

Abstract
Our objective was to assess the changes in protein abundance in the human sinoatrial node (SAN) compared with working cardiomyocytes to identify SAN‐specific protein signatures. Four pairs of samples (the SAN and working cardiomyocytes) were obtained postmortem from four human donors with no evidence of cardiovascular disease. We performed protein identification and quantitation using two‐dimensional chromatography‐tandem mass spectrometry with isobaric peptide labeling (iTRAQ). We identified 451 different proteins expressed in both the SAN and working cardiomyocytes, 166 of which were differentially regulated (110 were upregulated in the SAN and 56 in the working cardiomyocytes). We identified sarcomere structural proteins in both tissues, although they were differently distributed among the tested samples. For example, myosin light chain 4, myosin regulatory light chain 2‐atrial isoform, and tropomyosin alpha‐3 chain levels were twofold higher in the SAN than in working cardiomyocytes, and myosin light chain 3 and myosin regulatory light chain 2‐ventricular/cardiac muscle isoform levels were twofold higher in the ventricle tissue than in SAN. We identified many mitochondrial oxidative phosphorylation, β‐oxidation, and tricarboxylic acid cycle proteins that were predominantly associated with working cardiomyocytes tissue. We detected upregulation of the fatty acid omega activation pathway proteins in the SAN samples. Some proteins specific for smooth muscle tissue were highly upregulated in the SAN (e.g. transgelin), which indicates that the SAN tissue might act as the bridge between the working myocardium and the smooth muscle. Our results show possible implementation of proteomic strategies to identify in‐depth functional differences between various heart sub‐structures.
Keywords: cardiac system, electrical cardiac conduction system, heart proteome, omega‐oxidation, sinoatrial node artery, smooth muscle
Introduction
Profiling the proteome of various tissues is fundamental when studying the correlations between the molecular phenotypes of cells and the clinical characteristics of organs (Marx, 2014; Gedik et al. 2017). To date, the human heart proteome has not been completely explored, in particular the proteomic profile of particular cardiac areas and structures and their quantitative and qualitative relationships. One such example of this approach is identifying the protein signature of the two main components of the human heart: working cardiomyocytes and elements of the electrical cardiac conduction system (Atkinson et al. 2013).
The cardiac conduction system initiates and coordinates the electric signal that causes synchronized heart muscle contractions. The sinoatrial node (SAN), located at the junction of the superior vena cava and the right atrium, serves as the primary physiological pacemaker of the heart (Anderson et al. 2009). Working myocardium and myocardial cells of the conduction system have the same embryonic origin, which is the primitive pre‐cardiac mesoderm (Christoffels & Moorman, 2009). Differentiation of non‐migrating joint precursor cells into either a conducting or a working cardiomyocyte occurs locally within the developing heart (Pennisi et al. 2002). In the developing heart, the venosus pole consists of the sinus venous and the primitive atrium, which are separated by a sinoatrial fold. The entire sinus venosus originates from non‐cardiac progenitors. The sinus venosus, which comprises the SAN, is later incorporated into the right atrium (Gourdie et al. 2003; Christoffels et al. 2006).
Macroscopically, in humans, the conduction system components and the working cardiomyocytes cannot be distinguished from one another. Poor histological criteria have been used thus far to visualize the main conduction system components (Moorman et al. 2005). However, in recent decades, new molecular techniques have emerged that have enabled the identification of transcription factors involved in the development of the animal cardiac conduction system. These factors then served to identify the conduction tissues (Hoogaars et al. 2004; Atkinson et al. 2013). Using a proteomic approach to study the characteristics of the human cardiac conduction system might help to understand its origin and development as well as some regulatory physiological and even pathological mechanisms governing the cardiac tissue (Viganò et al. 2011; Martín‐Rojas et al. 2012; Ferreira et al. 2015).
In this study, we compare the abundance of various proteins in the human SAN and working cardiomyocytes to identify SAN‐specific protein signatures. This study might also serve as the proof‐of‐concept for a novel approach to map the conduction system and to explain the etiology of some types of arrhythmia.
Methods
Sample collection
The experimental protocol was approved by the Bioethical Committee of Jagiellonian University, Cracow, Poland (No. 122.6120.45.2016). We collected samples from deceased persons who did not express an objection when alive, or where the family did not express an objection. In accordance with Polish law, our Bioethical Committee waived the need for written or verbal informed consent. Four pairs of samples (the suspected SAN and working cardiomyocyte areas) were obtained postmortem from four young male human donors (age range, 28–35 years), in each of whom the cause of death was suicide or a traffic accident. There was no medical history of cardiovascular diseases and no macroscopic signs of cardiac pathologies during autopsy. The samples were obtained within 24 h of death. The heart, together with the great vessels, was removed from the chest in a routine manner. The superior vena cava and the SAN area were identified. The epicardium was carefully removed from the area of the terminal groove between the opening of the superior vena cava and right atrial appendage. The SAN artery was identified, and a block of surrounding tissue (5 × 10 mm) was micro‐dissected without the endocardium (the SAN sample). The ventricular muscle samples were collected from the left ventricle‐free wall, 3 cm above the heart apex. The epicardium was carefully removed from the ventricle surface, and the block of tissue (10 × 10 mm) was micro‐dissected without the endocardium (working cardiomyocyte sample; Fig. 1). All samples were washed three times using saline solution, immediately frozen, and stored at −80 °C until use. Through this approach, we obtained samples from structurally normal hearts; however, limitations regarding tissue autolysis after death may have significantly influenced our analysis and should be taken into consideration.
