

Glycogen synthase kinase-3 (GSK3), one of the busiest kinases in cells, phosphorylates more than 100 known substrates. Typical substrates of GSK3 contain a phosphorylated priming sequence, S/T-X-X-X-S/T(P), where the GSK3-targeted serine/threonine lies four residues N-terminal to a phosphorylated serine/threonine. The large number of GSK3 substrates and their location within different cellular compartments raises the question of how selectivity and specificity of GSK3/substrate interactions are achieved. In this issue, Wang et al demonstrate that S-nitrosylation of GSK3β inhibits its activity in the cytosol and that S-nitrosylated GSK3β (SNO-GSK3β) translocates to the nucleus to phosphorylate nuclear substrates1. Wang et al thus uncover novel interplay between S-nitrosylation and phosphorylation that subserves GSK3β targeting of substrates.
S-nitrosylation directly suppresses activity of protein kinases
In many cases, crosstalk between S-nitrosylation and phosphorylation is manifested by S-nitrosylation-mediated suppression of protein kinase activity (Figure A). G protein-coupled receptor kinase 2 (GRK2) is a prime example2. Specifically, GRK2-mediated phosphorylation of the β-adrenergic receptor (β-AR) at serine and threonine residues in β-AR’s arrestin binding domain promotes β-arrestin binding, which prevents further activation of G proteins and initiates receptor internalization and desensitization. β-adrenergic receptors do not, however, desensitize under normal physiological conditions because receptor-mediated kinase activation is followed by S-nitrosylation of a critical Cys (Cys340) within the activation loop of GRK2, which inhibits kinase activity2. β-AR-coupled, endothelial nitric oxide synthase (eNOS) mediated S-nitrosylation thereby prevents receptor inactivation3. There are many other examples of S-nitrosylation inhibiting phosphorylation-based signal transduction through induced conformational changes in kinases or by blocking their autophosphorylation: protein kinase C (PKC)4, protein kinase B (AKT)5, IκB kinase (IKK)6 and calcium/calmodulin-dependent protein kinase II (CaMKII)7 are to name but a few.
Figure.
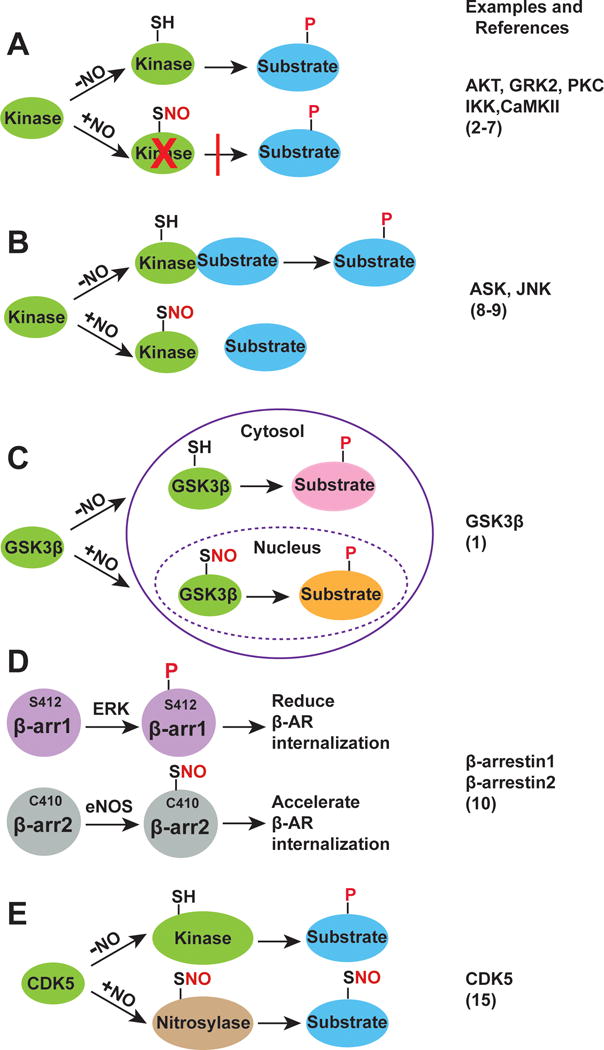
(a) S-nitrosylation directly inhibits kinase activity. SNO: S-nitrosylation; P: phosphorylation. (b) S-nitrosylation disrupts kinase-substrate interaction. (c) S-nitrosylation changes GSK3β localization and determines substrate specificity. (d) Phosphorylation and S-nitrosylation alternatively modify β-arrestin1 and β-arrestin2 to regulate GPCR internalization. (e) S-nitrosylation converts CDK5 from kinase to nitrosylase.
