

Abstract
Background and Purpose
Evidence suggests that exercise decreases nicotine withdrawal symptoms in humans; however, the mechanisms mediating this effect are unclear. We investigated, in a mouse model, the effect of exercise intensity during chronic nicotine exposure on nicotine withdrawal severity, binding of α4β2*, α7 nicotinic acetylcholine (nAChR), μ‐opioid (μ receptors) and D2 dopamine receptors and on brain‐derived neurotrophic factor (BDNF) and plasma corticosterone levels.
Experimental Approach
Male C57Bl/6J mice treated with nicotine (minipump, 24 mg·kg−1·day−1) or saline for 14 days underwent one of three concurrent exercise regimes: 24, 2 or 0 h·day−1 voluntary wheel running. Mecamylamine‐precipitated withdrawal symptoms were assessed on day 14. Quantitative autoradiography of α4β2*, α7 nAChRs, μ receptors and D2 receptor binding was performed in brain sections of these mice. Plasma corticosterone and brain BDNF levels were also measured.
Key Results
Nicotine‐treated mice undertaking 2 or 24 h·day−1 wheel running displayed a significant reduction in withdrawal symptom severity compared with the sedentary group. Wheel running induced a significant up‐regulation of α7 nAChR binding in the CA2/3 area of the hippocampus of nicotine‐treated mice. Neither exercise nor nicotine treatment affected μ or D2 receptor binding or BDNF levels. Nicotine withdrawal increased plasma corticosterone levels and α4β2* nAChR binding, irrespective of exercise regimen.
Conclusions and Implications
We demonstrated for the first time a profound effect of exercise on α7 nAChRs in nicotine‐dependent animals, irrespective of exercise intensity. These findings shed light onto the mechanism underlining the protective effect of exercise on the development of nicotine dependence.
Linked Articles
This article is part of a themed section on Nicotinic Acetylcholine Receptors. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.11/issuetoc
Abbreviations
- BDNF
brain‐derived neurotrophic factor
- CA1 or 2/3
regions of the hippocampus
- D2 receptor
dopamine D2 receptor
- DAMGO
D‐Ala2‐MePhe4‐Gly‐ol5 enkephalin
- μ receptor
μ‐opioid receptor
- nAChR
nicotinic acetylcholine receptor
- NSB
non‐specific binding
Introduction
More than 50% of attempts to quit smoking in the UK are not successful, which is thought to be at least partly due to the limited efficacy of the substitution pharmacotherapies currently available (Lifestyles Statistics Team, The Health and Social Care Information Centre, 2012). Exercise, however, has been shown to be of benefit as a non‐pharmacological aid for treating nicotine dependence. In particular, clinical and laboratory studies provide some evidence that exercise prior to smoking cessation and/or during smoking cessation can reduce the severity of nicotine withdrawal and craving following cessation of drug taking and might be protective against relapse (for reviews, see Taylor et al., 2007; Abrantes et al., 2009; Haasova et al., 2013). With regard to other drugs of abuse, in vivo animal studies showed that exercise can attenuate priming‐ and cue‐induced reinstatement of cocaine self‐administration (Smith et al., 2012; Thanos et al., 2013) and reduce morphine withdrawal symptoms (Balter and Dykstra, 2012 ; Miladi‐Gorji et al., 2012), further supporting the beneficial effect of exercise in reducing drug withdrawal symptoms and preventing relapse. Nonetheless, the frequency and intensity of exercise needed, as well as the neurobiological mechanisms underpinning these beneficial effects of exercise on reducing drug withdrawal and preventing relapse, remain unclear.
Since neuronal http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=76 (nAChRs) are the primary target of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2585 (Barik and Wonnacott, 2009), the reinforcing compound in cigarettes (Picciotto and Kenny, 2013), nAChRs are a central candidate system that may underlie the beneficial effect of exercise in reducing nicotine withdrawal symptoms. Previous studies have shown that mice lacking the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=465 or http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=472 subunits do not self‐administer nicotine (Picciotto et al., 1998; Marubio et al., 1999), while http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3990 (nAChR antagonist)‐precipitated withdrawal symptoms are absent in β2 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=468 knockout mice (Salas et al., 2007; Jackson et al., 2008), indicating α4β2* and α7 nAChRs as essential mediators of nicotine dependence and withdrawal. However, the effect of exercise on the nAChRs during chronic nicotine use and withdrawal has not yet been studied.
The endogenous opioid system, and more specifically the μ‐opioid system, has been implicated in the effects of exercise (e.g. de Oliveira et al., 2010), as well as during the different phases of nicotine addiction/withdrawal (see le Merrer et al., 2009). http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1643, an endogenous opioid ligand for the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=319 (μ receptor), is thought to mediate the mood‐enhancing effects of exercise via its actions on the μ receptor (de Oliveira et al., 2010), a concept referred to as ‘runner's high’. With regard to nicotine addiction, nicotine administration in mice lacking the μ receptor gene does not produce rewarding properties, and these mice have attenuated nicotine somatic withdrawal symptoms (Berrendero et al., 2002). Moreover, chronic nicotine administration results in higher expression of the μ receptor in the ventral tegmental area of the brain in mice (Walters et al., 2005), and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1638, an opioid receptor antagonist, triggers withdrawal symptoms in nicotine‐dependent rats (Malin et al., 1993) and in daily smokers (Krishnan‐Sarin et al., 1999). Although these findings clearly show a key role of the μ receptor system in the mediation of both the mood‐enhancing effects of exercise and the addiction‐related behavioural effects of nicotine administration and withdrawal, it is not clear if μ receptors are involved in the beneficial effects of exercise on nicotine dependence and abstinence. As a result, assessing if exercise in nicotine‐dependent individuals affects the regulation of μ receptors in the brain will shed light into the mechanisms underlining the beneficial effect of exercise on nicotine dependence and thus warrants further investigation.
Nicotine withdrawal is associated with a reduction of dopaminergic tone in the striatum (see Hadjiconstantinou et al., 2011), and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=215 are acutely down‐regulated during nicotine withdrawal in rats (Scott et al., 2007). Since there is clinical and preclinical evidence to suggest that exercise may be able to counteract the hypofunction of the dopaminergic system by specifically increasing brain D2 receptor levels in different psychiatric conditions (Vučcković et al., 2010; Fisher et al., 2013), we postulated that exercise during drug exposure might be exerting its beneficial effects against the development of nicotine dependence by up‐regulating striatal D2 receptors as well.
Another key mediator of drug addiction is http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4872 (BDNF). For example, BNDF levels are elevated in the ventral tegmental area and nucleus accumbens during withdrawal from chronic cocaine treatment (Tapia‐Arancibia et al., 2001) and in the hippocampus following alcohol cessation in ethanol‐dependent rats (Tapia‐Arancibia et al., 2001). Importantly, there is some evidence indicating that exercise decreases accumbal BDNF expression (Strickland et al., 2016), suggesting that exercise might be manifesting its beneficial properties against nicotine dependence by reducing elevated BDNF levels.
This study aimed to investigate the effect of three different intensities of exercise during chronic nicotine exposure on the development of physical dependence as measured by acute mecamylamine‐precipitated somatic withdrawal in mice and to assess the expression of α4β2* and α7 nAChRs, μ receptors, D2 receptors and BDNF in the brains of these mice. As there is some clinical evidence to suggest that exercise may be able to reduce nicotine withdrawal symptoms by attenuating the reduction in cortisol levels observed in temporarily abstinent smokers (Scerbo et al., 2010), we also measured plasma http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2869 levels in these mice.
Methods
Animal welfare and ethical statement
A total of 80 male C57Bl/6 mice (B&K Universal, UK) aged 8 weeks were individually housed in Macrolom Type II Long cages fitted with a 13‐cm‐diameter concentric free‐turning running wheel (ClockLab, Actimetrics, Wilmette, IL, USA) in light‐tight, sound‐attenuated cabinets. Mice were maintained in a 12:12 h light/dark cycle in an altered phase light protocol (lights off 11:00 h). Animals had ad libitum access to food and water throughout the experiment. Animal work procedures were carried out in accordance with the Animal (Scientific Procedures) Act 1986 Amendment Regulations (SI 2012/3039) under the project licence PPL 70/7203, approved on 17 February 2011, and animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). A mouse model was used in this study as it is commonly used to assess the neurobiological mechanisms underpinning nicotine addiction and exercise. The exact group size for each treatment/exercise group is provided for each experiment in Table 1. The experimenter who performed the minipump surgeries and injected the animals was aware of the pharmacological treatments and exercise regimen. Running wheel responses were registered by automated software, and the analyses of the behavioural and biochemical autoradiographic binding outcomes of the study were carried out by researchers who were blinded to the experimental/treatment groups. No animals were excluded from the analysis. However, three animals died following minipump implantation.
Table 1.
