

Abstract
The pathway of rubber (poly [cis-1,4-isoprene]) catabolism is well documented for Gram-positive rubber degraders but only little information exists for Gram-negative species. The first documented potent rubber degrading Gram-negative strain is Xanthomonas sp. strain 35Y that uses extracellular rubber oxygenases for the initial cleavage of the polyisoprene molecule. However, neither the exact phylogenetic position of Xanthomonas sp. strain 35Y nor the catabolic pathway of the primary polyisoprene cleavage products have been investigated. In this contribution, we started to address both these issues by a comprehensive taxonomic characterization and by the analysis of the draft genome sequence of strain 35Y. Evaluation of the 16S rRNA gene sequence pointed to a borderline taxonomic position of strain 35Y as a novel species of the genus Steroidobacter. Further, substantial differences in the genotypic properties of strain 35Y and the members of the genus Steroidobacter, including average nucleotide identity (ANI) and in silico DNA-DNA hybridization (DDH), resolved the taxonomic position of strain 35Y and suggested its positioning as a novel species of the genus Steroidobacter. This was further confirmed by comparative analysis of physiological and biochemical features of strain 35Y with other members of the genus Steroidobacter. Thus, we conclude that strain 35Y represents a novel species of the genus Steroidobacter, for which we propose the designation Steroidobacter cummioxidans sp. nov., strain 35YT. A comprehensive analysis of the draft genome of S. cummioxidans strain 35Y revealed similarities but also substantial differences to rubber degrading Gram-positive counterparts. In particular, the putative transporters for the uptake of polyisoprene cleavage products differ from Gram-positive rubber degrading species. The draft genome sequence of S. cummioxidans strain 35Y will be useful for researchers to experimentally verify the predicted similarities and differences in the pathways of polyisoprene catabolism in Gram-positive and Gram-negative rubber degrading species.
Introduction
Natural rubber (NR) is a hydrocarbon biopolymer of cis-1,4-isoprene and is widely used in many applications in household and industry. In 2015, the world production of rubber has been raised to 26.7 million tons (46% NR and 54% synthetic rubber) [1]. This huge use of NR for more than a century and the disposal of rubber waste materials in the environment has become a serious problem in all parts of the world. For example, the release of NR or its derivatives as effluents from NR-processing industries causes pollution of rivers and other aqueous ecosystems. The conventional NR waste-management strategies, including landfills and incineration are themselves a large source of environmental pollution. NR-recycling processes, such as pyrolysis and disposal of rubber wastes to the air, water, and soil lead to permanent pollution of the environment. NR-processing can also cause health hazards, including asthma and allergy [2]. All these unwanted consequences of the use of rubber rationalize the necessity for an efficient, economic, and eco-friendly process of NR waste-management.
Bio-remediation is a potential and an emerging alternative for the conventional waste-management strategies. A few microbes are already known for their ability to bio-degrade NR. Most of the known NR-degrading bacteria are Gram-positive Actinobacteria [3], including Streptomyces sp. K30 [4–6], Gordonia polyisoprenivorans VH2 [7], Nocardia nova SH22a [8, 9], and Rhodococcus rhodochrous RPK1 [10]. They synthesize a latex clearing protein (Lcp), which is the key enzyme in rubber degrading Gram-positive bacteria catalyzing the oxidative cleavage of polyisoprene double bonds. The typical incubation period of these bacteria under laboratory conditions on rubber-latex varies from 6 to 12 weeks [11], thus demonstrating a slow growth with polyisoprene as a sole carbon-source. The complete NR-degradation pathway has been well-characterised in Gram-positive bacteria [7, 9].