Figure 1.
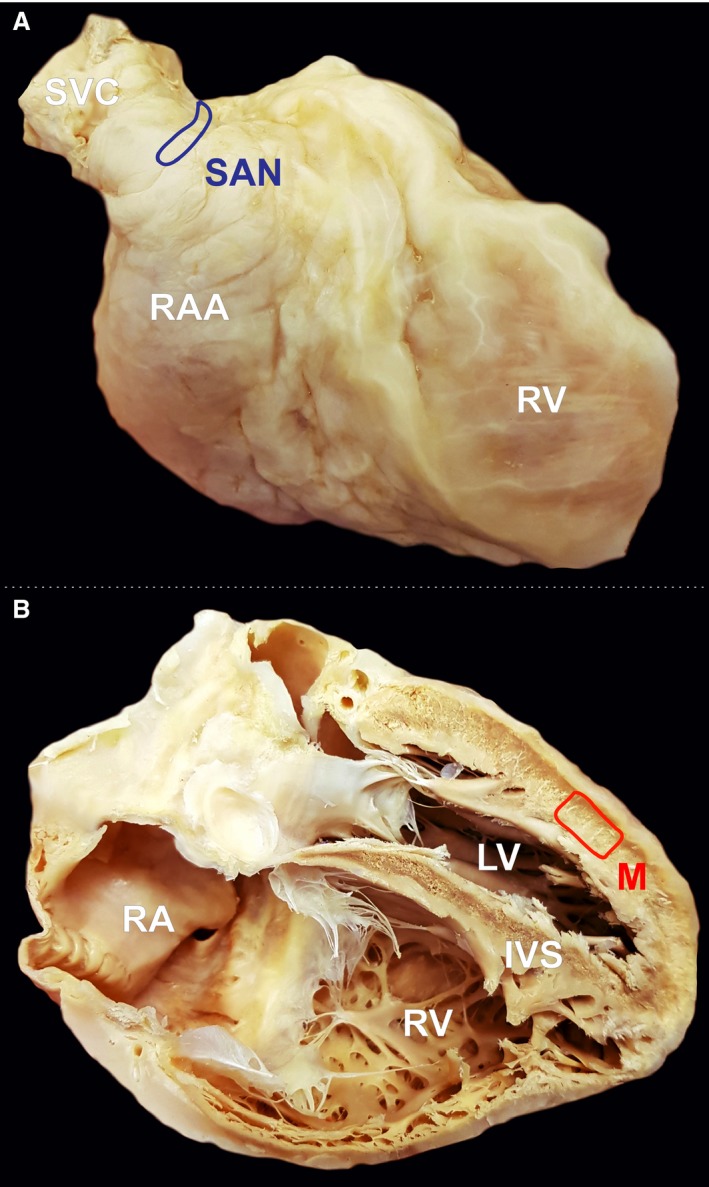
Photographic image of cadaveric heart specimen with two marked regions from where the samples were obtained: (A) sinoatrial node (SAN) area, (B) left ventricle free wall. IVS, interventricular septum; LV, left ventricle; M, working cardiomyocyte sample; RA, right atrium;RAA, right atrial appendage; RV, right ventricle; SVC, superior vena cava.
Sample preparation
The study design is shown in Fig. 2. All reagents used, unless stated otherwise, were purchased from Sigma‐Aldrich (Darmstadt, Germany). For each sample, approximately 100 mg of tissue was extracted with lysis buffer (20 mm HEPES pH 8, 1% CHAPS, 1 m urea) in a TissueLyser LT (Qiagen, Hilden, Germany). The samples were then centrifuged at 4 °C (30 min, 15 000 g) to remove cell debris. The protein concentrations were determined using a Coomassie Bradford protein assay kit (Thermo Scientific, Waltham, MA, USA). Protein (100 μg) from each sample was precipitated overnight using ice‐cold acetone (1 : 6, v : v), and the samples were centrifuged at 6000 g for 10 min at 4 °C. Acetone was carefully removed, and precipitates were dissolved in a 20‐μL dissolution buffer followed by addition of a 1‐μL denaturant solution that was reduced with reducing agent for 1 h at 60 °C and alkylated with a cysteine blocking reagent, as recommended by the isobaric tags for the relative and absolute quantitation (iTRAQ) protocol (ABSciex). Proteins were digested using trypsin (Thermo Scientific) overnight at a 1 : 50 (w : w) ratio at 37 °C. Samples were labeled with iTRAQ reagents, combined, and dried in a vacuum concentrator (Eppendorf, Hamburg, Germany). Peptides were then dissolved in 5% acetonitrile (ACN) and 0.1% trifluoroacetic acid and purified using C18 MacroSpin Columns (Harvard Apparatus, Holliston, MA, USA). The eluate was dried in a vacuum concentrator, reconstituted in 5% ACN and 0.1% formic acid (FA), and subjected to off‐line strong cation exchange fractionation. Before sample loading, SCX columns (Harvard Apparatus) were rehydrated using double‐distilled H2O (20 min). Peptide fractions were eluted by 11 consecutive salt plug injections (5, 10, 20, 40, 60, 80, 100, 150, 200, 300, and 500 mm ammonium acetate in solution with 5% ACN and 0.1% FA). Each fraction was dried in a vacuum concentrator (Eppendorf) and stored at −80 °C until liquid chromatography tandem‐mass spectrometry (LC‐MS/MS) analysis was conducted.
