

Abstract
Background and Purpose
In men with benign prostatic hyperplasia, increased smooth muscle tone in the prostate may lead to bladder outlet obstruction and subsequent lower urinary tract symptoms. Consequently, medical treatment aims to inhibit prostate smooth muscle contraction. However, the efficacy of the treatment options available is limited, and improved understanding of mechanisms of prostate smooth muscle contraction and identification of new targets for medical intervention are mandatory. Several studies suggest that LIM kinases (LIMKs) promote smooth muscle contraction; however, this has not yet been examined. Here, we studied effects of the LIMK inhibitors on prostate smooth muscle contraction.
Experimental Approach
Human prostate tissues were obtained from radical prostatectomy. Phosphorylation of cofilin, a LIMK substrate, was examined using a phospho‐specific antibody. Smooth muscle contractions were studied in organ bath experiments.
Key Results
Real‐time PCR, Western blot and immunofluorescence suggested LIMKs are expressed in smooth muscle cells of prostate tissues. Two different LIMK inhibitors, SR7826 (1 μM) and LIMKi3 (1 μM), inhibited contractions of prostate strips, which were induced by electrical field stimulation, α1‐adrenoceptor agonists phenylephrine and methoxamine and the TXA2 analogue, U46619. LIMK inhibition in prostate tissues and cultured stromal cells (WPMY‐1) was confirmed by cofilin phosphorylation, which was reduced by SR7826 and LIMKi3. In WPMY‐1 cells, SR7826 and LIMKi3 caused breakdown of actin filaments and reduced viability.
Conclusions and Implications
Smooth muscle tone in the hyperplastic human prostate may underlie the effects of LIMKs, which promote contraction. Contraction of prostate strips can be inhibited by small molecule LIMK inhibitors.
Abbreviations
- 4E‐BP1
eukaryotic translation initiation factor 4E‐binding protein 1
- BPH
benign prostatic hyperplasia
- CCK
cell counting kit
- ΔCP
Cttarget − CtGAPDH
- Ct
number of cycles
- ECL
enhanced chemiluminescence
- EFS
electric field stimulation
- IPSS
International Prostate Symptom Score
- LIMK
LIM domain kinase
- LIMKi3
N‐[5‐[1‐(2,6‐dichlorophenyl)‐3‐(difluoromethyl)‐1H‐pyrazol‐5‐yl]‐2‐thiazolyl]‐2‐methylpropanamide
- LUTS
lower urinary tract symptoms
- MLC
myosin light chain
- MW
molecular weight
- MYPT1
myosin light chain‐binding subunit of myosin light chain phosphatase
- PBS‐T
PBS containing 0.1% Tween 20
- PSA
prostate‐specific antigen
- ROCK
Rho kinase
- SR7826
N‐(2‐hydroxyethyl)‐N′‐[4,5‐methyl‐7H‐pyrrolo[2,3‐d]pyrimidine‐4‐yl)phenyl]‐N‐phenylurea
- STK16
serine/threonine kinase 16
- SYBR Green
N′,N′‐dimethyl‐N‐[4‐[(E)‐(3‐methyl‐1,3‐benzothiazol‐2 ylidene)methyl]‐1‐phenylquinolin‐1‐ium‐2‐yl]‐N‐propylpropane‐1,3‐diamine
- U46619
(Z)‐7‐[(1S,4R,5R,6S)‐5‐[(E,3S)‐3‐hydroxyoct‐1‐enyl]‐3 oxabicyclo[2.2.1]heptan‐6‐yl]hept‐5‐enoic acid
- WST‐8
2‐(2‐methoxy‐4‐nitrophenyl)‐3‐(4‐nitrophenyl)‐5‐(2,4‐disulfophenyl)‐2H‐tetrazolium monosodium salt
- Y27632
trans‐4‐[(1R)‐1‐aminoethyl]‐N‐4‐pyridinylcyclohexanecarboxamide dihydrochloride
Introduction
In men presenting with benign prostatic hyperplasia (BPH), obstruction of the prostatic urethra due to increased prostate smooth muscle tone and/or prostatic enlargement may impair bladder emptying, resulting in lower urinary tract symptoms (LUTS) (Hennenberg et al., 2014). Prostate smooth muscle contraction may be induced by activation of http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=22 (Hennenberg et al., 2014). Accordingly, α1‐adrenoceptors hold a key role in aetiology and medical treatment of male LUTS (Oelke et al., 2013; Hennenberg et al., 2014). In the clinic, routinely, α1‐adrenoceptor antagonists (α1‐blockers) are a preferred choice to relieve LUTS. Their mechanisms of action include prostate smooth muscle relaxation and subsequent improvement of urethral and bladder outlet obstruction (Oelke et al., 2013; Hennenberg et al., 2014).
Although α1‐blockers represent an essential medical option to improve LUTS suggestive of BPH, there are inherent limitations (Ventura et al., 2011; Oelke et al., 2013; Hennenberg et al., 2014). In fact, α1‐blockers improve symptom scores and urinary flow by no more than 50% in the majority of patients, resulting in disappointing or inadequate results. As a consequence, there are high rates of non‐responders, treatment discontinuation, hospitalization and BPH‐related surgeries (Chapple et al., 2011; Hennenberg et al., 2014; Cindolo et al., 2015). Together with demographic changes in the elderly population in Western countries, the development of new therapeutic options becomes more important than ever. This requires a precise understanding of prostate smooth muscle contraction. However, α1‐adrenoceptor‐mediated contraction has recently turned out to be much more complex than previously assumed, and intracellular mechanisms promoting prostate smooth muscle contraction are still not fully understood (Hennenberg et al., 2014).
http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=510 (LIMKs) are serine/threonine kinases, which occur in two isoforms (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2054&familyId=510&familyType=ENZYME and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2055&familyId=510&familyType=ENZYME) (Bernard, 2007). Both are known as important regulators of actin dynamics, which promote actin polymerization, filament organization and thus stress fibre formation (Yang et al., 1998; Bernard, 2007). LIMK‐dependent actin regulation is initiated by LIMK‐mediated phosphorylation of cofilin (Yang et al., 1998; Bernard, 2007). In smooth muscle, actin reorganization is an ultimate prerequisite for contraction (Hennenberg et al., 2008; Hennenberg et al., 2014). However, a role for LIMK1/2 in smooth muscle contraction has – to the best of our knowledge – not been reported yet. A potential role appears for LIMKs is possible, as it has been proposed that α1‐adrenoceptors in the cardiovascular system induce cofilin phosphorylation by activation of LIMKs (Dai et al., 2008; Du et al., 2010). Here, we studied effects of the LIMK inhibitors http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9840 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9839 on prostate smooth muscle contraction.
Methods
Human prostate tissues
Human prostate tissues were obtained from patients who underwent radical prostatectomy for prostate cancer (n = 174). Patients with previous transurethral resection of the prostate were excluded. This study was carried out in accordance with the Declaration of Helsinki of the World Medical Association and has been approved by the Ethics Committee of the Ludwig Maximilian University of Munich, Germany. Informed consent was obtained from all patients. All samples and data were collected and analysed anonymously. Prostates were collected immediately after surgery, followed by macroscopic examination by a uropathologist. Tissues were taken from the periurethral zone, as most prostate cancers arise in the peripheral zone (Pradidarcheep et al., 2011; Shaikhibrahim et al., 2012). Upon pathological evaluation, only tissue samples, which did not exhibit macroscopic signs of neoplasia, cancer or inflammation, were used for subsequent experiments. BPH is present in ~80% of patients with prostate cancer (Alcaraz et al., 2009; Orsted and Bojesen, 2013). Organ bath studies were performed immediately after sampling, while samples for molecular analyses were shock‐frozen in liquid nitrogen and stored at −80°C.
Real‐time PCR
RNA from frozen prostate tissues or cells was isolated using the RNeasy Mini kit (Qiagen, Hilden, Germany). For isolation from tissues, 30 mg of tissue were homogenized using the FastPrep®‐24 system with matrix A (MP Biomedicals, Illkirch‐Graffenstaden, France). RNA concentrations were measured spectrophotometrically. Reverse transcription to cDNA was performed with 1 μg of isolated RNA using the Reverse Transcription System (Promega, Madison, WI, USA). Real‐time PCR (RT‐PCR) for LIMK1, LIMK2 and GAPDH was performed with a Roche Light Cycler (Roche, Basel, Switzerland) using primers provided by Qiagen as ready‐to‐use mixes, based on the RefSeq accession numbers NM_001204426 for LIMK1, NM_001031801 for LIMK2 and NM_002046 for GAPDH. PCR reactions were performed in a volume of 25 μL containing 5 μL LightCycler® FastStart DNA MasterPlus SYBR Green I (Roche), 1 μL template, 1 μL primer and 18 μL water. Denaturation was performed for 10 min at 95°C and amplification with 45 cycles of 15 s at 95°C followed by 60 s at 60°C. The specificity of primers and amplification was demonstrated by subsequent analysis of melting points, which revealed single peaks for each target. Results are expressed using the ΔΔCP method, where number of cycles (Ct) at which the fluorescence signal exceeded a defined threshold for GAPDH was subtracted from Ct values for LIMK (CtLIMK − CtGAPDH = ΔCP), and values were calculated as 2−ΔCP and normalized to each other.
Western blot analysis
Frozen prostate tissues were homogenized in a buffer containing 25 mM Tris/HCl, 10 μM PMSF, 1 mM benzamidine and 10 μg·mL−1 leupeptine hemisulfate, using the FastPrep‐24 system with matrix A (MP Biomedicals). After centrifugation (20 000 g, 4 min), supernatants were assayed for protein concentration using the Dc‐Assay kit (Bio‐Rad, Munich, Germany) and boiled for 10 min with SDS sample buffer (Roth, Karlsruhe, Germany). Samples (20 μg per lane) were subjected to SDS‐PAGE, and proteins were blotted on Protran® nitrocellulose membranes (Schleicher & Schuell, Dassel, Germany). Membranes were blocked with PBS containing 5% milk powder (Roth) overnight and incubated with rabbit phospho‐LIMK1 (Thr508)/LIMK2 (Thr505) (#3841), rabbit anti‐LIMK1 (GTX10561‐50) (GeneTex, Irvine, CA, USA), rabbit anti‐LIMK2 (#3845), mouse anti‐phospho‐cofilin (p‐cofilin) 1 (sc‐365882) (Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit anti‐cofilin (D59), rabbit anti‐phospho‐myosin light chain (MLC)‐binding subunit of MLC phosphatase (MYPT1) (Thr696) (#5163), rabbit anti‐MYPT1 (#8574), mouse anti‐phospho‐MLC 2 (Ser19) (#3675), rabbit anti‐myosin light chain 2 (#8505), rabbit anti‐phospho‐eukaryotic translation initiation factor 4E‐binding protein 1 (4E‐BP1) (Thr37/46) (#9459), rabbit anti‐phospho‐4E‐BP1 (Ser65) (#13443), rabbit anti‐4E‐BP1 (#9644), mouse monoclonal anti‐pan‐cytokeratin (sc‐8018) (Santa Cruz Biotechnology), mouse monoclonal anti‐calponin 1/2/3 (sc‐136987) (Santa Cruz Biotechnology), mouse monoclonal anti‐prostate‐specific antigen (PSA) (sc‐7316) (Santa Cruz Biotechnology) or mouse monoclonal anti‐β‐actin antibody (sc‐47778) (Santa Cruz Biotechnology) (if not other stated, from Cell Signaling Technology, Danvers, MA, USA). Primary antibodies were diluted in PBS containing 0.1% Tween 20 (PBS‐T) and 5% milk powder. Subsequently, detection was continued using secondary biotinylated horse anti‐mouse or goat anti‐rabbit IgG (BA‐1000 and BA‐2000) (Vector Laboratories, Burlingame, CA, USA), followed by incubation with avidin and biotinylated HRP from the ‘Vectastain ABC kit’ (Vector Laboratories, Burlingame, CA, USA) both diluted 1:200 in PBS. Membranes were washed with PBS‐T after any incubation with primary or secondary antibodies or biotin‐HRP. Blots were developed with enhanced chemiluminescence (ECL) using ECL Hyperfilm (GE Healthcare, Freiburg, Germany). Intensities of resulting bands for LIMK1, LIMK2, cofilin and PSA were quantified densitometrically using ‘Image J’ (National Institutes of Health, Bethesda, MD, USA), and values (arbitrary units) were plotted against each other and subjected to Spearman's correlation analysis.