Only a few Gram-negative NR-degrading bacteria have been isolated and are described in the literature so far, the first being Xanthomonas sp. strain 35Y [12] and recently Rhizobacter gummiphilus NS21 [13, 14]. Xanthomonas sp. strain 35Y is able to cause a ~60% weight loss of solid NR pieces within one week of incubation [12], thus identifying this strain as one of the relatively faster NR-degrading microbes in comparison to other described NR-degraders. The initial cleavage of poly(cis-1,4-isoprene) by Xanthomonas sp. strain 35Y is catalyzed by two cooperatively acting extracellular rubber oxygenase enzymes (RoxA and RoxB) which cleave NR to a C15 compound, 12-oxo-4,8-dimethyl-trideca-4,8-diene-1-al (ODTD) as a major cleavage product [15, 16]. RoxA and RoxB of Xanthomonas sp. strain 35Y are both dihaem cytochrome c dioxygenase proteins whereas Lcps are b-type cytochromes. RoxAs and Lcps have been thoroughly investigated in the last decade [17, 18, 19, 20, 21], while RoxB has been discovered only recently [16]. The rubber-cleaving system of R. gummiphilus NS21 [13, 14] is similar to that of Xanthomonas sp. strain 35Y and also consists of two rubber oxygenases (RoxA and RoxB) (data unpublished). RoxA and RoxB (alternative designation of RoxB is LatA) of R. gummiphilus NS21 are highly similar in amino acid sequences to the respective RoxA and RoxB orthologs in Xanthomonas sp. strain 35Y. However, no significant similarity has been found between the sequences of RoxAs or RoxBs and Lcps. Nevertheless, RoxA, RoxB, and Lcp are all haem-dependent rubber oxygenases and catalyze an analogous reaction, i.e. the cleavage of NR. The complete NR-degradation process in Gram-negative bacteria remains unknown.
Currently, the application of NR-bioremediation is impeded due to the lack of a comprehensive understanding of the mechanistic details of the microbial NR-degradation process at the metabolic level. To unravel the complete NR-degradation pathway in Gram-negative rubber degraders, Xanthomonas sp. strain 35Y is an important candidate. However, the unavailability of its genome sequence has so far been a major hurdle in exploring its metabolic potentials. In addition, the exact taxonomic position of Xanthomonas sp. strain 35Y is obscure. The first and only publication describing the source and taxonomy of Xanthomonas sp. strain 35Y [12] states that Xanthomonas sp. strain 35Y was taken from the culture collection of the authors’ institute. A comprehensive taxonomic analysis was not performed for Xanthomonas sp. strain 35Y, except for the statement of a strict respiratory metabolism, the identification of the Gram status, the identification of an insoluble yellow pigment, and the presence of one or two flagella. From these data, strain 35Y was tentatively designated as Xanthomonas sp. strain 35Y. The detailed description of strain 35Y, that was announced in the publication from 1990 [12], to the best of our knowledge was never published.
The unavailability of the genome sequence and the unclear taxonomic position, but the otherwise great importance of Xanthomonas sp. strain 35Y for the understanding of the catabolism of rubber in Gram-negative bacteria, prompted us to perform the present study. To this end, we sequenced and annotated the draft genome of Xanthomonas sp. strain 35Y and performed a comprehensive analysis to gain insights into its general genomic features and metabolic potential with special emphasis on putative proteins involved in the catabolism of NR. Furthermore, we determined and evaluated several physiological and biochemical characteristics in combination with the analysis of the draft genome sequence which allowed us to perform a reliable taxonomic characterization of strain 35Y and to identify strain 35Y as a novel species of the genus Steroidobacter, for which we propose the designation Steroidobacter cummioxidans sp. nov., strain 35YT.
Materials and methods
Bacterial strains
S. cummioxidans strain 35Y was first described by Tsuchii and Takeda in 1990 [12] as Xanthomonas sp. strain 35Y and was obtained from their laboratory. It has been deposited to Leibniz Institute Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ) Germany (DSM 103114). The genome and protein sequences of other bacterial strains used for comparative genomics in this study (Table A in S1 Text) were retrieved from the GenBank database (http://www.ncbi.nlm.nih.gov/genbank/).
Bacterial growth
S. cummioxidans strain 35Y was grown in lysogeny broth (LB, 10 g tryptone, 10 g NaCl and 5 g yeast extract per liter) at 30°C, 100 rpm for 48 h.
DNA extraction
20 ml of S. cummioxidans strain 35Y culture was grown in LB medium (30°C, 100 rpm). The cells were centrifuged (10 minutes, 5000 g, 4°C) and re-suspended in potassium phosphate buffer (100 mM, pH 7.0). After a second centrifugation, cells were re-suspended in 1.7 ml saccharose-Tris (saccharose: 25 g; Tris 0.12 g; pH 8.0; 100 ml water) followed by the addition of 200 μl lysozyme solution (stock: 20 mg/ml). Then, 2.5 ml saccharose-Tris, 300 μl proteinase K solutions (2.5 mg/ml) and 2.5 ml sodium dodecyl sulfate solution (0.15 g in 100 ml water) were added and incubated for 2.5 h at 37°C. Subsequently, 1.88 ml sodium perchlorate solution (NaClO4: 70.32 g; water: 100 ml; acetic acid: pH 4.8) was added and mixed gently, then 2.5 ml phenol-chloroform-isoamylalcohol (25:24:1) was added and mixed gently. After centrifugation (10 minutes, 5000 g, 4°C), the aqueous phase was used for a second phenol-chloroform-isoamylalcohol extraction and subsequently, an extraction with 2.5 ml chloroform-isoamylalcohol (24:1). The aqueous phase was transferred into a new falcon tube, mixed with 25 μl RNAse solution (10 mg/ml) and incubated for 30 minutes at 37°C. The genomic DNA was extracted with 1.5 volumes of isopropanol and washed 3x5 minutes with 70% ethanol. The DNA was dried at 60°C and re-suspended in 2 ml water.