Figure 2.
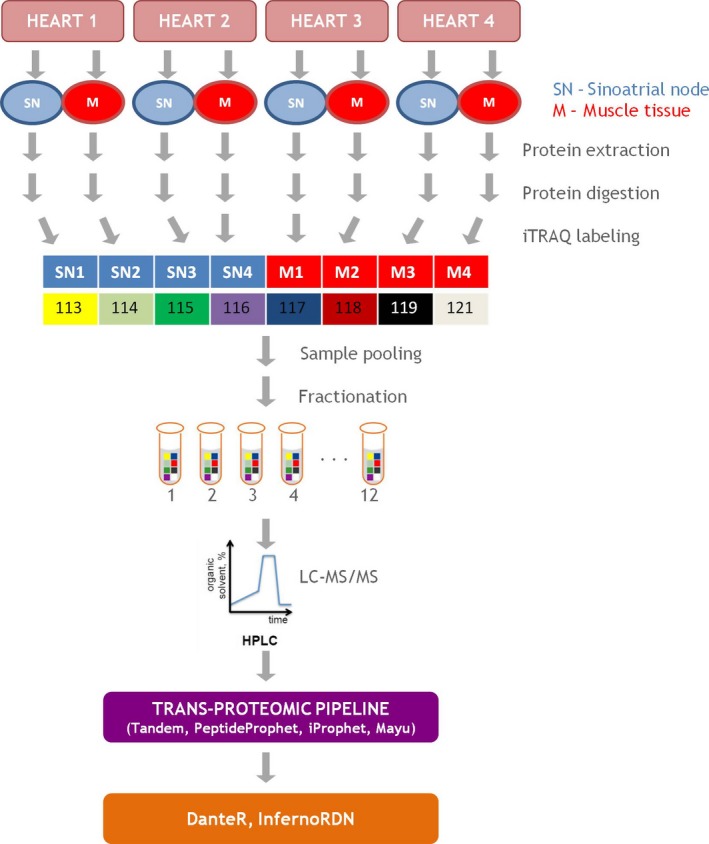
Quantitative comparison of protein profiles expressed in the sinoatrial node and ventricular muscle tissue.
LC‐MS/MS
Each peptide fraction (analyzed in two technical replicates) was injected onto a PepMap100 RP C18 75 μm i.d. × 50 cm column (Dionex, Thermo Scientific) via a trap column (PepMap100 RP C18 100 μm i.d. × 2 cm, Dionex, Thermo Scientific). The peptides were separated over 240 min using a 5–45% B phase linear gradient (A phase = 2% ACN and 0.1% FA; B phase = 80% ACN and 0.1% FA) with a flow rate of 300 nL min–1 using a UltiMate 3000 HPLC system (Dionex, Thermo Scientific) and applied on‐line to a Velos Pro (Thermo Scientific) dual‐pressure ion‐trap mass spectrometer. The main working nanoelectrospray ion source (Nanospray Flex, Thermo Scientific) parameters consisted of an ion spray voltage of 1.9 kV and a capillary temperature of 250 °C. Spectra were collected in the full scan mode (400–1500 Da), followed by a combination of collision induced dissociation (CID) higher‐energy collisional dissociation (HCD) MS/MS scans on the three most intense ions from the preceding survey full scan using dynamic exclusion criteria. The collected data were analyzed using a target‐decoy search strategy using the X!Tandem search engine (The GPM Organization) (Deutsch et al. 2010) and were statistically validated with peptideprophet using the trans‐proteomic pipeline suite of software (Institute for Systems Biology) (Ma et al. 2012). Search parameters were set as follows: concatenated target‐decoy database containing reviewed human protein sequences, UniProt release 2015_01; enzyme, trypsin; missed cleavage sites allowed, 2; fixed modification, methylthio(C); variable modifications, methionine oxidation(M), iTRAQ8plex(K), iTRAQ8plex(N‐term), and iTRAQ8plex(Y); parent mass error, −1.5 to +3.0 Da; and peptide fragment mass tolerance, 0.7 Da. peptideprophet results were combined with iprophet (Shteynberg et al. 2011) and validated using the Mayu algorithm (Reiter et al. 2009) to calculate peptide and protein false discovery rate (FDR). Peptide identification with FDR below 1.3% was considered to be a correct match (to ensure protein FDR below 5%). To minimize the protein quantification errors arising from the postmortem tissue, we only included the fully tryptic peptides in the analysis (to reduce the bias from the action of unspecific cellular proteases), and we performed the relative abundance measurements on distinct and unique peptides only, where the relative abundance ratio was a direct measure of its corresponding protein. Shared peptides, however, provide a weighted average of the abundance ratios that include all the corresponding proteins that arise from the absolute abundance of those proteins in the sample. Relative quantification based on shared peptides should be interpreted with caution (Nesvizhskii & Aebersold, 2005; Rauniyar & Yates, 2014). Thus, the replicate peptides were aggregated across iTRAQ channels to unique peptides, whereas the corresponding reporter ion intensities were added together (Figs S2 and S3). The dataset was then incorporated into danter and normalized using linear regression. Protein quantitation, based on iTRAQ reporter intensities, was performed using danter analysis of variance (anova) at the peptide level. This required at least two measurements to compare SAN with the working cardiomyocyte peptides using a linear model (Taverner et al. 2012). The minimum and maximum number of peptides was set to 2 and 500, respectively. Finally, P‐values were adjusted using the Benjamini & Hochberg FDR correction. Regulated proteins were determined using an adjusted P‐value cut‐off of 0.05. The mass spectrometry proteomics data were deposited into the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD004109 (Vizcaíno et al. 2014).
