

Abstract
Background and Purpose
Agonists at μ‐opioid receptors (μ‐receptors) are used for pain management but produce adverse effects including tolerance, dependence and euphoria. The co‐administration of a μ‐receptor agonist with a δ‐opioid receptor (δ‐receptor) antagonist has been shown to produce antinociception with reduced development of some side effects. We characterized the effects of three μ‐receptor agonist/δ‐receptor antagonist peptidomimetics in vivo after acute and repeated administration to determine if this profile provides a viable alternative to traditional opioid analgesics.
Experimental Approach
Three μ‐receptor agonist / δ‐receptor antagonist peptidomimetics, AAH8, AMB46 and AMB47, and morphine were evaluated for the development of tolerance and dependence after 5 days of twice daily treatment with escalating doses of drug (10–50 mg·kg−1). Antinociceptive effects were measured in the warm water tail withdrawal assay before and after repeated drug treatment. Physical dependence was evaluated by naltrexone‐precipitated withdrawal jumping. The rewarding effects of AAH8 were evaluated using a conditioned place preference (CPP) assay with twice daily conditioning sessions performed for 5 days.
Key Results
Morphine, AAH8, AMB47 and AMB46 all demonstrated acute antinociceptive effects, but repeated administration only produced tolerance in animals treated with morphine and AMB46. Injection of naltrexone precipitated fewer jumps in mice treated repeatedly with AAH8 as compared with morphine, AMB47 or AMB46. Conditioning with morphine, but not AAH8, produced significant CPP.
Conclusions and Implications
AAH8 may be a better alternative than traditional opioid analgesics, producing antinociception with less development of tolerance and dependence and may be less rewarding than morphine.
Abbreviations
- BID
twice daily
- CPP
conditioned place preference
- DAMGO
[d‐Ala2,N‐MePhe4,Gly‐ol]‐enkephalin
- DPDPE
[d‐Pen2,5]‐enkephalin
- MPE
maximum possible effect
- Ke
potency of a pure antagonist
- Ki
inhibition constant for a ligand
- TFA
trifluroacetic acid
- TST
tail suspension test
- w/v
weight to volume
- w/w
weight to weight
- WWTW
warm water tail withdrawal
- +/+
wild‐type
- −/−
homozygous knockout
Introduction
While opioid drugs have significant clinical utility in treating pain, there are drawbacks associated with their chronic use, including tolerance, dependence, constipation and addiction liability (Benyamin et al., 2008). The development of tolerance to the analgesic effects of opioids often leads to dose escalation, which may contribute to opioid misuse and abuse (Ballantyne and LaForge, 2007). Further, individuals who are dependent on opioids may misuse them to prevent withdrawal (Ross and Peselow, 2009; Bailey and Connor, 2005; Johnston et al., 2009; Ballantyne and LaForge, 2007). Opioid compounds that produce robust analgesia with limited development of tolerance and dependence could address a significant unmet medical need and provide an alternative to traditional opioid analgesics to prevent opioid misuse and abuse.
Opioids produce both their pain‐relieving and adverse effects through stimulation of the μ‐opioid receptor (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=319); therefore, creating more selective ligands for the μ‐receptor is unlikely to reduce the incidence of adverse events. There are reports that the co‐administration of μ‐receptor agonist with a http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=317 antagonist produces μ‐receptor‐mediated antinociception with reduced tolerance and dependence liabilities (Abdelhamid et al., 1991; Fundytus et al., 1995; Hepburn et al., 1997; Purington et al., 2009; Schiller, 2009; Anand et al., 2016; Váradi et al., 2016), and similar results have been found in δ‐receptor knockout (KO) animals (Zhu et al., 1999). As a result, the development of μ‐receptor agonist/δ‐receptor antagonist compounds – mixed efficacy ligands – has been explored, and several peptide (Purington et al., 2011; Purington et al., 2009; Schiller et al., 1999; Schmidt et al., 1994; Anand et al., 2012; Cai et al., 2014), peptide‐like (Balboni et al., 2002b; Balboni et al., 2002a; Salvadori et al., 1999; Lee et al., 2011; Bender et al., 2015; Mosberg et al., 2013) and alkaloid (Anathan et al., 1999; Anathan et al., 2004; Horan et al., 1993; Healy et al., 2013) compounds have been described. Noteworthy ligands are the peptides DIPPψNH2 (Schiller et al., 1999) and VRP26 (Anand et al., 2016), the bivalent ligand MDAN‐21 (Lenard et al., 2007) and the multifunctional opioid alkaloid UMB425 (Healy et al., 2013). All show some improvement over http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1627, but both DIPPψNH2 and UMB425 produce significant tolerance and dependence after chronic administration, and MDAN‐21 was effective in some (Aceto et al., 2012; Daniels et al., 2005), but not all (Aceto et al., 2012), measures of antinociception. We previously reported that VRP26 produces no antinociceptive tolerance or physical dependence after 7 days of continuous administration and produces little conditioned place preference (CPP) (Anand et al., 2016); however, VRP26 is difficult to synthesize and purify and therefore makes a poor drug candidate. While these μ‐receptor agonist/δ‐receptor antagonist compounds display promising effects in vivo, there is still room for improvement.
We have previously described a series of peptidomimetics that display μ‐receptor agonist/δ‐receptor antagonist characteristics in vitro and produce opioid‐mediated anti‐nociception in vivo after peripheral administration (Bender et al., 2015; Harland et al., 2015; Mosberg et al., 2013). In this report, we evaluated the acute and chronic effects of these compounds after repeated escalating doses for 5 days and the role of δ‐receptors in the development of μ‐receptor‐mediated tolerance and dependence.
Methods
In vitro characterization of compounds
Cell lines and membrane preparations
C6‐rat glioma cells stably transfected with a rat μ (C6‐μ‐receptor) or rat δ (C6‐δ‐receptor)‐opioid receptor (Lee et al., 1999) and CHO cells stably expressing a human κ‐opioid receptor (Husbands et al., 2005) were used for all in vitro assays. Cells were cultured, and membranes were prepared as previously described (Anand et al., 2012).
Radioligand binding assays
Radioligand binding assays were performed as previously described (Anand et al., 2012). In brief, assays were performed using competitive displacement of 0.2 nM http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1612 (250 μCi, 1.85 TBq·mmol−1) by the test compound from membrane preparations containing opioid receptors. The assay mixture, containing membranes (approximately 20 μg protein per tube) in 50 mM Tris–HCl buffer (pH 7.4), [3H]diprenorphine and various concentrations of test compound, was incubated on a shaker at room temperature for 1 h to allow binding to reach equilibrium. The samples were filtered through Whatman GF/C filters and washed five times with 50 mM Tris–HCl buffer (pH 7.4). The radioactivity retained on dried filters was determined by liquid scintillation counting after saturation with EcoLume liquid scintillation cocktail in a Wallac 1450 MicroBeta (PerkinElmer, Waltham, MA). Non‐specific binding was determined using 10 μM naloxone. The results presented are the mean ± SEM from three individual assays performed on three different days. Each individual assay is performed in duplicate and then averaged.
Radioligand binding assays in sodium‐containing Tris buffer
Binding assays were performed by competitive displacement of [3H]diprenorphine (250 μCi, 1.85 TBq·mmol−1) by test compounds. The assay mixture contained the following components: assay buffer (50 mM Tris–HCl, 100 mM NaCl, 5 mM MgCl2 and 1 mM EDTA, pH 7.4), various concentrations of test compound diluted in buffer, 0.2 nM [3H]diprenorphine and membrane preparations containing opioid receptors (approximately 20 μg protein per tube) supplemented with 10 μM GTPγS. Non‐specific binding was determined using 10 μM naloxone. The assay plate was incubated at room temperature on a shaker for 75 min to allow binding to reach equilibrium. The mixture was then vacuum filtered through Whatman GF/C filters using a Brandel harvester (Brandel, Gaithersburg, MD, USA) and washed five times with 50 mM Tris–HCl buffer. Retained radioactivity was measured as described above. The results presented are the mean ± SEM from three individual assays performed on three different days. Each individual assay is performed in duplicate and then averaged.
Stimulation of [35S]GTPγ binding
Agonist stimulation of [35S]http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4207 (1250 Ci, 46.2 TBq·mmol−1) binding was measured as described previously (Anand et al., 2012). Briefly, membranes (10–20 μg of protein per tube) were incubated 1 h at room temperature in GTPγS buffer (50 mM Tris–HCl, 100 mM NaCl and 5 mM MgCl2, pH 7.4) containing 0.1 nM [35S]GTPγS, 30 μM GDP and varying concentrations of test compound. Test compound stimulation of [35S]GTPγS was compared with 10 μM standard compounds [d‐Ala2,N‐MePhe4,Gly‐ol]‐enkephalin (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1647) at μ‐receptors and [d‐Pen2,5]‐enkephalin (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1608) at δ‐receptors. The reaction was terminated by rapidly filtering through GF/C filters and washing 10 times with GTPγS buffer, and retained radioactivity was measured as described above. The results presented are the mean ± SEM from three individual assays performed on three different days. Each individual assay is performed in duplicate and then averaged; maximal stimulation was determined using non‐linear regression analysis to fit a logistic equation to the competition binding.
Determination of K e
Agonist stimulation of [35S]GTPγS binding by the known standard agonist DPDPE at δ‐receptor was measured as described above. This was then compared with [35S]GTPγS binding stimulated by DPDPE in the presence of test compound. Both conditions produced 100% stimulation relative to DPDPE. The difference between the EC50 of DPDPE alone and in the presence of test antagonist is the shift in dose response. The potency of a pure antagonist (K e) was then calculated as K e = (concentration of compound)/(dose–response shift − 1). The results presented are the mean ± SEM from three individual assays performed on three different days. Each individual assay is performed in duplicate and then averaged; maximal stimulation was determined using non‐linear regression analysis to fit a logistic equation to the competition binding data.
Calculation of relative efficacy at μ‐receptors
Agonist efficacy was calculated based on the ability to stimulate [35S]GTPγS according to the equation: efficacy = 0.5 × (E max,test/ E max,std) × (1 + (K itest/EC50test)), where E max,test is the maximum stimulation by test agonist, E max,std is the maximum stimulation by DAMGO, K itest is the affinity of test agonist in buffer containing sodium and EC50test is the potency of test agonist. Hill slopes for all of the binding and functional data were not significantly different from one, allowing use of the Ehlert equation (Quock et al., 1999).