Genome sequencing and assembly
Genomic shotgun libraries were sequenced by the Goettingen Genomics Laboratory (G2L, http://www.appmibio.uni-goettingen.de/index.php?sec=g2l/) by a combination of 454 pyrosequencing and Illumina sequencing technologies. The 454 shotgun library was generated using the GS FLX Rapid Library Prep Kit (454 Roche, Branford) and the genome was sequenced on the 454 FLX instrument (454 Roche, Branford) using XL+ chemistry. The shotgun library for Illumina sequencing was done by Nextera DNA Sample preparation kit (Illumina, San Diego, USA) following the manufacturer’s instructions and the genome was sequenced on the Illumina GA IIx instrument using a 112 bp paired end single indexed run technology. The number of reads generated by the 454 pyrosequencing and Illumina sequencing technologies were 101,250 and 6,494,354, respectively. Hybrid genome assembly was done using default parameters of the MIRA (Mimicking Intelligent Read Assembly) assembler (version as of February, 2013) [22] which resulted in 127 contigs in total, exhibiting an average coverage of 34.47X. Upon screening of the resultant assembly for contaminations, including PhiX sequences using BLASTN (version 2.2.29+), 126 contigs were obtained.
Basic genome analysis
Open reading frames (ORFs) and proteins of 126 contigs were predicted by IMG/ER (http://www.img.jgi.doe.gov/cgi-bin/mer/main.cgi/). ORFs were annotated using BLASTP (version 2.2.29+) against the NCBI NR protein database (version as of October 20, 2014) with a bit-score of 50 and an e-value cut-off of 10−6. All contigs were used for the prediction of tRNA and rRNA genes using tRNA and hmm_rRNA prediction modules of WebMGA server (http://weizhong-lab.ucsd.edu/metagenomic-analysis/). For comparative analysis, assignment of orthologs, and similarity searches, a threshold of 30% amino acid sequence identity and 60% coverage of both query and hit in the alignment were used. In order to find orthologs, predicted protein sequences of S. cummioxidans strain 35Y were blasted (BLASTP version 2.2.29+) against bacterial strains used in this study (Table A in S1 Text) using an e-value cut-off of 10−4 and the best hits were selected.
Functional annotation of all predicted protein sequences was done using Clusters of Orthologous Groups (COGs) module of WebMGA server (http://hweizhong-lab.ucsd.edu/metagenomic-analysis/server/cog/) with an e-value cut-off of 10−4. Pathway mapping was done using BlastKOALA module of Kyoto Encyclopedia of Genes and Genomes (KEGG) online server (http://www.kegg.jp/blastkoala/) using prokaryotes as the taxonomy group and genus_prokaryotes as the KEGG genes database. PSORTb v3.0.2 server (http://www.psort.org/psortb/) was used for the prediction of the subcellular-localization (parameters used, organism: bacteria; Gram-stain: negative). SecretomeP v2.0 was used for the prediction of non-classically secreted proteins (http://www.cbs.dtu.dk/services/SecretomeP/). Transporter proteins were predicted using Transporter Classification Database (TCDB) database (http://www.tcdb.org/, version as of March 25, 2015) by performing BLASTP (version 2.2.29+) with an e-value cut-off of 10−10. To predict the carbohydrate active enzymes, the standalone version of Database for Automated Carbohydrate-Active Enzyme Annotation (dbCAN HMMs v5.0) (http://csbl.bmb.uga.edu/dbCAN/) was used. The results were parsed with the following two parameters recommended for bacteria (i) if alignment >80 amino acids, use an e-value < 10−18, otherwise use e-value < 10−3 and (ii) covered fraction of HMM > 0.35. Promoter sequences were predicted on 126 contigs using Neural Network Promoter Prediction (NNPP) v2.2 server (http://www.fruitfly.org/seq_tools/promoter.html/) using a minimum promoter score of 0.8 and, including reverse strands.