Statistical analyses
| Sample size (figure order) | Factorial effects | Interaction effects | |||||
|---|---|---|---|---|---|---|---|
| Overall effects for Figure 1 | |||||||
| Running wheel activity | – | Factor ‘treatment’ | Factor ‘time (days)’ | Factor ‘treatment’ × ‘time’ | |||
| Habituation | n = 14,12,13,13 | F [1,46] = 0.784; | P > 0.05* | F [6,276] = 1.350 | P > 0.05 | F [6,276] = 0.590 | P > 0.05 |
| Factor ‘exercise’ | Factor ‘exercise’ × ‘time’ | Factor ‘treatment’ × ‘exercise’ × ‘time’ | |||||
| F [1,46] = 304.1; | P < 0.05* | F [1,46] = 0.001 | P > 0.05 | F [6,276] = 0.515 | P > 0.05 | ||
| Treatment | n = 14,12,13,13 | Factor ‘treatment’ | Factor ‘time (days)’ | Factor ‘treatment’ × ‘time’ | |||
| F [1,47] = 0.010; | P > 0.05* | F [13,611] = 7.392 | P < 0.05* | F [13,611] = 0.246 | P > 0.05 | ||
| Factor ‘exercise’ | Factor ‘exercise’ × ‘time’ | Factor ‘treatment’ × ‘exercise’ × ‘time’ | |||||
| F [1,47] = 204.1; | P < 0.05* | F [13,611] = 0.708 | P > 0.05 | F [13,611] = 0.365 | P > 0.05 | ||
| Withdrawal score | n = 13,14,13,12,12,13 | H[2] = 8.940a | P < 0.05* | – | – | – | – |
| Overall effects for Figure 2 | |||||||
| a4β2* nAChR binding | – | Factor ‘treatment’ | Factor ‘exercise regimen’ | Factor ‘treatment’ × ‘exercise regimen’ | |||
| PrL | n = 5,5,6,5,5,6 | F [1,25] = 5.362 | P < 0.05* | F [2,25] = 0.575 | P > 0.05 | F [2,25] = 0.134 | P > 0.05 |
| CgCx | n = 5,5,6,5,5,6 | F [1,25] = 9.144 | P < 0.05* | F [2,25] = 0.738 | P > 0.05 | F [2,25] = 0.971 | P > 0.05 |
| AcbC | n = 5,5,6,5,5,6 | F [1,25] = 2.415 | P > 0.05 | F [2,25] = 1.017 | P > 0.05 | F [2,25] = 0.044 | P > 0.05 |
| AcbSh | n = 5,5,6,5,5,6 | F [1,25] = 5.199 | P < 0.05* | F [2,25] = 1.391 | P > 0.05 | F [2,25] = 0.168 | P > 0.05 |
| Th | n = 5,5,6,5,5,6 | F [1,25] = 1.101 | P > 0.05 | F [2,25] = 2.243 | P > 0.05 | F [2,25] = 0.027 | P > 0.05 |
| Hyp | n = 5,5,6,5,5,6 | F [1,25] = 21.10 | P < 0.05* | F [2,25] = 1.051 | P > 0.05 | F [2,25] = 0.007 | P > 0.05 |
| Hip | n = 5,5,6,5,5,6 | F [1,25] = 4.198 | P = 0.05 | F [2,25] = 0.158 | P > 0.05 | F [2,25] = 0.816 | P > 0.05 |
| SNc | n = 5,5,6,5,5,6 | F [1,25] = 11.51 | P < 0.05* | F [2,25] = 0.881 | P > 0.05 | F [2,25] = 0.202 | P > 0.05 |
| VTA | n = 5,5,6,5,5,6 | F [1,25] = 39.09 | P < 0.05* | F [2,25] = 0.546 | P > 0.05 | F [2,25] = 0.572 | P > 0.05 |
| Overall effects for Figure 3 | |||||||
| a7 nAChR binding | – | Factor ‘treatment’ | Factor ‘exercise regimen’ | Factor ‘treatment’ × ‘exercise regimen’ | |||
| CgCx | n = 5,5,6,5,5,6 | F [1,25] = 6.588 | P < 0.05* | F [2,25] = 4.357 | P < 0.05* | F [2,25] = 3.020 | P > 0.05 |
| DEn | n = 5,5,6,5,5,6 | F [1,25] = 7.059 | P < 0.05* | F [2,25] = 1.719 | P > 0.05 | F [2,25] = 0.629 | P > 0.05 |
| MtCx | n = 5,5,6,5,5,6 | F [1,25] = 1.618 | P > 0.05 | F [2,25] = 1.740 | P > 0.05 | F [2,25] = 5.178 | P < 0.05* |
| Cl | n = 5,5,6,5,5,6 | F [1,25] = 6.661 | P < 0.05* | F [2,25] = 0.141 | P > 0.05 | F [2,25] = 1.218 | P > 0.05 |
| CA1 | n = 5,5,6,5,5,6 | F [1,25] = 13.30 | P < 0.05* | F [2,25] = 8.299 | P < 0.05* | F [2,25] = 2.447 | P > 0.05 |
| CA2/3 | n = 5,5,6,5,5,6 | F [1,25] = 36.63 | P < 0.05* | F [2,25] = 21.80 | P < 0.05* | F [2,25] = 4.005 | P < 0.05* |
| Amy | n = 5,5,6,5,5,6 | F [1,25] = 20.61 | P < 0.05* | F [2,25] = 2.138 | P > 0.05 | F [2,25] = 0.022 | P > 0.05 |
| Hyp | n = 5,5,6,5,5,6 | F [1,25] = 8.764 | P < 0.05* | F [2,25] = 2.475 | P > 0.05 | F [2,25] = 0.480 | P > 0.05 |
| Overall effects for Figure 4 | |||||||
| μ receptor binding | – | Factor ‘treatment’ | Factor ‘exercise regimen’ | Factor ‘treatment’ × ‘exercise regimen’ | |||
| AcbC | n = 5,5,5,5,5,6 | F [1,24] = 0.0002 | P > 0.05 | F [2,24] = 0.190 | P > 0.05 | F [2,24] = 0.323 | P > 0.05 |
| AcbSh | n = 5,5,5,5,5,6 | F [1,24] = 1.094 | P > 0.05 | F [2,24] = 0.807 | P > 0.05 | F [2,24] = 0.660 | P > 0.05 |
| CPu | n = 5,5,5,5,5,6 | F [1,24] = 0.105 | P > 0.05 | F [2,24] = 0.892 | P > 0.05 | F [2,24] = 0.156 | P > 0.05 |
| DEn | n = 5,5,5,5,5,6 | F [1,24] = 0.013 | P > 0.05 | F [2,24] = 0.539 | P > 0.05 | F [2,24] = 0.820 | P > 0.05 |
| Hip | n = 5,5,5,5,5,6 | F [1,24] = 0.160 | P > 0.05 | F [2,24] = 0.422 | P > 0.05 | F [2,24] = 0.527 | P > 0.05 |
| MHb | n = 5,5,5,5,5,6 | F [1,24] = 0.647 | P > 0.05 | F [2,24] = 1.329 | P > 0.05 | F [2,24] = 0.019 | P > 0.05 |
| CL | n = 5,5,5,5,5,6 | F [1,24] = 0.953 | P > 0.05 | F [2,24] = 0.626 | P > 0.05 | F [2,24] = 0.129 | P > 0.05 |
| CM | n = 5,5,5,5,5,6 | F [1,24] = 0.057 | P > 0.05 | F [2,24] = 0.602 | P > 0.05 | F [2,24] = 0.719 | P > 0.05 |
| IMD | n = 5,5,5,5,5,6 | F [1,24] = 2.465 | P > 0.05 | F [2,24] = 1.891 | P > 0.05 | F [2,24] = 0.243 | P > 0.05 |
| Amy | n = 5,5,5,5,5,6 | F [1,24] = 0.251 | P > 0.05 | F [2,24] = 0.027 | P > 0.05 | F [2,24] = 0.075 | P > 0.05 |
| Hyp | n = 5,5,5,5,5,6 | F [1,24] = 0.070 | P > 0.05 | F [2,24] = 3.205 | P > 0.05 | F [2,24] = 1.229 | P > 0.05 |
| D2 receptor binding | – | Factor ‘treatment’ | Factor ‘exercise regimen’ | Factor ‘treatment’ × ‘exercise regimen’ | |||
| AcbC | n = 5,5,6,5,5,6 | F [1,25] = 0.097 | P > 0.05 | F [2,25] = 0.102 | P > 0.05 | F [2,25] = 1.414 | P > 0.05 |
| AcbSh | n = 5,5,5,5,5,6 | F [1,25] = 0.744 | P > 0.05 | F [2,25] = 0.320 | P > 0.05 | F [2,25] = 0.792 | P > 0.05 |
| Tu | n = 5,5,5,5,5,6 | F [1,25] = 0.328 | P > 0.05 | F [2,25] = 0.017 | P > 0.05 | F [2,25] = 0.229 | P > 0.05 |
| CPu | n = 5,5,5,5,5,6 | F [1,25] = 0.043 | P > 0.05 | F [2,25] = 0.066 | P > 0.05 | F [2,25] = 0.370 | P > 0.05 |
| Overall effects for Figure 5 | |||||||
| BDNF levels | – | Factor ‘treatment’ | Factor ‘exercise regimen’ | Factor ‘treatment’ × ‘exercise regimen’ | |||
| PFC | n = 8,7,9,8,7,7 | F [1,40] = 0.0003 | P > 0.05 | F [2,40] = 0.089 | P > 0.05 | F [2,40] = 0.811 | P > 0.05 |
| Str | n = 8,7,9,8,7,7 | F [1,40] = 0.089 | P > 0.05 | F [2,40] = 0.796 | P > 0.05 | F [2,40] = 0.088 | P > 0.05 |
| Hip | n = 8,7,9,8,7,7 | F [1,40] = 0.003 | P > 0.05 | F [2,40] = 0.335 | P > 0.05 | F [2,40] = 1.219 | P > 0.05 |
| Corticosterone levels | |||||||
| Plasma | n = 7,8,7,7,7,8 | F [1,34] = 9.757 | P < 0.05* | F [2,34] = 1.429 | P > 0.05 | F [2,25] = 0.514 | P > 0.05 |
| Overall effects for Supporting Information Figure S1 | |||||||
| a4β2* nAChR binding | – | Factor ‘treatment’ | Factor ‘exercise regimen’ | Factor ‘treatment’ × ‘exercise regimen’ | |||
| MHb (cytisine‐sensitive) | n = 5,5,5,5,5,6 | F [1,26] = 0.388 | P > 0.05 | F [2,26] = 1.848 | P > 0.05 | F [2,26] = 0.644 | P > 0.05 |
| MHb (cytisine‐resistant) | n = 5,5,5,5,5,6 | F [1,52] = 0.060 | P > 0.05 | F [2,52] = 0.0690 | P > 0.05 | F [2,52] = 0.960 | P > 0.05 |
| Overall effects for Supporting Information Table S1 | |||||||
| a4β2* nAChR binding | |||||||
| FrA | n = 5,5,6,5,5,6 | F [1,25] = 25.34 | P < 0.05* | F [2,25] = 0.063 | P > 0.05 | F [2,25] = 1.479 | P > 0.05 |
| MtCx | n = 5,5,6,5,5,6 | F [1,25] = 13.23 | P < 0.05* | F [2,25] = 1.304 | P > 0.05 | F [2,25] = 0.287 | P > 0.05 |
| SS | n = 5,5,6,5,5,6 | F [1,25] = 24.82 | P < 0.05* | F [2,25] = 0.589 | P > 0.05 | F [2,25] = 0.150 | P > 0.05 |
| Pir | n = 5,5,6,5,5,6 | F [1,25] = 29.42 | P < 0.05* | F [2,25] = 0.504 | P > 0.05 | F [2,25] = 0.185 | P > 0.05 |
| RS | n = 5,5,6,5,5,6 | F [1,25] = 6.512 | P < 0.05* | F [2,25] = 0.589 | P > 0.05 | F [2,25] = 0.778 | P > 0.05 |
| CPu | n = 5,5,6,5,5,6 | F [1,25] = 7.022 | P < 0.05* | F [2,25] = 0.624 | P > 0.05 | F [2,25] = 0.010 | P > 0.05 |
| MS | n = 5,5,6,5,5,6 | F [1,25] = 16.50 | P < 0.05* | F [2,25] = 0.709 | P > 0.05 | F [2,25] = 0.651 | P > 0.05 |
| VDB | n = 5,5,6,5,5,6 | F [1,25] = 15.93 | P < 0.05* | F [2,25] = 0.266 | P > 0.05 | F [2,25] = 0.650 | P > 0.05 |
| AuCx | n = 5,5,6,5,5,6 | F [1,25] = 24.89 | P < 0.05* | F [2,25] = 0.470 | P > 0.05 | F [2,25] = 0.236 | P > 0.05 |
| Overall effects for Supporting Information Table S2 | |||||||
| a7 nAChR binding | |||||||
| FrA | n = 5,5,6,5,5,6 | F [1,25] = 2.145 | P > 0.05 | F [2,25] = 0.547 | P > 0.05 | F [2,25] = 0.165 | P > 0.05 |
| CPu | n = 5,5,6,5,5,6 | F [1,25] = 1.564 | P > 0.05 | F [2,25] = 1.327 | P > 0.05 | F [2,25] = 0.501 | P > 0.05 |
| ZI | n = 5,5,6,5,5,6 | F [1,25] = 2.275 | P > 0.05 | F [2,25] = 1.264 | P > 0.05 | F [2,25] = 0.718 | P > 0.05 |
| VLG | n = 5,5,6,5,5,6 | F [1,25] = 3.710 | P > 0.05 | F [2,25] = 1.302 | P > 0.05 | F [2,25] = 2.215 | P > 0.05 |
Data were analysed with ANOVA unless otherwise indicated, with significance threshold of *P < 0.05.