S-nitrosylation affects kinase substrate specificity
Productive interactions between kinases and their substrates identify bona fide phosphorylation sites (among many thousands of candidate sites in the proteome). S-nitrosylation of protein kinases can influence these kinase/substrate interactions. This operating principle is illustrated in the cases of apoptosis signal-regulating kinase-1 (ASK1) and Jun N-terminal kinases (JNK) (Figure B), which undergo S-nitrosylation in response to apoptotic stimuli8,9. For example, interferon-γ-induced S-nitrosylation of ASK1 at Cys869 inhibits the binding of ASK1 to its principal downstream effectors MKK3 and MKK6, attenuating apoptotic cell death8. S-nitrosylation of Cys116 in JNK inhibits JNK-mediated phosphorylation and transactivation of c-Jun by disrupting the interaction between JNK and c-Jun9. Thus, S-nitrosylation provides a mechanism to regulate substrate recognition in physiological context.
Subcellular compartmentalization provides another commonly exploited mechanism to achieve substrate specificity; compartmentalization restricts kinases to a subset of substrates and increases local kinase concentrations. The present study illustrates elegantly how S-nitrosylation determines substrate specificity by changing the localization of GSK3β1 (Figure C). GSK3β is traditionally considered to be a cytosolic protein; however, GSK3β is also found in the mitochondria and nucleus, as well as in other subcellular compartments. Although numerous nuclear substrates of GSK3β, including transcriptional factors and epigenetic regulators have been identified, regulation of GSK3β nuclear localization is poorly understood. Wang et al found that S-nitrosylation of GSK3β causes GSK3β translocation to the nucleus where it phosphorylates nuclear substrates1. Interestingly, many nuclear substrates of GSK3β contain a S/T-P motif rather than the prototypical priming sequence S/T-X-X-X-S/T(P). This nuance may be due to a S-nitrosylation-induced conformational change in GSK3β that shifts structure-based recognition from the canonical priming site to an alternate phospho-targeting motif. More studies are needed to determine if S-nitrosylation of GSK3β has evolved to provide a mechanism for recognition of the nuclear S/T-P motif.
S-nitrosylation and phosphorylation selectively modify different protein isoforms
Protein isoforms provide a natural mechanism for divergence within signaling networks. Differential modification of protein isoforms by S-nitrosylation versus phosphorylation can fine-tune cellular signaling. Post-translational modification of β-arrestins illustrates this principle (Figure D). β-arrestins are versatile, multifunctional scaffold proteins that are best known for their ability to internalize and desensitize G protein-coupled receptors (GPCRs). β-arrestin1 and β-arrestin2 can be both phosphorylated (Ser412 in β-arrestin1 and Thr276, Ser361 and Thr383 in β-arrestin2) and nitrosylated (Cys251 in β-arrestin1 and Cys253, Cys410 in β-arrestin2) at multiple loci. Stimulus-coupled dephosphorylation of Ser412 in β-arrestin1 is requisite for clathrin-mediated β2-AR internalization. Interestingly, Ser412 in β-arrestin1 is replaced by a highly conserved Cys410 in β-arrestin2, and S-nitrosylation by eNOS of Cys410 in β-arrestin2 promotes its binding to clathrin, thereby accelerating β2-AR internalization10. Thus, phosphorylation and nitrosylation subserve classic clathrin-mediated internalization of GPCRs by β-arrestin1 and β-arrestin2, respectively. Inasmuch as β-arrestin1 and β-arrestin2 are ubiquitously distributed, these data support the idea of functional crosstalk between nitrosylation and phosphorylation in wide-ranging cellular functions.