MvirDB database (http://www.mvirdb.llnl.gov/, version as of February 19, 2015) was used for predicting the virulence factors using BLASTP (version 2.2.29+) with an e-value cut-off of 10−10. To predict the antibiotic resistant genes, Resistance Gene Identifier (RGI) module of Comprehensive Antibiotic Resistance Database (CARD) database (https://card.mcmaster.ca/) was used with default parameters. To predict secreted proteins and protein secretion systems, EffectiveDB server (http://www.effectivedb.org/) was used with enabled “genome mode” and prediction module “EffectiveS346”. PHAge Search Tool (PHAST) (http://phast.wishartlab.com/) and Phage Classification Tool Set (PHACTS) (http://www.phantome.org/PHACTS) servers were used on 126 contigs of S. cummioxidans strain 35Y for the prediction of prophages. ISfinder server (https://www-is.biotoul.fr/) was used with default parameters for the prediction of insertion elements. CRISPRfinder server (http://crispr.i2bc.paris-saclay.fr/Server/) was used with default parameters to identify the putative clustered regularly interspaced short palindromic repeats (CRISPRs).
In silico analysis of rubber oxygenases
For the functional analysis of rubber oxygenases and their homologs by classifying them into families and predicting domains and important sites, InterPro (https://www.ebi.ac.uk/interpro/) server was used.
Taxonomy analysis of strain 35Y
The taxonomic positioning of strain 35Y was determined using polyphasic taxonomic approaches, including (i) 16S rRNA gene based method, (ii) average nucleotide identity (ANI) based method, (iii) in silico DNA-DNA hybridization (DDH) estimate based method, and (iv) physiological and biochemical characterization.
(i) 16S rRNA gene based method
The 16S rRNA gene sequence of strain 35Y (STC_rRNA4) was used for online BLASTN (version 2.8.0+) analysis against the 16S ribosomal RNA sequences (Bacteria and Archaea) database of NCBI webserver (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastSearch, version as of February 05, 2018) which revealed high identity with members of the class Gammaproteobacteria. For further analysis, 16S rRNA gene sequence from at least one member from each genus (except for unclassified groups and environment samples) under the class Gammaproteobacteria was taken for the phylogenetic study. The 16S rRNA gene sequence of strain 35Y was showing highest identity with that of the genus Steroidobacter under the family Sinobacteraceae, hence all the available members of this family were used for this analysis. Evolutionary analyses were conducted in Molecular Evolutionary Genetics Analysis 6 (MEGA6) [23] software. The sequences were aligned using ClustalW module of MEGA6 with default parameters. The evolutionary history was inferred by using the maximum likelihood method based on the Kimura 2-parameter model [24]. Robustness of the phylogenetic trees was evaluated using the bootstrap resampling method of Felsenstein [25]. The bootstrap consensus tree inferred from 1000 replicates is taken to represent the evolutionary history of the taxa analyzed. Initial trees for the heuristic search were obtained automatically by applying neighbor-join and BioNJ algorithms to a matrix of pairwise distances estimated using the maximum composite likelihood approach and then selecting the topology with superior log likelihood value. Nitrosomonas aestuarii Nm69 was used as the outgroup. The tree was drawn to scale, with branch lengths measured in the number of substitutions per site. The analysis involved 58 nucleotide sequences. All positions containing gaps and missing data were eliminated. There were a total of 1,146 positions in the final dataset. In addition, BLASTN (version 2.2.29+) was used to calculate the percent identity between the 16S rRNA gene sequences for a given pair of bacteria. FigTree v1.4.2 [26] was used for tree-visualization.
(ii) Average nucleotide identity based method
The complete and draft quality genomes of S. denitrificans DSM 18526 and strain 35Y, respectively were used for the calculation of the ANI value [27]. Towards this, ANIb module of standalone version of JSpecies v1.2.1 [28] was used with default parameters.
(iii) In silico DNA-DNA hybridization estimate based method
Genome-to-Genome Distance Calculator (GGDC) v2.1 server (http://ggdc.dsmz.de/distcalc2.php) was used to calculate genome to genome distance of strain 35Y with respect to S. denitrificans DSM 18526 in order to determine in silico DDH estimate.
(iv) Physiological and biochemical characterization
A comprehensive literature survey was carried out for the physiological and biochemical characterization of strain 35Y.