Fluorescence staining
Human prostate specimens, embedded in optimal cutting temperature compound, were snap‐frozen in liquid nitrogen and kept at −80°C. Sections (8 μm) were cut in a cryostat and collected on Superfrost® microscope slides. Sections were post‐fixed in methanol at −20°C and blocked in 1% BSA before incubation with primary antibody overnight at room temperature. For double labelling, the following primary antibodies were used: rabbit anti‐LIMK1 (GTX10561‐50) (GeneTex, Irvine, CA, USA), rabbit anti‐p‐cofilin (serine 3) (77G2) (Cell Signaling Technology), rabbit anti‐cofilin (D59) (Cell Signaling Technology), mouse anti‐pan‐cytokeratin (sc‐8018) or mouse anti‐calponin 1/2/3 (sc‐136987) (if not stated otherwise, compounds were obtained from Santa Cruz Biotechnology). Binding sites were visualized using Cy3‐conjugated goat anti‐mouse IgG (AP124C), FITC‐conjugated rabbit anti‐goat IgG (AP106F) (both from Millipore, Billerica, MA, USA) and Cy5‐conjugated goat anti‐rabbit IgG (ab6564) (Abcam, Cambridge, UK). Nuclei were counterstained with DAPI (Invitrogen, Camarillo, CA, USA). Immunolabelled sections were analysed using a laser scanning microscope (Leica SP2, Wetzlar, Germany). Fluorescence was recorded with separate detectors. Control stainings without primary antibodies did not yield any signals.
Tension measurements
Prostate strips (6 × 3 × 3 mm) were mounted in 10 mL aerated (95% O2 and 5% CO2) tissue baths (Danish Myotechnology, Aarhus, Denmark) with four chambers, containing Krebs–Henseleit solution (37°C, pH 7.4). Preparations were stretched to 4.9 mN and left to equilibrate for 45 min. In the initial phase of the equilibration period, spontaneous decreases in tone are usually observed. Therefore, tension was adjusted three times during the equilibration period, until a stable resting tone of 4.9 mN was attained. After the equilibration period, maximum contraction induced by 80 mM potassium chloride (KCl) was assessed. Subsequently, chambers were washed three times with Krebs–Henseleit solution for a total of 30 min. Cumulative concentration–response curves for http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=505, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=485, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=483, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=989 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1888 or frequency‐response curves induced by electrical field stimulation (EFS) were created after addition of inhibitors or DMSO for controls. Effects of SR7826 and LIMKi3 were assessed in separate series of experiments, using corresponding controls from the same prostate in each experiment. Thus, from each prostate, samples were allocated to the control and inhibitor groups within the same experiment. Consequently, both groups in each series had identical group sizes. This allocation was randomized. Moreover, application of DMSO (two chambers) and inhibitor (two chambers) to chambers was changed for each experiment, that is, again randomized. As two chambers were run for controls and two other for inhibitors in each experiment, all values of one independent experiment were determined in duplicate. In a separate series of experiments, samples from each prostate were allocated to groups containing LIMK inhibitor or a combination of LIMK inhibitor with http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5290. The application of EFS simulates action potentials, resulting in the release of endogenous neurotransmitters, including noradrenaline. For the calculation of agonist‐ or EFS‐induced contractions, tensions were expressed as % of KCl‐induced contractions, as this corrects for different stromal/epithelial ratios, different smooth muscle content, varying degree of BPH or any other heterogeneity between prostate samples and patients (Kenakin, 2008; Kunit et al., 2014; Wang et al., 2016a).
Cell culture
WPMY‐1 is an immortalized cell line from human prostate stroma without malignant transformation (Webber et al., 1999). Cells were purchased from American Type Culture Collection (Manassas, VA, USA) and grown in RPMI 1640 (Gibco, Carlsbad, CA, USA) supplemented with 10% FCS and 1% penicillin/streptomycin at 37°C with 5% CO2. Before addition of SR7826 or LIMKi3, the medium was changed to a FCS‐free medium. For Western blot analysis, cells were lysed using RIPA buffer (Sigma‐Aldrich, St. Louis, MO, USA) and removed from flasks after 15 min of incubation on ice. Cell debris was removed by centrifugation (10 000× g, 10 min, 4°C), and different aliquots of supernatants were either subjected to protein determination or boiled with SDS sample buffer.
Phosphorylation studies
Tissues from each prostate were cut into several small strips (6 × 1 × 1 mm), which were then allocated to two samples (control and inhibitor or control and agonist). Consequently, both groups in each series had identical group sizes. Incubation of samples with inhibitors, agonists and solvent (controls) was performed in six‐well plates filled with custodiol solution. After an equilibration period of 20 min, inhibitors, agonists and solvent were added, and plates were kept at 37°C under continuous shaking for indicated periods. Following incubations, tissues were shock frozen with liquid nitrogen and subjected to Western blot analysis for phospho‐LIMK, LIMK, p‐cofilin, cofilin, phospho‐MYPT1, MYPT1, phospho‐MLC, MLC, phospho‐4E‐BP1, 4E‐BP1 and β‐actin. Each setting was repeated in several independent experiments using different prostates, as indicated. For phosphorylation analyses in WMPY‐1 cells, cells were grown in T75 flasks, and inhibitors or DMSO were added after 48 h. After 2 h of incubation, cells were lysed with RIPA buffer and removed from flasks after 15 min of incubation on ice. Cell debris was removed by centrifugation (10 000× g, 10 min, 4°C), and supernatants were subjected to Western blot analysis.
The intensities of the resulting bands were quantified densitometrically using ‘Image J’ (National Institutes of Health). For semiquantitative calculation, values of each sample were normalized to the mean of the corresponding control group. This normalization is inevitable due to large variations in control values obtained by densitometric quantification (providing ‘arbitrary units’). These variations mostly result from detection conditions (slight differences in incubation periods and exposure times) and from different adjustments during digitization of blots (required due to varying background or diverging LIMK and cofilin content). These variations in control values cannot be avoided in practice; therefore, normalization to controls was required.
Phalloidin staining
For fluorescence staining of polymerized actin with phalloidin, cells were plated on Lab‐Tek chamber slides (Thermo Fisher, Waltham, MA, USA) and covered with inhibitors or solvent. Staining was performed using 100 μM FITC‐labelled phalloidin (Sigma‐Aldrich, Munich, Germany), according to the manufacturer's instruction. Labelled cells were analysed using a laser scanning microscope (Leica SP2, Wetzlar, Germany). Finally, stainings were quantified using ‘Image J’ (National Institutes of Health).
Viability assay
Effects of LIMK inhibitors on viability of WPMY‐1 cells were assessed using the Cell Counting Kit‐8 (CCK‐8) (Sigma‐Aldrich, St. Louis, MO, USA). Cells were grown in 96‐well plates (20 000 cells per well) for 24 h, before inhibitors or solvent were added in the concentrations indicated. Subsequently, cells were grown for different time periods (24, 48 or 72 h). Separate control experiments were performed for each period. At the end of this period, 10 μL of [2‐(2‐methoxy‐4‐nitrophenyl)‐3‐(4‐nitrophenyl)‐5‐(2,4‐disulfophenyl)‐2H‐tetrazolium monosodium salt (WST‐8) from CCK‐8 was added, and absorbance in each well was measured at 450 nm after incubation for 2 h at 37°C.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Data are presented as means ± SEM with the indicated number (n) of independent experiments. Multivariate ANOVA and one‐way ANOVA were used for paired or unpaired observations and performed using SPSS® version 20 (IBM SPSS Statistics, IBM Corporation, Armonk, NY, USA). P values <0.05 were considered statistically significant. Spearman's correlation analysis was performed using GraphPad Prism 6 (Statcon, Witzenhausen, Germany). All groups subjected to statistical analyses were based on five or more independent experiments or included tissues from five or more patients. Thus, minimum group sizes of all groups subjected to statistical tests were n = 5. Moreover, all groups being compared with each other by statistical tests showed identical group sizes; consequently, any statistical comparisons between groups of different sample sizes were not performed.
Results in phosphorylation analyses of agonist‐stimulated prostate tissues (phenylephrine, U46619) were not subjected to statistical analysis, as it was obvious that no effect could be expected from agonist stimulation. According to the exploratory character of these series and considering that no statistical test was applied and, finally, because it was obvious after three experiments that no effect could be expected, one of these series was stopped after three independent experiments; in fact, such exceptions are in line with the ‘British Journal of Pharmacology's recommendations on experimental design and analysis in pharmacology’ (Curtis et al., 2015).
Randomization was performed as described above for organ bath experiments. Assignment to groups (control or inhibitor) and analysis were not blinded, because this study was started before the new guidance for publication in the British Journal of Pharmacology was published, and was submitted before August 2017, when blinding became a negotiable requirement for studies published in the British Journal of Pharmacology (Curtis et al., 2015). At this time, blinding was in fact not routinely performed in our studies.
Calculations of group sizes for organ bath experiments were carried out using GLIMMPSE (version 2.2.7, http://glimmpse.samplesizeshop.org), an open‐source online tool for calculating power and sample size, on the basis of own previous experimental data (Hennenberg et al., 2013; Kreidler et al., 2013; Wang et al., 2015; 2016a). Calculations were performed separately for each agonist or EFS. The type I error rate was set to 0.05, while the desired statistic power was set to 0.8. Other input variables include mean and variability, which were extracted from our own previously published data series. The results are given in Table 1.
Table 1.
Calculation of group sizes for organ bath experiments
| Stimulus | Calculated group size |
|---|---|
| Noradrenaline | n = 9 |
| Phenylephrine | n = 5 |
| U46619 | n = 5 |
| Endothelin‐1 | n = 5 |
| EFS | n = 5 |
Numbers indicated for group sizes are numbers for the control and inhibitor groups, which were compared with each other for each stimulus (i.e. each agonist or EFS), and tissue from the same prostate was allocated to both the control and inhibitor group.
Principally, we adhered to these calculated groups sizes. If it became obvious during the study that an inhibitor has an effect, but without reaching significance with the calculated group size, group sizes were optimized (increased) until statistical significance was attained, that is, we opted for safety rather than just adhering to the bare minimum. A major reason could be that the human prostate tissues used in our study are characterized by highly divergent degree of BPH, together with additional overall individual variations, including different content and condition of smooth muscle and different stromal/glandular ratio. This has been demonstrated recently (Kunit et al., 2014; Wang et al., 2015) and was confirmed again in this study. Thus, adherence to the bare minimum of calculated group sizes instead of flexible design may yield disappointing results in organ bath experiments and may consequently not reflect the reality. According to the ‘new guidance for publication in BJP’, large variations may indeed impede such power analyses (Curtis et al., 2015). Consequently, it has been recommended that selecting the group sizes may be preferred in the interests of flexibility, rather than adhering to the bare minimum (Curtis et al., 2015). No data or experiments were excluded from analyses.
For methoxamine, we did not have our own previous results, so that our group size calculation for this agonist was based on previous experiments with phenylephrine. This appears feasible as both agonists show similar pharmacological properties (Alexander et al., 2015).
Materials and drugs
SR7826 is a LIMK family‐specific inhibitor, showing IC50 values of 43, 5536 and 6565 nM for LIMK1, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1503 (ROCK1) and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1504 (ROCK2) respectively (Yin et al., 2015). LIMKi3 (alternatively termed BMS 5) is a LIMK inhibitor showing IC50 values of 7 and 8 nM for LIMK1 and LIMK2 respectively (Ross‐Macdonald et al., 2008). Y27632 is a commonly used inhibitor of Rho kinase (Uehata et al., 1997). Stock solutions (10 mM) were prepared with DMSO and kept at −20°C until use. Phenylephrine and methoxamine are selective agonists for α1‐adrenoceptors. U46619 is an analogue of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4482 and frequently used as an agonist for TXA2 receptors. Aqueous stock solutions of phenylephrine, methoxamine hydrochloride and noradrenaline (10 mM) were freshly prepared before each experiment. Stock solutions of U46619 were prepared in ethanol and stored at −80°C until use. Aqueous stock solutions of endothelin‐1 were stored at −20°C until used. SR7826 and LIMKi3 were obtained from Tocris (Bristol, UK), phenylephrine and noradrenaline were obtained from Sigma (Munich, Germany), U46619 was obtained from Santa Cruz Biotechnology and endothelins from Enzo Life Sciences (Lörrach, Germany).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b,c).