Data normalization
Data for all in vitro competition binding assays are normalized such that basal (in the presence of 10 μM naloxone) and total binding (in the absence of any drug) are set to 0 and 100% binding respectively. Data for all in vitro [35S]GTPγS assays are normalized such that basal (in the absence of drug) and total (in the presence of 10 μM standard agonist) are set to 0 and 100% stimulation respectively. This normalization is used to account for variation between membrane preparations or assays.
In vivo characterization of compounds
Drug preparation
All compounds were administered by i.p. or s.c. injection in a volume of 10 mL·kg−1 of body weight. Morphine sulfate, AMB47 trifluroacetic acid (TFA) salt, AMB46 TFA and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1639 HCl (Tocris Bioscience, Minneapolis, MN, USA) were dissolved in sterile saline (0.9% NaCl w/v). AAH8 TFA was dissolved in 10:10:80 ethanol : Alkamuls 620 (Solvay, St. Louis, MO, USA) : sterile water. http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=summary&ligandId=1611 was dissolved in 1 M HCl and brought to a final concentration of 3% HCl (v/v) with sterile water. http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1641 HCl (Tocris Bioscience) was prepared in sterile water.
Animals
All animal care and experimental protocols were in accordance with US National Research Council's Guide for the Care and Use of Laboratory Animals (Council, 2011) and were approved by the University of Michigan Institutional Animal Care and Use Committee. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Male and female C57BL/6 δ‐receptor KO mice (B6.129S2‐Oprd1 tm1Kff/J stock number 007557; Jackson Laboratory, Sacramento, CA, USA) and their wild‐type littermates, C57BL/6 μ‐receptor knock out mice (B6.129S2‐Oprm1 tm1Kff/J stock number 007559; Jackson Laboratory), or C57BL/6 wild‐type mice (stock number 000664; Jackson Laboratory) weighing between 20 and 30 g at 8–16 weeks old, were used for behavioural experiments. KO animals were bred in‐house from heterozygous breeding pairs or trios. Mice were group housed with a maximum of five animals per cage in clear polypropylene cages with corn cob bedding and nestlets as enrichment. Mice had free access to food and water at all times, except during measurements of faecal boli production. Animals were housed in pathogen‐free rooms maintained between 20 and 26°C and with 30 ‐ 70% humidity with a 12 h light/dark cycle with lights on at 07:00 h.
Experiments were conducted in the housing room during the light cycle. Each mouse was used in only one experiment for acute antinociception, tolerance, physical dependence, tail suspension test (TST), CPP or measurement of constipating effects. C57BL/6 mice are the background strain for all the genetic KO strains used in this study. C57BL/6 mice were used for all studies as this species is commonly used in pharmacological and behavioural research. For antinociception and constipation assays, experiments were not blinded to drug conditions due to the complication of multiple drug doses required for escalating doses. However, there were limited a priori expectations for drug effects as most compounds tested are novel entities.
Antinociception
Antinociceptive effects were evaluated in the mouse warm water tail withdrawal (WWTW) assay. Withdrawal latencies were determined by briefly placing a mouse into a cylindrical plastic restrainer and immersing 2–3 cm of the tail tip into a water bath maintained at either 50 or 55°C. The latency to tail withdrawal or rapidly flicking the tail back and forth was recorded with a maximum cut‐off time of 20 s (50°C) or 15 s (55°C) to prevent tissue damage; baseline latencies were consistent for each assay: 3–5 s for 50°C and 2–3 s for 55°C.
Acute antinociceptive effects were determined using a cumulative dosing procedure (n = 6 animals per treatment group). Each mouse received an injection of saline i.p., and then 30 min later, baseline withdrawal latencies were recorded; mice were then given an i.p. injection of either saline or 1 mg·kg−1 naltrexone, and withdrawal latencies were recorded 30 min later. Following baseline determinations, cumulative doses of the test compound were given i.p. at 30 min intervals. Thirty minutes after each injection, the tail withdrawal latency was measured as described above.
For antinociceptive tests, agonist‐stimulated antinociception is expressed as a percentage of maximum possible effect (% MPE), where % MPE = (post‐drug latency − baseline latency) ÷ (cut‐off latency − baseline latency) × 100. Data are normalized to illustrate the difference in ED50 values as basal and maximal values vary based on temperature. Experiments were run by two separate individuals across several days.
Tolerance experimental design
Antinociceptive dose–effect curves for AAH8 (n = 12), AMB46 (n = 12), AMB47 (n = 12) and morphine (n = 12) were established on the morning of day 1 in wild‐type C57BL/6 mice using the 50°C WWTW assay described above. Each group was then randomly divided such that six mice were assigned to receive repeated drug injections and six mice were assigned to receive repeated saline injections.
On the morning of day 1, a dose–response curve for the test compound was established up to 10 mg·kg−1 i.p., and mice were then given an injection of 10 mg·kg−1 test compound i.p. at 19:00 h on the evening of day 1. For the remainder of the experiment, mice were given twice daily injections at 07:00 and 19:00 h; an escalating drug regimen was used such that mice received 20 mg·kg−1 test compound twice daily (BID) on day 2, 30 mg·kg−1 test compound BID on day 3, 40 mg·kg−1 test compound BID on day 4 and 50 mg·kg−1 test compound BID on day 5. Cumulative dose effect curves were established for all mice on the morning of day 6 for their respective test compounds. Data are presented as mean ± SEM for each treatment group before and after repeated treatment.
To determine agonist potency before and after repeated treatment with drug or vehicle, dose–response curves and ED50 values were calculated for each mouse and then averaged within each chronic treatment group. To calculate ED50 values for each mouse in the WWTW assay, the 50% level of maximum effect was determined from a linear regression analysis of individual latency to tail flick data, using only the linear portion of the curve and including only one dose that produced <10% of the baseline latency and one dose that produced >90% of the maximum latency (Jutkiewicz et al., 2011). ED50 values from each mouse were then averaged for each treatment group (mean ± SEM). Statistical comparisons between ED50 values were made using a repeated measures, factorial ANOVA for each compound.
In a separate experiment, antinociceptive dose–response curves for morphine were established using the mouse WWTW assay in δ‐receptor KO mice (n = 6) and their wild‐type littermates (n = 6). Mice were given twice daily escalating doses of morphine, and dose–response curves pre‐escalating and post‐escalating doses were performed, as described above. Experiments were run by two separate individuals across multiple sessions.
Physical dependence experimental design
Wild‐type C57BL/6 (n = 6) or δ‐receptor KO (n = 6) mice were treated for 5 days with either saline or escalating doses of test compound twice daily as described above. On the morning of day 6, mice were given 50 mg·kg−1 test compound, morphine or saline i.p. and then returned to their home cages. Two hours later, mice were given 10 mg·kg−1 naltrexone i.p. and placed individually in clear plastic observation cages (10 × 6 × 8 in.) without bedding. Mice were observed for jumps as a sign of opioid withdrawal for 30 min after naltrexone injection. Statistical comparisons of the number of jumps recorded were assessed using a one‐way ANOVA. Experiments were run by two separate individuals across multiple sessions.
Tail suspension test
Mice were pretreated with vehicle (n = 6), 3.2 mg·kg−1naltrindole (n = 6), a single dose (1–10 mg·kg−1) of test compound (n = 6 per dose) or 10 mg·kg−1 morphine (n = 6) s.c. either 30 min prior to injection of 3.2 mg·kg−1 SNC80 or vehicle s.c. Thirty minutes after SNC80 (or vehicle) injection, mice were suspended by their tail from a height of ~40 cm using tape for 6 min, and behaviour was recorded using a Sony HDR‐CX220 digital camcorder. Videos were scored by observers blind to the test condition, and the total time mice spent immobile was summed for each animal and then averaged within each treatment group. Immobility was defined as the animal remaining motionless or making only minor, non‐escape‐related movements. Statistical comparisons in immobility were made using a two‐way ANOVA. TST videos were scored by a separate individual who did not run the assay and was blinded to experimental conditions.
Conditioned place preference and locomotor activity
Apparatus
A two‐compartment place‐conditioning apparatus (MedAssociates, Inc. St. Albans, VT) was used for all CPP studies. The compartmentalized box was divided into two equal size sections (8 × 5 × 5 in.), accessed through a single, manual, guillotine door. The compartments differed in the wall colour and floor texture (black walls with rod flooring vs. white walls with mesh flooring). Time spent in each chamber, number of beam breaks (used as a measure of locomotor activity) and number of entrances to each side were recorded using IR photobeam detectors.
Conditioned place preference protocol
Experiments consisted of three phases: bias evaluation (2 days), conditioning (5 days) and testing (1 day).
Bias evaluation of CPP
Wild‐type mice were placed randomly into one chamber on day 1 and the opposite chamber on day 2 and then allowed to freely explore the apparatus for 30 min. If mice exhibited a greater than 70% preconditioning compartment bias, they were discarded from the study; no mice were discarded based on this criterion.
Conditioning phase of CPP
Mice were randomly assigned to be conditioned with 10 mg·kg−1 AAH8 (n = 6), 10 mg·kg−1 morphine (n = 6) or saline (n = 6) in either the black or white chamber. During conditioning, mice were given a saline injection (i.p.) and immediately placed in the saline‐paired chamber for 30 min; 6 h later, mice were given an injection of AAH8, morphine or saline (i.p.) and immediately placed in the drug‐paired chamber for 30 min. During all conditioning sessions, movement and activity were recorded.
Test day of CPP
Test day was always performed the day after the final conditioning session. Mice were randomly placed in either compartment and allowed to roam freely for 30 min. No injection was given on test day. Time spent in each chamber, beam breaks and entrances to each side were recorded. CPP scores were calculated as the difference between time spent on the drug‐paired side on test day and the average of time spent on the future drug‐paired side on the two bias evaluations.
Experiments were run by two individuals across multiple sessions.