Detection of polyhydroxyalkanoate granules
Morphological analysis and fluorescence microscopy were performed on a Nikon Ti-E microscope (MEA53100) equipped with a Hamamatsu Orca Flash 4.0 sCMOS camera. The formation of polyhydroxyalkanoate (PHA) granules was followed after staining the cells (grown on basal broth medium supplemented with glucose) with Nile red [29] (1–10 μg/ml in DMSO or ethanol) and a filter set with extinction at 562/40 nm and emission at 594 LP. To further validate our results, the gas chromatography analysis was carried out after acidic methanolysis of lyophilized cells under PHA permissive cultivation conditions as per standard protocol [30].
Detection of polyphosphate granules
Polyphosphate (polyP) granules of bacterial cells (grown on nutrient broth medium supplemented with glucose) were stained with 4’,6-diamino-2-phenylindole 2HCl (DAPI) [31] (DAPI, concentration 60 μg/ml) and detected with the aid of a DAPI-polyP specific filter set (ex: 415/20 nm, em: 520/60 nm). Ralstonia eutropha H16 cells were used as control. This species is well known for its ability to synthesize both PHA and polyP granules.
Results and discussion
Determination of the taxonomic position of Xanthomonas sp. strain 35Y
The previous taxonomic classification of strain 35Y as a member of the genus Xanthomonas was based on the presence of a yellow pigment and strict aerobic respiratory metabolism of the Gram-negative and motile cells of strain 35Y [12]. To determine the taxonomic position of strain 35Y with more reliability, we first performed 16S rRNA gene sequence analysis. The gene sequence of the complete 16S rRNA was a by-product of the determination of the draft genome sequence that is described in detail below. Strain 35Y demonstrated considerably low 16S rRNA gene sequence identities with the members of the genus Xanthomonas; the highest value was for Xanthomonas campestris ATCC 33913 (85.14%). These data suggest that strain 35Y is not a species of the genus Xanthomonas. Furthermore, comparison of the 16S rRNA gene sequence of strain 35Y with the 16S ribosomal RNA sequences (Bacteria and Archaea) database revealed high identities with the members of the genus Steroidobacter; the highest value was found for S. flavus CPCC 100154 (98.27%) followed by S. agariperforans KA5-B (98.19%) (Table B in S1 Text). The 16S rRNA gene sequence identity with S. denitrificans DSM 18526T, the only member of the genus Steroidobacter for which the genome sequence is available, was 97.78%. These data suggest that strain 35Y is a species of the genus Steroidobacter within the family Sinobacteraceae. The phylogenetic position of strain 35Y in the family Sinobacteraceae is shown in Fig 1.
Fig 1. Phylogenetic relationships between Steroidobacter cummioxidans sp. nov., strain 35Y and members of the family Sinobacteraceae on the basis of 16S rRNA gene sequences.

The phylogenetic tree was constructed by using the maximum likelihood method, and the 16S rRNA gene sequence of Nitrosomonas aestuarii Nm69 was used as the outgroup. Numbers at nodes indicate levels of bootstrap support (%) based on a maximum likelihood analysis of 1000 resampled datasets; values of less than 50 % are not shown. Bar, 0.02 nucleotide substitutions per site. Solid lines represent the lengths of branches; dotted lines are used to align the tip labels for better visualization; circle represents the node.
In order to delineate the species, Kim et al. proposed a threshold of 98.65% 16S rRNA gene sequence identity on a large collection of bacterial species [32]. A 16S rRNA sequence identity of 98.27% with S. flavus CPCC 100154 presents strain 35Y as a borderline case for species demarcation on the basis of this feature alone. Thus in order to delineate the species of strain 35Y in the genus Steroidobacter, we have adopted polyphasic taxonomic approaches. The first approach is based on in silico genotypic characterisation and the second approach is based on thorough literature evidence for various physiological and biochemical features.
In the in silico based approach, we have used two standard methods of species demarcation, including ANI analysis and in silico DDH analysis. An ANI threshold range of 95–96% is proposed to demarcate the species boundary [32, 33, 34]. The ANI value of strain 35Y with S. denitrificans DSM 18526 was found to be 72.23%, which is too low to consider it as a known species of the genus Steroidobacter. The recommended threshold value of species demarcation as per in silico DDH based method is 70% [35]. This value was calculated to be 19.8% between strain 35Y and S. denitrificans DSM 18526 and thus this method also could not classify strain 35Y as a known species of the genus Steroidobacter. The genotypic data obtained using three independent approaches, including 16S rRNA gene sequence identity based-, ANI value based-, and in silico DDH value based-methods showed that strain 35Y should be considered as a novel species of the genus Steroidobacter.