Cluster/Group comparison by ClueGO and pathway analysis
ClueGO, a Cytoscape plug‐in (Bindea et al. 2009), was used to visualize the gene ontology (GO) annotation and pathways in the constructed protein network under the cytoscape 3.3.0 software environment (Shannon et al. 2003) or the enrichment of biological terms and groups, and the two‐sided (enrichment/depletion) tests were used based on the hyper‐geometric distribution. The statistical significance was set to 0.05 (P ≤ 0.05) with the Bonferroni step‐down adjustment to correct the P‐value for the terms and the groups created by ClueGo. Fusion criteria (GO Term Fusion) were used to diminish the redundancy of the terms shared by similar associated proteins, which allowed us to maintain the most representative parent or child terms. The kappa‐statistics score was set to 0.4, which refers to the quantitative measurement of the degree of agreement on how proteins share similar annotation of GO terms. The GO level intervals were set between 2 and 4, and the minimum number and percent of associated proteins were set to 8 and 3% for cluster 1, and 6 and 3% for cluster 2, respectively. The GO term specificity for the cluster was set to 60%. The leading group was based on highest significance, and the group merge was set to 50%. For pathway enrichment, ClueGO was set for Wikipathways, and the network specificity was set to medium. The kappa‐statistics score was set to 0.4, and the minimum number and percent of associated proteins for both clusters were set to 3 and 3%, respectively. The Wikipathways term specificity for the cluster was set to 60%. Other analysis parameters were the same as in the GO enrichment. We also performed additional pathway analysis using the PANTHER tool (Thomas et al. 2006) and the Reactome pathway database (Fabregat et al. 2016; Mi et al. 2017). Briefly, we submitted overexpressed proteins into the online Gene List Analysis PANTHER tool and selected the statistical over‐representation test with default settings. We then annotated the dataset using Reactome pathways with Bonferroni correction for multiple testing.
Results
Protein identification and quantitation
Collectively, we identified 451 different proteins in both SAN and working cardiomyocytes, among which 166 were differentially regulated (110 were upregulated in SAN and 56 in working cardiomyocytes; Fig. 3A,B). In 16 cases, specific protein isoforms could not be distinguished in the identified peptides. All identification and quantitation details are provided in Supporting Information Tables S1 and S2. Despite the high degree of biological variability between donors, mainly in the SAN samples, the unsupervised analysis (based on the quantitative data of the unique peptide) enabled the distinction of tissue specimens from principal component analysis plots (Fig. 3C). Thus, the approach used here is suitable for differentiation between tissues from the same organ. However, these results should be interpreted with caution because human postmortem tissue was used and there was a lack of iTRAQ confirmation with different, more specific methods.
Figure 3.
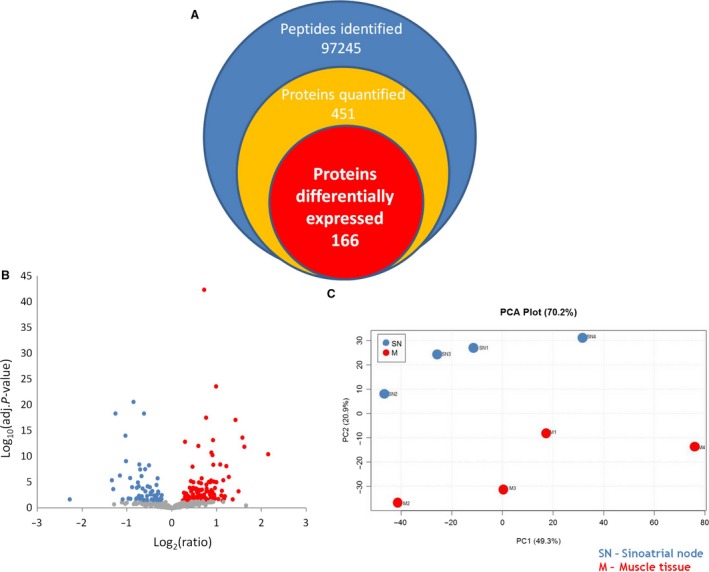
Schematic representation of the proteins identified in the iTRAQ experiment. Despite the high degree of variation in protein abundance between biological replicates, the depth of the proteome assessed in the analysis (A) and the global quantitative differences (B) allowed for a correct and principal component analysis (C) to distinguish between tissue specimens.
GO‐term enrichment analysis
For the enrichment analysis in ClueGO, we divided the identified proteins into two groups to illustrate their functional differences and reflect the relationship between the biological terms based on the similarity of their linked proteins. The first group contained proteins that were more abundant in the SAN sample, and the second group contained proteins that were more abundant in working cardiomyocytes. The two protein groups were uploaded in ClueGO as two separate clusters and were compared by applying the appropriate settings (see Cluster/Group comparison by ClueGO in Methods section).