Results
Detection of LIMK and cofilin in human prostate tissues
Analysis by RT‐PCR suggested mRNA expression of LIMK1 and LIMK2 in human prostate tissues and in WPMY‐1 cells (Figure 1A). Western blot analysis of prostate tissues and WPMY‐1 cells using antibodies raised again LIMK1 or LIMK2 revealed bands with sizes of the expected molecular weight (MW) of LIMK1 and LIMK2 (72 kDa) (Figure 1B, C). These bands were observed in each prostate sample included to this analysis, despite obvious variations in intensity between bands obtained with samples from different patients (Figure 1B). Western blot analysis of prostate tissues and WPMY‐1 cells using an antibody raised against cofilin 1 revealed bands with a size of the expected MW of cofilin 1 (18.5 kDa) (Figure 1B, C). These bands were observed in each prostate sample included to this analysis, with similar intensity between samples of different patients (Figure 1B).
Figure 1.
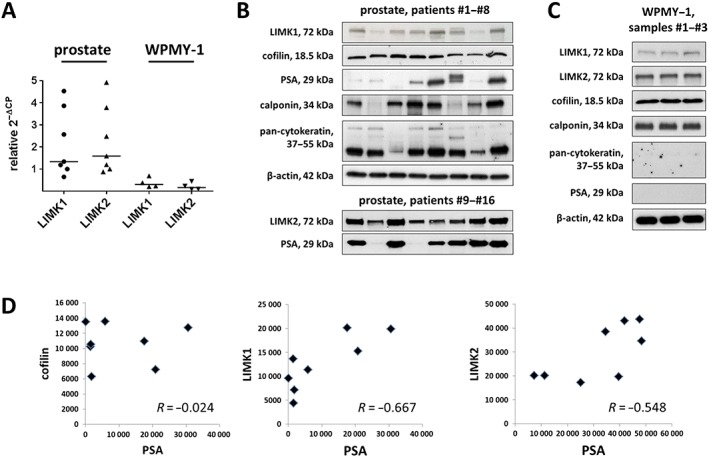
Detection of LIMK and cofilin in human prostate tissues. Samples of prostate tissues or WPMY‐1 cells were subjected to (A) RT‐PCR (tissues from n = 7 patients and cells from n = 4 independent experiments) or (B–D) Western blot analysis (n = 8 patients in each blot from a total of n = 16 patients or cells from n = 3 independent experiments). Data in (A) are ΔΔCP values (2^‐(Cttarget − CtGAPDH), normalized to each other) and median values (bar). In (B) and (C), bands for all samples included are shown, with sizes matching the expected and indicated MWs of proteins. Western blot analysis included calponin as a marker for smooth muscle cells, pan‐cytokeratin as a marker of endothelial cells (glands) and PSA as a marker for BPH. Because the limited sample sizes of prostate tissues did not allow analyses for all antigens in the same samples, different sample sets were used. In (D), values (arbitrary units) after densitometric quantification of Western blots were plotted in diagrams and subjected to Spearman's correlation analysis.
Bands for calponin and pan‐cytokeratin as markers for smooth muscle and epithelial glandular cells, respectively, were observed using all prostate samples included to these analyses but with varying intensities between samples from different patients (Figure 1B). Similarly, PSA was detected in almost all prostate samples included to these analyses, with very obvious variations of PSA content between prostates from different patients (Figure 1B). Together, this may reflect individual variations, and/or different degree of hyperplasia, as recently described (Kunit et al., 2014; Wang et al., 2015; 2016a,b). Calponin, but not pan‐cytokeratin, was detectable in WPMY‐1 cells (Figure 1C), confirming the smooth muscle‐like phenotype of these cells (Wang et al., 2015).
Following semiquantitative evaluation of bands obtained with prostate tissues, correlation analysis was performed for cofilin, LIMK1, LIMK2 and PSA. No correlation was observed between intensities of assumed cofilin and PSA bands (R = −0.02) (Figure 1D). In contrast, the correlation coefficient resulting from analysis of assumed bands for LIMK1 and PSA indicated a possible correlation between LIMK1 and PSA content, suggesting that expression of LIMK1 increases with the degree of BPH (R = 0.667) (Figure 1D). A similar, but slightly weaker correlation, may be suggested between expression of LIMK2 and PSA (R = 0.548) (Figure 1D).
Histological sections of prostate tissues from five different patients showed some individual variation in tissue architecture but clearly shared the typical composition of glands (with pan‐cytokeratin‐positive glandular epithelia) and stroma (with calponin‐positive smooth muscle cells) (Figure 2). Following fluorescence staining of prostate sections with an antibody raised again LIMK1, immunoreactivity was observed in the stroma and to limited extent in the glandular epithelium (Figure 2A). Colocalization with immunoreactivity for calponin suggested localization of the putative LIMK1 immunoreactivity in smooth muscle cells (Figure 2A). Immunoreactivity following fluorescence staining with an antibody raised against cofilin 1 was observed in the stroma and to lesser extent in the glandular epithelium (Figure 2B). Colocalization with immunoreactivity for calponin suggested localization of the putative cofilin immunoreactivity in smooth muscle cells (Figure 2B).
Figure 2.
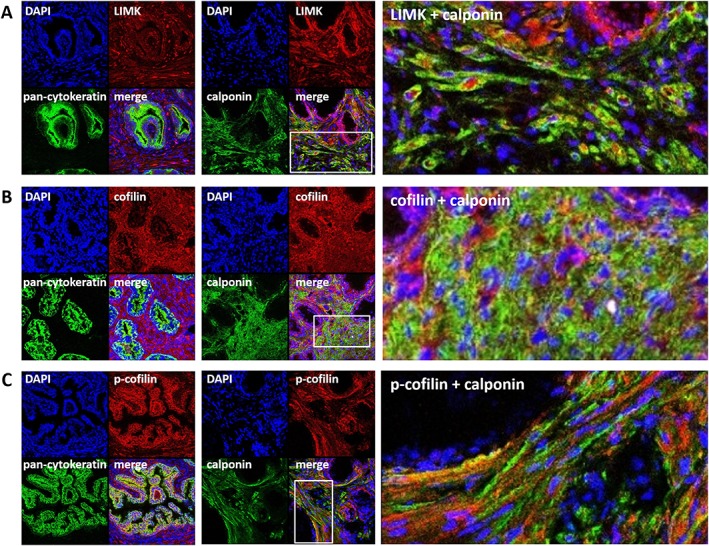
Immunofluorescence staining of human prostate tissues. Sections were double labelled with antibodies for (A) LIMK1, (B) cofilin or (C) p‐cofilin, together with antibodies for calponin (marker for smooth muscle cells) or pan‐cytokeratin (marker for glandular epithelial cells). Yellow colour in merged pictures indicates colocalization of targets. Regions of cut‐outs shown in the right panels are marked by white rectangles in the middle panels. Shown are representative stainings from a series of tissues from n = 5 patients for each combination.
Effects of SR7826 and LIMKi3 on EFS‐induced contractions
EFS (2–32 Hz) induced frequence‐dependent contractions of prostate strips, which were inhibited by SR7826 (1 μM) and LIMKi3 (1 μM) (Figure 3A). For both inhibitors, inhibition was significant after multivariate analysis at 8, 16 and 32 Hz (Figure 3A). Two‐way ANOVA was conducted to compare inhibitor and control groups, indicating significant inhibition of EFS‐induced contractions by SR7826 (P < 0.05) and LIMKi3 (P < 0.05). Using lower inhibitor concentrations of 500 nM, no effects of SR7826 and LIMKi3 on EFS‐induced contractions were observed, with the exception of a non‐significant trend towards an inhibition at 32 Hz by SR7826 (Figure 3B).
Figure 3.
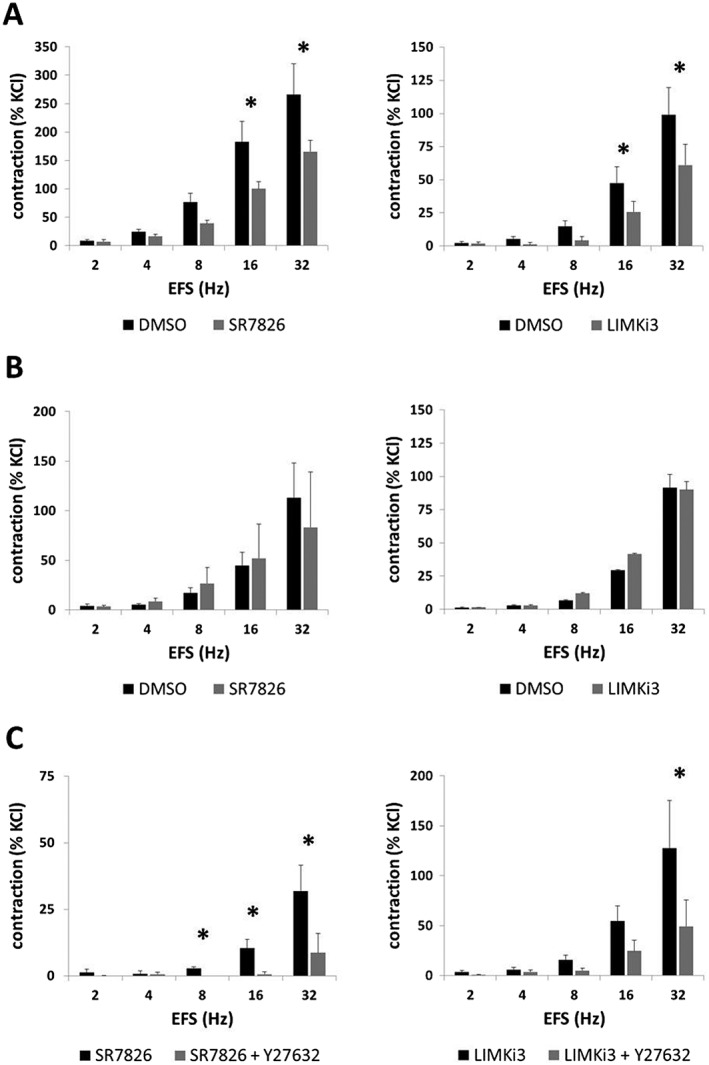
Effects of SR7826 and LIMKi3 on EFS‐induced contractions of human prostate strips. In an organ bath, contractions of human prostate strips were induced by EFS. Effects of the LIMK inhibitors SR7826 or LIMKi3 on contractions were tested in concentrations of (A) 1 μM or (B) 500 nM or in combination with the (C) Rho kinase inhibitor Y27632 (30 μM). Inhibitor groups were compared with corresponding controls (DMSO) in (A) and (B). In (C), contractions were compared in the presence of LIMK inhibitor (1 μM) alone or in the presence of a combination of LIMK inhibitor (1 μM) with Y27632 (30 μM). To eliminate heterogeneities due to individual variations, different degrees of BPH or other varying smooth muscle content (compare Figure 1), tensions have been expressed as % of contraction induced by high molar KCl, being assessed before application of inhibitors or solvent. Data are means ± SEM from series with tissues from n = 10 patients for 1 μM SR7826, n = 5 for 1 μM LIMKi3, n = 5 for 500 nM SR7826, n = 5 for 500 nM LIMKi3, n = 5 for SR7826 versus SR7825 + Y27632 and n = 8 for LIMKi3 versus LIMKi3 + Y27632 (* P < 0.05 for control vs. inhibitor). Samples from each patient were allocated to both groups within one diagram, so that both groups in each diagram had identical group sizes. Statistical comparisons were performed between both groups in each diagram but not between groups across different diagrams.