Non‐parametric Kruskal–Wallis test, with significance threshold of *P < 0.05.
AcbC, nucleus accumbens core; AcbSh, nucleus accumbens shell; Amy, amygdala; AuCx, auditory cotex; CgCx, cingulate cortex; Cl, clostrum; CL, centrolateral thalamic nuclei; CM, centromedial thalamic nuclei; CPu, caudate‐putamen; DEn, dorsal endopiriform; FrA, frontal association; Hip, hippocampus; Hyp, hypothalamus; IMD, intermediate thalamic nuclei; MHb, medial habenula; MS, medial septum; MtCx, motor cortex; Pir, piriform cortex; PrL, prelimbic cortex; RS, retrosplenial cotex; SNc, substantia nigra pars compacta; SS, somatosensory cortex; Th, thalamus; Tu, olfactory tubercle; VDB, vertical limb of the diagonal band of Broca; VLG, ventral lateral geniculate; VTA, ventral tegmental area; ZI, zona incerta.
Assessment of running wheel activity
Mice were randomly assigned to one of three running wheel conditions and treated with either nicotine or saline: wheels unlocked 24 h·day−1 (n = 13), wheels unlocked 2 h·day−1 (n = 12–14) and wheels unlocked 0 h·day−1 (sedentary group, n = 12–13). In the 2 h·day−1 group, wheels were unlocked at 13–15 zeitgeber time, as this is the peak activity time for C57Bl/6J male mice on a 12:12 h light/dark cycle (Hasan et al., 2011). To determine profiles of average running wheel activity, the total number of wheel revolutions ·day−1 was converted into distance run for the 7 days of habituation and 14 days of treatment (nicotine or saline delivered via minipumps).
Minipump preparation and implantation
After habituating the mice in their running wheel condition for 7 days (see above), mice were treated with a chronic, 14 day, nicotine administration regimen, as previously described (Zanos et al., 2015), with minor modifications. Briefly, mice were surgically implanted with s.c. osmotic minipumps (Model 2002, Alzet®, Cupertino, CA, USA) containing saline or (−)‐nicotine hydrogen tartrate (24 mg·kg−1·day−1; Sigma Aldrich, Poole, UK) in sterile saline delivering a constant flow at a rate of 0.5 μL·h−1 for a period of 14 days. All nicotine concentrations are expressed as nicotine free base. Drug dose was selected to achieve blood nicotine levels comparable to the physiologically relevant concentrations measured in plasma of human smokers (see Matta et al., 2007). For minipump implantation, animals were anaesthetized with a volatile isoflurane anaesthetic (4.0%) (Isoflo, Abbott Laboratories Ltd., Kent, UK), which was vapourized in 95% O2/5% CO2 gas and delivered by a U400 anaesthetic unit (Univentor, Royem Scientific, Luton, UK) at a flow rate of approximately 450 mL·min−1 http://topics.sciencedirect.com/topics/page/Isoflurane/oxygen vapour mixture (3.5–4.5%; Isoflo, Abbott Laboratories Ltd, Maidenhead, Berkshire, UK). The animals were placed in the anaesthetic chamber for 1 min until the righting reflex was lost and were subsequently placed under a mask delivering anaesthesia throughout the surgery. Mice were injected with a non‐opioid analgesic (Metacam, 1.5 mg·kg−1, s.c.). A single incision along the midline of the back of each animal was made and osmotic mini‐pumps were placed in a parallel position to the spine. The flow operator was pointing away from the incision site. The incision was closed using 2–3 Michele clips (11 × 2.5 mm). Upon completion of the surgical procedure, mice were allowed to recover in heated‐recovery chambers until their righting reflex returned and were then placed back in their home cages.
Assessment of nicotine withdrawal severity
Fourteen days after minipump implantation, all animals were injected with mecamylamine (3 mg·kg−1, s.c.; Sigma Aldrich, Poole, UK) (Damaj et al., 2003) and immediately assessed for nicotine somatic withdrawal symptoms. Mice were videotaped and observed for 30 min in clear plastic activity cages for somatic withdrawal symptoms, according to the scale developed by Castañé et al. (2002). The following abstinence signs were evaluated during a 30 min period after mecamylamine injection: body tremor, ptosis, wet dog shakes, rearing, teeth chattering, paw tremor, scratching, genital licks, sniffing and piloerection. A global withdrawal score was calculated for each animal by giving each individual symptom a relative weight: 0.5 for each episode of wet dog shake, front paw tremor, sniffing, rearing and scratching; and 1 for appearance or 0 for non‐appearance within each 5 min bin for the presence of ptosis, genital licks, tremor, piloerection and teeth chattering. A composite of all these individual withdrawal symptoms was calculated to make up a global withdrawal symptom score. Scoring of behaviour was carried out by two independent observers blind to the treatment protocol.
Thirty minutes after the end of withdrawal assessment, mice were killed by being exposed to CO2 for 20 s, and trunk blood was collected, following decapitation, in EDTA‐containing Eppendorf tubes. Brains were excised and immediately frozen in isopentane (−20°C) and then stored at −80°C for autoradiography or BDNF measurements. Trunk blood was centrifuged (240× g at 4°C for 15 min) and the plasma stored at −20°C for subsequent analysis of corticosterone content.
Quantitative receptor autoradiography
Brains from some animals used for the behavioural studies (for exact number, see Table 1) were sectioned in a cryostat (Zeiss Hyrax C 25, Carl Zeiss AG, Oberkochen, Germany), at −21°C; 20 μm coronal sections were cut at 300 μm intervals, from rostral to caudal levels, and thaw‐mounted onto gelatine‐coated ice‐cold microscope slides and processed for autoradiography. Adjacent sections were cut for determination of total and non‐specific binding. Sections were stored at −20°C prior to radioligand binding.
Quantitative autoradiography was performed on brain sections for α4β2*, α7, μ receptors and D2 receptors using [125I]‐epibatidine (100 pM ± 20 nM http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5347), [125I]α‐bungarotoxin (α‐Bgtx; 3 nM), http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3815 (DAMGO; 4 nM) and [3H]‐raclopride (4 nM), respectively, according to established protocols (Metaxas et al., 2013; Georgiou et al., 2016; Wright et al., 2016), with minor modifications (see Supporting Information).
Plasma corticosterone and brain BDNF measurements
Plasma corticosterone levels
Plasma samples from trunk blood were assayed for corticosterone content using a rat/mouse [125I]‐corticosterone radioimmunoassay kit (MP Biomedicals, New York, NY, USA), according to the manufacturer's instructions.
Brain BDNF levels
Brains from some animals used for behavioural studies (for exact number, see Table 1) were defrosted in distilled water and the frontal cortex, striatum (i.e. nucleus accumbens and caudate putamen) and hippocampus dissected and weighed. The key role of BDNF in these brain regions has been extensively demonstrated in the drug addiction field (Li and Wolf, 2015). These brain regions were selected based on previous evidence for alterations of BDNF following chronic drug use (see McGinty et al., 2010). Each sample was homogenized by ultrasonification in lysis buffer containing 100 mM PIPES, 500 mM NaCl, 15 mM NaN3, 20% BSA, 2.5 mM EDTA, 0.2% TRITON X‐100 and EDTA‐free protease inhibitor cocktail (P8340, Sigma Aldrich, Poole, UK), pH 7 at room temperature. Total BDNF protein levels in homogenates were determined using the Promega BDNF Emax® ImmunoAssay System with acid treatment according to the manufacturer's instructions (Promega, Madison, WI, USA).
Data analysis and statistical procedures
All data are presented as mean ± SEM and were analysed using Statistica (STATsoft, Inc., version 10, Tulsa, OK, USA). ANOVAs were followed by Bonferroni post hoc tests where significance was achieved (P < 0.05). Withdrawal data were analysed using non‐parametric tests followed by post hoc tests where significance was P < 0.05. For details on statistical analyses, see Supporting Information. ANOVA results and precise sample sizes are detailed in Table 1. All the data and statistical analyses comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Materials
(−)‐Nicotine hydrogen tartrate, mecamylamine and cytisine were purchased from Sigma Aldrich, Poole, UK. BDNF kits and corticosterone kits were purchased from Promega (Madison, WI, USA) and MP Biomedicals (New York, NY, USA) respectively. [125I]‐epibatidine (specific activity 2200 Ci·mmol−1), [125I]‐α‐bungarotoxin (specific activity 108.8 Ci·mmol−1), [3H]‐DAMGO (specific activity 51.5 Ci·mmol−1) and [3H]‐raclopride (specific activity 60 Ci·mmol−1) used for autoradiographic binding experiments were purchased from PerkinElmer (Waltham, MA, USA).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b).