Measurement of faecal bolus production
Tinted food was prepared by combining 25 g of regular chow with 40 mL of water and 0.25 mL of blue food dye. The food pellets were allowed to soften (approximately 2 h) and were mixed so that the food colouring was evenly distributed through the food paste. Mice were given 24 h access to tinted chow 1 week prior to an experiment in order to habituate them to the novel food preparation and then returned to regular chow. For experiments, mice were single housed in cages free of bedding and were food deprived overnight; mice had ad libitum access to water for the duration of the experiment. In the morning of the experiment, mice were given free access to tinted chow for 1 h. The tinted food was then removed, the cages wiped down and the mice were given an injection of either drug or vehicle i.p. and access to approximately 3 g of normal chow for the remainder of the experiment. The weight of both the normal chow and the tinted chow was recorded both before and after the experiment. The time to first tinted faecal bolus and the number of tinted faecal boli were recorded every hour for 6 h.
Experiments were run by two individuals across multiple sessions.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Data analysis was performed using GraphPad Prism version 6.02 (GraphPad, San Diego, CA, USA) or SPSS v22 with Tukey's post hoc tests to correct for multiple comparisons. Inhibition constant for a ligand (K i) and EC50 values were calculated using non‐linear regression analysis to fit a logistic equation to the competition binding data. ED50 values were calculated using GraphPad Prism version 6.02 by extrapolating the 50% maximum effect from the straight‐line analysis of the individual dose–effect curves (Jutkiewicz et al., 2011), and then ED50 values were averaged within a treatment group. For in vivo experiments, six mice per experimental condition (e.g. per drug and per genotype) were used. For statistical tests, post hoc tests were run only when F achieved P < 0.05 (α level was set to 0.05). There was no exclusion of any data in any studies. Treatment conditions were randomized across cages of mice and across at least three independent experiments. For in vivo studies, power analysis (α = 0.05; 1‐β = 0.9) revealed that for a calculated effect size of 1–3 (Cohen's d), depending on the experiment that a sample size of 4–6 mice per experimental condition would be needed (G*Power 3.1.9.2, Faul et al., 2007).
Materials
AAH8, AMB46 and AMB47 were synthesized using the route previously described (Bender et al., 2015; Harland et al., 2015). All other reagents and solvents were purchased from commercial sources and used without further purification. All chemicals and biochemicals were purchased from Sigma‐Aldrich (St. Louis, MO) or Fisher Scientific (Hudson, NH), unless otherwise noted. All tissue culture reagents were purchased from Gibco Life Sciences (Grand Island, NY). Radioactive compounds were purchased from PerkinElmer.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
Results
In vitro results
Affinity
As previously reported, AAH8 (Harland et al., 2015), AMB47 and AMB46 (Bender et al., 2015) all display low nanomolar binding affinity for μ‐receptors and δ‐receptors when binding assays are conducted in Tris buffer. Morphine, a prototypical μ‐receptor ligand, binds with low nanomolar affinity to μ‐receptors, preferring μ‐receptors 50‐fold over δ‐receptors (Table 1). As it has been demonstrated that sodium ions can alter the affinity of opioid ligands for their receptors (Simon and Groth, 1975; Pert et al., 1973; Selley et al., 2000), we assessed the affinity of AAH8, AMB47, AMB46 and morphine for μ‐ and δ‐receptors in the presence of sodium. Sodium ions decrease the affinity at μ‐ and δ‐receptors for all compounds tested, though the fold change in affinity is different for different compounds at each receptor (Table 1).
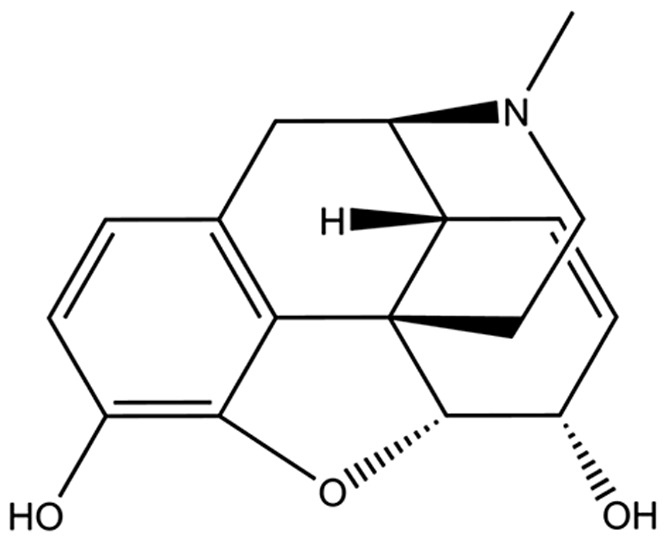
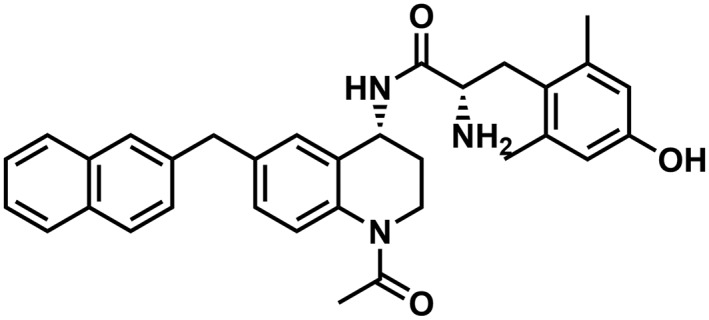
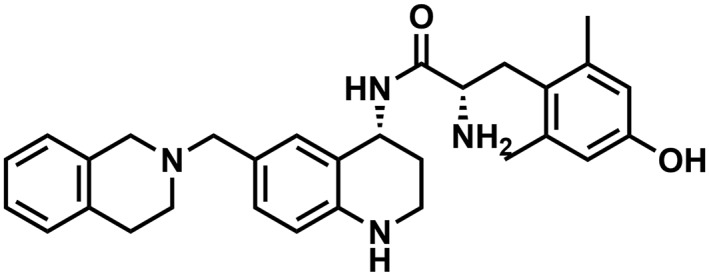
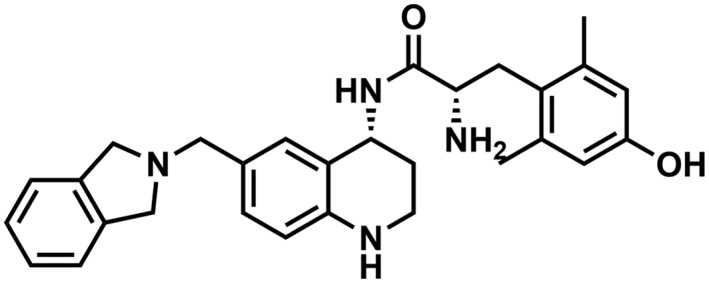
Table 1.
Binding affinity and efficacy data for the peptidomimetics
| Compound | Structure | K i in Tris, nM | K i in Tris + 100 mM NaCl, nM | GTPγS | Relative efficacy at μ | K e, δ | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| μ | δ | k | μ : δ ratio | μ | δ | μ : δ ratio | μ % stimulation | μ EC50 (nM) | δ % stimulation | δ EC50 (nM) | κ % stimulation | κ EC50 (nM) | ||||
| Morphine |
|
1.3 (0.3) | 103 (4) | 1:80 | 149 (66) | 433 (43) | 1:3 | 57 (2) | 152 (36) | 28 (2) | 1200 (600) | 0.56 | n/a | |||
| AAH8 |
|
0.04 (0.01)a | 0.2 (0.02)a | 50 (18)a | 1:5 | 1.7 (0.7) | 1.1 (0.4) | 1:1 | 87 (3)a | 0.9 (0.2)a | dnsa | n/aa | dnsa | n/aa | 1.26 | 1.8 (0.1) |
| AMB47 |
|
0.19 (0.08)b | 0.9 (0.2)b | 0.8 (0.1)b | 1:5 | 0.4 (0.1) | 3.5 (0.4) | 1:9 | 96 (4)b | 6 (3)b | dnsb | n/ab | 40 (8)b | >1000b | 0.51 | 4.4 (0.4) |
| AMB46 |
|
0.15 (0.08)b | 15 (5)b | 2 (1)b | 1:100 | 1.6 (0.2) | 83 (11) | 1:52 | 96 (4)b | 2.6 (1.5)b | dnsb | n/ab | 15 (2)b | 15 (9)b | 0.78 | 95 (17) |
Binding affinities (K i) were obtained by competitive displacement of [3H]‐diprenorphine in the presence or absence of sodium chloride.
Efficacy data were obtained using [35S]GTPγS binding assay. Efficacy is represented as percent maximal stimulation relative to standard agonists DAMGO (μ), DPDPE (δ) or U69,593 (κ) at 10 μM concentrations. Relative efficacy at μ was calculated using the Ehlert equation. K e values at δ were determined by shifting the dose ‐response curve for DPDPE, a standard δ agonist. All values are expressed as mean (SEM) of three separate assays performed in duplicate. n = 3 for all experiments. n/a, not applicable; nd, not determined; dns, does not stimulate.
Data previously published in Harland et al., 2015.
Data previously published in Bender et al., 2015.
Efficacy
AAH8, AMB47 and AMB46 are full agonists in the [35S]GTPγS assay at the μ‐receptor compared with DAMGO, with low nanomolar EC50 values, whereas morphine is a partial agonist, compared with DAMGO. The relative efficacy of these compounds in vitro is as follows: AAH8 > AMB46 > AMB47 > morphine. AAH8, AMB47 and AMB46 are antagonists at the δ‐receptor as they attenuate DPDPE‐stimulated [35S]GTPγS binding, with pA2 values (K e) in the nanomolar range; in this assay, naltrindole, a known δ‐receptor antagonist, displays a K e value of 0.13 ± 0.03 nM. Morphine is a low‐affinity, partial agonist at the δ‐receptor and as such does not shift the dose–response curve for DPDPE and does not produce a measurable K e value in vitro (Table 1).