This conclusion was further supported by a comprehensive literature evidence-based evaluation of various physiological and biochemical features of strain 35Y and the other members of the genus Steroidobacter (Table 1). As shown in Table 1, all members of the genus Steroidobacter, including strain 35Y, are non-spore-forming Gram-negative rods. In addition, they are also negative for the hydrolysis of gelatine. Strain 35Y can be distinguished from the close species neighbours by several traits, including being able to hydrolyse casein, growth on complex media, secretion of xanthomonadin pigment, the inability for nitrate-reduction, and being catalase negative. In addition, strain 35Y is also distinguished from the known species of the genus Steroidobacter based upon significant differences among their genome sizes and temperature ranges for growth. Taken together, the polyphasic taxonomy analysis indicated strain 35Y as a novel species in the genus Steroidobacter. We propose to designate this novel species as Steroidobacter cummioxidans to show its rubber oxidizing capacity. Strain 35Y is currently the only member of the species and represents the type strain (35YT).
Table 1. Characteristics of Steroidobacter cummioxidans strain 35Y (35Y), Steroidobacter flavus CPCC 100154 (CPCC 100154), Steroidobacter agariperforans KA5-B (KA5-B), Steroidobacter denitrificansis DSM 18526 (DSM 18526).
| Characteristics | 35Y [12, 50, this study] | CPCC 100154 [52] | KA5-B [53] | DSM 18526 [54] |
|---|---|---|---|---|
| Flagellar arrangement | Motile by 1 (or 2) polar flagellum | Motile by single polar flagellum | Non-motile | Motile by single polar flagellum |
| Cell size width by length (μm) | 0.4–0.6 by 2.0–5.0 | 0.6–0.8 by 1.5–1.8 | 0.4–0.6 by 1.0–2.1 | 0.3–0.5 by 0.6–1.6 |
| Cell morphology | Rod | Rod | Straight to slightly curved rod | Slightly curved rods |
| Sporulation | - | - | - | - |
| Gram-stain | - | - | - | - |
| Oxidase | + | - | + | + |
| Catalase | - | + | + | + |
| Utilization as sole carbon source: | ||||
| D-fructose | + | - | + | - |
| D-maltose | + | - | + | - |
| Dextrin | + | - | + | - |
| D-galactose | + | - | + | + |
| D-rhamnose | ND | - | + | - |
| D-trehalose | + | + | - | - |
| D-turanose | + | - | + | - |
| Tween 40 | + | - | + | - |
| α-D-lactose | + | - | + | - |
| Hydrolysis of: | ||||
| Casein | + | - | ND | ND |
| Gelatine | - | - | ND | ND |
| Starch | + | - | + | ND |
| Anaerobic growth | - | ND | - | + |
| Nitrate reduction | - | ND | ND | + |
| Growth on complex media | + | ND | ND | - |
| Utilization of agar as a C-source | - | ND | + | - |
| Temperature range for growth (°C) | 25–41 | 20–37 | 15–37 | 20–38 |
| DNA G+C content (mol %) | 60.94 | 64.4 | 62.9 | 61.9 |
| 16S rRNA gene identity (%) | 100 | 98.28 | 98.2 | 97.59 |
| Genome Size (Mbp) | 7.9 | ND | ND | 3.5 |
| Xanthomonadin pigment | + C27H28Br2O2 [47] |
ND | ND | ND |
| Xanthomonadin biosynthetic gene cluster [55] | A cluster of 14 genes | ND | ND | Only 4 genes* |
ND: not determined or not available; +: positive; -: negative.
* Details are given in S1 Text.
Description of Steroidobacter cummioxidans sp. nov.
Steroidobacter cummioxidans sp. nov. (cum.mi.o'xi.dans. L. n. cummi, rubber sap; N.L. part. adj. oxidans, oxidizing; N.L. masc. adj. cummioxidans, oxidizing rubber. The ability to utilize natural rubber (poly [cis-1, 4-isoprene]) as a carbon source and to form clearing zones on opaque latex agar is eponymous for the species. The cellular dimensions of S. cummioxidans strain 35Y are 0.4 to 0.6 μm in width and 2 to 5 μm in length but cells occasionally can become considerably longer (10 μm). The cells are motile rods by one or few polar flagella. The strains of S. cummioxidans possess a Gram-negative cell wall structure and form yellow-orange colored colonies due to the synthesis of bromine-containing pigments (xanthomonadins). However, because of low 16S rRNA gene identities of S. cummioxidans strain 35YT to true Xanthomonas species, this strain belongs to the genus Steroidobacter despite the formation of xanthomonadins. The strains of S. cummioxidans rely on a strictly respiratoric metabolism. They are prototrophic and can use a wide range of compounds for growth, including sugars, fatty acids, low molecular weight hydrocarbons, amino acids, and diverse complex media at moderate conditions. Strains of S. cummioxidans have a temperature range of 25–41°C for growth, thus are mesophilic in nature.