Results
Activity profiles of saline‐ and nicotine‐treated mice
As expected, no wheel‐running activity was recorded for the 0 h·day−1 wheel‐running group. Total activity per day was determined for animals in the 2 and 24 h·day−1 wheel‐running groups throughout the habituation and treatment periods in order to assess whether mice reached a steady state of activity (Figure 1A). Three‐way repeated measures ANOVA revealed a significant effect of exercise; the 24 h·day−1 group showed higher activity throughout the habituation and treatment phases of the experiment. There was no significant effect of nicotine treatment on wheel‐running activity (Figure 1A; see Table 1).
Figure 1.

Effect of wheel‐running exercise regimen on severity of nicotine withdrawal syndrome. Mice underwent one of three exercise regimes: 0, 2 or 24 h·day−1 running wheel access. Withdrawal was precipitated by mecamylamine (3 mg·kg−1, s.c.) following 14 days of either saline or nicotine (24 mg·kg−1·day−1) treatment via s.c. minipumps. (A) Total wheel‐running activity during habituation and treatment phases of the experiment. Wheel‐running activity was recorded and converted into distance run day−1 during the 7 day habituation and 14 day treatment periods. (B) Data for individual withdrawal symptoms were combined to give a total withdrawal measure. Data are presented as mean ± SEM. *P < 0.05. Precise group sizes are reported in Table 1.
Effect of different exercise regimes on severity of nicotine withdrawal syndrome
Individual withdrawal symptoms were analysed, and a composite total withdrawal factor was calculated (Figure 1B). Non‐parametric Kruskal–Wallis test revealed a significant effect of exercise on withdrawal in nicotine‐treated mice. Multiple Mann–Whitney U‐tests showed that precipitated withdrawal induced significantly higher withdrawal symptoms in nicotine‐treated mice in the 0 h·day−1 group only compared with the saline‐treated controls (U = 27.50, z = −2.75, P = 0.003, one‐tailed) but showed no difference between saline‐ and nicotine‐treated mice within the 2 or 24 h·day−1 groups. Moreover, mecamylamine administration induced higher severity of withdrawal symptoms in nicotine‐treated mice in the sedentary group compared with nicotine‐treated mice in the 2 or 24 h·day−1 wheel access groups (U = 26.50, z = 2.63, P = 0.004 and U = 32.00, z = 2.50, P = 0.006, one‐tailed, respectively; Dunn's corrected α‐level = 0.025). There was also no difference in severity of withdrawal between nicotine‐treated mice in 2 and 24 h·day−1 wheel access groups (Figure 1B; see Table 1). Interestingly, when different components of the withdrawal symptoms were analysed, mecamylamine‐precipitated withdrawal in nicotine‐treated sedentary animals induced an increase of paw tremors, sniffing and rearing which was absent in the groups exposed to exercise regimes (Supporting Information Figure S1).
Effect of exercise on α4β2* nAChR binding in nicotine‐treated mice
Levels of α4β2* nAChR binding were determined using cytisine‐sensitive [125I]‐epibatidine binding in brain regions of mecamylamine‐precipitated saline‐ or nicotine‐treated mice with 0, 2 and 24 h·day−1 running wheel access (Figure 2A,B; Supporting Information Table S1). Cytisine‐resistant binding was only present in the medial habenula for all groups (Supporting Information Figure S2), indicating a high level of non‐α4β2* (most likely http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=464 http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=474*) heteromeric nAChR binding in that region. A two‐way ANOVA found no significant nicotine (P > 0.05) or exercise (P > 0.05) effects within that region (Table 1). Two‐way ANOVA followed by Bonferroni post hoc in each region revealed significant, nicotine‐induced up‐regulation of cytisine‐sensitive [125I]‐epibatidine binding in the frontal association, as well as the prelimbic cortex, motor cortex, cingulate cortex, nucleus accumbens core and shell, hypothalamus, substantia nigra pars compacta and ventral tegmental area irrespective of exercise regimen (Figure 2B). α4β2* nAChR binding was also up‐regulated in the motor, somatosensory, piriform, retrosplenial and auditory cortices, as well as the medial septum, ventral limb of the diagonal band of Broca, olfactory tubercle and subiculum of nicotine‐treated animals compared with saline controls irrespective of exercise regimen (Supporting Information Table S1). No significant treatment effect was observed in the nucleus accumbens core, thalamus or the hippocampus. There were no exercise or interaction effects in any of the brain regions analysed (see Table 1 and Supporting Information Table S1).
Figure 2.
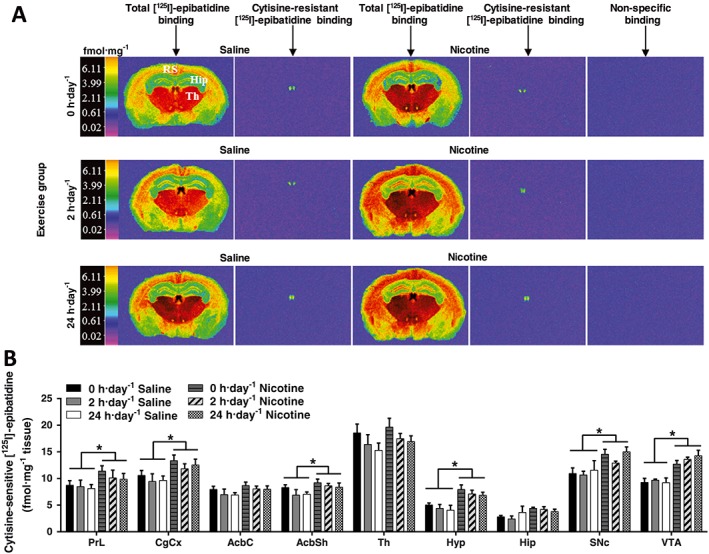
Effect of exercise on α4β2* nAChR binding in saline‐ and nicotine‐withdrawn mice. (A) Computer‐enhanced colour autoradiograms of total and cytisine‐resistant [125I]‐epibatidine binding in coronal brain sections of C57Bl/6 mice treated with saline or nicotine (24 mg·kg−1·day−1) via s.c. minipumps for 14 days, followed by mecamylamine‐precipitated (3 mg·kg−1) withdrawal. Mice underwent one of three exercise regimes: 0, 2 or 24 h·day−1 running wheel access in their home cage. Coronal brain sections are shown cut at the level of the dorsal hippocampus and thalamus (Bregma −1.46 mm). The calibration bar presents pseudo‐colour interpretation of black and white film images in fmol·mg−1 tissue equivalent. (B) Cytisine‐sensitive [125I]‐epibatidine binding in saline‐ and nicotine‐withdrawn mice undergoing different exercise regimes in cortical brain regions. Data are presented as mean ± SEM. *P < 0.05. Precise group sizes are reported in Table 1. AcbC, nucleus accumbens core; AcbSh, nucleus accumbens shell; CgCx, cingulate cortex; Hip, hippocampus; Hyp, hypothalamus; SNc, substantia nigra pars compacta, Th, thalamus; VTA, ventral tegmental area.
Effect of exercise on α7 nAChR binding in nicotine‐treated mice
α7 nAChR density was determined by [125I]‐α‐bungarotoxin binding in the brain of mecamylamine‐precipitated saline‐ or nicotine‐treated mice that were permitted 0, 2 and 24 h·day−1 running wheel access (Figure 3A,B; Supporting Information Table S2). Two‐way ANOVA in each brain region revealed a significant treatment effect in the cingulate cortex, endopiriform nucleus, motor cortex, clostrum, CA1 region of the hippocampus, amygdala and hypothalamus (Figure 3B). In the motor cortex, where a significant ANOVA interaction between treatment and exercise was identified (see Table 1), we demonstrated a significant decrease in α7 binding in the 24 h·day−1 exercise saline‐treated group compared with their sedentary controls (P < 0.05), which was absent in nicotine‐treated animals (Figure 3B). Moreover, saline‐treated mice that were permitted 24 h·day−1 running wheel access showed a significantly lower α7 binding compared to nicotine‐treated mice which were permitted the same exercise schedule (Figure 3B).
Figure 3.
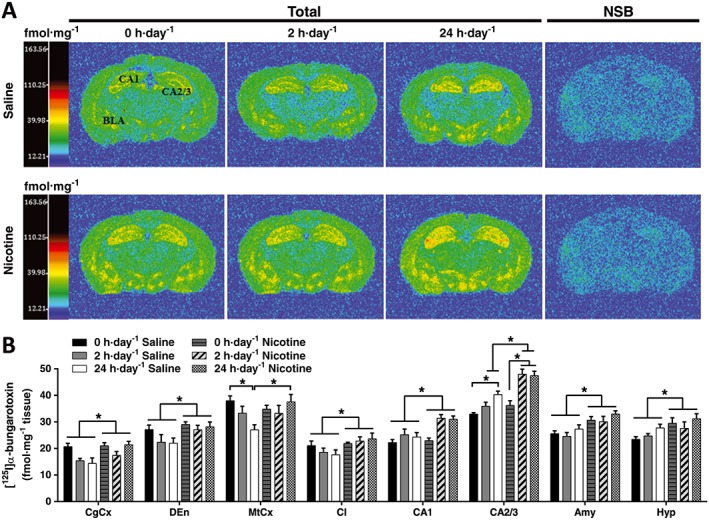
Effect of exercise on α7nAChR binding in saline‐ and nicotine‐withdrawn mice. (A) Computer‐enhanced colour autoradiograms of total [125I]‐α‐bungarotoxin and NSB in coronal brain sections of C57Bl/6 mice treated with chronic saline or nicotine via s.c. minipumps, followed by mecamylamine‐precipitated (3 mg·kg−1) withdrawal. Mice underwent one of three exercise regimes: 0, 2 or 24 h·day−1 running wheel access in their home cage. Coronal brain sections are shown cut at the level of the dorsal hippocampus and thalamus (Bregma −1.46 mm). The calibration bar presents pseudo‐colour interpretation of black and white film images in fmol·mg−1 tissue equivalent. (B) Quantitative [125I]‐α‐bungarotoxin binding in saline‐ and nicotine‐withdrawn mice undergoing different exercise regimes. Data are presented as mean ± SEM. *P < 0.05. Precise group sizes are reported in Table 1. Amy, amygdala; CgCx, cingulate cortex; CA2/3, CA2 and CA3 layers of the hippocampus; Cl, claustrum; DEn, dorsal endopiriform; Hyp, hypothalamus; MtCx, motor cortex; NSB, non‐specific binding.