In vivo results
μ‐Receptor‐mediated acute antinociceptive effects
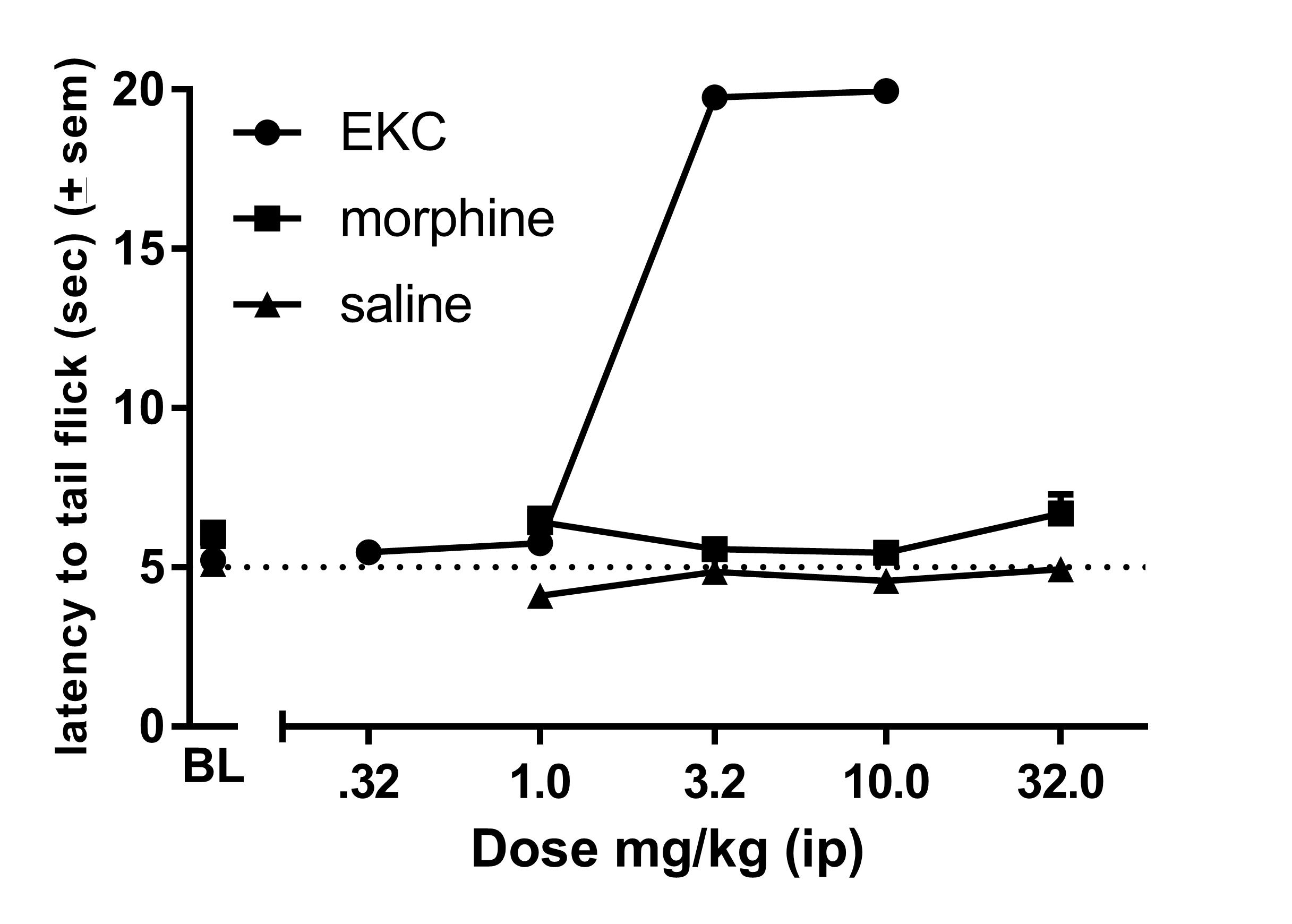
The anti‐nociceptive effects of AAH8, AMB47, AMB46 and morphine were assessed using the 50°C WWTW assay in wild‐type C57BL6/J mice pretreated with either saline or 1 mg·kg−1 naltrexone, a non‐selective opioid antagonist, to determine if the antinociceptive effects are opioid‐mediated in vivo. Consistent with earlier results (Bender et al., 2015; Harland et al., 2015), all compounds produce maximal anti‐nociceptive effects at 10 mg·kg−1 after i.p. injection in mice pretreated with saline (Figure 1). Pretreatment with 1 mg·kg−1 naltrexone i.p. produces an approximate threefold parallel rightward shift in the dose–response curves for AAH8, AMB47, AMB46 and morphine (Table 2). All compounds were then tested in μ‐receptor KO mice, to determine if the antinociceptive effects were μ‐receptor‐mediated. Consistent with in vitro results, neither the test peptidomimetics nor morphine produced any antinociception in μ‐receptor KO mice (Figure 1). http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1602, a known κ‐receptor agonist (used as a positive control), produced dose‐dependent antinociception in these KO mice (Supporting Information Figure S1).
Figure 1.
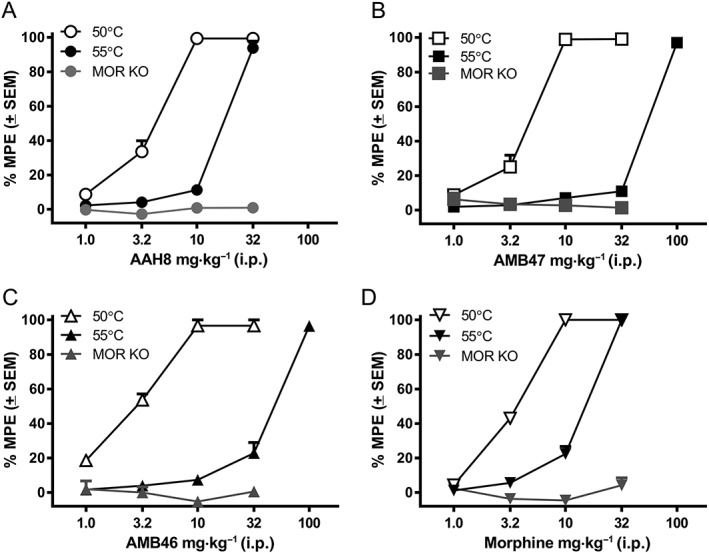
Cumulative dose–response curves for (A) AAH8, (B) AMB47, (C) AMB46 and (D) morphine in the mouse WWTW assay at 50°C or 55°C in wild‐type mice or at 50°C in μ‐receptor KO mice. Data shown are means ± SEM for all groups (n = 6 for each group).
Table 2.
ED50 values for peptidomimetics and morphine tested in the WWTW assay with either saline (50 and 55°C) or 1 mg·kg−1 naltrexone (NTX; 50°C) pretreatment
| Compound | ED50 (SEM) mg·kg−1 i.p. | ||
|---|---|---|---|
| Saline pretreatment (50°C) | 1 mg·kg−1 NTX pretreatment (50°C) | Saline pretreatment (55°C) | |
| AAH8 | 4.4 (0.4) | 13.7 (1.6) | 21.0 (0.8) |
| AMB47 | 5.3 (0.3) | 14.7 (0.6) | 64.5 (1.5) |
| AMB46 | 4.7 (0.2) | 12.9 (1.5) | 55.0 (1.3) |
| Morphine | 4.7 (0.05) | 15.2 (0.1) | 16.4 (0.9) |
ED50 values were calculated using a linear regression fit for the cumulative dose–response data from each individual mouse then averaged to get an ED50 value for each treatment group (n = 6).
While the peptidomimetics are equipotent in the 50°C WWTW assay, when tested at 55°C, differences in ED50 between the compounds are observed, even though they are all still fully effective (Figure 1 and Table 2). One‐way ANOVA of ED50 values (F(3, 40) = 398.9) shows a main effect of drug, demonstrating that AAH8 and morphine are significantly more potent than either AMB47 or AMB46 (F(3, 40) = 41.8).
In vivo acute δ‐receptor antagonist effects
To investigate whether AAH8, AMB47 and AMB46 function as centrally active δ‐receptor antagonists in vivo, we examined their ability to block the antidepressant‐like effects of a δ‐receptor agonist, SNC80, in the TST as compared with the prototypic δ‐receptor antagonist naltrindole (Figure 2A). Two‐way ANOVA of the data shows a significant main effect of SNC80 dose (F(1, 30) = 101.1) and of pretreatment (naltrindole, morphine or vehicle, F(2, 30) = 45.83) and an interaction of pretreatment × SNC80 dose (F(2, 30) = 14.10). Mice treated with 3.2 mg·kg−1 SNC80 (s.c.) alone display a significant decrease in immobility, compared with vehicle‐treated mice. This SNC80‐induced decrease in immobility is blocked by pretreatment with 3.2 mg·kg−1naltrindole (s.c.). Naltrindole‐pretreated mice have immobility scores that are not statistically different from immobility scores in vehicle‐treated mice. Pretreatment with morphine produces small, though not statistically significant, decreases in immobility scores in vehicle‐treated mice.
Figure 2.
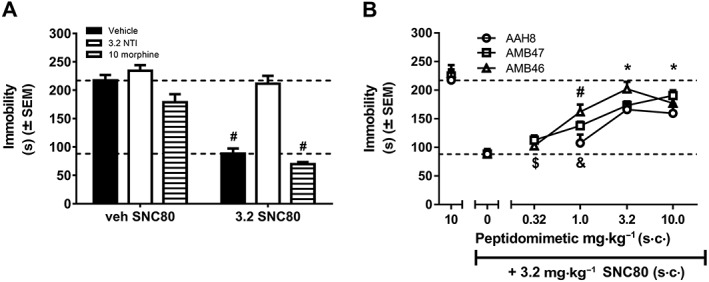
(A) Immobility scores in the mouse TST for animals pretreated with vehicle, 3.2 mg·kg−1 naltrindole or 10 mg·kg−1 morphine 30 min before 3.2 mg·kg−1 SNC80. Pretreatment with naltrindole attenuates SNC80‐induced antidepressant‐like effects, as expected for a δ‐antagonist. Morphine does not alter the effects of SNC80 in the TST. # P<0.05, significantly different from vehicle. (B) Dose–response curves for AAH8, AMB47 and AMB46 in the mouse TST. * P<0.05, all peptidomimetics significantly different from SNC80 alone; # P<0.05, AMB47 and AMB46 significantly different from SNC80 alone; & P<0.05, all peptidomimetics significantly different from 10 mg·kg−1 peptidomimetic alone; $ P<0.05, AMB47 and AMB46 significantly different from 10 mg·kg−1 peptidomimetic alone. Data shown means ± SEM for all groups (n = 6 for each group).