Basic features of the draft genome of S. cummioxidans strain 35Y
The draft genome of S. cummioxidans strain 35Y has a size of almost 8 Mbp (7,936,208 bp) with a DNA G+C content of 60.94 mol% (Table 2). The draft genome has a capacity for 6,906 protein encoding genes as well as 47 and 7 tRNAs and rRNAs, respectively. Further details of the predicted genes and their putative functions are given in Table 2, S1 Dataset and S1 Text.
Table 2. Basic genome features of Steroidobacter cummioxidans strain 35Y.
| Number of Contigs | 126 (smallest: 3,181 bp, largest: 336,437 bp, average: ~62,986 bp) | ||
| Genome Size | 7,936,208 (bp) | ||
| G+C Content (mol%) | 60.94 | ||
| Number of Genes | 6,960 | Protein encoding genes (PEGs) | 6,906 |
| tRNA | 47 | ||
| rRNA | 7 | ||
| Proteins with Predicted Functions | 5,475 | ||
| Hypothetical or Uncharacterized Proteins | 1,431 | ||
| Proteins with COG Annotations | 2,637 | Metabolism | 35.61% |
| Cellular processes and signalling | 17.29% | ||
| Information storage and processing | 15.24% | ||
| Poorly characterized | 18.54% | ||
| Multiple classes | 13.31% | ||
| Proteins with KEGG Annotations | 2,892 | ||
| Membrane Transporters | 479 | ||
| Virulence Factors | 909 | ||
| Subcellular Localization | Cytoplasmic | 37.85% | |
| Cytoplasmic membrane | 20.50% | ||
| Extracellular | 1.51% | ||
| Outer membrane | 3.82% | ||
| Periplasmic | 2.43% | ||
| Unclassified | 33.88% | ||
| Prophages | 2 (incomplete) | 10.1 kb | 8 PEGs |
| 9.4 kb | 9 PEGs | ||
| Insertion Elements | 90 | ||
| CRISPR Loci | 3 | ||
| Antibiotic Resistant Genes | 50 | ||
| Carbohydrate-Active Enzymes | 269 | Auxiliary Activity | 5.20% |
| Carbohydrate Esterase | 14.50% | ||
| Carbohydrate-Binding Module | 15.99% | ||
| Glycoside Hydrolase | 39.03% | ||
| Glycosyl Transferase | 21.56% | ||
| Polysaccharide Lyase | 3.72% | ||
NR-degradation potential of S. cummioxidans strain 35Y
NR-degradation related genes were predicted in the draft genome of S. cummioxidans strain 35Y using comparative analyses with other known NR-degrading bacteria, such as G. polyisoprenivorans VH2 [7] and N. nova SH22a [8, 9]. The NR-degradation process can be categorized into five major phases (Fig 2), including (1) biosynthesis of rubber oxygenases and extracellular oxidative cleavage of polyisoprene chains, (2) import of oligoisoprenes, (3) β-oxidation, (4) acetyl-CoA and propionyl-CoA metabolism, and (5) anaplerotic reactions and gluconeogenesis. In the following sections, the main features of the five phases are discussed in detail.
Fig 2. Predicted natural rubber degradation pathway of Steroidobacter cummioxidans strain 35Y.

The pathway comprises of five steps: (1) biosynthesis of rubber oxygenases and extracellular oxidative cleavage of polyisoprene chains, (2) import of oligoisoprenes, (3) β-oxidation, (4) acetyl-CoA and propionyl-CoA metabolism, and (5) anaplerotic reactions and gluconeogenesis. RoxA: Rubber oxygenase A, RoxB: Rubber oxygenase B, ODTD: 12-oxo-4,8-dimethyltrideca-4,8-diene-1-al, Mce: Mammalian cell entry protein, Mla: An ABC transport system that maintains lipid asymmetry, TBDRs: TonB-dependent outer membrane receptors, FACS: fatty acyl coenzyme A (CoA) synthetase: FadL: It encodes outer membrane proteins/ transporters involved in long-chain fatty acid, TCA: Tricarboxylic acid cycle, PEP: Phosphoenolpyruvate.