Two‐way ANOVA revealed a significant exercise effect (P < 0.05), a significant treatment effect (P < 0.05) and a significant exercise × treatment interaction effect (P < 0.05) in the CA2/3, clearly demonstrating an interaction effect of nicotine and exercise on α7 nAChR up‐regulation in the CA2/3. Nicotine treatment elicited higher levels of α7 binding in the CA2/3 hippocampal area of mice exposed to 2 or 24 h·day−1 running wheel access (P < 0.05), compared to nicotine‐treated sedentary animals and compared to their saline, exercise‐matching controls. Mice with 24 h·day−1 access to a running‐wheel also displayed higher levels of α7 nAChR binding in the CA2/3 of saline‐treated mice compared with mice in the 0 h·day−1 saline‐treated group (Figure 3B; Table 1).
Effect of exercise on μ receptor binding in nicotine‐treated mice
Binding of the μ receptor was determined by [3H]‐DAMGO binding in brain regions of mecamylamine‐precipitated saline‐ or nicotine‐treated mice permitted 0, 2 and 24 h·day−1 running wheel access (Figure 4A). Two‐way ANOVA for each brain region did not reveal any effect of treatment or exercise, nor interactions between these factors (Figure 4A; Table 1).
Figure 4.
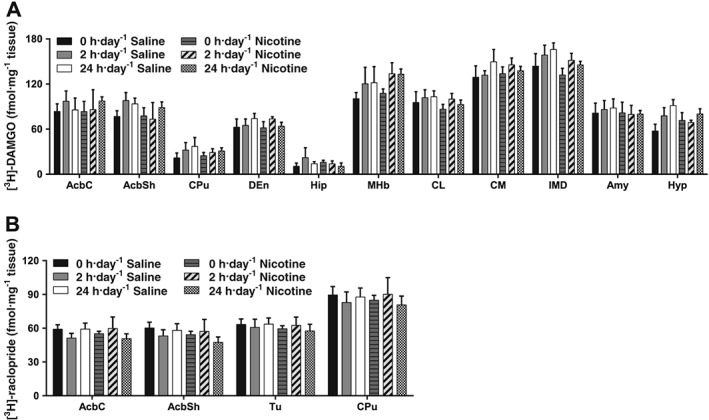
Effect of exercise on μ and D2 receptor binding in saline‐ and nicotine‐withdrawn mice. (A) [3H]‐DAMGO binding in non‐cortical regions of saline‐ and nicotine‐withdrawn mice undergoing different exercise regimes: 0, 2 or 24 h·day−1 running wheel access in their home cage. (B) [3H]‐raclopride binding of saline‐ and nicotine‐withdrawn mice undergoing different exercise regimes: 0, 2 or 24 h·day−1 running wheel access in their home cage. Data are presented as mean ± SEM. Precise group sizes are reported in Table 1. AcbC, nucleus accumbens core; AcbSh, nucleus accumbens shell; Amy, amygdala; CL, centrolateral thalamic nuclei; CM, centromedial thalamic nuclei; CPu, caudate putamen; DEn, dorsal endopiriform; Hip, hippocampus; Hyp, hypothalamus; IMD, intermediate thalamic nuclei; MHb, medial habenula; Tu, tubercle.
Effect of exercise on D2 receptor binding in nicotine‐treated mice
Binding of D2 receptors was determined by [3H]‐raclopride binding in brain regions of mecamylamine‐precipitated saline‐ or nicotine‐treated mice permitted 0, 2 and 24 h·day−1 running wheel access (Figure 4B). Two‐way ANOVA for each brain region revealed no significant changes in [3H]‐raclopride binding in any of the regions analysed (Figure 4B; Table 1).
Effect of exercise on brain BDNF in nicotine‐treated mice
The level of free BDNF in the prefrontal cortex, striatum and hippocampus of mecamylamine‐precipitated saline‐ or nicotine‐treated permitted 0, 2 and 24 h·day−1 running wheel access was determined using an ELISA. Two‐way ANOVA for each brain region showed no significant changes in any of the regions analysed (Figure 5A; Table 1).
Figure 5.

Effect of exercise on brain BDNF and plasma corticosterone levels in saline‐ and nicotine‐withdrawn mice. (A) BDNF levels in saline‐ and nicotine‐withdrawn mice undergoing different exercise regimes. Total BDNF levels from acid‐withdrawn samples were determined using an enzyme‐linked radioimmunoassay for nicotine‐ and saline‐withdrawn mice undergoing 0, 2 or 24 h·day−1 running wheel access. (B) Plasma corticosterone levels in saline‐ and nicotine‐withdrawn mice undergoing different exercise regimes. Plasma corticosterone content was determined using a 125I radioimmunoassay for nicotine‐ and saline‐withdrawn mice undergoing 0, 2 or 24 h·day−1. Data are presented as mean ± SEM. *P < 0.05. Precise group sizes are reported in Table 1. Hip, hippocampus; PFC, prefrontal cortex; Str, striatum.
Effect of exercise on plasma corticosterone in nicotine‐treated mice
Plasma corticosterone levels were determined by radioimmunoassay of mecamylamine‐precipitated saline‐ or nicotine‐treated mice permitted 0, 2 and 24 h·day−1 running wheel access (Figure 5B). Two‐way ANOVA revealed a significant increase of plasma corticosterone levels induced by nicotine treatment (treatment effect; Table 1) irrespective of exercise regimen. No effects of exercise were found.
Discussion
The present study highlights the beneficial effect of exercise during nicotine exposure in markedly reducing the severity of nicotine somatic withdrawal symptoms, an effect that is accompanied by an up‐regulation of the hippocampal α7 nAChRs. These findings support the protective effect of exercise preceding smoking cessation against the development of physical dependence, which may aid smoking cessation by reducing withdrawal symptom severity. Moreover, we propose a novel mechanism of action of exercise involving hippocampal α7 nAChRs.
Two hours a day access to a running wheel was equally effective in attenuating nicotine withdrawal symptoms as continuous 24 h·day−1 access. This is consistent with human clinical studies showing that just 10 min of moderate intensity exercise during smoking cessation is sufficient to reduce cigarette craving, withdrawal symptoms and cue‐induced cravings (Ussher et al., 2001; Taylor and Katomeri, 2007; Scerbo et al., 2010), supporting the translational validity of our mouse model. In rodent models, 2 h·day−1 access to running wheels during a period of abstinence from nicotine self‐administration decreased subsequent nicotine‐seeking in rats (Sanchez et al., 2013), demonstrating a beneficial effect of exercise on nicotine craving during abstinence. However, 2 h daily exercise failed to prevent cue‐induced reinstatement of nicotine‐seeking (Sanchez et al., 2013). This effect does not preclude the possibility that a more intense exercise schedule could have prevented reinstatement of nicotine‐seeking after extinction; however, this hypothesis needs to be investigated further. Here, we show that exercise exposure concurrent with nicotine administration is able to significantly reduce physical symptoms of withdrawal, which might underlie its ability to reduce nicotine craving during abstinence. It is important to note that, based on our results, it is not possible to ascertain if exercise during the withdrawal phase (irrespective of exercise during the nicotine exposure phase) would be sufficient to decrease withdrawal severity, as mice were not exposed to an exercise regime during the withdrawal phase. Studies assessing the effects of exercise during un‐precipitated nicotine withdrawal are warranted to address this question. Nonetheless, the data clearly suggest that exercise preceding smoking cessation might be able to increase the chances of abstinence from smoking by reducing acute physical withdrawal symptom severity. We also aimed to identify possible neurobiological mechanisms underlying this effect.
α4β2* nAChR up‐regulation was observed in most brain regions of mice exposed to chronic nicotine administration followed by mecamylamine‐precipitated withdrawal, irrespective of exercise regimen, demonstrating that exercise does not influence nicotine‐induced α4β2* nAChR up‐regulation. Up‐regulation of α4β2* nAChR following prolonged exposure to nicotine has been consistently shown in cigarette smokers (Breese et al., 1997; Cosgrove et al., 2009) and animal models of nicotine administration (e.g. Metaxas et al., 2013) and was associated with increased self‐administration of the drug (Hambsch et al., 2014). The up‐regulation is almost certainly due to chronic nicotine treatment not ‘mecamylamine‐precipitated withdrawal’ indicating that this α4β2* nAChR up‐regulation persists at least following acute precipitated withdrawal. The present results demonstrate that exercise does not influence nicotine‐induced α4β2* nAChR up‐regulation and thus is unlikely to be involved in the mechanism underlying the beneficial effect of exercise during nicotine exposure on nicotine withdrawal symptoms.
Moreover, we showed that α7 nAChRs are almost globally up‐regulated in most of the brain regions analysed in chronically nicotine‐treated mice undergoing mecamylamine‐precipitated withdrawal compared with saline‐treated controls. This finding is in line with previous studies showing that α7 nAChRs are up‐regulated in response to chronic nicotine exposure (Metaxas et al., 2013), indicating that this up‐regulation persists during acute precipitated withdrawal. Importantly, we demonstrated that hippocampal α7 nAChR binding is regulated by exercise since 2 or 24 h·day−1, but not 0 h·day−1 running wheel access induced a significant up‐regulation of α7 nAChR binding in the CA2/3 region of the hippocampus irrespective of nicotine/saline treatment schedule, suggesting the presence of a specific exercise‐induced effect on α7 nAChRs.