Dose–response curves as δ‐receptor antagonists were established for AAH8, AMB47 and AMB46, as pretreatments to SNC80 (Figure 2B). Analysis of these peptidomimetic dose–response curves using the mouse TST, comparing each dose to SNC80 alone and peptidomimetic alone (control conditions) showed a significant effect of treatment: AAH8 [F(4, 25) = 12.88], AMB47 [F(5, 30) = 36.47] and AMB46 [F(5, 30) = 24.62]. The high dose of each peptidomimetic alone (10 mg·kg−1) produces immobility levels similar to that observed with no drug conditions (Figure 2A), and SNC80 alone significantly decreases immobility. Mice pretreated with the lowest doses tested of AAH8 (1 mg·kg−1), AMB47 (0.32 mg·kg−1) and AMB46 (0.32 mg·kg−1) display immobility scores similar to those produced by SNC80 alone. However, pretreatment with higher doses of AAH8 (3.2 and 10 mg·kg−1), AMB47 (1, 3.2 and 10 mg·kg−1) and AMB46 (1, 3.2 and 10 mg·kg−1) prior to SNC80 significantly attenuated the SNC80‐induced decreases in immobility, and these scores were not statistically different from treatment with peptidomimetic alone. IC50 values derived from the peptidomimetic dose–effect curves show that AAH8, AMB47 and AMB46 have similar δ‐receptor antagonist potencies in vivo (IC50 2.06 mg·kg−1, 1.66 mg·kg−1 and 1.61 mg·kg−1 respectively). Naltrindole is reported to have an IC50 of 2 mg·kg−1 in the mouse TST in male C57BL6 mice (Naidu et al., 2007).
Development of tolerance to antinociceptive action
AAH8
A factorial ANOVA of the AAH8 dose–effect curves before and after repeated treatment shows no interaction between factors (AAH8 dose × day × repeated treatment). There is a main effect of AAH8 dose (F(4, 40) = 510.28), demonstrating that AAH8 produces dose‐dependent increases in antinociceptive effects, but there is no effect of day (day 1 vs. day 6) or repeated treatment (saline vs. AAH8). A separate two‐way, repeated measures ANOVA of the ED50 values only also demonstrated that there was no significant shift in the dose–response curves for AAH8 before and after repeated treatment in either saline‐treated or AAH8‐treated groups (day 1 saline‐treated group: 4.73 ± 0.002 mg·kg−1, day 1 AAH8‐treated group: 4.74 ± 0.02, day 6 saline‐treated group: 4.73 ± 0.002 mg·kg−1 and day 6 AAH8‐treated group: 4.74 ± 0.0001; Figure 3A).
Figure 3.
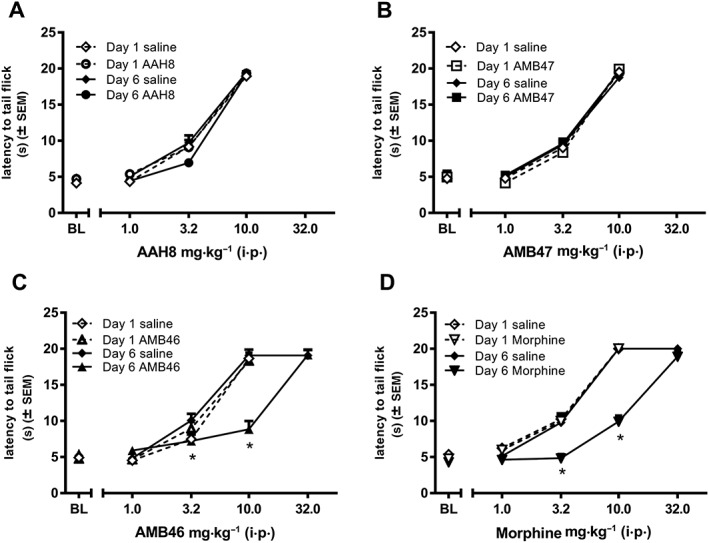
Five days of chronic escalating treatment with (A) AAH8 or (B) AMB47 (10–50 mg·kg−1 i.p., twice daily) treatment i.p. (n = 6) produces no shift in the dose–effect curve in wild‐type BL6 mice. Five days of chronic escalating (D) morphine or (C) AMB46 (10–50 mg·kg−1 i.p., twice daily) treatment i.p. (n = 6), but not saline (n = 6) produces a significant threefold rightward shift in the dose–effect curve in wild‐type BL6 mice. * P<0.05, significantly different from data from day 1. Data shown are mean ± SEM for all groups (n = 6 for each group).
AMB47
Similar to AAH8, a factorial ANOVA of the AMB47 dose–effect curves before and after repeated treatment shows no interaction between factors (AMB47 dose × day × repeated treatment). There is a main effect of AMB47 dose (F(4, 40) = 1129.71), demonstrating that AMB47 produces dose‐dependent increases in antinociceptive effects, but there is no effect of day (day 1 vs. day 6) or repeated treatment (saline vs. AMB47). A separate two‐way, repeated measures ANOVA of the ED50 values also demonstrated that there was no significant shift in the dose–response curves for AMB47 before and after repeated treatment in either saline‐treated or AMB47‐treated groups (day 1 saline‐treated group: 4.73 ± 0.19 mg·kg−1, day 1 AMB47‐treated group: 4.64 ± 0.09, day 6 saline‐treated group: 4.95 ± 0.24 mg·kg−1 and day 6 AMB47‐treated group 4.73 ± 0.14; Figure 3B).
AMB46
A factorial ANOVA comparing AMB46 dose–effect curves before and after repeated treatment shows a significant interaction (AMB46 dose × day × repeated treatment; F(4, 40) = 23.245) and significant main effects of AMB46 dose (F(4, 40) = 1096.44), day (1 vs. 6 F(1, 10) = 12.71) and repeated treatment (saline vs. AMB46, F(1, 10) = 8.60). Repeated treatment with AMB46, but not saline, produces a threefold, rightward, parallel shift in the AMB46 dose–response curve (repeated treatment × day interaction F(1, 10) = 51.71). After 5 days of treatment with escalating doses of AMB46, the ED50 of the AMB46 dose–effect curve is more than 3.5‐fold larger on day 6 (17.04 ± 1.25 mg·kg−1) as compared with day 1 (4.63 ± 1.06 mg·kg−1). The AMB46 dose–effect curves in mice treated with saline are not different on days 1 and 6 (day 1: 5.18 ± 0.31 mg·kg−1 vs. day 6: 4.73 ± 0.006 mg·kg−1). A separate two‐way, repeated measures ANOVA of the ED50 values shows significant main effects of repeated treatment (F(1, 10) = 78.25) and day (F(1, 10) = 89.68)) and an interaction of repeated treatment × day (F(1, 10) = 103.8; Figure 3C).
Morphine
A factorial ANOVA comparing morphine dose–effect curves before and after repeated treatment shows a significant interaction (morphine dose × day × repeated treatment; F(4, 40) = 25.07) and significant main effects of morphine dose (F(4, 40) = 1008.61), day (1 vs. 6; F(1, 10) = 51.62) and repeated treatment (saline vs. morphine; F(1, 0) = 35.71). Repeated morphine, but not repeated saline, treatment produces a threefold, rightward, parallel shift in the morphine dose–response curve (treatment × day interaction F(1, 10) = 31.79). After 5 days of treatment with escalating doses of morphine, the ED50 of the morphine dose–effect curve is more than threefold larger on day 6 (14.72 ± 1.39 mg·kg−1) as compared with day 1 (4.74 ± 0.11 mg·kg−1, F(1, 10) = 9.881). The morphine dose–effect curves in mice treated with saline are not different on days 1 and 6 (day 1: 4.93 ± 0.32 mg·kg−1 vs. day 6: 4.53 ± 0.26 mg·kg−1). A separate two‐way, repeated measures ANOVA of ED50 values shows a significant effect of repeated treatment (F(1, 10) = 45.56) and day (F(1, 10) = 44.96)) and an interaction of chronic treatment × day (F(1, 10) = 52.83; Figure 3D).
δ‐Receptor knockout mice and their wild‐type littermates
A factorial ANOVA comparing morphine dose–effect curves in δ‐receptor KO mice and their wild‐type littermates before and after repeated morphine treatment shows a significant interaction (morphine dose × day × genotype; F(4, 40) = 32.89) and significant main effects of morphine dose (F(4, 40) = 962.39), day tested (1 vs. 6; F(1, 10) = 4.14) and genotype (δ‐receptor KO vs. wild type; F(1, 0) = 46.03). Repeated morphine administration in wild‐type mice, but not δ‐receptor KO mice, produces a threefold, rightward, parallel shift in the morphine dose–response curve (genotype × day interaction F(1, 10) = 33.28). After repeated treatment with escalating doses of morphine, the ED50 of the morphine dose–effect curve in wild‐type littermates is threefold larger on day 6 (14.73 ± 0.79 mg·kg−1) as compared with day 1 (4.63 ± 0.26 mg·kg−1). The morphine dose–effect curves in δ‐receptor KO mice are not different on days 1 and 6 (day 1: 4.73 ± 0.13 mg·kg−1 vs. day 6: 4.74 ± 0.24 mg·kg−1; Figure 5A). A separate two‐way, repeated measures ANOVA of ED50 values shows a significant effect of genotype [F(1, 10) = 196] and day [F(1, 10) = 97.9] and an interaction of chronic treatment × day [F(1, 10) = 97.8].
Figure 5.