There are two isoforms of glycogen synthase kinase-3 (GSK3α and GSK3β) in mammals. Although GSK3α and GSK3β are ubiquitously expressed and share numerous substrates, they mediate different biological functions in response to cellular stimuli. GSK3β is modified by S-nitrosylation at Cys76, Cys199 and Cys317; GSK3α contains Cys199 and Cys317 but lacks Cys761. This is somewhat akin to β-arrestin1 and β-arrestin2, which share one S-nitrosylation site (Cys251/253), but not a second (C410 is present only in β-arrestin2). This raises the question as to whether S-nitrosylation can selectively modify GSK3α and GSK3β to mediate distinct cellular functions.
S-nitrosylation converts a protein kinase into a protein nitrosylase
The machinery for enzymatic S-nitrosylation involves three classes of enzymes operating coordinately: NO synthases, SNO synthases and transnitrosylases11. Transnitrosylases mediate thiol-based transfer of NO groups between proteins. Most transnitrosylases have canonical functions apart from their transnitrosylase activity. S-nitrosylation is thus a prerequisite to convert proteins into nascent nitrosylases; this principle is illustrated by glyceraldehyde-3-phosphate dehydrogenase (GAPDH), a prototypic transnitrosylase12. S-nitrosylation of GAPDH abolishes its catalytic activity and promotes binding to the E3-ubiquitin-ligase Siah, which subsequently facilitates translocation of SNO-GAPDH to the nucleus. In the nucleus, SNO-GAPDH acts as a transnitrosylase for numerous nuclear proteins, including the deacetylating enzyme sirtuin-1 (SIRT1), histone deacetylase-2 (HDAC2) and DNA-activated protein kinase (DNA-PK)12, thereby regulating the transcriptional and metabolic activity of the cell.
Cyclin-dependent kinase 5 (CDK5) is another elegant example of this principle. CDK5 is a proline-directed kinase that phosphorylates serines/threonines immediately upstream of proline residues. CDK5 is implicated in neuronal injury through activation of Dynamin-1-like protein (Drp1), which induces mitochondrial fission. Interestingly, Drp1 can be activated by both phosphorylation and S-nitrosylation. But whereas S-nitrosylation of Drp1-Cys644 mediates mitochondrial fission in Alzheimer (and Huntington) models13, Ser585 phosphorylation mediates mitochondrial fission in NMDA-induced injury14. Notably, in Alzheimer disease, CDK5 is S-nitrosylated at an essential Cys83, which resides within the ATP-binding pocket of the kinase thereby inhibiting kinase activity; SNO-CDK5 is thus converted from a kinase into a Drp-1 transnitrosylase, possibly mediating synaptic damage15 (Figure E).
Wang et al show here that S-nitrosylation of GSK3β promotes its trafficking into the nucleus1, and it is well-known that multiple nuclear phospho-substrates of GSK3β, including NF-κB, p53, STAT3 and Sirt1, can also be modified by S-nitrosylation. It seems likely that SNO-GSK3β may act as a transnitrosylase in the nucleus to mediate the S-nitrosylation of at least some of these substrates.
Acknowledgments
Sources of Funding
This work was supported by National Institutes of Health grants HL075443, HL128192 and HL126900.