Three major networks of GO terms related to ‘biological processes’ and ‘cellular components’ were highly enriched (Fig. 4A–C). The first group, which is related to the generation of precursor metabolites and energy, the oxidation‐reduction process, and mitochondrial portions (Fig. 4A), was characteristic of working cardiomyocytes. The second group, which is related to blood microparticles and extracellular space (Fig. 4B), consisted mainly of proteins overexpressed in the SAN samples. The third group, which is related to contractile fiber portions (Fig. 4C), contained proteins found in both the SAN and working cardiomyocytes samples. Other small networks (hydrogen peroxide catabolic process, response to an inorganic substance, and focal adhesion) or single nodes (cytoplasmic region, extracellular exosome, and response to unfolded protein) contained proteins mainly upregulated in the SAN group (Fig. 4B). Details regarding Fig. 4, including the adjusted Term/Group P‐values and a list of proteins specific for each node and cluster, are presented in Supporting Information Table S3.
Figure 4.
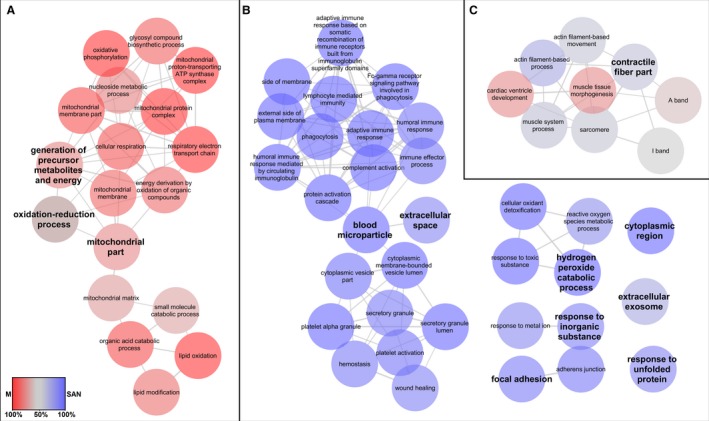
Enriched GO network related to ‘biological processes’ and ‘cellular components’ in the sinoatrial node and ventricular muscle. The most significant parent or child term per cluster is shown in the ClueGO grouped network in the following groups: (A) GO terms specific for ventricular muscle; (B) GO terms specific for the sinoatrial node; (C) GO terms with mixed specificity. Biological processes and cellular components (GO categories) of the identified over‐ and under‐synthesized proteins in the sinoatrial node and ventricular muscle were visualized with ClueGO (kappa score ≥ 0.4) as a functional grouped network; only the most significant interactions are shown. Each node represents a GO term. The enrichment significance of the GO terms is reflected by the size of the nodes. Edges represent connections between the nodes. The specific functional terms that were used to generate the network in cytoscape resulted from comparison of the two clusters (blue, sinoatrial node; red, ventricular muscle). Node color intensity is equivalent to a percentage of cluster‐specific proteins. Details are available in Table S3.
Collectively, these data indicate which biological processes and cellular compartments were differentially expressed in the analyzed samples. Additionally, some processes or compartments were described by a similar number of different proteins, which might suggest altered biological processes or cellular compartments in the SAN or working cardiomyocytes.
Pathway enrichment analysis
To examine which biological pathways are apparently altered in the SAN and working cardiomyocytes, we next performed a pathway analysis. Differentially represented proteins in the SAN and working cardiomyocytes were searched against the Wikipathways database using the ClueGO app in cytoscape. The two protein groups were uploaded into ClueGO as two separate clusters and were compared by applying the appropriate settings (see Cluster/Group comparison by ClueGO in the Methods section). Enriched pathways were grouped into several categories based on the biological processes in which they participate (Table 1). Details regarding Table 1, including enriched networks, are listed in Supporting Information Fig. S1. Additional information from the pathway analysis using the PANTHER tool were included in Table 1 and Supporting Information Table S4. The majority of the enriched pathways related to general metabolic processes were exclusive to working cardiomyocytes, including the electron transport chain, oxidative phosphorylation, fatty acid biosynthesis, and mitochondrial long‐chain fatty acid beta‐oxidation. The results were consistent between the Wikipathways and Reactome pathway databases. Only one pathway of metabolic origin, the fatty acid omega activation pathway, was exclusive to the SAN, but it was only shown in Wikipathways. The same proteins in the Reactome pathway database were assigned to a different pathway: ethanol oxidation. Additionally, a similar number of different proteins from both tissue types participate in several pathways, including the tricarboxylic acid cycle (TCA cycle), pyruvate dehydrogenase complex (PDHc), and glycolysis and gluconeogenesis. Unfortunately, these results from Wikipathways were true only for working cardiomyocytes in the Reactome analysis. We also observed an enriched pathway related to striated muscle contraction, which was composed of different proteins in the SAN and working cardiomyocytes. In the working myocardium, myosin 7, myomesin 1, myosin binding protein C, myosin light chain 2, troponin C, titin, and nebulette were upregulated, whereas myosin light chain 4, myosin regulatory light chain 2, tropomyosin α1, tropomyosin α3, tropomyosin β, actin 1, actin α smooth muscle, actin α skeletal muscle, actinin α1, myosin 6, moesin, transgelin 1, transgelin 2, gelsolin, vimentin, and desmin were over‐represented in the SAN samples. A similar observation was discovered in the Reactome pathways. Several enriched Wikipathways belonging exclusively to the SAN were also described, including the complement and coagulation cascade, pathogenic Escherichia coli infection, the FAS pathway, stress induction of heat shock protein regulation, and a selenium micronutrient network. The Reactome pathways only overlapped with the coagulation cascade pathway; the others were not presented.
Table 1.