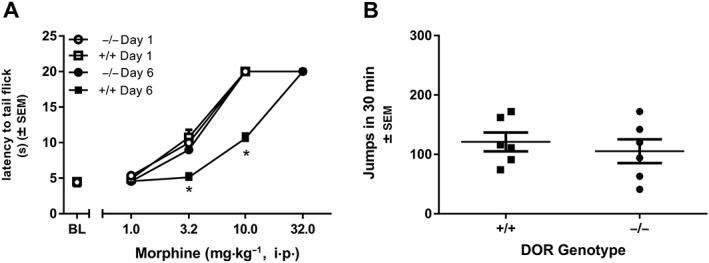
(A) Five days of chronic escalating treatment with morphine (10–50 mg·kg−1 i.p., twice daily) in δ‐KO mice (−/−) and their wild‐type littermates (+/+) produces no shift in the dose–effect curve in δ‐KO mice but produces a threefold rightward shift in wild‐type mice. (B) Wild‐type (+/+) and δ KO (−/−) mice were treated for 5 days with escalating doses of morphine (10–50 mg·kg−1 i.p., twice daily). Withdrawal was precipitated with 10 mg·kg−1 naltrexone. There was no significant difference in the number of jumps observed across genotype. * P<0.05, significantly different from data from day 1. Data shown are means ± SEM for all groups (n = 6 for each group).
Physical dependence
In wild‐type mice treated repeatedly with increasing doses of morphine, AMB47 or AMB46 for 5 days, naltrexone precipitated jumping behaviour [F(4, 25) = 8.15; Figure 4]. In morphine and AMB46‐treated mice, naltrexone elicits significantly more jumps than in mice treated with saline or AAH8. The number of naltrexone‐precipitated jumps in AMB47‐treated mice is significantly larger than in saline‐treated mice but not AAH8‐treated mice. There was no difference between mice treated chronically with saline or AAH8. After 5 days of escalating morphine doses, naltrexone precipitated a similar number of withdrawal jumps in δ‐receptor KO mice and wild‐type littermates (Figure 5B).
Figure 4.
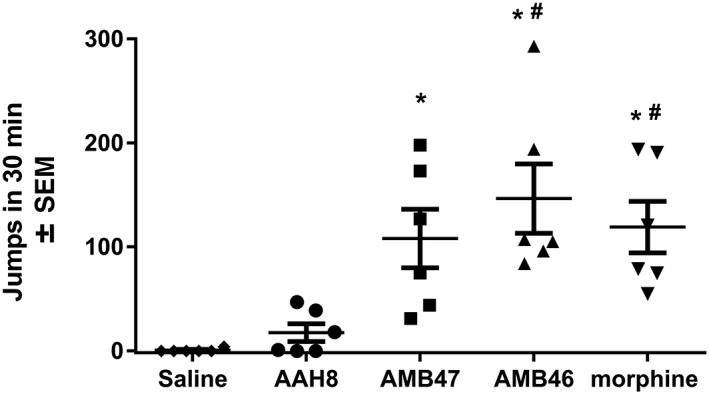
Wild‐type mice were treated for 5 days with either saline or escalating doses of AAH8, AMB47, AMB46 or morphine (10–50 mg·kg−1 i.p., twice daily). Withdrawal was precipitated with 10 mg·kg−1 naltrexone i.p., and a number of jumps were counted. Animals treated chronically with AMB47, AMB46 and morphine experienced more naltrexone‐precipitated withdrawal jumps than animals treated chronically with saline or AAH8. * P<0.05, significantly different from saline; # P<0.05, significantly different from AAH8. Data shown are means ± SEM for all groups (n = 6 for each group).
Conditioned place preference
The rewarding effects of both morphine and AAH8 were explored using the CPP assay [Figure 6A; F(2, 15) = 6.382]. Conditioning with morphine produces a significant increase in time spent on the morphine‐paired side of the apparatus compared with conditioning with saline or AAH8 (Figure 6A). Conditioning with AAH8 did not increase time spent on the AAH8‐paired side of the apparatus compared with saline conditioning.
Figure 6.
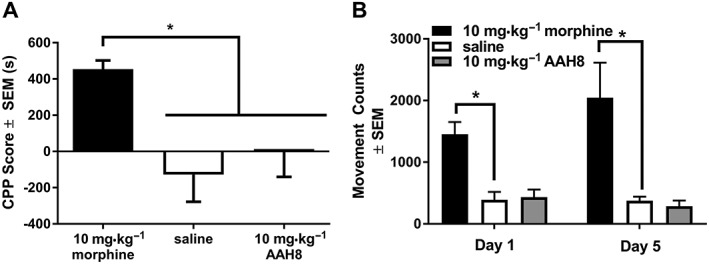
CPP scores for animals trained for 5 days on 10 mg·kg−1 morphine, 10 mg·kg−1 AAH8 or saline for 5 days i.p. CPP scores are defined as the difference between time spent on drug‐paired side preconditioning and post‐conditioning measured in seconds. (A) Animals conditioned with morphine spent more time on the drug‐paired side of the CPP apparatus than those trained to either AAH8 or saline. (B) Locomotor activity over 30 min for 10 mg·kg−1 morphine, 10 mg·kg−1 AAH8 and saline on days 1 and 5. Data shown are means ± SEM for all groups (n = 6 for each group). * P<0.05, significantly different as indicated.
Locomotor activity was recorded during all conditioning sessions. A two‐way, repeated measures ANOVA showed a significant main effect of drug (F(2, 15) = 12.10), but no effect of day and no significant interaction. Morphine produced a significant increase in locomotor activity as compared with saline on both day 1 and day 5. AAH8 did not increase locomotor activity on either day 1 or day 5 (Figure 6B).
Production of faecal boli
Mice treated with saline produce significantly more tinted faecal boli than those treated with AAH8, AMB47, AMB46 or morphine [F(4, 27) = 30.77; Figure 7], and there was no difference in number of tinted faecal boli between mice treated with AAH8, AMB47, AMB46 and morphine. One‐way ANOVA showed that mice treated with saline produced tinted faecal boli significantly earlier than those treated with AAH8, AMB47, AMB46 or morphine [F(4, 35) = 49.14; Supporting Information Figure S1]. The time to first tinted faecal bolus was not statistically different in mice treated with AAH8, AMB47, AMB46 and morphine (Supporting Information Figure S1).
Figure 7.
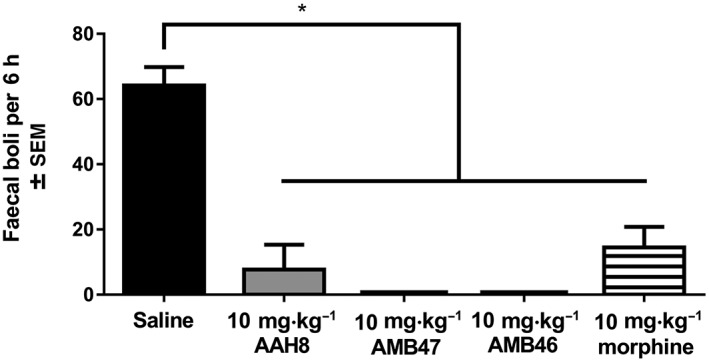
Acute treatment with 10 mg·kg−1 AAH8, AMB47, AMB46 and morphine all significantly reduce the number of faecal boli produced over 6 h as compared with saline controls. Data shown are means ± SEM for all groups (n = 6 for each group). * P<0.05, significantly different as indicated.
Discussion
The data described in this report demonstrate that the structurally related, mixed efficacy opioid ligands AAH8, AMB47 and AMB46 produce similar effects in vivo after acute administration but have different profiles of activity following repeated administration. Consistent with their in vitro profile, these compounds act as μ‐receptor agonists and δ‐receptor antagonists in vivo (Figures 1 and 2). They produce dose‐dependent antinociceptive effects with similar potencies and are fully effective, compared with morphine, in the 50°C WWTW assay. Naltrexone attenuates the antinociceptive effects to a similar extent as shown by equivalent shifts in their ED50s (Table 2), suggesting that these antinociceptive effects are opioid receptor‐mediated. Further, the antinociceptive effects of these peptidomimetics are completely attenuated in μ‐receptor KO mice, demonstrating that the antinociception is μ‐receptor‐mediated (Figure 1). Doses that are fully effective in a 50°C WWTW assay also decrease the production of faecal boli produced over a 6 h window (Figure 7), consistent with the effects of morphine. As δ‐receptor antagonists, these compounds attenuate SNC80‐induced decreases in immobility in the mouse TST (Figure 2) with IC50s similar to naltrindole (Naidu et al., 2007). These compounds may be slightly less effective than the known δ‐receptor antagonist naltrindole, which could be due to their μ‐receptor agonist activity, consistent with the small, non‐significant decreases in immobility produced by morphine alone (Figure 2). Overall, these data demonstrate that AAH8, AMB47 and AMB46 simultaneously function as μ‐receptor agonists and δ‐receptor antagonists in vivo. Further, the δ‐receptor antagonist properties of these compounds do not alter their acute antinociceptive or constipating effects.
The acute behavioural effects of these compounds are consistent with their in vitro profile as μ‐receptor agonists. All three peptidomimetics display high‐affinity μ‐receptor binding affinities in the absence of sodium. In the presence of sodium ions, the affinity of these ligands for μ‐receptors is decreased, as expected since sodium ions stabilize inactive receptor states and alter agonist affinity (Pert et al., 1973; Selley et al., 2000; Simon and Groth, 1975). However, these compounds still have K i values in the nanomolar range and demonstrate higher μ‐receptor affinity than morphine under these conditions. In addition, these ligands are more efficacious than morphine in vitro. Consistent with this idea, their calculated relative efficacies using the Ehlert equation (Quock et al., 1999) can be rank ordered: AAH8 > AMB46 > AMB47 ≈ morphine. However, these in vitro data do not effectively predict their potency and efficacy in vivo. For example, in a 50°C WWTW assay, these compounds demonstrate similar potency to morphine and produce a maximal response at similar doses, but under conditions requiring higher efficacy (55°C WWTW), the dose–effect curves for AMB46 and AMB47 are shifted to a greater extent than those for AAH8 and morphine. These findings would suggest that AAH8 and morphine are higher efficacy agonists in vivo than AMB46 and AMB47, which is not entirely consistent with their in vitro profile. It is possible that some unidentified pharmacokinetic parameter is responsible for the differences between these ligands in vivo. It is also possible that differential plasma protein binding, metabolism or distribution to the active site, presumably the CNS, leads to different local concentrations of the peptidomimetics, which may explain the discrepancies between in vitro and in vivo potencies and efficacies. Future work will explore how the pharmacokinetic properties of compounds in this series affect their acute and chronic effects.