Effects of a combination of SR7826 with Y27632 on EFS‐induced contractions
In a separate series of experiments, EFS‐induced contractions of prostate strips were induced after addition of SR7826 (1 μM) alone or of a combination containing SR7826 (1 μM) and Y27632 (30 μM) (Figure 3C). In these experiments, residual contractions in the presence of SR7826 were further suppressed by Y2732, which was significant at 8, 16 and 32 Hz (Figure 3C). Two‐way ANOVA was conducted to compare the SR7826 with the SR7826 + Y27632 group, indicating that the additional suppression of residual contractions by Y27632 was significant (P < 0.05).
In a similar series of experiments, EFS‐induced contractions were induced after addition of LIMKi3 (1 μM) alone or of a combination containing LIMKi3 (1 μM) and Y27632 (30 μM) (Figure 3C). Contractions in the presence of LIMKi3 were further suppressed by Y27632, which was significant at 32 Hz (Figure 3C). Two‐way ANOVA was conducted to compare the LIMKi3 with the LIMKi3 + Y27632 group, indicating that the additional suppression of residual contractions by Y27632 was significant (P < 0.05).
Effects of SR7826 and LIMKi3 on phenylephrine‐induced contractions
Phenylephrine (0.1–100 μM) induced concentration‐dependent contractions of prostate strips, which were inhibited by SR7826 (1 μM) and LIMKi3 (1 μM) (Figure 4A). For SR7826, inhibition was significant after multivariate analysis at 10, 30 and 100 μM phenylephrine (Figure 4A). For LIMKi3, inhibition was significant after multivariate analysis at 3, 10 and 30 μM phenylephrine (Figure 4A). Two‐way ANOVA was conducted to compare inhibitor and control groups, indicating significant inhibition of phenylephrine‐induced contractions by SR7826 (P < 0.05) and LIMKi3 (P < 0.05).
Figure 4.
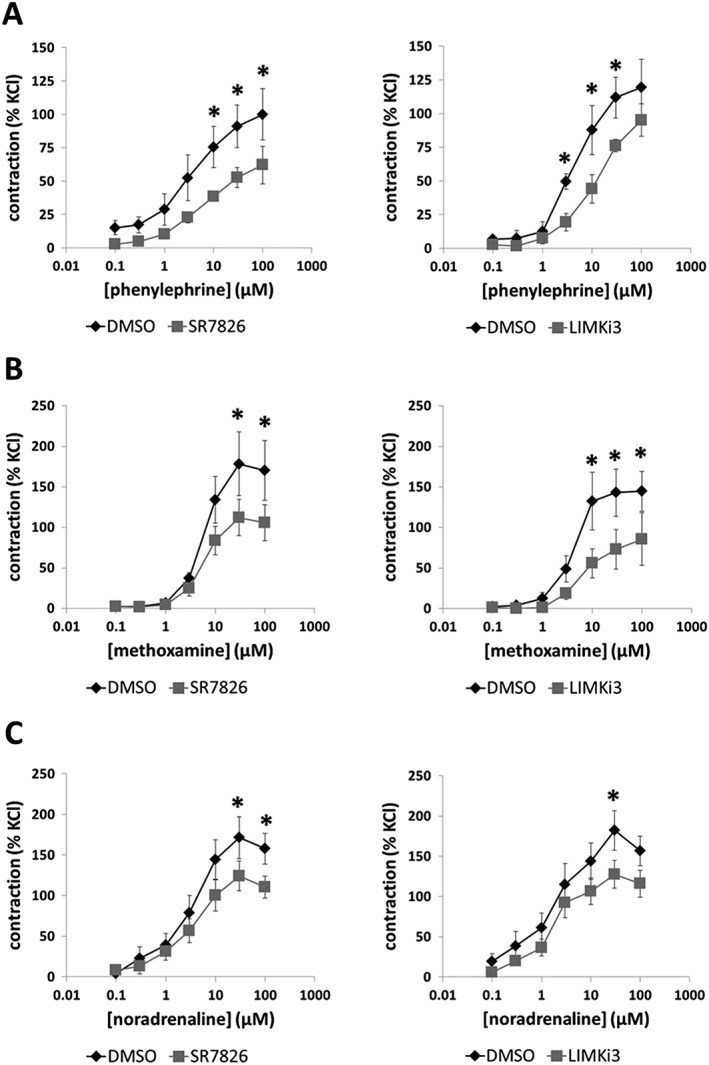
Effects of SR7826 and LIMKi3 on adrenergic contractions of human prostate strips. In an organ bath, contractions of human prostate strips were induced by the α1‐adrenoceptor agonists (A) phenylephrine, (B) methoxamine or (C) noradrenaline. Effects of the LIMK inhibitors SR7826 (1 μM) or LIMKi3 (1 μM) on contractions were compared with corresponding controls (DMSO) in separate sets of experiments. To eliminate heterogeneities due to individual variations, different degrees of BPH or varying smooth muscle content (compare Figure 1), tensions have been expressed as % of contraction induced by high molar KCl, being assessed before application of inhibitors or solvent. Data are means ± SEM from a series of tissues from n = 9 patients for phenylephrine/SR7826, n = 5 for phenylephrine/LIMKi3, n = 6 for methoxamine/SR7826, n = 5 for methoxamine/LIMKi3, n = 8 for noradrenaline/SR7826 and n = 9 for noradrenaline/LIMKi3 (* P < 0.05 for control vs. inhibitor). Samples from each patient were allocated to both groups within one diagram, so that both groups in each diagram had identical group sizes. Statistical comparisons were performed between both groups in each diagram but not between groups across different diagrams.
Effects of SR7826 and LIMKi3 on methoxamine‐induced contractions
Methoxamine (0.1–100 μM) induced concentration‐dependent contractions of prostate strips, which were inhibited by SR7826 (1 M) and LIMKi3 (1 μM) (Figure 4B). For SR7826, inhibition was significant after multivariate analysis at 30 and 100 μM methoxamine (Figure 4B). For LIMKi3, inhibition was significant after multivariate analysis at 10, 30 and 100 μM methoxamine (Figure 4B). Two‐way ANOVA was conducted to compare inhibitor and control groups, indicating significant inhibition of methoxamine‐induced contractions by SR7826 (P < 0.05) and LIMKi3 (P < 0.05).
Effects of SR7826 and LIMKi3 on noradrenaline‐induced contractions
Noradrenaline (0.1–100 μM) induced concentration‐dependent contractions of prostate strips, which were inhibited by SR7826 (1 M) and LIMKi3 (1 μM) (Figure 4C). For SR7826, inhibition was significant after multivariate analysis at 30 and 100 μM noradrenaline (Figure 4C). For LIMKi3, inhibition was significant after multivariate analysis at 30 μM noradrenaline (Figure 4C). Two‐way ANOVA was conducted to compare inhibitor and control groups, indicating significant inhibition of noradrenaline‐induced contractions by SR7826 (P < 0.05) and LIMKi3 (P < 0.05).
Effects of SR7826 and LIMKi3 on U46619‐induced contractions
U46619 (0.01–30 μM) induced concentration‐dependent contractions of prostate strips, which were inhibited by SR7826 (1 μM) and LIMKi3 (1 μM) (Figure 5A). For SR7826, inhibition was significant after multivariate analysis at 1, 3 and 10 μM U46619 (Figure 5A). For LIMKi3, inhibition was significant after multivariate analysis at 1, 3, 10 and 30 μM U46619 (Figure 5A). Two‐way ANOVA was conducted to compare inhibitor and control groups, indicating significant inhibition of U46619‐induced contractions by SR7826 (P < 0.05) and LIMKi3 (P < 0.05).
Figure 5.
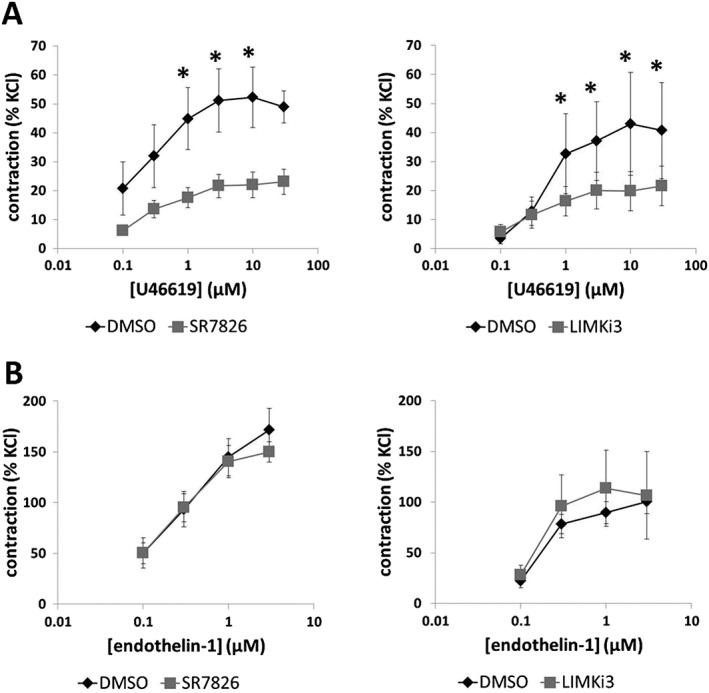
Effects of SR7826 and LIMKi3 on U46619‐ and endothelin‐1‐induced contractions of human prostate strips. In an organ bath, contractions of human prostate strips were induced by the TXA2 analogue (A) U46619 or (B) endothelin‐1. Effects of the LIMK inhibitors SR7826 (1 μM) or LIMKi3 (1 μM) on contractions were compared with corresponding controls (DMSO) in separate sets of experiments. To eliminate heterogeneities due to individual variations, different degrees of BPH or varying smooth muscle content (compare Figure 1), tensions have been expressed as % of contraction induced by high molar KCl, being assessed before application of inhibitors or solvent. Data are means ± SEM from a series of tissues from n = 7 patients for U46619/SR7826, n = 5 for U46619/LIMKi3, n = 5 for endothelin‐1/SR7826 and n = 5 for endothelin‐1/LIMKi3 (* P < 0.05 for control vs. inhibitor). Samples from each patient were allocated to both groups within one diagram, so that both groups in each diagram had identical group sizes. Statistical comparisons were performed between both groups in each diagram but not between groups across different diagrams.
Effects of SR7826 and LIMKi3 on endothelin‐1‐induced contractions
Endothelin‐1 (0.1–3 μM) induced concentration‐dependent contractions of prostate strips. These endothelin‐1‐induced contractions were neither inhibited by SR7826 (1 μM) nor by LIMKi3 (1 μM) (Figure 5B).
Effects of SR7826 and LIMKi3 on cofilin and LIMK phosphorylation in prostate tissues
Using site‐specific and phospho‐specific antibodies, serine‐3‐phosphorylated cofilin and threonine‐508/505 LIMK1/2 were detectable by Western blot analyses of prostate tissues with bands matching the expected MWs of cofilin and LIMK1/2 (Figures 6 and 7). The content of p‐cofilin in prostate tissues was reduced with similar efficacy by SR7826 and LIMKi3 (Figures 6 and 7): SR7826 reduced the p‐cofilin content by 59 ± 15%, while LIMKi3 reduced the p‐cofilin content by 56 ± 13%. In contrast, the contents of total cofilin, β‐actin, phospho‐LIMK, LIMK1 and −2, and β‐actin remained unchanged by both inhibitors (Figures 6 and 7).
Figure 6.