Enriched pathways in sinoatrial node and ventricular muscle
| Pathway name | Gene symbols of over‐represented proteins in sinoatrial node | Gene symbols of over‐represented proteins in ventricular muscle | Adjusted P‐value |
|---|---|---|---|
| Tryptophan metabolism | ALDH1A1 ALDH2 HSD17B10 | ACAT1 ECHS1 HADH OGDH | 1.7e‐3 |
| TCA cycle | IDH3A SUCLG2 | CS DLD DLST MDH2 OGDH | 1.4e‐6 |
| TCA cycle and PDHc | IDH3A SUCLG2 | CS DLD DLST MDH1 OGDH | 8.8e‐7 |
| Electron transport chain | ATP5A1 ATP5B ATP5C1 ATP5F1 ATP5H ATP5L ATP5O COX4I1 COX5B NDUFB6 NDUFV2 SLC25A4 UQCRC1 | 1.6e‐5 | |
| Oxidative phosphorylation | ATP5A1 ATP5B ATP5F1 ATP5H ATP5L ATP5O NDUFB6 NDUFV2 | 1.4e‐3 | |
| Fatty acid biosynthesis | ACAA2 DECR1 ECH1 ECHS1 HADH | 2.3e‐3 | |
| Fatty acid beta oxidation | ACADS ACADVL ACAT1 DECR1 DLD ECHS1 HADH HADHA HADHB | 1.3e‐6 | |
| Mitochondrial LC‐fatty acid beta‐oxidation | ACADS ACADVL HADH HADHA | 8.2e‐3 | |
| Fatty acid omega oxidation | ADH1A ADH1B ALDH1A1 ALDH2 | 6.8e‐3 | |
| Glycolysis and gluconeogenesis | ENO1 PGAM1 PGAM2 PKM | DLD ENO3 GAPDH GOT1 GPI LDHB MDH1 MDH2 | 2.2e‐8 |
| Striated muscle contraction | ACTA1 ACTA2 DES MYH6 MYL4 TPM1 TPM2 TPM3 VIM | MYBPC3 MYL2 MYL3 MYOM1 TNNC1 TTN | 5.2e‐14 |
| Complement and coagulation cascades | APOA2 C3 C4B CFB FGB KNG1 SERPINA1 SERPING1 | 1.3e‐3 | |
| SIDS susceptibility pathways | C4B HSP90B1 HSPD1 SPTBN1 TF YWHAE YWHAZ | GAPDH HADHA HADHB SLC25A4 | 1.7e‐2 |
| Pathogenic Escherichia coli infection | ACTB CDC42 TUBA1A TUBB TUBB2A TUBB4B YWHAZ | 4.8e‐3 | |
| FAS pathway and stress induction of HSP regulation | ACTA1 ACTB HSPB1 LMNA SPTAN1 | 3.3e‐2 | |
| Selenium micronutrient Network | ALB APOA1 GPX3 HBA1 HBB PRDX1 PRDX5 SERPINA3 | 1.6e‐2 |
Differently represented proteins in sinoatrial node and ventricular muscle were subjected to pathway analysis by querying the Wikipathways database using ClueGo app in the cytoscape software.
Proteins were characterized by cluster and assigned to each pathway; they are listed by protein‐coding gene (Human Genome Organization gene symbol).
Adjusted P‐value corresponds to the P‐value adjusted by the Bonferroni step‐down correction during the enrichment test.
Proteins overlapping with the Reactome pathway database are presented in bold.
Discussion
Metabolic pathways
Our study indicates that some proteins are differentially represented in SAN and working cardiomyocytes, which may suggest that these tissues have different biological processes and cellular compartments. The generation of precursor metabolites and energy, the oxidation‐reduction process, and the mitochondrial functions were characteristic for working cardiomyocytes. The majority of the enriched pathways related to general metabolic processes were exclusive for working cardiomyocytes (electron transport chain, oxidative phosphorylation, fatty acid biosynthesis, and mitochondrial long‐chain fatty acid beta‐oxidation). Only one pathway of metabolic origin (the fatty acid omega activation pathway) was exclusive to SAN, but it was found only in the Wikipathways database. To show the predominance of the fatty acid omega activation pathway, additional experiments are required (e.g. a SAN cell culture with different source of energy).
Nodal artery
Proteins related to blood microparticles and the extracellular space were upregulated in the SAN samples compared with the working cardiomyocyte samples. We suggest that this is the direct result of the inherent difficulties in the precise preparation of uniform SAN tissue and the presence of the SAN artery supplying the node. In further studies, the SAN artery should be removed from samples to avoid sample contamination by blood.
Is the SAN the smooth muscle?
Proteins related to contractile fibers were found in both SAN and working cardiomyocyte samples, but these tissues were composed of different proteins components. In the nodal tissue, except for the presence of myosin, tropomyosin, and actin, the following smooth muscle markers were found to be upregulated: alpha 2 smooth muscle actin (ACTA2), transgelin, vimentin, and desmin. The troponins were not expressed in SAN tissue. This protein profile is similar to the smooth muscle proteome.
The contractile units of smooth muscle do not contain the troponin complex, but they are associated with two muscle‐specific regulatory proteins: caldesmon and calponin (Gimona et al. 2003). In the SAN tissue samples, transgelin, an actin‐binding protein of the calponin family, was upregulated relative to working cardiomyocytes. The transgelin protein contains a C‐terminal calponin‐like module and an upstream positively charged amino acid region required for actin binding. Transgelin is ubiquitous in vascular and visceral smooth muscle and is an early marker of smooth muscle differentiation (Assinder et al. 2009; Zhang et al. 2014). For many years, the function of transgelin was unclear. However, more recently, several functions of transgelin have been elucidated, including organization of actin distribution, inhibition of the phenotypic modulation of smooth muscle cells from contractile to synthetic/proliferative cells, and regulation of calcium‐independent smooth muscle cell contraction, proliferation, cell migration, and tumor suppression (Gimona et al. 2003; Je & Sohn, 2007; Yang et al. 2007; Assinder et al. 2009).