While these compounds are μ‐receptor agonists in vitro, they do not stimulate δ‐receptor‐mediated [35S]GTPγS binding in cells and attenuate δ‐receptor agonist‐stimulated G protein activation, suggesting they are δ‐receptor antagonists. Notably, these ligands differ in their affinity for δ‐receptors in vitro over an 80‐fold range. In the absence of sodium, these ligands have low nanomolar or subnanomolar affinity for δ‐receptors with a rank order of AAH8 > AMB47 > AMB46. In the presence of sodium, the rank order for affinity at δ‐receptors did not change, but the K i values did shift, inconsistent with neutral antagonist activity. These findings suggest that these compounds could potentially be low‐efficacy δ‐receptor agonists (below the threshold for this assay). Again, these in vitro data do not correlate well with in vivo δ‐receptor antagonist activity, as the three peptidomimetics display similar δ‐receptor antagonist‐like activity in vivo with equal potency (Figure 2).
Although these compounds have similar μ‐receptor and δ‐receptor activity following acute administration in vivo, their behavioural effects differed following repeated administration. For example, tolerance, as demonstrated by rightward shifts in the dose–effect curves, was observed following repeated administration of morphine or AMB46, but not with repeated AAH8 and AMB47 (Figure 3). Naltrexone precipitated withdrawal in mice treated with repeated morphine, AMB46 and AMB47, but significantly fewer signs of withdrawal were observed in mice that receive repeated AAH8. Considering the in vivo effects of these three compounds evaluated in the current study, the rank order of most favourable profile is AAH8 > AMB47 > AMB46 ≈ morphine. Overall, the compound with the most promising profile is AAH8 as it produced less tolerance and physical dependence, compared with morphine under the same conditions. In addition, AAH8 also failed to produce CPP at a dose that provided significant antinociception (Figure 6). These findings suggest that AAH8 is less rewarding than morphine and, therefore, may be a safer analgesic than traditional opioids.
While this study identifies a promising candidate, it also highlights that the combination of a μ‐receptor agonist with δ‐receptor antagonist is not sufficient to prevent tolerance development, as all of these compounds were δ‐receptor antagonists in vivo. To further probe the disparities between these compounds in terms of tolerance development, we considered whether differences in (i) μ‐receptor efficacy, (ii) δ‐receptor affinity and/or (iii) μ‐receptor : δ‐receptor affinity ratio would correlate with the rank order of favourable profiles (AAH8 > AMB47 > AMB46 ≈ morphine). In terms of μ‐receptor efficacy, we hypothesized that high‐efficacy μ‐receptor agonists would be less likely to produce tolerance due to a larger receptor reserve. In vitro relative efficacy calculations at μ‐receptor orders the compounds: AAH8 > AMB46 > AMB47 ≈ morphine, but in vivo, we observe a different rank order under the higher efficacy conditions such that AAH8 = morphine > AMB47 = AMB46. Therefore, in vitro relative efficacy does not appear to predict in vivo efficacy requirement, and compound efficacy in vitro or in vivo does not correlate with the lack of tolerance development.
While δ‐receptor antagonist activity alone is not sufficient to prevent tolerance, it is likely that action at δ‐receptors played a significant role, as demonstrated by less tolerance development in δ‐receptor knockout mice. Some compound properties that do correlate with the lack of tolerance development under these conditions include (i) δ‐receptor affinity in both binding assay conditions or as determined K e values and (ii) μ‐receptor : δ‐receptor affinity ratios, such that high‐affinity binding at δ‐receptors may protect against tolerance, and possibly, dependence. However, δ‐receptor expression and/or signalling may be less relevant to the mechanisms involved in physical dependence, as precipitated withdrawal is similar in wild‐type and δ‐receptor KO mice. Further studies will probe the role of δ‐receptors in the effects of chronic administration of mixed efficacy opioid ligands.
Furthermore, a single characteristic alone may not account for the lack of tolerance development with some of these ligands, but a combination of several features may be required to produce some preferred pharmacological profile, such as a combination of high‐efficacy μ‐receptor agonist activity and high‐affinity binding to δ‐receptor. Still other mechanisms, not considered here, may be important in preventing tolerance development. For instance, activity at the κ‐receptor may play an important role. All three peptidomimetics bind the κ‐receptor with nanomolar affinity, and both AMB47 and AMB46 display some κ‐receptor activation in the GTPγS assay. It is possible that chronic activation of κ‐receptors may play a role in the development of adverse effects associated with opioid use. Another possible factor to consider is that these peptidomimetics may activate distinct intracellular signalling pathways and may exhibit biased signalling at one or more of the opioid receptors. It has been proposed that developing biased μ‐receptor agonists that favour G‐protein signalling over arrestin‐3 signalling might provide pain relief without the development of adverse effects (Kelly, 2013; Raehal et al., 2011). However, the loss of arrestin‐3 does not attenuate the development of adverse effects for all opioid agonists, suggesting that agonists produce adverse effects through different mechanisms or that other factors mediate adverse effects (Raehal and Bohn, 2011). Further, the theory is not supported by studies of the G‐protein‐biased, μ‐receptor agonist http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7334 (Altarifi et al., 2017). Future work will explore these possibilities to determine what role, if any, they play in the development of adverse effects to opioid analgesics.
Overall, this report has identified a promising opioid ligand that produced antinociception without development of tolerance or dependence under the conditions tested. Moreover, our lead compound, AAH8, was less rewarding than morphine. However, it also highlights that the combination of μ‐receptor agonist activity with δ‐receptor antagonist activity is not sufficient to prevent the development of tolerance or physical dependence, as all of these compounds were δ‐receptor antagonists in vivo. Future studies will test AAH8 over longer periods of administration and in chronic pain models. Finally, we will continue to probe the mechanisms by which δ‐receptor antagonist activity modifies tolerance development to μ‐receptor agonists in order to better understand how these mixed efficacy ligands differ in their in vivo effects following repeated administration.
Author contributions
J.P.A. contributed to the experimental design and data collection for Figures 1, 2, 3, 4, 5, 6, 7 and data for Table 2; K.E.K. to the data collection for Figures 1, 2, 3, 4, 5, 6, 7 and data for Table 2; A.F.N. by the synthesis of novel compounds AMB47 and AMB46; D.M. by the synthesis of the novel compound AAH8; N.W.G. to the data in Table 1. J.R.T. H.I.M. and E.M.J. contributed to the experimental design.
Conflict of interest
E.M.J. consulted for Trevena, Inc., in 2011–2012 with compensation. The other authors have no other conflicts of interest to disclose.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Cumulative dose response curves for morphine or the κ‐receptor agonist ethylketocyclazocine (EKC) (or repeated saline injection) in the 50 °C mouse warm water tail withdrawal assay at 50 °C in μ‐receptor knockout mice. Data are plotted as mean ± SEM for all groups (n = 6 for each group).
{kind=link}
Figure S2 Acute treatment with 10 mg kg−1 AAH8, AMB47, AMB46 and morphine all significantly increase the time to first tinted faecal bolus as compared with saline controls. Data are plotted as mean ± SEM for all groups (n = 6 for each group).
{kind=link}
Acknowledgements
The authors thank Dr Kenner Rice for his generous gift of SNC80 and Dr. Aubrie A. Harland for chemical synthesis assistance. This work was funded by NIH grants DA003910 and DA041565. J.P.A. was supported by the National Institutes of Health under Ruth L. Kirschstein National Research Service Award T32 DA007267. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Anand, J. P. , Kochan, K. E. , Nastase, A. F. , Montgomery, D. , Griggs, N. W. , Traynor, J. R. , Mosberg, H. I. , and Jutkiewicz, E. M. (2018) In vivo effects of μ‐opioid receptor agonist/δ‐opioid receptor antagonist peptidomimetics following acute and repeated administration. British Journal of Pharmacology, 175: 2013–2027. doi: 10.1111/bph.14148.
Contributor Information
Henry I Mosberg, Email: him@umich.ed.
Emily M Jutkiewicz, Email: ejutkiew@umich.edu.