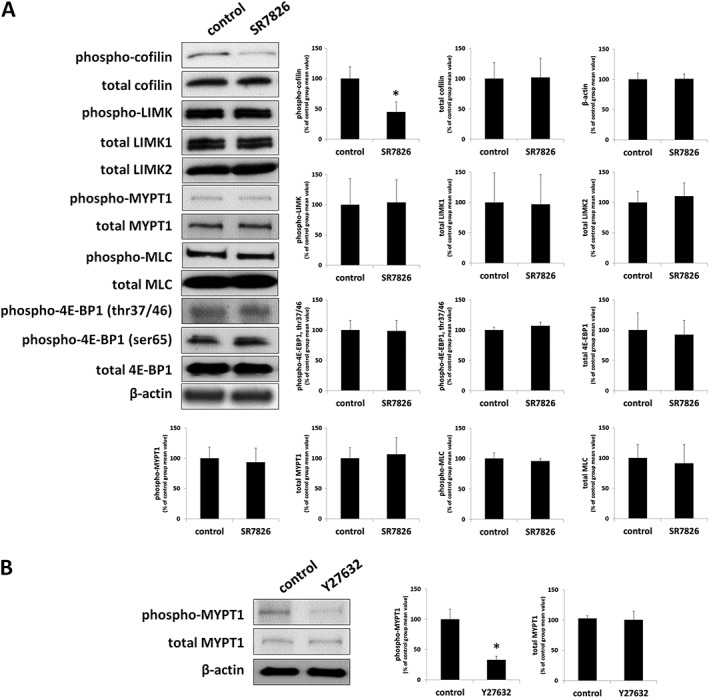
Effects of SR7826 and Y27632 on phosphorylations in human prostate tissues. In separate series of experiments, prostate tissues were incubated with (A) SR7826 (1 μM, 2 h) or DMSO (control) or with (B) Y27632 (30 μM, 1 h) or DMSO. Subsequently, the phosphorylation states of cofilin, LIMK and MYPT1 (threonine 696), MLC and 4E‐BP1 (threonine 37/46, serine 65) were semiquantitatively compared between inhibitor and control groups by Western blot analyses using site‐ and phospho‐specific antibodies; values for each sample were normalized to the mean of the corresponding control group. Shown are representative blots, and means ± SEM from a series of tissues from n = 7 (SR7826), and n = 5 patients (Y27632) (* P < 0.05 for control vs. inhibitor). Samples from each patient were allocated to both groups within each series, so that both groups in each series had identical group sizes.
Figure 7.
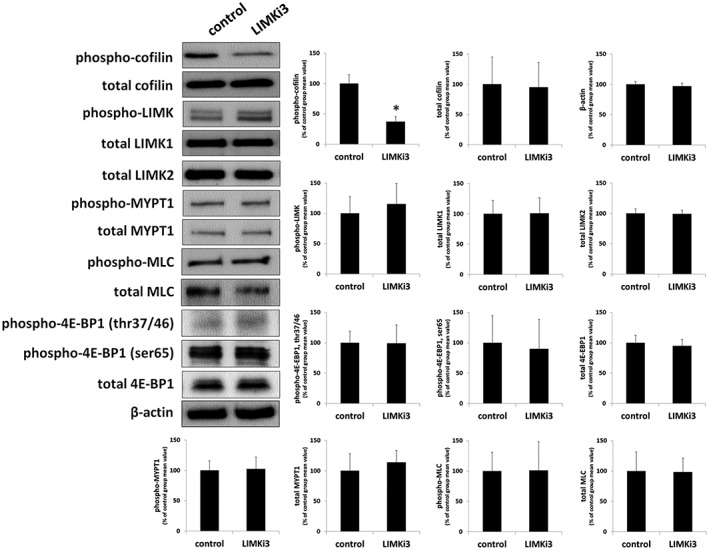
Effects of LIMKi3 on phosphorylations in human prostate tissues. In separate series of experiments, prostate tissues were incubated with LIMKi3 (1 μM, 2 h) or DMSO. Subsequently, the phosphorylation states of cofilin, LIMK and MYPT1 (threonine 696), MLC and 4E‐BP1 (threonine 37/46, serine 65) were semiquantitatively compared between inhibitor and control groups by Western blot analyses using site‐ and phospho‐specific antibodies; values for each sample were normalized to the mean of the corresponding control group. Shown are representative blots, and means ± SEM from a series of tissues from n = 6 (LIMKi3), and n = 5 patients (Y27632). Samples from each patient were allocated to both groups within each series, so that both groups in each series had identical group sizes.
Following fluorescence staining of prostate sections with an antibody raised again p‐cofilin, immunoreactivity was observed in the stroma and in the glandular epithelium (Figure 2C). Colocalization with immunoreactivity for calponin suggested putative localization of the p‐cofilin immunoreactivity in smooth muscle cells (Figure 2C).
Effects of SR7826 and LIMKi3 on MYPT1 phosphorylation in prostate tissues
Using a site‐ and phospho‐specific antibody, threonine‐696‐phosphorylated MYPT1 was detectable by Western blot analyses of prostate tissues with bands matching the expected MW of MYPT1 (115 kDa) (Figures 6 and 7). MYPT1 phosphorylation at this position is mediated by Rho kinase (Kimura et al., 1996; Khromov et al., 2009). The phospho‐MYPT1 concentration in prostate tissues was not changed by either SR7826 or LIMKi3 (Figures 6A and 7). Similarly, total MYPT1 concentration, which was detected with bands showing the size of the expected MW of MYPT1, did not differ between control and inhibitor groups (Figures 6A and 7).
To confirm that monitoring of threonine‐696 phosphorylation of MYPT1 in prostate tissues by Western blot analysis is sensitive to Rho kinase inhibition, we examined effects of the Rho kinase inhibitor Y27632 on phosphorylation of this position. Y27632 reduced the content of threonine‐696‐phosphorylated MYPT1 by 67 ± 6% (Figure 6B). In contrast, the contents of total MYPT1 and β‐actin remained unchanged by Y27632 (Figure 6B).
Effects of SR7826 and LIMKi3 on MLC phosphorylation in prostate tissues
Using a site‐ and phospho‐specific antibody, serine‐19‐phosphorylated MLCs were detectable by Western blot analyses of prostate tissues with bands matching the expected MW of MLC (18.7 kDa) (Figures 6 and 7). The content of phospho‐MLC in prostate tissues was neither changed by SR7826 nor by LIMKi3 (Figures 6 and 7). Similarly, the content of total MLCs, which was detected with bands showing the size of the expected MW of MLC, did not differ between control and inhibitor groups (Figures 6 and 7).
Effects of SR7826 and LIMKi3 on 4E‐BP1 phosphorylation in prostate tissues
Using site‐specific and phospho‐specific antibodies, threonine‐37/46‐phosphorylated and serine‐65‐phosphorylated 4E‐BP1 were detectable by Western blot analyses of prostate tissues with bands matching the expected MW of 4E‐BP1 (12.6 kDa) (Figures 6 and 7). Phosphorylation of 4E‐BP1 is may be mediated by http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2213 (STK16 also known as MPSK1) (Liu et al., 2016; Liu et al., 2017). The content of phospho‐4E‐BP1 in prostate tissues was not changed by either SR7826 or LIMKi3 (Figures 6 and 7). Similarly, the content of total 4E‐BP1, which was detected with bands showing the size of the expected MW of 4E‐BP1, did not differ between control and inhibitor groups (Figures 6 and 7).
Effects of phenylephrine and U46619 on LIMK and cofilin phosphorylation in prostate tissues
Effects of different contractile stimuli on the content of phospho‐LIMK, LIMK, p‐cofilin, cofilin and β‐actin were assessed by Western blot analysis (Figure 8). Neither stimulation with phenylephrine (30 μM) for 10 min nor with U46619 (30 μM) for 1 h, resulted in a change in the content of phospho‐LIMK, LIMK, p‐cofilin, cofilin or β‐actin in prostate tissues (Figure 8). Similarly, a preliminary series of experiments did not provide an informative basis to assume that stimulation with phenylephrine for 45 min (30 μM) increased the phosphorylation state of cofilin or LIMK in prostate tissues, so this series was stopped after three independent experiments (data not shown).
Figure 8.
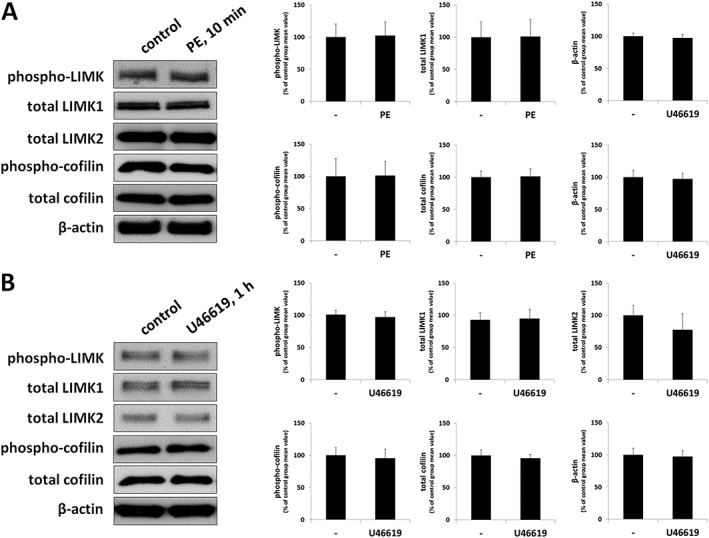
Effects of contractile stimuli on LIMK and cofilin phosphorylation in human prostate tissues. In separate series of experiments, prostate tissues were incubated with (A) phenylephrine (PE) (30 μM) for 10 min or with (B) U46619 (30 μM) for 1 h, or with solvent (ethanol for U46619) as required. Subsequently, the phosphorylation states of LIMK and cofilin were semiquantitatively compared between agonist and control groups by Western blot analyses using site‐ and phospho‐specific antibodies; values for each sample were normalized to the mean of the corresponding control group (i.e. without agonist). Shown are representative blots, and means ± SEM from a series of tissues from n = 5 (phenylephrine 10 min), n = 3 (phenylephrine 45 min) or n = 7 patients (U46619). Another, preliminary series did not provide an informative basis to assume that stimulation with phenylephrine for 45 min (30 μM) resulted in changes in LIMKs or cofilin phosphorylation, so this series was not continued (data not shown).
Effects of SR7826 and LIMKi3 on viability of WPMY‐1 cells
A viability assay revealed concentration‐ and time‐dependent effects of SR7826 and LIMKi3 in WPMY‐1 cells. At a concentration of 1 μM (which inhibited smooth muscle contraction of prostate tissues), SR7826 and LIMKi3 showed minor effects on viability after 24, 48 or 72 h of exposure (Figure 9A). At 5 μM, both inhibitors markedly reduced viability in cells after 24, 48 or 72 h of exposure (Figure 9A). Therefore, the following experiments in WPMY‐1 cells were performed using the concentration of 5 μM.
Figure 9.
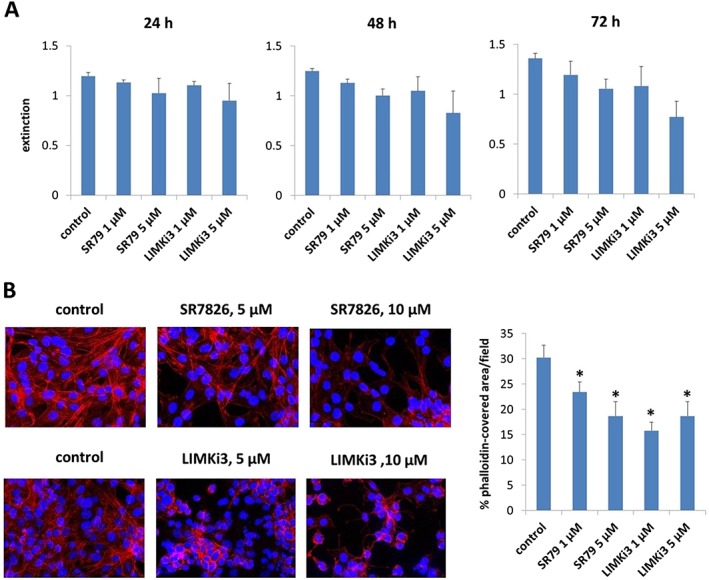
Concentration‐dependent effects of SR7826 and LIMKi3 in WPMY‐1 cells. WPMY‐1 cells were exposed to SR7826 and LIMKi3 at different concentrations and for different periods and subjected to a (A) viability assay or (B) phalloidin staining to visualize polymerized actin. In (A), cells were exposed to 1 or 5 μM of SR7826 or LIMKi3 for 24, 48 or 72 h or remained without inhibitors under the same conditions (= controls). In (B), cells were exposed to 5 or 10 μM of SR7826 (upper panels) or LIMKi3 (lower panels) for 24 h or remained without inhibitors under the same conditions (= controls). Shown are means ± SD from n = 5 independent experiments in (A), representative pictures from n = 5 independent experiments in (B) and means ± SD from quantification of phalloidin staining in n = 5 independent experiments in (B) (right panel).