While exercise increases α7 nAChR binding in saline‐ and nicotine‐treated animals, the up‐regulation in the exercise plus nicotine group was found to be significantly higher than the saline plus exercise group, indicating an exercise × nicotine interaction on α7 nAChR up‐regulation in the CA2/3 of the hippocampus. This up‐regulation is concomitant with the complete abolition of somatic nicotine withdrawal symptoms in chronically nicotine treated mice exposed to exercise. Although, on the basis of the present data alone it would be presumptuous to assume any causal relationship between the protective effect of exercise on somatic withdrawal symptoms and α7 nAChR hippocampal up‐regulation, there is considerable evidence linking α7 nAChRs with at least some of the somatic symptoms of mecamylamine induced nicotine withdrawal. Salas et al. (2007) reported a decrease of shaking and scratching but not wet dog shakes and head nods in nicotine‐treated α7 knockout mice undergoing mecamylamine‐precipitated withdrawal. Interestingly, we also show in the present study an abolition of mecamylamine‐induced withdrawal paw shakes in nicotine‐treated mice exposed to exercise, an effect which was concomitant to a hippocampal CA2/3 α7 nAChR up‐regulation, suggesting that there may be a link between α7 nAChR up‐regulation and the protective effect of exercise on nicotine withdrawal symptoms. Moreover, the selective α7 nAChR agonist PNU282987 and the high α7/low α4β2* efficacy agonist varenicline have shown good efficacy in decreasing motivation to consume nicotine (Brunzell et al., 2010; Harmey et al., 2012) and in reducing withdrawal symptoms and craving (Rankin and Jones, 2011). It is important to note that α7 nAChRs have also been implicated in nicotine withdrawal‐associated anhedonia (Stoker et al., 2012), which is clinically relevant as it constitutes a motivational trigger to relapse. Even more intriguingly, recent data points specifically to the hippocampal α7 nAChRs as key modulators of negative affect (Mineur et al., 2018) which makes our hypothesis for a direct link between the protective effect of exercise on the negative consequences of nicotine abstinence and α7 hippocampal up‐regulation even more appealing. It is of course impossible to know based on the current study whether those up‐regulated receptors are desensitized or active and if these lead to downstream adaptations that may protect the development of physical dependence. Future studies should focus on the biological significance of this up‐regulation in order to test this hypothesis.
Interestingly, although α4β2* nAChRs have been recognized to play a key role in the cognitive impairment associated with nicotine withdrawal (Simmons and Gould, 2014), α7 nAChR activation has been shown to improve cognition, which is impaired during nicotine withdrawal in both mice and humans (Parrott et al., 1996; Dajas‐Bailador and Wonnacott, 2004; Wilkinson and Gould, 2013). Administration of varenicline, a partial α4β2* agonist and a full α7/α3β4 nAChR agonist, has been found to attenuate contextual fear conditioning during nicotine withdrawal (Raybuck et al., 2008). In addition, up‐regulation of α7 nAChRs has been associated with the pro‐cognitive effects of α7 agonists (Christensen et al., 2010). Therefore, given the key role of the hippocampus as a brain region involved in nicotine withdrawal mechanisms related to cognitive effects, future studies are warranted to directly investigate whether exercise exerts its beneficial effect in attenuating the cognitive deficits induced by nicotine withdrawal via an enhancement of hippocampal α7 nAChRs. It is important to note that many other nAChR subtypes have been implicated in somatic symptoms of withdrawal including α3, α5, α4 and α2 nAChR subtypes (see review by Jackson et al., 2015). Of particular interest is the emergence of the habenula‐interpenduncular nucleus and cytisine resistant α3β4* nAChRs in the manifestation of somatic withdrawal symptoms (Salas et al., 2009; Baldwin et al., 2011). Nonetheless, no nicotine nor exercise effect was observed in habenular cytisine resistant [125I]‐epibatidine binding sites which most likely represent α3β4* nAChRs (Supporting Information Figure S2). This finding plausibly suggests that exercise is unlikely to affect α3β4* nAChR density in the habenula and thus may not play a key role in the protective effect of exercise on nicotine dependence. Future research should be directed in the investigation of α3 and α5 in the effect of exercise in decreasing nicotine withdrawal symptoms.
BDNF, which has been shown to be increased in the hippocampus following exercise (Fuss et al., 2010) and chronic nicotine treatment (Czubak et al., 2009; Kenny, 2009; Aydin et al., 2012), has also been shown to specifically up‐regulate the intracellular pool of α7‐, but not β2*‐, containing nAChRs in cultured hippocampal neurons (Zhou et al., 2004; Massey et al., 2006). As a result, we postulated that the observed exercise‐induced region‐specific up‐regulation of α7 nAChR in the brain of nicotine‐treated mice might be mediated by an elevation of BDNF levels. However, in the present study, neither voluntary wheel‐running nor nicotine treatment had any effect on BDNF levels in the hippocampus, striatum or prefrontal cortex. This discrepancy with the literature may be due to different species, exercise regimens, treatment period and nicotine doses tested. For instance, while nicotine down‐regulates BDNF in the short‐term (2–24 h), there is a positive correlation between the amount of exercise and BDNF production (for review, see Erickson et al., 2011). The species differences between the published studies and our findings might also explain these discrepancies as environmental enrichment, including use of running wheels, increased hippocampal BDNF in rats (Ickes et al., 2000), but not in mice (Rueda et al., 2012). Nonetheless, our data do not support the hypothesis that exercise during nicotine exposure might affect nicotine withdrawal symptoms via a mechanism involving hippocampal, striatal or cortical BDNF up‐regulation nor that changes in BDNF levels in these brain regions are involved in the observed exercise‐induced up‐regulation of hippocampal α7 nAChRs following nicotine treatment.
Exercise has previously been shown to up‐regulate D2 receptors in humans (Fisher et al., 2013) and rodents (Vučcković et al., 2010); however, the present study found no change in D2 receptor binding following either exercise or nicotine withdrawal. The reason behind this discrepancy may lie in the fact that exercise‐induced D2 receptor up‐regulation was previously observed in a mouse model of Parkinsons' disease (Vučcković et al., 2010), indicating that exercise‐induced up‐regulation only occurs in compensation for loss of dopaminergic tone; this loss does not appear to happen in our mouse model of precipitated nicotine withdrawal. Nevertheless, our findings do not preclude the possibility that changes in the downstream D2 receptor signalling pathway, or functional changes at the receptor, might be involved in the mechanism underpinning the effects of exercise during nicotine exposure on acute somatic withdrawal symptoms and warrants further investigation.
In contrast to α7 nAChRs, no exercise or nicotine treatment interaction effects were observed in μ receptor binding in any of the regions analysed, suggesting that changes in this receptor system is unlikely to be part of the mechanism underpinning the beneficial effect of exercise during nicotine exposure on reducing nicotine withdrawal symptoms. This is somewhat surprising considering the plethora of evidence demonstrating a key role of the endogenous opioid system in the mechanism underlying the rewarding effect of exercise and nicotine (Berrendero et al., 2002; de Oliveira et al., 2010). Nonetheless, the findings from our study clearly suggest that any involvement of the opioid system is likely to be at the receptor signalling and/or the opioid peptide level rather than at the receptor expression level.
Although exercise has been suggested to influence nicotine withdrawal and craving via a possible modulation of the hypothalamic–pituitary adrenal axis activity (Scerbo et al., 2010), here, we show that exercise during nicotine exposure had no effect on corticosterone levels in saline‐ or nicotine‐treated mice, indicating that its protective effects on nicotine dependence are not mediated by its actions on the hypothalamic–pituitary adrenal axis level. However, consistent with the high levels of plasma cortisol observed in regular smokers (Field et al., 1994; al'Absi et al., 2003), we found an elevation of corticosterone levels in mecamylamine‐precipitated nicotine‐withdrawn mice irrespective of exercise regimen, supporting the translational validity of our mouse model of chronic nicotine administration.
Other than the opioid, dopaminergic and, nicotinic systems all investigated in this study, the endocannabinoid system may also play a key role in the mechanism underlining the effect of exercise in reducing nicotine withdrawal symptoms. Stimulation of the endogenous http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=56 receptors is a prerequisite for voluntary running in mice (Dubreucq et al., 2012), and enhancement of two endogenous endocanabinoids (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2364 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=729), by inhibition of their metabolic enzyme FAAH, was shown to reduce nicotine withdrawal symptoms in rats (Cippitelli et al., 2011). Investigation of the role of the endocannabinoid system in the beneficial effect of exercise in nicotine dependence would be an important and interesting concept for future investigation.
In conclusion, our results demonstrate the effectiveness of even a moderate amount of exercise during nicotine exposure in attenuating nicotine withdrawal symptoms and point towards the hippocampal α7 nAChR system as a potential mechanism underlining this effect. These findings may also have implications for the development of targeted interventions prior to smoking cessation, which may increase the chances of smoking cessation.
Author contributions
H.K., A.B., M.C., Y.C. and I.K. conceived and designed the experiments. H.K. and A.V.R. performed the experiments. H.K., P.G., P.Z. and A.B. analysed the data. A.V.R. and R.C. contributed reagents/materials/analysis tools. H.K., A.B., M.C., P.G. and P.Z. wrote the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Effect of wheel‐running exercise regimen on individual nicotine withdrawal symptoms. Mice underwent one of three exercise regimes: 0, 2 or 24 h·day−1 running‐wheel access. Withdrawal was precipitated by mecamylamine (3 mg·kg−1, s.c.) following 14 days of either saline or nicotine (24 mg·kg−1 day−1) treatment via subcutaneous minipumps. Abstinence signs were evaluated during a 30‐min period after mecamylamine injection. (A) Front paw tremors were assigned a value of 0.5 for each episode. (B) Sniffing was assigned a value of 0.5 for each episode. (C) Scratches were assigned a value of 0.5 for each episode. (D) Rearing was assigned a value of 0.5 for each episode. (E) Genital licks were assigned a value of 1 for appearance within each 5‐min bin. Data are presented as mean ± SEM. *P < 0.05. Precise group sizes are reported in Table 1.
Figure S2 Effect of exercise on cytisine‐resistant and cytisine‐sensitive epibatidine binding in saline‐ and nicotine‐withdrawn mice undergoing mecamylamine‐precipitated withdrawal. Cytisine‐resistant (A) and cytisine‐sensitive (B) [125I]‐epibatidine binding in the medial habenula of saline‐ and nicotine‐withdrawn mice undergoing different exercise regimes. Data are presented as mean ± SEM. *P < 0.05. Precise group sizes are reported in Table 1. Abbreviations: MHb, medial habenula.