References
- 1.Wang S, Venkatraman V, Crowgey EL, Liu T, Fu Z, Holewinski R, Ranek M, Kass DA, O’Rourke B, EyK JEV. Protein S-Nitrosylation Controls Glycogen Synthase Kinase 3β Function Independent of its Phosphorylation State. Circ Res. 2018 doi: 10.1161/CIRCRESAHA.118.312789. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whalen EJ, Foster MW, Matsumoto A, Ozawa K, Violin JD, Que LG, Nelson CD, Benhar M, Keys JR, Rockman HA, Koch WJ, Daaka Y, Lefkowitz RJ, Stamler JS. Regulation of beta-adrenergic receptor signaling by S-nitrosylation of G-protein-coupled receptor kinase 2. Cell. 2007;129:511–22. doi: 10.1016/j.cell.2007.02.046. [DOI] [PubMed] [Google Scholar]
- 3.Huang ZM, Gao E, Fonseca FV, Hayashi H, Shang X, Hoffman NE, Chuprun JK, Tian X, Tilley DG, Madesh M, Lefer DJ, Stamler JS, Koch WJ. Convergence of G protein-coupled receptor and S-nitrosylation signaling determines the outcome to cardiac ischemic injury. Sci Signal. 2013;6:ra95. doi: 10.1126/scisignal.2004225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choi H, Tostes RC, Webb RC. S-nitrosylation Inhibits protein kinase C-mediated contraction in mouse aorta. J Cardiovasc Pharmacol. 2011;57:65–71. doi: 10.1097/FJC.0b013e3181fef9cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yasukawa T, Tokunaga E, Ota H, Sugita H, Martyn JA, Kaneki M. S-nitrosylation-dependent inactivation of Akt/protein kinase B in insulin resistance. J Biol Chem. 2005;280:7511–8. doi: 10.1074/jbc.M411871200. [DOI] [PubMed] [Google Scholar]
- 6.Reynaert NL, Ckless K, Korn SH, Vos N, Guala AS, Wouters EF, van der Vliet A, Janssen-Heininger YM. Nitric oxide represses inhibitory kappaB kinase through S-nitrosylation. Proc Natl Acad Sci U S A. 2004;101:8945–50. doi: 10.1073/pnas.0400588101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coultrap SJ, Bayer KU. Nitric oxide induces Ca2+-independent activity of the Ca2+/calmodulin-dependent protein kinase II (CaMKII) J Biol Chem. 2014;289:19458–65. doi: 10.1074/jbc.M114.558254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park HS, Yu JW, Cho JH, Kim MS, Huh SH, Ryoo K, Choi EJ. Inhibition of apoptosis signal-regulating kinase 1 by nitric oxide through a thiol redox mechanism. J Biol Chem. 2004;279:7584–90. doi: 10.1074/jbc.M304183200. [DOI] [PubMed] [Google Scholar]
- 9.Park HS, Huh SH, Kim MS, Lee SH, Choi EJ. Nitric oxide negatively regulates c-Jun N-terminal kinase/stress-activated protein kinase by means of S-nitrosylation. Proc Natl Acad Sci U S A. 2000;97:14382–7. doi: 10.1073/pnas.97.26.14382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ozawa K, Whalen EJ, Nelson CD, Mu Y, Hess DT, Lefkowitz RJ, Stamler JS. S-nitrosylation of beta-arrestin regulates beta-adrenergic receptor trafficking. Mol Cell. 2008;31:395–405. doi: 10.1016/j.molcel.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seth D, Hess DT, Hausladen A, Wang L, Wang YJ, Stamler JS. A Multiplex Enzymatic machinery for cellular protein S-nitrosylation. Mol Cell. 2018;69:451–464 e6. doi: 10.1016/j.molcel.2017.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kornberg MD, Sen N, Hara MR, Juluri KR, Nguyen JV, Snowman AM, Law L, Hester LD, Snyder SH. GAPDH mediates nitrosylation of nuclear proteins. Nat Cell Biol. 2010;12:1094–100. doi: 10.1038/ncb2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–5. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jahani-Asl A, Huang E, Irrcher I, Rashidian J, Ishihara N, Lagace DC, Slack RS, Park DS. CDK5 phosphorylates DRP1 and drives mitochondrial defects in NMDA-induced neuronal death. Hum Mol Genet. 2015;24:4573–83. doi: 10.1093/hmg/ddv188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qu J, Nakamura T, Cao G, Holland EA, McKercher SR, Lipton SA. S-Nitrosylation activates Cdk5 and contributes to synaptic spine loss induced by beta-amyloid peptide. Proc Natl Acad Sci U S A. 2011;108:14330–5. doi: 10.1073/pnas.1105172108. [DOI] [PMC free article] [PubMed] [Google Scholar]