Effects of SR7826 and LIMKi3 on actin organization in WPMY‐1 cells
Phalloidin staining visualized actin organization in WPMY‐1 cells. In control cells without inhibitor treatment, actin was organized to long and thin filaments, being formed by most of the cells, and with filamentous protrusions of different cells overlapping each other (Figure 9B). Exposure to SR7826 for 24 h revealed concentration‐dependent effects. Thus, exposure to 5 μM SR7826 reduced the length of filaments in some but not all cells (Figure 9B). Cells with actin organization similar to control cells were still visible but obviously at a smaller number (Figure 9B). Exposure to 10 μM SR7826 resulted in extensive breakdown of actin organization, that is, phalloidin‐stained actin disappeared from most cells, and any remaining filaments were obviously shorter (Figure 9B). Similarly, exposure to 5 or 10 μM LIMKi3 for 24 h caused again extensive breakdown of actin organization (Figure 9B). Thus, actin filaments disappeared virtually completely after exposure to LIMKi3, so that any remaining phalloidin‐stained actin was almost completely restricted to the region surrounding some of the nuclei (Figure 9B). These changes were confirmed by quantification of phalloidin staining, which suggested the polymerized actin concentration was decreased after incubation with SR7826 and LIMKi3 (Figure 9B).
Effects of SR7826 and LIMKi3 on cofilin and LIMK phosphorylation in WPMY‐1 cells
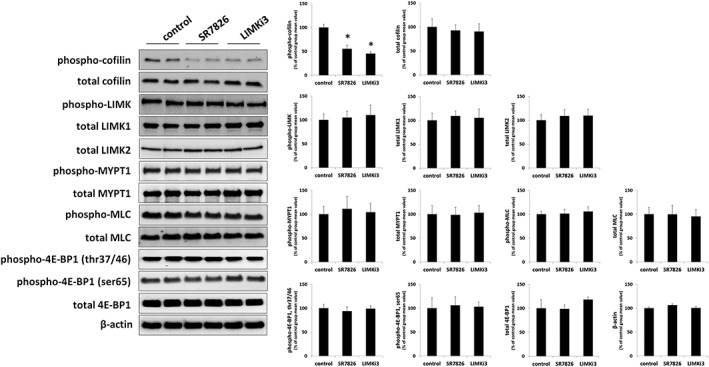
Western blot analysis of WPMY1‐cells using site‐ and phospho‐specific antibodies, serine‐3‐revealed that phosphorylated cofilin and threonine‐508/505 LIMK1/2 were detectable with bands matching the expected MWs of cofilin (Figure 10). The content of p‐cofilin in WPMY‐1 cells was reduced with similar efficacy by SR7826 and LIMKi3 (Figure 10): SR7826 reduced the p‐cofilin content by 45 ± 6%, while LIMKi3 reduced the p‐cofilin content by 54 ± 4%. In contrast, the contents of total cofilin, phospho‐LIMK, total LIMK1 and LIMK2, and β‐actin were unchanged in the presence of either inhibitor (Figure 10).
Figure 10.
Effects of SR7826 and LIMKi3 on phosphorylations in WPMY‐1 cells. WPMY‐1 cells were incubated with SR7826 (5 μM, 2 h), LIMKi3 (5 μM, 2 h) or DMSO (control, 2 h). Subsequently, the phosphorylation states of cofilin, LIMK and MYPT1 (threonine 696), MLC and 4E‐BP1 (threonine 37/46, serine 65) were semiquantitatively compared between inhibitor and control groups by Western blot analyses using site‐ and phospho‐specific antibodies; values for each sample were normalized to the mean of the corresponding control group. Shown are representative blots, and means ± SEM from a series of five independent experiments (* P < 0.05 for control vs. inhibitor).
Effects of SR7826 and LIMKi3 on MYPT1 phosphorylation in WPMY‐1 cells
Using a site‐ and phospho‐specific antibody, threonine‐696‐phosphorylated MYPT1 was detectable by Western blot analyses of WPMY‐1 cells with bands matching the expected MW of MYPT1 (Figure 10). The content of phospho‐MYPT1 in WPMY‐1 cells was not changed by either SR7826 or LIMKi3 (Figure 10). Similarly, the content of total MYPT1 did not differ between control and inhibitor groups (Figure 10).
Effects of SR7826 and LIMKi3 on MLC phosphorylation in WPMY‐1 cells
Using a site‐ and phospho‐specific antibody, serine‐19‐phosphorylated MLCs were detectable by Western blot analyses of WPMY‐1 cells with bands matching the expected MW of MLC (Figure 10). The content of phospho‐MLCs in WPMY‐1 cells was not changed by either SR7826 or LIMKi3 (Figure 10). Similarly, the content of total MLCs did not differ between control and inhibitor groups (Figure 10).
Effects of SR7826 and LIMKi3 on 4E‐BP1 phosphorylation in WPMY‐1 cells
Using site‐ and phospho‐specific antibodies, threonine‐37/46‐phosphorylated and serine‐65‐phosphorylated 4E‐BP1 were detectable by Western blot analyses of WPMY‐1 cells with bands matching the expected MW of 4E‐BP1 (Figure 10). The content of phospho‐4E‐BP1 in WPMY‐1 cells was not changed by either SR7826 or LIMKi3 (Figure 10). Similarly, the content of total 4E‐BP1 did not differ between control and inhibitor groups (Figure 10).
Discussion
Our findings suggest that two structurally unrelated LIMK inhibitors inhibited contractions of human prostate tissues. To the best of our knowledge, this study is the first one reporting inhibition of smooth muscle contraction by small molecule LIMK inhibitors in any organ. Prostate smooth muscle contraction is of particular interest, as smooth muscle tone in the hyperplastic prostate may contribute to urethral compression, resulting in bladder outlet obstruction and LUTS (Hennenberg et al., 2014). Due to limited efficacy of available options for medical treatment of LUTS suggestive of BPH, new medications are needed (Hennenberg et al., 2014). This requires a more precise understanding of the molecular mechanisms involved in prostate smooth muscle contraction. However, the recent discovery of previously unknown mechanisms demonstrated that prostate smooth muscle contraction is still insufficiently understood (Strittmatter et al., 2012; Hennenberg et al., 2014; Kunit et al., 2014; Wang et al., 2015; Wang et al., 2016a,b). Thus, our present findings may contribute to a better understanding of smooth muscle contraction in the prostate.
SR7826 and LIMKi3 are small molecule inhibitors, which inhibit LIMK1 and LIMK2 with high potency. In biochemical kinase assays, where inhibition was assessed using recombinant kinases in vitro, LIMK1 was inhibited by SR7826 with an IC50 value of 43 nM, while LIMK1 and −2 were inhibited with IC50 values of 7–8 nM by LIMKi3 (Ross‐Macdonald et al., 2008; Yin et al., 2015). Considering that IC50 values for other kinases were much higher, this may confer high specificity. Thus, the IC50 value of SR7826 for Rho kinase II (which may also mediate smooth muscle contraction) exceeded 6.5 μM, while IC50 values for a panel of 61 other kinases were even higher with the exception of inhibition of STK16, which was inhibited to 80% by 1 μM SR7826 (Yin et al., 2015). For LIMKi3, data describing inhibition of other kinases have to the best of our knowledge not been reported (Ross‐Macdonald et al., 2008). In prostate tissues or in WPMY‐1 cells, we did not observe any impact of SR7826 or LIMKi3 on phosphorylations of the Rho kinase substrate MYPT1 or of the STK16 substrate 4E‐EBP1 under our conditions but inhibition of cofilin phosphorylation by both inhibitors. In principle, this may suggest that both inhibitors acted by inhibition of LIMK in our models, whereas actions due to unspecific kinase inhibition appear unlikely or may have been low. On the other hand, the number of substrates of putative off‐target kinases examined in our study was low, so that unspecific inhibition may not be completely ruled out, especially regarding STK16. Conclusions whether effects of SR7826 were solely attributed to LIMK1 or included LIMK2 may not be possible, as screening of SR7826 included a panel of more than 60 kinases but not LIMK2 (Yin et al., 2015).
EC50 values in organ bath experiments and in intact tissues may be higher than IC50 values in biochemical assays, for example, due to divergent access of inhibitors to their targets resulting from barriers like membranes or connective tissue (Vauquelin et al., 2002; Swinney, 2004; Kunit et al., 2014; Wang et al., 2016b). In fact, EC50 values for biological effects of LIMK inhibitors in previous studies were higher than IC50 values in kinase assays, so that migration of prostate cancer cells was inhibited by 100 nM SR7826 by 16% and by 74% by 1 μM (Yin et al., 2015). For LIMKi3, the EC50 value for inhibition of proliferation in a cell culture model ranged around 10 μM (Ross‐Macdonald et al., 2008). We applied both inhibitors in concentrations of 1 μM to prostate tissues or up to 5 μM to WPMY‐1 cells, which exceeded the IC50 values for LIMKs but were still below IC50 values for all other screened kinases in biochemical assays with the exception of STK16 (Ross‐Macdonald et al., 2008; Yin et al., 2015). It appears possible that inhibition of prostate smooth muscle contraction and effects in WPMY‐1 cells were caused by inhibition of LIMK1, as our findings suggested no inhibition of STK16 or Rho kinase under our conditions.
Inhibition of LIMK1/2 in prostate tissues and WPMY‐1 cells was confirmed by the effects of SR7832 and LIMKi3 on the content of p‐cofilin, which was substantially reduced by both inhibitors. In fact, cofilin is an important substrate of LIMK1/2 (Arber et al., 1998; Yang et al., 1998; Bernard, 2007). Consequently, inhibition of cofilin phosphorylation in our tissues and cells was most likely attributed to LIMK inhibition but limited to 45–60%. Whether this reflects incomplete LIMK inhibition or may result from LIMK‐independent cofilin phosphorylation may not be concluded from our data; to the best of our knowledge, LIMK‐independent cofilin phosphorylation has not been reported. LIMK itself may be phosphorylated at threonine 508/505, which increases its kinase activity and may be critical for LIMK activation by upstream signalling pathways (Arber et al., 1998; Edwards et al., 1999; Ohashi et al., 2000). As we observed no decreases in LIMK phosphorylation at this position by SR7826 or LIMKi3, we conclude that this site is no substrate for LIMK autophosphorylation in the prostate.
To the best of our knowledge, previous investigations addressing LIMK in the prostate were limited to an oncological context (Misra et al., 2005; Mardilovich et al., 2015b). Our findings suggest LIMK1 and −2 are expressed in the hyperplastic human prostate, and this may increase with the degree of BPH. Apart from a role for LIMK1/2 in cancer of the prostate and other organs, the primary function of LIMK may be the regulation of actin organization (Yang et al., 1998; Bernard, 2007). LIMK1 promotes actin polymerization and filament assembly and thus stress fibre formation in a range of cell types, including vascular smooth muscle cells (Yang et al., 1998; Bernard, 2007; San Martin et al., 2008). This LIMK‐dependent actin organization requires the phosphorylation of cofilin by LIMK (Yang et al., 1998; Maekawa et al., 1999; Mouneimne et al., 2006; Bernard, 2007; Scott et al., 2010; Mardilovich et al., 2015a). Consequently, LIMK has been reported to regulate several actin‐dependent functions, including migration, motility and chemotaxis, as well as proliferation (Mouneimne et al., 2006; Li et al., 2014; Yin et al., 2015; Mardilovich et al., 2015a,b; Kapur et al., 2016). As smooth muscle contraction requires actin organization and LIMK‐dependent actin organization has been reported in vascular smooth muscle cells, it is assumed LIMK is involved in smooth muscle contraction (San Martin et al., 2008; Hennenberg et al., 2014). Surprisingly and to the best of our knowledge, this mechanism, including the application of small molecule LIMK inhibitors to intact tissues, has not yet been considered or investigated. Previous investigations addressing the role of LIMKs in contraction were limited to isolated cancer‐associated fibroblasts in cell culture, in which LIMK inhibition reduced collagen matrix contractions (Scott et al., 2010). Thus, it may be speculated that findings similar to ours may be obtained with smooth muscle preparations from other organs, for example, vessels or airways.