Table S1 Effect of exercise on α4β2* nAChR binding in saline‐ and nicotine‐withdrawn mice.
Table S2 Effect of exercise on α7 nAChR binding in saline‐ and nicotine‐withdrawn mice.
Acknowledgements
This work was supported by a UK funded Medical Research Council‐Economic and Social Research Council studentship grant (H.K., I.K., Y.C. and M.C.; grant number 681120), FAPESP‐University of Surrey Mobility grant (H.K. and A.B.; grant number 12/50207‐8) and Horizon 2020 EU grant (A.B. SmokeFreeBrain, grant number 681120).
Keyworth, H. , Georgiou, P. , Zanos, P. , Rueda, A. V. , Chen, Y. , Kitchen, I. , Camarini, R. , Cropley, M. , and Bailey, A. (2018) Wheel running during chronic nicotine exposure is protective against mecamylamine‐precipitated withdrawal and up‐regulates hippocampal α7 nACh receptors in mice. British Journal of Pharmacology, 175: 1928–1943. doi: 10.1111/bph.14068.
References
- Abrantes AM, Strong DR, Lloyd‐Richardson EE, Niaura R, Kahler CW, Brown RA (2009). Regular exercise as a protective factor in relapse following smoking cessation treatment. Am J Addict 18: 100–101. [DOI] [PubMed] [Google Scholar]
- al'Absi M, Wittmers LE, Erickson J, Hatsukami D, Crouse B (2003). Attenuated adrenocortical and blood pressure responses to psychological stress in ad libitum and abstinent smokers. Pharmacol Biochem Behav 74: 401–410. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide To PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion NV, Faccenda E, Harding SD et al (2017b). The Concise Guide To PHARMACOLOGY 2017/18: Ligand‐gated ion channels. Br J Pharmacol 174: S130–S159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aydin C, Oztan O, Isgor C (2012). Nicotine‐induced anxiety‐like behavior in a rat model of the novelty‐seeking phenotype is associated with long‐lasting neuropeptidergic and neuroplastic adaptations in the amygdala: effects of the cannabinoid receptor 1 antagonist AM251. Neuropharmacology 63: 1335–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin PR, Alanis R, Salas R (2011). The role of the habenula in nicotine addiction. J Addict Res Ther S1 pii: 002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balter RE, Dykstra LA (2012). The effect of environmental factors on morphine withdrawal in C57BL/6J mice: running wheel access and group housing. Psychopharmacology (Berl) 224: 91–100. [DOI] [PubMed] [Google Scholar]
- Barik J, Wonnacott S (2009). Molecular and cellular mechanisms of action of nicotine in the CNS In: Henningfield JE, London ED, Pogun S. (eds). Nicotine Psychopharmacology, Vol. 192 Springer: Berlin Heidelberg, pp. 173–207. [DOI] [PubMed] [Google Scholar]
- Berrendero F, Kieffer BL, Maldonado R (2002). Attenuation of nicotine‐induced antinociception, rewarding effects, and dependence in mu‐opioid receptor knock‐out mice. J Neurosci 22: 10935–10940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breese CR, Marks MJ, Logel J, Adams CE, Sullivan B, Collins AC et al (1997). Effect of smoking history on [3H]nicotine binding in human postmortem brain. J Pharmacol Exp Ther 282: 7–13. [PubMed] [Google Scholar]
- Brunzell DH, Boschen KE, Hendrick ES, Beardsley PM, McIntosh JM (2010). Alpha‐conotoxin MII‐sensitive nicotinic acetylcholine receptors in the nucleus accumbens shell regulate progressive ratio responding maintained by nicotine. Neuropsychopharmacology 35: 665–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castañé A, Valjent E, Ledent C, Parmentier M, Maldonado R, Valverde O (2002). Lack of CB1 cannabinoid receptors modifies nicotine behavioural responses, but not nicotine abstinence. Neuropharmacology 43: 857–867. [DOI] [PubMed] [Google Scholar]
- Christensen DZ, Mikkelsen JD, Hansen HH, Thomsen MS (2010). Repeated administration of α7 nicotinic acetylcholine receptor (nAChR) agonists, but not positive allosteric modulators, increases α7 nAChR levels in the brain. J Neurochem 114: 1205–1216. [DOI] [PubMed] [Google Scholar]
- Cippitelli A, Astarita G, Duranti A, Caprioli G, Ubaldi M, Stopponi S et al (2011). Endocannabinoid regulation of acute and protracted nicotine withdrawal: effect of FAAH inhibition. PLoS One 6: e28142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgrove KP, Batis J, Bois F, Maciejewski PK, Esterlis I, Kloczynski T et al (2009). Beta2‐nicotinic acetylcholine receptor availability during acute and prolonged abstinence from tobacco smoking. Arch Gen Psychiatry 66: 666–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czubak A, Nowakowska E, Kus K, Burda K, Metelska J, Baer‐Dubowska W et al (2009). Influences of chronic venlafaxine, olanzapine and nicotine on the hippocampal and cortical concentrations of brain‐derived neurotrophic factor (BDNF). Pharmacol Rep 61: 1017–1023. [DOI] [PubMed] [Google Scholar]
- Dajas‐Bailador F, Wonnacott S (2004). Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol Sci 25: 317–324. [DOI] [PubMed] [Google Scholar]
- Damaj MI, Kao W, Martin BR (2003). Characterization of spontaneous and precipitated nicotine withdrawal in the mouse. J Pharmacol Exp Ther 307: 526–534. [DOI] [PubMed] [Google Scholar]
- Dubreucq S, Durand A, Matias I, Bénard G, Richard E, Soria‐Gomez E et al (2012). Ventral tegmental area cannabinoid type‐1 receptors control voluntary exercise performance. Biol Psychiatry 73: 895–903. [DOI] [PubMed] [Google Scholar]
- Erickson KI, Voss MW, Prakash RS, Basak C, Szabo A, Chaddock L et al (2011). Exercise training increases size of hippocampus and improves memory. Proc Natl Acad Sci U S A 108: 3017–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field AE, Colditz GA, Willett WC, Longcope C, McKinlay JB (1994). The relation of smoking, age, relative weight, and dietary intake to serum adrenal steroids, sex hormones, and sex hormone‐binding globulin in middle‐aged men. J Clin Endocrinol Metab 79: 1310–1316. [DOI] [PubMed] [Google Scholar]
- Fisher BE, Li Q, Nacca A, Salem GJ, Song J, Yip J et al (2013). Treadmill exercise elevates striatal dopamine D2 receptor binding potential in patients with early Parkinson's disease. Neuroreport 24: 509–514. [DOI] [PubMed] [Google Scholar]
- Fuss J, Ben Abdallah NMB, Vogt MA, Touma C, Pacifici PG, Palme R et al (2010). Voluntary exercise induces anxiety‐like behavior in adult C57BL/6J mice correlating with hippocampal neurogenesis. Hippocampus 20: 364–376. [DOI] [PubMed] [Google Scholar]
- Georgiou P, Zanos P, Garcia‐Carmona JA, Hourani S, Kitchen I, Laorden ML et al (2016). Methamphetamine abstinence induces changes in mu‐opioid receptor, oxytocin and CRF systems: association with an anxiogenic phenotype. Neuropharmacology 105: 520–532. [DOI] [PubMed] [Google Scholar]
- Haasova M, Warren FC, Ussher M, Janse Van Rensburg K, Faulkner G, Cropley M et al (2013). The acute effects of physical activity on cigarette cravings: systematic review and meta‐analysis with individual participant data. Addiction 108: 26–37. [DOI] [PubMed] [Google Scholar]
- Hadjiconstantinou M, Duchemin AM, Zhang H, Neff NH (2011). Enhanced dopamine transporter function in striatum during nicotine withdrawal. Synapse 65: 91–98. [DOI] [PubMed] [Google Scholar]
- Hambsch B, Keyworth H, Lind J, Otte DM, Racz I, Kitchen I et al (2014). Chronic nicotine improves short‐term memory selectively in a G72 mouse model of schizophrenia. Br J Pharmacol 171: 1758–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmey D, Griffin PR, Kenny PJ (2012). Development of novel pharmacotherapeutics for tobacco dependence: progress and future directions. Nicotine Tob Res 14: 1300–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan S, van der Veen DR, Winsky‐Sommerer R, Dijk D‐J, Archer SN (2011). Altered sleep and behavioral activity phenotypes in PER3‐deficient mice. Am J Physiol Regul Integr Comp Physiol 301: R1821–R1830. [DOI] [PubMed] [Google Scholar]
- Ickes BR, Pham TM, Sanders LA, Albeck DS, Mohammed AH, Granholm AC (2000). Long‐term environmental enrichment leads to regional increases in neurotrophin levels in rat brain. Exp Neurol 164: 45–52. [DOI] [PubMed] [Google Scholar]
- Jackson KJ, Martin BR, Changeux JP, Damaj MI (2008). Differential role of nicotinic acetylcholine receptor subunits in physical and affective nicotine withdrawal signs. J Pharmacol Exp Ther 325: 302–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson KJ, Muldoon PP, De Biasi M, Damaj MI (2015). New mechanisms and perspectives in nicotine withdrawal. Neuropharmacology 96 (Pt B): 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny PJ (2009). Emerging therapeutic targets for the treatment of nicotine addiction. Expert Rev Clin Pharmacol 2: 221–225. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan‐Sarin S, Rosen MI, O'Malley SS (1999). Naloxone challenge in smokers. Preliminary evidence of an opioid component in nicotine dependence. Arch Gen Psychiatry 56: 663–668. [DOI] [PubMed] [Google Scholar]
- Li X, Wolf ME (2015). Multiple faces of BDNF in cocaine addiction. Behav Brain Res 279: 240–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifestyles Statistics Team, The Health and Social Care Information Centre (2012). Statistics on NHS stop smoking services: England, April 2011–March 2012. Department of Health: Leeds, UK. [Google Scholar]
- Malin DH, Lake JR, Carter VA, Cunningham JS, Wilson OB (1993). Naloxone precipitates nicotine abstinence syndrome in the rat. Psychopharmacology (Berl) 112: 339–342. [DOI] [PubMed] [Google Scholar]