Future research
We detected SAN‐specific proteins in our investigation. Some of the differentially expressed proteins detected in this work might shed new light on the embryological origin of individual cardiac components. However, all hypotheses derived from this study should be confirmed in more focused studies using separate targeted techniques. The next stage of our research will be to compare the SAN with the atrial musculature and superior vena cava wall using proteomic techniques, to more determine precisely the proteomic SAN signature. A similar comparison will be conducted for the atrioventricular node. This approach will allow the creation of a detailed protein map of the human atria and, in particular, will identify the physiological conduction pathways and ectopic clusters of conductive tissue.
Study limitations
This study has some limitations. First, we analyzed human autopsied material collected within 24 h of death. This may result in autolysis of some unstable proteins and proteins expressed at low levels. However, this is the only available source of structurally normal human heart tissue. Secondly, localizing the SAN and its dissection are extremely difficult and crucial steps. It is possible that the sample contains tissue other than SAN tissue, and the margin of the surrounding tissue should not be used. This limitation might be reduced by proteomic multilateral comparison of the SAN, superior vena cava, and right atrial tissue. Thirdly, to avoid contamination by intra‐vessel components, the SAN artery should be carefully removed from the samples before the iTRAQ analysis. Finally, additional validation of the sample quality and the proteomic results was not performed in this study because of the lack of available material, especially from the SAN. Performing replicate high‐resolution 2D‐LC separations required the use of large‐scale, sample‐consuming measurements, which absorbed all of our collected SAN sample. We were not able to verify experimentally the quality of our postmortem material to assess precisely the influence of sample processing (inter alia protein extraction, digestion performance), quantification variability by mass spectrometry, and biological variability of the final reported protein differential abundance. Thus, we restricted our analysis to fully tryptic, unique, and distinct peptides to minimize quantification error at the expense of the loss of additional information derived from semi‐tryptic and shared peptides. Therefore, the potential limitations associated with the use of human postmortem tissues discussed above should be taken into consideration and the data interpreted with caution. These limitations should be addressed in future studies.
Conclusions
We showed comparative analysis of protein abundance in the human SAN and working cardiomyocytes. Our results show possible implementation of proteomic strategies to identify in‐depth functional differences between various heart sub‐structures.
Author contributions
W.K‐P. and P.P.W.: conceived and designed the experiments, analyzed the data, contributed reagents/materials/analysis tools, wrote the paper, reviewed drafts of the paper. A.K‐O.: conceived and designed the experiments, analyzed the data, wrote the paper, reviewed drafts of the paper. M.S. and P.K.: conceived and designed the experiments, performed the experiments, analyzed the data, wrote the paper, prepared Figures and/or Tables, reviewed drafts of the paper. M.K.H.: conceived and designed the experiments, analyzed the data, contributed reagents/materials/analysis tools, wrote the paper, prepared Figures and/or Tables, reviewed drafts of the paper.
Conflict of interest
All authors declare no conflicts of interest.
Supporting information
Fig. S1. Enriched Wikipathways network in the sinoatrial node and ventricular muscle.
Fig. S2. Technical replicates correlation plots. Biological replicates are represented by the assigned reporter ions. Technical replicates are depicted by run number.
Fig. S3. Mean log‐intensity plots of all experiments. Mean log‐intensity of all biological and technical replicates was correlated separately across the observed log‐intensity for all experiments included in the quantitative analysis.
Table S1. Quantitation details of the proteins identified in the iTRAQ experiment.
Table S2. Peptide identification and quantitation details.
Table S3. Enriched GO terms in the sinoatrial node and ventricular muscle (over‐represented and uniquely identified proteins).
Table S4. Results of additional pathway analysis using the PANTHER tool.
Acknowledgements
Dr. Mateusz K. Hołda was supported by the Foundation for Polish Science (FNP).