It has been reported that contractile agonists, including noradrenaline, induce LIMK activation and cofilin phosphorylation in intact vessels (Dai et al., 2008). Similarly, the α1‐adrenoceptor agonist phenylephrine induced LIMK activation, cofilin phosphorylation and actin polymerization in vascular smooth muscle cells (Du et al., 2010). We assumed that contractile agonists may activate LIMK in the prostate as well and therefore tested effects of different contractile stimuli on LIMK and cofilin phosphorylation. However, neither phenylephrine nor U46619 changed the content of phospho‐LIMK or p‐cofilin, suggesting that neither α1‐adrenoceptors nor TXA2 receptors activate LIMK in the human prostate. On the other hand, we cannot fully rule out that a transient activation ranging out of our time frames may take place, although we tested different stimulation periods. We detected immunoreactivity for p‐cofilin by immunofluorescence in untreated prostate tissues. This finding may suggest a pool of active LIMK in these cells; however, no conclusion on whether this reflects a constitutive or inducible activity is yet possible. Contrary to other pathways including RhoA/Rho kinase, PLC/inositol‐1,4,5‐trisphosphate/calcium or DAG/PKC, LIMK/cofilin may not be activated by α1‐adrenoceptors or TXA2 receptors to induce contraction but regulates contraction independently of these receptors. Together, LIMK may be a critical intracellular regulator of prostate smooth muscle contractility. Unlike other pathways, it may not be involved in signal transduction from receptors to contraction in the human prostate itself but could maintain smooth muscle cells in a contractile state, and its activators in the prostate remain to be identified.
Our findings obtained with WPMY‐1 cells suggest that SR7826 and LIMKi3 cause a breakdown of the actin cytoskeleton in prostate smooth muscle cells. This resembles previous findings, which demonstrated a role for LIMK in the regulation of actin organization in other cell types (Yang et al., 1998; Bernard, 2007; San Martin et al., 2008). Actin organization, that is, polymerization of actin and its organization to filaments, is an ultimate prerequisite for smooth muscle contraction, besides MLC phosphorylation (Hennenberg et al., 2014). A breakdown of this organization (as observed in WPMY‐1 cells) reduces smooth muscle contractility and may account for the inhibition of contraction in prostate tissues by SR7826 and LIMKi3. In contrast, MLC phosphorylation was not affected by SR7826 or LIMKi3. Thus, it appears unlikely that inhibition of contraction by SR7826 and LIMKi3 was based on a reduction in MLC phosphorylation. Moreover, the possibility that LIMK works in concert with Rho kinase to induce prostate smooth muscle contraction may be excluded, as our findings using a combination of SR7826 with Y27632 suggest separate actions of both kinases. In fact, activation of Rho kinase is crucial in agonist‐induced smooth muscle contraction in the prostate and other organs (Uehata et al., 1997; Hennenberg et al., 2008; 2014) but works obviously independently from LIMK in the prostate. At present, we cannot explain why SR7826 and LIMKi3 inhibited α1‐adrenoceptor and TXA2‐induced, but not endothelin‐induced contractions, although all should depend on actin organization. This shows that prostate smooth muscle contraction is still incompletely understood and includes different mechanisms underlying regulation of contractions by α1‐adrenoceptors, endothelin and Tx.
In CCK‐8 assays performed in WPMY‐1 cells, 1 μM of SR7826 and LIMKi3 induced only moderate effects, while effects on viability became clearly obvious at 5 μM. Therefore, we used this concentration in following experiments performed in WPMY‐1 cells. Our findings did not suggest an unspecific inhibition of Rho kinase or STK16 in WPMY‐1 cells by these conditions, as phosphorylation of their substrates MYPT1 and 4E‐BP1 remained unaffected by 5 μM of SR7826 and LIMKi3. Notably, 5 μM was sufficient to induce a breakdown of actin organization and induced low decreases in viability. Again, these findings are in line with previous studies, reporting about reduced viability due to LIMK deficiency or following application of small molecule LIMK inhibitors in different cell types (Park et al., 2014; Petrilli et al., 2014).
We observed that small molecule LIMK inhibitors inhibited neurogenic contractions, which are assumed to be mediated by release of endogenous neurotransmitters including noradrenaline and subsequent activation of postsynaptic α1‐adrenoceptors on smooth muscle cells in tissue preparations. This inhibition ranged around 50%, what may resemble or even exceed inhibition of EFS‐induced smooth muscle contractions by α1‐blockers in previous studies using human prostate tissues (Oger et al., 2009; 2010; Buono et al., 2014). Besides inhibition of neurogenic contractions, we observed inhibition of agonist‐induced contractions induced by the TXA2 analogue U46619. As SR7826 and LIMKi3 inhibited TXA2 in parallel with α1‐adrenoceptor or neurogenic contractions, it may be hypothesized that these inhibitors induce urodynamic effects in vivo, which may even exceed the effects of α1‐blockers. However, any effect in vivo still needs to be confirmed in further studies, including animal models with experimentally‐induced LUTS. To the best of our knowledge, both compounds have not been tested in vivo to date. Probably, side effects will limit clinical studies or the use of LIMK inhibitors in patients, as the tolerability of kinase inhibitors is usually low.
LUTS suggestive of BPH is routinely treated by α1‐blockers, because (i) they inhibit α1‐adrenoceptor‐induced prostate smooth muscle contraction and (ii) prostate smooth muscle contraction contributes to urethral obstruction and LUTS (Oelke et al., 2013; Hennenberg et al., 2014). However, their effects are limited: reductions in International Prostate Symptom Scores (IPSS) or improvements in urinary flow (Qmax) do not exceed 50%, while effects of placebos range up to 30% (Kortmann et al., 2003; Madersbacher et al., 2007; Oelke et al., 2013; Hennenberg et al., 2014). In 30–35% of patients, decreases in IPSS will be restricted to 25% or less, so that α1‐blockers are inadequately effective in up to 69% of patients (Chapple et al., 2011; Fullhase et al., 2013; Matsukawa et al., 2013; Lee et al., 2015). These restrictions may be attributed to non‐adrenergic mediators, which contribute to prostate smooth muscle tone in parallel with α1‐adrenoceptors, but which are not affected by α1‐blockers (Strittmatter et al., 2011; Hennenberg et al., 2013; 2017). Therefore, it would make sense that future therapies address adrenergic and non‐adrenergic contractions simultaneously, to obtain higher efficacy in medical LUTS treatment.
The high popularity of α1‐blockers may be reflected by worldwide annual expenses ranging around 3.5 billion USD being spent for LUTS treatment with α1‐blockers (Ventura et al., 2011). This is contrasted by clinical problems arising from insufficient efficacy. Thus, disappointing results may contribute to high discontinuation rates: 12 months after being first prescribed α1‐blockers, only 35% of patients still continue with their medication (Cindolo et al., 2015). This low adherence to medical therapy results in hospitalization and in high numbers of surgery due to BPH (Cindolo et al., 2015). Therefore, novel options with higher efficacy may be appreciated for medical treatment of male LUTS. The development of such options requires a precise understanding of prostate smooth muscle contraction and the identification of new targets for possible pharmacological interventions.
Based on our findings, it is possible that LIMKs promote smooth muscle contraction in the human prostate by phosphorylation of cofilin and subsequent actin organization in favour of the contractile state. Thereby, LIMKs could be involved in urethral obstruction and bladder outlet obstruction in BPH. These conclusions may be obvious but need to be considered with caution, as unspecific inhibition may still not be completely ruled out, in particular for STK16 (MPSK1). It is hypothesized that SR7826, LIMKi3 or other LIMK inhibitors show urodynamic effects in vivo or improve LUTS. To date, tolerability studies for SR7826 or LIMKi3 are lacking, and side effects are expected and may limit their application in clinical studies, common for kinase inhibitors. Nevertheless, our findings shed new light on the pharmacology and intracellular mechanisms of prostate smooth muscle contraction and may allow new models to demonstrate its regulation and the underlying mechanisms.
Conclusions
Small molecule LIMK inhibitors inhibit smooth muscle contraction in the human prostate. Our findings suggest that LIMKs promote prostate smooth muscle contraction by phosphorylation of cofilin and subsequent actin organization. Therefore, LIMKs may have a role in urethral obstruction and bladder outlet obstruction in BPH patients. A similar role in the regulation of smooth muscle contraction in other organs is also possible.
Author contributions
Q.Y. contributed to the acquisition, analysis and interpretation of data for the work and drafting of the manuscript. C.G. and C.G.S. contributed to the conception of the work and acquisition of data for the work and critically revised the manuscript for important intellectual content. Y.W. contributed to the acquisition and analysis of data for the work and critically revised the manuscript for important intellectual content. A.H., C.M.S. and F.S. contributed to the acquisition of data for the work and critically revised the manuscript for important intellectual content. B.R. and X.W. contributed to the acquisition and analyses of data for the work and critically revised the manuscript for important intellectual content. M.H. contributed to the conception and design of the work, acquisition, analysis and interpretation of data and drafting of the manuscript. All authors approved the final version of the manuscript and provided agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
We thank Professor Dr E. Noessner and her co‐workers (Institute of Molecular Immunology, Helmholtz Center, Munich) for their support with immunofluorescence microscopy. We thank Professor Dr T. Kirchner (Institute of Pathology, Ludwig Maximilian University of Munich) and his co‐workers Dr V. Mai and Dr C. Faber for providing the tissue samples from prostates. This work was supported by grants from the Deutsche Forschungsgemeinschaft (grants HE 5825/2‐1 and GR 3333/2‐1) and the Friedrich‐Baur‐Stiftung (grant 71/16).
Yu, Q. , Gratzke, C. , Wang, Y. , Herlemann, A. , Sterr, C. M. , Rutz, B. , Ciotkowska, A. , Wang, X. , Strittmatter, F. , Stief, C. G. , and Hennenberg, M. (2018) Inhibition of human prostate smooth muscle contraction by the LIM kinase inhibitors, SR7826 and LIMKi3. British Journal of Pharmacology, 175: 2077–2096. doi: 10.1111/bph.14201.