References
- Abdelhamid EE, Sultana M, Portoghese PS, Takemori AE (1991). Selective blockage of the δ opioid receptors prevents the development of morphine tolerance and dependence in mice. J Pharmacol Exp Ther 258: 299–303. [PubMed] [Google Scholar]
- Aceto MD, Harris LS, Negus SS, Banks ML, Hughes LD, Akgun E et al (2012). MDAN‐21: a bivalent opioid ligand containing μ‐agonist and δ‐antagonist pharmacophores and its effects in rhesus monkeys. International Journal of Medicinal Chemistry 2012: 327257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altarifi AA, David B, Muchhala KH, Blough BE, Akbarali H, Negus SS (2017). Effects of acute and repeated treatment with the biased μ opioid receptor agonist TRV130 (oliceridine) on measures of antinociception, gastrointestinal function, and abuse liability in rodents. J Psychopharmacol 31: 730–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand JP, Boyer BT, Mosberg HI, Jutkiewicz EM (2016). The behavioral effects of a mixed efficacy antinociceptive peptide, VRP26, following chronic administration in mice. Psychopharmacology (Berl) 233: 2479–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand JP, Purington LC, Pogozheva ID, Traynor JR, Mosberg HI (2012). Modulation of opioid receptor ligand affinity and efficacy using active and inactive state receptor models. Chem Biol Drug Des 80: 763–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anathan S, Kezar HS, Carter RL, Saini SK, Rice KC, Wells JL et al (1999). Synthesis, opioid receptor binding, and biological activities of naltrexone‐derived pyrido‐ and pyrimidomorphans. J Med Chem 42: 3527–3538. [DOI] [PubMed] [Google Scholar]
- Anathan S, Khare NK, Saini SK, Seitz LE, Bartlett JL, Davis P et al (2004). Identification of opioid ligands possessing mixed μ agonist/δ antagonist activity among pyridomorphans derived from naloxone, oxymorphone, and hydromorphone. J Med Chem 47: 1400–1412. [DOI] [PubMed] [Google Scholar]
- Bailey CP, Connor M (2005). Opioids: cellular mechanisms of tolerance and physical dependence. Curr Opin Pharmacol 5: 60–68. [DOI] [PubMed] [Google Scholar]
- Balboni G, Guerrini R, Salvadori S, Bianchi C, Rizzi D, Bryant SD et al (2002a). Evaluation of the Dmt–Tic pharmacophore: conversion of a potent δ‐opioid receptor antagonist into a potent δ agonist and ligands with mixed properties. J Med Chem 45: 713–720. [DOI] [PubMed] [Google Scholar]
- Balboni G, Salvadori S, Guerrini R, Negri L, Giannini E, Jinsmaa Y et al (2002b). Potent δ‐opioid receptor agonists containing the Dmt–Tic pharmacophore. J Med Chem 45: 5556–5563. [DOI] [PubMed] [Google Scholar]
- Ballantyne JC, LaForge KS (2007). Opioid dependence and addiction during opioid treatment of chronic pain. Pain 129: 235–255. [DOI] [PubMed] [Google Scholar]
- Bender AM, Griggs NW, Anand JP, Traynor JR, Jutkiewicz EM, Mosberg HI (2015). Asymmetric synthesis and in vitro and in vivo activity of tetrahydroquinolines featuring a diverse set of polar substitutions at the 6 position as mixed‐efficacy μ opioid receptor/δ opioid receptor ligands. ACS Chem Neurosci 6: 1428–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benyamin R, Trescot AM, Datta S, BeunaventurA R, Adlaka R, Sehgal N et al (2008). Opioid complications and side effects. Pain Physicaian 11: S105–S120. [PubMed] [Google Scholar]
- Cai J, Song B, Cai Y, Ma Y, Lam A‐L, Magiera J et al (2014). Endomorphin analogues with mixed μ‐opioid (MOP) receptor agonism/δ‐opioid (DOP) receptor antagonism and lacking β‐arrestin2 recruitment activity. Bioorg Med Chem 22: 2208–2219. [DOI] [PubMed] [Google Scholar]
- Council NR (2011). Guide for the Care and Use of Laboratroy Animals. National Academies Press: Washington DC. [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels DJ, Lenard NR, Etienne CL, Law PY, Roerig SC, Portoghese PS (2005). Opioid‐induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proc Natl Acad Sci U S A 102: 19208–19213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faul F, Erdfelder E, Lang AG, Buchner A (2007). G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods 39: 175–191. [DOI] [PubMed] [Google Scholar]
- Fundytus ME, Schiller PW, Shapiro M, Weltrowska H, Coderre TJ (1995). Attenuation of morphine tolerance and dependence with the highly selective δ opioid receptor antagonist TIPP(psi). Eur J Pharmacol 286: 105–108. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res. 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harland AA, Yeomans L, Griggs NW, Anand JP, Pogozheva ID, Jutkiewicz EM et al (2015). Further optimization and evaluation of bioavailable, mixed‐efficacy μ‐opioid receptor (MOR) agonists/δ‐opioid receptor (DOR) antagonists: balancing MOR and DOR affinities. J Med Chem 58: 8952–8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy JR, Bezawada P, Shim J, Jones JW, Kane MA, MacKerell AD Jr et al (2013). Synthesis, modeling, and pharmacological evaluation of UMB 425, a mixed μ agonist/δ antagonist opioid analgesic with reduced tolerance liabilities. ACS Chem Nerosci 4: 1256–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepburn MJ, Little PJ, Gringas J, Khun CM (1997). Differential effects of naltrindole on morphine‐induced tolerance and physical dependence in rats. J Pharmacol Exp Ther 281: 1350–1356. [PubMed] [Google Scholar]
- Horan PJ, Mattia A, Bilsky EJ, Weber S, Davis TP, Yamamura HI et al (1993). Antinociceptive profile of biphalin, a dimeric enkephalin analog. J Phamacol Exp Ther 265: 1446–1454. [PubMed] [Google Scholar]
- Husbands SM, Neilan CL, Broadbear J, Grundt P, Breeden S, Aceto MD et al (2005). BU74, a complex oripavine derivative with potent κ opioid receptor agonism and delayed opioid antagonism. European Journal of Pharmacology 509: 117–135. [DOI] [PubMed] [Google Scholar]
- Johnston LD, O'malley PM, Bachman JG, Schulenberg JE (2009). Monitoring the future: national survey results on drug use. Natl Inst Drug Abuse 1: 1–721. [Google Scholar]
- Jutkiewicz EM, Brooks EA, Kynaston AD, Rice KC, Woods JH (2011). Patterns of nicotinic receptor antagonism: nicotine discrimination studies. J Pharmacol Exp Ther 339: 194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly E (2013). Efficacy and ligand bias at the μ‐opioid receptor. Br J Pharmacol 169: 1430–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KO, Akil H, Woods JH, Traynor JR (1999). Differential binding properties of oripavines at cloned μ‐ and δ‐opioid receptors. Eur J Pharmacol 378: 323–330. [DOI] [PubMed] [Google Scholar]
- Lee YS, Kulkarani V, Cowell SM, Ma SW, Davis P, Hanlon KE et al (2011). Development of potent μ and δ opioid agonsits with high lipophilicity. J Med Chem 54: 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenard NR, Daniels DJ, Portoghese PS, Roerig SC (2007). Absence of conditioned place preference or reinstatement with bivalent ligands containing μ‐opioid receptor agonist and δ‐opioid receptor antagonist pharmacophores. Eur J Pharmacol 566: 75–82. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosberg HI, Yeomans L, Harland AA, Bender AM, Sobczyk‐Kojiro K, Anand JP et al (2013). Opioid peptidomimetics: leads for the design of bioavailable mixed efficacy μ opioid receptor (MOR) agonist/δ opioid receptor (DOR) antagonist ligands. J Med Chem 56: 2139–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naidu PS, Lichtman AH, Archer CC, May EL, Harris LS, Aceto MD (2007). NIH 11082 produces antidepressant‐like activity in the mouse tail suspension test through a δ opioid receptor mechanism of action. Eur J Pharmacol 566: 132–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pert CB, Pasternak G, Snyder SH (1973). Opiate agonists and antagonists discriminated by receptor binding in brain. Science 182: 1359–1361. [DOI] [PubMed] [Google Scholar]
- Purington LC, Pohozheva ID, Traynor JR, Mosberg HI (2009). Pentapeptides displaying μ opioid receptor agonist and δ opioid receptor partial agonist/antagonist properties. J Med Chem 52: 7724–7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purington LC, Sobczyk‐Kojiro K, Pogozheva ID, Traynor JR, Mosberg HI (2011). Development and in vitro characterization of a novel bifunctional μ‐agonist/δ‐antagonist opioid tetrapeptide. ACS Chem Biol 6: 1375–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quock RM, Burkey TH, Varga E, Hosohata Y, Hosohata K, Cowell SM et al (1999). The δ opioid receptor: molecular pharmacology, signal transduction, and the determination of drug efficacy. Pharmacol Rev 51: 503–532. [PubMed] [Google Scholar]
- Raehal KM, Bohn LM (2011). The role of β‐arrestin2 in the severity of antinociceptive tolerance and physical dependence induced by different opioid pain therapeutics. Neuropharmacology 60: 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raehal KM, Schmid CL, Groer CE, Bohn LM (2011). Functional selectivity at the μ‐opioid receptor: implications for understanding opioid analgesia and tolerance. Pharmacol Rev 63: 1001–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross S, Peselow E (2009). The neurobiology of addicitive disorders. Clin Neuropharmacol 32: 269–276. [DOI] [PubMed] [Google Scholar]
- Salvadori S, Guerrini R, Balboni G, Bianchi C, Bryant SD, Cooper PS et al (1999). Further studies on the Dmt–Tic pharmacophore: hydrophobic substituents at the C‐terminus endow δ antagonists to manifest μ agonism or μ antagonism. J Med Chem 42: 5010–5019. [DOI] [PubMed] [Google Scholar]
- Schiller PW (2009). Bi‐ or multifunctional opioid peptide drugs. Life Sci 86: 598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller PW, Fundytus ME, Merovitz L, Weltrowska G, Nguten TM‐D, Lemieux C et al (1999). The opioid μ agonist/δ antagonist DIPP‐NH2(psi) produces a potent analgesic effect, no physical dependence and less tolerance than morphine in rats. J Med Chem 42: 3520–3526. [DOI] [PubMed] [Google Scholar]
- Schmidt R, Vogel D, Mrestani‐Klaus C, Brandt W, Neubert K, Chung NN et al (1994). Cyclic β‐casomorphin analgoues with mixed μ agonist/δ antagonist properties: synthesis, pharmacological characterization and conformational aspects. J Med Chem 37: 1136–1144. [DOI] [PubMed] [Google Scholar]
- Selley DE, Cao CC, Liu Q, Childers SR (2000). Effects of sodium on agonist efficacy for G‐protein activation in μ‐opioid receptor‐transfected CHO cells and rat thalamus. Br J Pharmacol 130: 987–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon EJ, Groth J (1975). Kinetics of opiate receptor inactivation by sulfhydryl reagents: evidence for conformational change in presence of sodium ions. Proc Natl Acad Sci U S A 72: 2404–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Váradi A, Marrone GF, Palmer TC, Narayan A, Szabó MR, Le Rouzic V et al (2016). Mitragynine/corynantheidine pseudoindoxyls as opioid analgesics with μ agonism and δ antagonism, which do not recruit β‐arrestin‐2. J Med Chem 59: 8381–8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, King MA, Schuller AGP, Nitsche JF, Reidl M, Elde RP et al (1999). Retention of supraspinal δ‐like analgesia and loss of morphine tolerance in δ opioid receptor knockout mice. Neuron 24: 243–252. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Cumulative dose response curves for morphine or the κ‐receptor agonist ethylketocyclazocine (EKC) (or repeated saline injection) in the 50 °C mouse warm water tail withdrawal assay at 50 °C in μ‐receptor knockout mice. Data are plotted as mean ± SEM for all groups (n = 6 for each group).
Figure S2 Acute treatment with 10 mg kg−1 AAH8, AMB47, AMB46 and morphine all significantly increase the time to first tinted faecal bolus as compared with saline controls. Data are plotted as mean ± SEM for all groups (n = 6 for each group).