- Marubio LM, del Mar Arroyo‐Jimenez M, Cordero‐Erausquin M, Lena C, Le Novere N, de Kerchove d'Exaerde A et al (1999). Reduced antinociception in mice lacking neuronal nicotinic receptor subunits. Nature 398: 805–810. [DOI] [PubMed] [Google Scholar]
- Massey KA, Zago WM, Berg DK (2006). BDNF up‐regulates α7 nicotinic acetylcholine receptor levels on subpopulations of hippocampal interneurons. Mol Cell Neurosci 33: 381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta S, Balfour DJ, Benowitz NL, Boyd R, Buccafusco J, Caggiula AR et al (2007). Guidelines on nicotine dose selection for in vivo research. Psychopharmacology (Berl) 190: 269–319. [DOI] [PubMed] [Google Scholar]
- McGinty JF, Whitfield TW Jr, Berglind WJ (2010). Brain‐derived neurotrophic factor and cocaine addiction. Brain Res 1314: 183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- le Merrer J, Becker JA, Befort K, Kieffer BL (2009). Reward processing by the opioid system in the brain. Physiol Rev 89: 1379–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metaxas A, Al‐Hasani R, Farshim P, Tubby K, Berwick A, Ledent C et al (2013). Genetic deletion of the adenosine A(2A) receptor prevents nicotine‐induced upregulation of alpha7, but not alpha4beta2* nicotinic acetylcholine receptor binding in the brain. Neuropharmacology 71: 228–236. [DOI] [PubMed] [Google Scholar]
- Miladi‐Gorji H, Rashidy‐Pour A, Fathollahi Y (2012). Anxiety profile in morphine‐dependent and withdrawn rats: effect of voluntary exercise. Physiol Behav 105: 195–202. [DOI] [PubMed] [Google Scholar]
- Mineur YS, Mose TN, Blakeman S, Picciotto MR (2018). Hippocampal α7 nicotinic ACh receptors contribute to modulation of depression‐like behaviour in C57BL/6J mice. Br J Pharmacol 175: 1903–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira MSR, da Silva Fernandes MJ, Scorza FA, Persike DS, Scorza CA, da Ponte JB et al (2010). Acute and chronic exercise modulates the expression of MOR opioid receptors in the hippocampal formation of rats. Brain Res Bull 83: 278–283. [DOI] [PubMed] [Google Scholar]
- Parrott AC, Garnham NJ, Wesnes K, Pincock C (1996). Cigarette smoking and abstinence: comparative effects upon cognitive task performance and mood state over 24 hours. Hum Psychopharmacol Clin Exp 11: 391–400. [Google Scholar]
- Picciotto MR, Kenny PJ (2013). Molecular mechanisms underlying behaviors related to nicotine addiction. Cold Spring Harb Perspect Med 3: a012112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciotto MR, Zoli M, Rimondini R, Lena C, Marubio LM, Pich EM et al (1998). Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature 391: 173–177. [DOI] [PubMed] [Google Scholar]
- Rankin KV, Jones DL (2011). Treatment of nicotine dependence with Chantix (varenicline). Tex Dent J 128: 457–461. [PubMed] [Google Scholar]
- Raybuck JD, Portugal GS, Lerman C, Gould TJ (2008). Varenicline ameliorates nicotine withdrawal‐induced learning deficits in C57BL/6 mice. Behav Neurosci 122: 1166–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda AV, Teixeira AM, Yonamine M, Camarini R (2012). Environmental enrichment blocks ethanol‐induced locomotor sensitization and decreases BDNF levels in the prefrontal cortex in mice. Addict Biol 17: 736–745. [DOI] [PubMed] [Google Scholar]
- Salas R, Main A, Gangitano D, De Biasi M (2007). Decreased withdrawal symptoms but normal tolerance to nicotine in mice null for the alpha7 nicotinic acetylcholine receptor subunit. Neuropharmacology 53: 863–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas R, Sturm R, Boulter J, De Biasi M (2009). Nicotinic receptors in the habenulo‐interpeduncular system are necessary for nicotine withdrawal in mice. J Neurosci 29: 3014–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez V, Moore C, Brunzell D, Lynch W (2013). Effect of wheel‐running during abstinence on subsequent nicotine‐seeking in rats. Psychopharmacology (Berl) 227: 403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scerbo F, Faulkner G, Taylor A, Thomas S (2010). Effects of exercise on cravings to smoke: the role of exercise intensity and cortisol. J Sports Sci 28: 11–19. [DOI] [PubMed] [Google Scholar]
- Scott DJ, Domino EF, Heitzeg MM, Koeppe RA, Ni L, Guthrie S et al (2007). Smoking modulation of mu‐opioid and dopamine D2 receptor‐mediated neurotransmission in humans. Neuropsychopharmacology 32: 450–457. [DOI] [PubMed] [Google Scholar]
- Simmons SJ, Gould TJ (2014). Involvement of neuronal β2 subunit‐containing nicotinic acetylcholine receptors in nicotine reward and withdrawal: implications for pharmacotherapies. J Clin Pharm Ther 39: 457–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Pennock MM, Walker KL, Lang KC (2012). Access to a running wheel decreases cocaine‐primed and cue‐induced reinstatement in male and female rats. Drug Alcohol Depend 121: 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoker AK, Olivier B, Markou A (2012). Role of α7‐ and β4‐containing nicotinic acetylcholine receptors in the affective and somatic aspects of nicotine withdrawal: studies in knockout mice. Behav Genet 42: 423–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickland JC, Abel JM, Lacy RT, Beckmann JS, Witte MA, Lynch WJ et al (2016). The effects of resistance exercise on cocaine self‐administration, muscle hypertrophy, and BDNF expression in the nucleus accumbens. Drug Alcohol Depend 163: 186–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia‐Arancibia L, Rage F, Givalois L, Dingeon P, Arancibia S, Beauge F (2001). Effects of alcohol on brain‐derived neurotrophic factor mRNA expression in discrete regions of the rat hippocampus and hypothalamus. J Neurosci Res 63: 200–208. [DOI] [PubMed] [Google Scholar]
- Taylor A, Katomeri M (2007). Walking reduces cue‐elicited cigarette cravings and withdrawal symptoms, and delays ad libitum smoking. Nicotine Tob Res 9: 1183–1190. [DOI] [PubMed] [Google Scholar]
- Taylor A, Ussher M, Faulkner G (2007). The acute effects of exercise on cigarette cravings, withdrawal symptoms, affect and smoking behaviour: a systematic review. Addiction 102: 534–543. [DOI] [PubMed] [Google Scholar]
- Thanos PK, Stamos J, Robison LS, Heyman G, Tucci A, Wang GJ et al (2013). Daily treadmill exercise attenuates cocaine cue‐induced reinstatement and cocaine induced locomotor response but increases cocaine‐primed reinstatement. Behav Brain Res 239: 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ussher M, Nunziata P, Cropley M, West R (2001). Effect of a short bout of exercise on tobacco withdrawal symptoms and desire to smoke. Psychopharmacology (Berl) 158: 66–72. [DOI] [PubMed] [Google Scholar]
- Vučcković MG, Li Q, Fisher B, Nacca A, Leahy RM, Walsh JP et al (2010). Exercise elevates dopamine D2 receptor in a mouse model of Parkinson's disease: in vivo imaging with [18F]fallypride. Mov Disord 25: 2777–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters CL, Cleck JN, Kuo YC, Blendy JA (2005). Mu‐opioid receptor and CREB activation are required for nicotine reward. Neuron 46: 933–943. [DOI] [PubMed] [Google Scholar]
- Wilkinson DS, Gould TJ (2013). Withdrawal from chronic nicotine and subsequent sensitivity to nicotine challenge on contextual learning. Behav Brain Res 250: 58–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright SR, Zanos P, Georgiou P, Yoo JH, Ledent C, Hourani SM et al (2016). A critical role of striatal A2A R‐mGlu5 R interactions in modulating the psychomotor and drug‐seeking effects of methamphetamine. Addict Biol 21: 811–825. [DOI] [PubMed] [Google Scholar]
- Zanos P, Georgiou P, Metaxas A, Kitchen I, Winsky‐Sommerer R, Bailey A (2015). Region‐specific up‐regulation of oxytocin receptor binding in the brain of mice following chronic nicotine administration. Neurosci Lett 600: 33–37. [DOI] [PubMed] [Google Scholar]
- Zhou X, Nai Q, Chen M, Dittus JD, Howard MJ, Margiotta JF (2004). Brain‐derived neurotrophic factor and trkB signaling in parasympathetic neurons: relevance to regulating α7‐containing nicotinic receptors and synaptic function. J Neurosci 24: 4340–4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effect of wheel‐running exercise regimen on individual nicotine withdrawal symptoms. Mice underwent one of three exercise regimes: 0, 2 or 24 h·day−1 running‐wheel access. Withdrawal was precipitated by mecamylamine (3 mg·kg−1, s.c.) following 14 days of either saline or nicotine (24 mg·kg−1 day−1) treatment via subcutaneous minipumps. Abstinence signs were evaluated during a 30‐min period after mecamylamine injection. (A) Front paw tremors were assigned a value of 0.5 for each episode. (B) Sniffing was assigned a value of 0.5 for each episode. (C) Scratches were assigned a value of 0.5 for each episode. (D) Rearing was assigned a value of 0.5 for each episode. (E) Genital licks were assigned a value of 1 for appearance within each 5‐min bin. Data are presented as mean ± SEM. *P < 0.05. Precise group sizes are reported in Table 1.
Figure S2 Effect of exercise on cytisine‐resistant and cytisine‐sensitive epibatidine binding in saline‐ and nicotine‐withdrawn mice undergoing mecamylamine‐precipitated withdrawal. Cytisine‐resistant (A) and cytisine‐sensitive (B) [125I]‐epibatidine binding in the medial habenula of saline‐ and nicotine‐withdrawn mice undergoing different exercise regimes. Data are presented as mean ± SEM. *P < 0.05. Precise group sizes are reported in Table 1. Abbreviations: MHb, medial habenula.
Table S1 Effect of exercise on α4β2* nAChR binding in saline‐ and nicotine‐withdrawn mice.
Table S2 Effect of exercise on α7 nAChR binding in saline‐ and nicotine‐withdrawn mice.