References
- Alcaraz A, Hammerer P, Tubaro A, Schroder FH, Castro R (2009). Is there evidence of a relationship between benign prostatic hyperplasia and prostate cancer? Findings of a literature review. Eur Urol 55: 864–873. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al (2015). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174 (Suppl 1): S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174 (Suppl 1): S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Overview. Br J Pharmacol 174 (Suppl 1): S1–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O et al (1998). Regulation of actin dynamics through phosphorylation of cofilin by LIM‐kinase. Nature 393: 805–809. [DOI] [PubMed] [Google Scholar]
- Bernard O (2007). Lim kinases, regulators of actin dynamics. Int J Biochem Cell Biol 39: 1071–1076. [DOI] [PubMed] [Google Scholar]
- Buono R, Briganti A, Freschi M, Villa L, La Croce G, Moschini M et al (2014). Silodosin and tadalafil have synergistic inhibitory effects on nerve‐mediated contractions of human and rat isolated prostates. Eur J Pharmacol 744: 42–51. [DOI] [PubMed] [Google Scholar]
- Chapple CR, Montorsi F, Tammela TL, Wirth M, Koldewijn E, Fernandez Fernandez E et al (2011). Silodosin therapy for lower urinary tract symptoms in men with suspected benign prostatic hyperplasia: results of an international, randomized, double‐blind, placebo‐ and active‐controlled clinical trial performed in Europe. Eur Urol 59: 342–352. [DOI] [PubMed] [Google Scholar]
- Cindolo L, Pirozzi L, Fanizza C, Romero M, Tubaro A, Autorino R et al (2015). Drug adherence and clinical outcomes for patients under pharmacological therapy for lower urinary tract symptoms related to benign prostatic hyperplasia: population‐based cohort study. Eur Urol 68: 418–425. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai YP, Bongalon S, Mutafova‐Yambolieva VN, & Yamboliev IA (2008). Distinct effects of contraction agonists on the phosphorylation state of cofilin in pulmonary artery smooth muscle. Adv Pharmacol Sci 2008: 362741, 1, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Wang X, Wu J, Qian Q (2010). Phenylephrine induces elevated RhoA activation and smooth muscle alpha‐actin expression in Pkd2+/− vascular smooth muscle cells. Hypertens Res 33: 37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DC, Sanders LC, Bokoch GM, Gill GN (1999). Activation of LIM‐kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol 1: 253–259. [DOI] [PubMed] [Google Scholar]
- Fullhase C, Chapple C, Cornu JN, De Nunzio C, Gratzke C, Kaplan SA et al (2013). Systematic review of combination drug therapy for non‐neurogenic male lower urinary tract symptoms. Eur Urol 64: 228–243. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennenberg M, Acevedo A, Wiemer N, Kan A, Tamalunas A, Wang Y, et al (2017). Non‐adrenergic, tamsulosin‐insensitive smooth muscle contraction is sufficient to replace alpha1‐adrenergic tension in the human prostate. Prostate 77: 697–707. [DOI] [PubMed] [Google Scholar]
- Hennenberg M, Miljak M, Herrmann D, Strittmatter F, Walther S, Rutz B et al (2013). The receptor antagonist picotamide inhibits adrenergic and thromboxane‐induced contraction of hyperplastic human prostate smooth muscle. Am J Physiol Renal Physiol 305: F1383–F1390. [DOI] [PubMed] [Google Scholar]
- Hennenberg M, Stief CG, Gratzke C (2014). Prostatic alpha1‐adrenoceptors: new concepts of function, regulation, and intracellular signaling. NeurourolUrodyn 33: 1074–1085. [DOI] [PubMed] [Google Scholar]
- Hennenberg M, Trebicka J, Sauerbruch T, Heller J (2008). Mechanisms of extrahepatic vasodilation in portal hypertension. Gut 57: 1300–1314. [DOI] [PubMed] [Google Scholar]
- Kapur R, Shi J, Ghosh J, Munugalavadla V, Sims E, Martin H et al (2016). ROCK1 via LIM kinase regulates growth, maturation and actin based functions in mast cells. Oncotarget 7: 16936–16947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T (2008). Overview of receptor interactions of agonists and antagonists. Curr Protoc Pharmacol Chapter 4: Unit 4 1. [DOI] [PubMed] [Google Scholar]
- Khromov A, Choudhury N, Stevenson AS, Somlyo AV, Eto M (2009). Phosphorylation‐dependent autoinhibition of myosin light chain phosphatase accounts for Ca2+ sensitization force of smooth muscle contraction. J Biol Chem 284: 21569–21579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M et al (1996). Regulation of myosin phosphatase by Rho and Rho‐associated kinase (Rho‐kinase). Science 273: 245–248. [DOI] [PubMed] [Google Scholar]
- Kortmann BB, Floratos DL, Kiemeney LA, Wijkstra H, de la Rosette JJ (2003). Urodynamic effects of alpha‐adrenoceptor blockers: a review of clinical trials. Urology 62: 1–9. [DOI] [PubMed] [Google Scholar]
- Kreidler SM, Muller KE, Grunwald GK, Ringham BM, Coker‐Dukowitz ZT, Sakhadeo UR et al (2013). GLIMMPSE: online power computation for linear models with and without a baseline covariate. J Stat Softw 54: i10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunit T, Gratzke C, Schreiber A, Strittmatter F, Waidelich R, Rutz B et al (2014). Inhibition of smooth muscle force generation by focal adhesion kinase inhibitors in the hyperplastic human prostate. Am J Physiol Renal Physiol 307: F823–F832. [DOI] [PubMed] [Google Scholar]
- Lee HN, Lee KS, Kim JC, Chung BH, Kim CS, Lee JG et al (2015). Rate and associated factors of solifenacin add‐on after tamsulosin monotherapy in men with voiding and storage lower urinary tract symptoms. Int J Clin Pract 69: 444–453. [DOI] [PubMed] [Google Scholar]
- Li Y, Hu F, Chen HJ, Du YJ, Xie ZY, Zhang Y et al (2014). LIMK‐dependent actin polymerization in primary sensory neurons promotes the development of inflammatory heat hyperalgesia in rats. Sci Signal 7: ra61. [DOI] [PubMed] [Google Scholar]
- Liu F, Wang J, Yang X, Li B, Wu H, Qi S et al (2016). Discovery of a highly selective STK16 kinase inhibitor. ACS Chem Biol 11: 1537–1543. [DOI] [PubMed] [Google Scholar]
- Liu J, Yang X, Li B, Wang J, Wang W, Liu J et al (2017). STK16 regulates actin dynamics to control Golgi organization and cell cycle. Sci Rep 7: 44607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madersbacher S, Marszalek M, Lackner J, Berger P, Schatzl G (2007). The long‐term outcome of medical therapy for BPH. Eur Urol 51: 1522–1533. [DOI] [PubMed] [Google Scholar]
- Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A et al (1999). Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM‐kinase. Science 285: 895–898. [DOI] [PubMed] [Google Scholar]
- Mardilovich K, Baugh M, Crighton D, Kowalczyk D, Gabrielsen M, Munro J et al (2015a). LIM kinase inhibitors disrupt mitotic microtubule organization and impair tumor cell proliferation. Oncotarget 6: 38469–38486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardilovich K, Gabrielsen M, McGarry L, Orange C, Patel R, Shanks E et al (2015b). Elevated LIM kinase 1 in nonmetastatic prostate cancer reflects its role in facilitating androgen receptor nuclear translocation. Mol Cancer Ther 14: 246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsukawa Y, Gotoh M, Komatsu T, Funahashi Y, Sassa N, Hattori R (2013). Efficacy of silodosin for relieving benign prostatic obstruction: prospective pressure flow study. J Urol 189: S117–S121. [DOI] [PubMed] [Google Scholar]
- Misra UK, Deedwania R, Pizzo SV (2005). Binding of activated alpha2‐macroglobulin to its cell surface receptor GRP78 in 1‐LN prostate cancer cells regulates PAK‐2‐dependent activation of LIMK. J Biol Chem 280: 26278–26286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouneimne G, DesMarais V, Sidani M, Scemes E, Wang W, Song X et al (2006). Spatial and temporal control of cofilin activity is required for directional sensing during chemotaxis. Curr Biol 16: 2193–2205. [DOI] [PubMed] [Google Scholar]
- Oelke M, Bachmann A, Descazeaud A, Emberton M, Gravas S, Michel MC et al (2013). EAU guidelines on the treatment and follow‐up of non‐neurogenic male lower urinary tract symptoms including benign prostatic obstruction. Eur Urol 64: 118–140. [DOI] [PubMed] [Google Scholar]
- Oger S, Behr‐Roussel D, Gorny D, Lebret T, Denoux Y, Alexandre L et al (2010). Combination of alfuzosin and tadalafil exerts an additive relaxant effect on human detrusor and prostatic tissues in vitro. Eur Urol 57: 699–707. [DOI] [PubMed] [Google Scholar]
- Oger S, Behr‐Roussel D, Gorny D, Lecoz O, Lebret T, Denoux Y et al (2009). Combination of doxazosin and sildenafil exerts an additive relaxing effect compared with each compound alone on human cavernosal and prostatic tissue. J Sex Med 6: 836–847. [DOI] [PubMed] [Google Scholar]
- Ohashi K, Nagata K, Maekawa M, Ishizaki T, Narumiya S, Mizuno K (2000). Rho‐associated kinase ROCK activates LIM‐kinase 1 by phosphorylation at threonine 508 within the activation loop. J Biol Chem 275: 3577–3582. [DOI] [PubMed] [Google Scholar]
- Orsted DD, Bojesen SE (2013). The link between benign prostatic hyperplasia and prostate cancer. Nat Rev Urol 10: 49–54. [DOI] [PubMed] [Google Scholar]
- Park JB, Agnihotri S, Golbourn B, Bertrand KC, Luck A, Sabha N et al (2014). Transcriptional profiling of GBM invasion genes identifies effective inhibitors of the LIM kinase–cofilin pathway. Oncotarget 5: 9382–9395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrilli A, Copik A, Posadas M, Chang LS, Welling DB, Giovannini M et al (2014). LIM domain kinases as potential therapeutic targets for neurofibromatosis type 2. Oncogene 33: 3571–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradidarcheep W, Wallner C, Dabhoiwala NF, Lamers WH (2011). Anatomy and histology of the lower urinary tract. Handb Exp Pharmacol : 117–148. [DOI] [PubMed] [Google Scholar]
- Ross‐Macdonald P, de Silva H, Guo Q, Xiao H, Hung CY, Penhallow B et al (2008). Identification of a nonkinase target mediating cytotoxicity of novel kinase inhibitors. Mol Cancer Ther 7: 3490–3498. [DOI] [PubMed] [Google Scholar]
- San Martin A, Lee MY, Williams HC, Mizuno K, Lassegue B, Griendling KK (2008). Dual regulation of cofilin activity by LIM kinase and Slingshot‐1L phosphatase controls platelet‐derived growth factor‐induced migration of human aortic smooth muscle cells. Circ Res 102: 432–438. [DOI] [PubMed] [Google Scholar]
- Scott RW, Hooper S, Crighton D, Li A, Konig I, Munro J et al (2010). LIM kinases are required for invasive path generation by tumor and tumor‐associated stromal cells. J Cell Biol 191: 169–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikhibrahim Z, Lindstrot A, Ellinger J, Rogenhofer S, Buettner R, Perner S et al (2012). The peripheral zone of the prostate is more prone to tumor development than the transitional zone: is the ETS family the key? Mol Med Rep 5: 313–316. [DOI] [PubMed] [Google Scholar]
- Strittmatter F, Gratzke C, Weinhold P, Steib CJ, Hartmann AC, Schlenker B et al (2011). Thromboxane A2 induces contraction of human prostate smooth muscle by Rho kinase‐ and calmodulin‐dependent mechanisms. Eur J Pharmacol 650: 650–655. [DOI] [PubMed] [Google Scholar]
- Strittmatter F, Walther S, Gratzke C, Gottinger J, Beckmann C, Roosen A et al (2012). Inhibition of adrenergic human prostate smooth muscle contraction by the inhibitors of c‐Jun N‐terminal kinase, SP600125 and BI‐78D3. Br J Pharmacol 166: 1926–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinney DC (2004). Biochemical mechanisms of drug action: what does it take for success? Nat Rev Drug Discov 3: 801–808. [DOI] [PubMed] [Google Scholar]
- Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T et al (1997). Calcium sensitization of smooth muscle mediated by a Rho‐associated protein kinase in hypertension. Nature 389: 990–994. [DOI] [PubMed] [Google Scholar]
- Vauquelin G, Van Liefde I, Vanderheyden P (2002). Models and methods for studying insurmountable antagonism. Trends Pharmacol Sci 23: 514–518. [DOI] [PubMed] [Google Scholar]
- Ventura S, Oliver V, White CW, Xie JH, Haynes JM, Exintaris B (2011). Novel drug targets for the pharmacotherapy of benign prostatic hyperplasia (BPH). Br J Pharmacol 163: 891–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Gratzke C, Tamalunas A, Rutz B, Ciotkowska A, Strittmatter F et al (2016a). Smooth muscle contraction and growth of stromal cells in the human prostate are both inhibited by the Src family kinase inhibitors, AZM475271 and PP2. Br J Pharmacol 173: 3342–3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Gratzke C, Tamalunas A, Wiemer N, Ciotkowska A, Rutz B et al (2016b). P21‐activated kinase inhibitors FRAX486 and IPA3: inhibition of prostate stromal cell growth and effects on smooth muscle contraction in the human prostate. PLoS One 11: e0153312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kunit T, Ciotkowska A, Rutz B, Schreiber A, Strittmatter F et al (2015). Inhibition of prostate smooth muscle contraction and prostate stromal cell growth by the inhibitors of Rac, NSC23766 and EHT1864. Br J Pharmacol 172: 2905–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webber MM, Trakul N, Thraves PS, Bello‐DeOcampo D, Chu WW, Storto PD et al (1999). A human prostatic stromal myofibroblast cell line WPMY‐1: a model for stromal–epithelial interactions in prostatic neoplasia. Carcinogenesis 20: 1185–1192. [DOI] [PubMed] [Google Scholar]
- Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K et al (1998). Cofilin phosphorylation by LIM‐kinase 1 and its role in Rac‐mediated actin reorganization. Nature 393: 809–812. [DOI] [PubMed] [Google Scholar]
- Yin Y, Zheng K, Eid N, Howard S, Jeong JH, Yi F et al (2015). Bis‐aryl urea derivatives as potent and selective LIM kinase (Limk) inhibitors. J Med Chem 58: 1846–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]