References
- Anderson RH, Yanni J, Boyett MR, et al. (2009) The anatomy of the cardiac conduction system. Clin Anat 22, 99–113. [DOI] [PubMed] [Google Scholar]
- Assinder SJ, Stanton JA, Prasad PD (2009) Transgelin: an actin‐binding protein and tumour suppressor. Int J Biochem Cell Biol 41, 482–486. [DOI] [PubMed] [Google Scholar]
- Atkinson AJ, Logantha SJ, Hao G, et al. (2013) Functional, anatomical, and molecular investigation of the cardiac conduction system and arrhythmogenic atrioventricular ring tissue in the rat heart. J Am Heart Assoc 2, e000246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Hackl H, et al. (2009) ClueGO: a Cytoscape plug‐in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffels VM, Moorman AF (2009) Development of the cardiac conduction system: why are some regions of the heart more arrhythmogenic than others? Circ Arrhythm Electrophysiol 2, 195–207. [DOI] [PubMed] [Google Scholar]
- Christoffels VM, Mommersteeg MT, Trowe MO, et al. (2006) Formation of the venous pole of the heart from an Nkx2‐5‐negative precursor population requires Tbx18. Circ Res 98, 1555–1563. [DOI] [PubMed] [Google Scholar]
- Deutsch EW, Mendoza L, Shteynberg D, et al. (2010) A guided tour of the trans‐proteomic pipeline. Proteomics 10, 1150–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabregat A, Sidiropoulos K, Garapati P, et al. (2016) The Reactome pathway Knowledgebase. Nucleic Acids Res 44, D481–D487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira R, Moreira‐Gonçalves D, Azevedo AL, et al. (2015) Unraveling the exercise‐related proteome signature in heart. Basic Res Cardiol 110, 454. [DOI] [PubMed] [Google Scholar]
- Gedik N, Krüger M, Thielmann M, et al. (2017) Proteomics/phosphoproteomics of left ventricular biopsies from patients with surgical coronary revascularization and pigs with coronary occlusion/reperfusion: remote ischemic preconditioning. Sci Rep 7, 7629 https://doi.org/10.1038/s41598-017-07883-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimona M, Kaverina I, Resch GP, et al. (2003) Calponin repeats regulate actin filament stability and formation of podosomes in smooth muscle cells. Mol Biol Cell 14, 2482–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourdie RG, Harris BS, Bond J, et al. (2003) Development of the cardiac pacemaking and conduction system. Birth Defects Res C Embryo Today 69, 46–57. [DOI] [PubMed] [Google Scholar]
- Hoogaars WM, Tessari A, Moorman AF, et al. (2004) The transcriptional repressor Tbx3 delineates the developing central conduction system of the heart. Cardiovasc Res 62, 489–499. [DOI] [PubMed] [Google Scholar]
- Je HD, Sohn UD (2007) SM22α is required for agonist‐induced regulation of contractility: evidence from SM22α knockout mice. Mol Cells 23, 175–181. [PubMed] [Google Scholar]
- Ma K, Vitek O, Nesvizhskii AI (2012) A statistical model‐building perspective to identification of MS/MS spectra with PeptideProphet. BMC Bioinformatics 13(Suppl 1), S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín‐Rojas T, Gil‐Dones F, Lopez‐Almodovar LF, et al. (2012) Proteomic profile of human aortic stenosis: insights into the degenerative process. J Proteome Res 11, 1537–1550. [DOI] [PubMed] [Google Scholar]
- Marx V (2014) Proteomics: an atlas of expression. Nature 509, 645–649. [DOI] [PubMed] [Google Scholar]
- Mi H, Huang X, Muruganujan A, et al. (2017) PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res 45, D183–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorman AF, Christoffels VM, Anderson RH (2005) Anatomic substrates for cardiac conduction. Heart Rhythm 2, 875–886. [DOI] [PubMed] [Google Scholar]
- Nesvizhskii AI, Aebersold R (2005) Interpretation of shotgun proteomic data. Mol Cell Proteomics 4, 1419–1440. [DOI] [PubMed] [Google Scholar]
- Pennisi DJ, Rentschler S, Gourdie RG, et al. (2002) Induction and patterning of the cardiac conduction system. Int J Dev Biol 46, 765–775. [PubMed] [Google Scholar]
- Rauniyar N, Yates JR (2014) Isobaric labeling‐based relative quantification in shotgun proteomics. J Proteome Res 13, 5293–5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter L, Claassen M, Schrimpf SP, et al. (2009) Protein identification false discovery rates for very large proteomics data sets generated by tandem mass spectrometry. Mol Cell Proteomics 8, 2405–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, et al. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shteynberg D, Deutsch EW, Lam H, et al. (2011) iProphet: multi‐level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol Cell Proteomics 10(M111), 007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverner T, Karpievitch YV, Polpitiya AD, et al. (2012) DanteR: an extensible R‐based tool for quantitative analysis of ‐omics data. Bioinformatics 28, 2404–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PD, Kejariwal A, Guo N, et al. (2006) Applications for protein sequence‐function evolution data: mRNA/protein expression analysis and coding SNP scoring tools. Nucleic Acids Res 34, W645–W650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viganò A, Vasso M, Caretti A, et al. (2011) Protein modulation in mouse heart under acute and chronic hypoxia. Proteomics 11, 4202–4217. [DOI] [PubMed] [Google Scholar]
- Vizcaíno JA, Deutsch EW, Wang R, et al. (2014) ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol 32, 223–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Chang YJ, Miyamoto H, et al. (2007) Transgelin functions as a suppressor via inhibition of ARA54‐enhanced androgen receptor transactivation and prostate cancer cell growth. Mol Endocrinol 21, 343–358. [DOI] [PubMed] [Google Scholar]
- Zhang R, Shi L, Zhou L, et al. (2014) Transgelin as a therapeutic target to prevent hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 306, L574–L583. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Enriched Wikipathways network in the sinoatrial node and ventricular muscle.
Fig. S2. Technical replicates correlation plots. Biological replicates are represented by the assigned reporter ions. Technical replicates are depicted by run number.
Fig. S3. Mean log‐intensity plots of all experiments. Mean log‐intensity of all biological and technical replicates was correlated separately across the observed log‐intensity for all experiments included in the quantitative analysis.
Table S1. Quantitation details of the proteins identified in the iTRAQ experiment.
Table S2. Peptide identification and quantitation details.
Table S3. Enriched GO terms in the sinoatrial node and ventricular muscle (over‐represented and uniquely identified proteins).
Table S4. Results of additional pathway analysis using the PANTHER tool.