I, Yasui Y, Tabuchi M, Kishi F. Chaperone protein involved in transmembrane transport of iron. Biochem J. 2014;462:25–37. doi: 10.1042/BJ20140225. [DOI] [PubMed] [Google Scholar]
- 26.Asano T, Komatsu M, Yamaguchi-Iwai Y, Ishikawa F, Mizushima N, Iwai K. Distinct mechanisms of ferritin delivery to lysosomes in iron-depleted and iron-replete cells. Mol Cell Biol. 2011;31:2040–2052. doi: 10.1128/MCB.01437-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yeh S, Chang C. Cloning and characterization of a specific coactivator, ARA70, for the androgen receptor in human prostate cells. Proc Natl Acad Sci U S A. 1996;93:5517–5521. doi: 10.1073/pnas.93.11.5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, Triantafellow E, Menon S, Wang Z, Honda A, Pardee G, et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol. 2014;16:1069–1079. doi: 10.1038/ncb3053. [DOI] [PubMed] [Google Scholar]
- 29.Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509:105–109. doi: 10.1038/nature13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30*.Mancias JD, Pontano Vaites L, Nissim S, Biancur DE, Kim AJ, Wang X, Liu Y, Goessling W, Kimmelman AC, Harper JW. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife. 2015:4. doi: 10.7554/eLife.10308. Ferritin iron trafficking is found to be important in erythrocyte development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31*.Bellelli R, Federico G, Matte A, Colecchia D, Iolascon A, Chiariello M, Santoro M, De Franceschi L, Carlomagno F. NCOA4 Deficiency Impairs Systemic Iron Homeostasis. Cell Rep. 2016;14:411–421. doi: 10.1016/j.celrep.2015.12.065. NCOA4-dependent ferritin iron trafficking is required for iron homeostasis in mice. [DOI] [PubMed] [Google Scholar]
- 32.Tabuchi M, Yoshimori T, Yamaguchi K, Yoshida T, Kishi F. Human NRAMP2/DMT1, which mediates iron transport across endosomal membranes, is localized to late endosomes and lysosomes in HEp-2 cells. J Biol Chem. 2000;275:22220–22228. doi: 10.1074/jbc.M001478200. [DOI] [PubMed] [Google Scholar]
- 33.Esposito BP, Epsztejn S, Breuer W, Cabantchik ZI. A review of fluorescence methods for assessing labile iron in cells and biological fluids. Anal Biochem. 2002;304:1–18. doi: 10.1006/abio.2002.5611. [DOI] [PubMed] [Google Scholar]
- 34.Petrat F, de Groot H, Sustmann R, Rauen U. The chelatable iron pool in living cells: a methodically defined quantity. Biol Chem. 2002;383:489–502. doi: 10.1515/BC.2002.051. [DOI] [PubMed] [Google Scholar]
- 35.Zhang DL, Hughes RM, Ollivierre-Wilson H, Ghosh MC, Rouault TA. A ferroportin transcript that lacks an iron-responsive element enables duodenal and erythroid precursor cells to evade translational repression. Cell Metab. 2009;9:461–473. doi: 10.1016/j.cmet.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang DL, Senecal T, Ghosh MC, Ollivierre-Wilson H, Tu T, Rouault TA. Hepcidin regulates ferroportin expression and intracellular iron homeostasis of erythroblasts. Blood. 2011;118:2868–2877. doi: 10.1182/blood-2011-01-330241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dong XP, Cheng X, Mills E, Delling M, Wang F, Kurz T, Xu H. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature. 2008;455:992–996. doi: 10.1038/nature07311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daniele T, Hurbain I, Vago R, Casari G, Raposo G, Tacchetti C, Schiaffino MV. Mitochondria and melanosomes establish physical contacts modulated by Mfn2 and involved in organelle biogenesis. Curr Biol. 2014;24:393–403. doi: 10.1016/j.cub.2014.01.007. [DOI] [PubMed] [Google Scholar]