(1) Biosynthesis of rubber oxygenases and extracellular oxidative cleavage of polyisoprene chains
Rubber degrading bacteria must first cleave the water-insoluble high molecular weight polyisoprene molecules into low molecular weight products outside the cells. To this end, S. cummioxidans strain 35Y secretes two extracellular NR-degrading enzymes [16, 19], rubber oxygenases RoxA and RoxB. RoxB cleaves rubber into a mixture of C20 and higher oligoisoprenoids in an endo-type fashion thereby increasing the number of chain-ends. RoxA cleaves poly (cis-1, 4-isoprene) as well as the oligoisoprenoids derived from RoxB cleavage in an exo-type manner to the tri-isoprenoid ODTD. ODTD can be taken up by the bacterial cells directly or after oxidation of the terminal aldehyde group to a carboxylic acid group.
RoxA and RoxB harbour two haem-binding motifs (CXXCH) in the primary amino acid sequences [36] that enable a covalent attachment of the haem groups by the cytochrome c maturation proteins (see below) to the RoxA (RoxB) apo-peptides resulting in the mature holo-RoxA (holo-RoxB) enzymes (c-cytochromes). The presence of two c-type haem groups has already been confirmed by biophysical characterization of isolated RoxA protein [17] and by structure determination [18]. The published gene sequences of RoxA (GenBank accession number: KC980911.1) and RoxB (GenBank accession number: KY498024.1) enzymes were used to identify their locations in the draft genome of S. cummioxidans strain 35Y (roxA: STC_00358 (2,034 bp) and roxB: STC_02518 (2,043 bp)). The roxA and roxB genes had been identified and DNA-sequenced, previously [16, 36]. RoxA and RoxB exhibited high identities to the orthologous gene products of R. gummiphilus, i.e. RoxA (67%) and RoxB (LatA) (83%), respectively. However, the similarity of RoxA to RoxB was only moderate (30–40% identity) both for the S. cummioxidans strain 35Y and R. gummiphilus proteins. The biochemical properties of purified RoxA and RoxB of R. gummiphilus corresponded well with the homologous proteins of S. cummioxidans strain 35Y (data unpublished).
ODTD is the main end product of the RoxA/RoxB catalyzed extracellular cleavage of NR. The aldehyde group of ODTD must be oxidized to the corresponding acid extracellularly due to the high reactivity and toxicity of free aldehydes to living cells. The key enzyme for this step remains unknown [7, 8, 9] (Fig 2). However, numerous putative oxidoreductases were present in the draft genome of S. cummioxidans strain 35Y, among which only three (STC_01691, STC_05669, STC_06528) (S1 Dataset) were predicted to be extracellular (SecP score ≥ 0.5, non-classically secreted without any signal peptide) and might be responsible for this function.
The incorporation of covalently attached haem groups during the maturation of c-type cytochromes, such as RoxA and RoxB or haem proteins of the respiratory chain is carried out by cytochrome c maturation (Ccm) proteins and DsbD [37]. We predicted a cluster of ccm genes, ccmABC_EFGHI with one hypothetical gene (STC_04494, between ccmC and ccmE) in S. cummioxidans strain 35Y (Table C and Figure A in S1 Text) using comparative genomics. STC_04494 showed high similarity (44% identity over 79% of its length) with putative haem exporter protein CcmD of Methylococcus capsulatus Bath (GenBank accession number: NC_002977.6). We therefore assume that STC_04494 represents the haem exporter protein (CcmD). In addition, a dsbD gene was also predicted in the draft genome sequence but was not part of the ccm gene cluster. DsbD proteins (thiol: disulfide interchange proteins) are involved in the assembly of functional c-type cytochromes in the periplasm of Gram-negative bacteria. Furthermore, we identified a putative promoter containing a transcriptional start site 36 bp upstream of the ccm gene cluster which presumably is responsible for the regulation of the expression of the Ccm proteins. Our data suggests that all nine Ccm proteins are encoded by one transcriptional unit (ccmABCDEFGHI).
(2) Import of oligoisoprenes
Subsequent to the extracellular cleavage of polyisoprene via RoxA and RoxB and oxidation of ODTD to the corresponding fatty acid, the C15-tri-isoprenoid derivate has to be transported into the cells. The cell wall and cell membranes of Gram-positive and Gram-negative bacteria largely differ; moreover, the Lcp-derived primary degradation products of polyisoprene in Gram-positive rubber degrading bacteria are substantially larger (C20, C25 and higher oligoisoprenoids). Therefore, the transport proteins for the uptake of the primary rubber cleavage products might be different in Gram-positive and Gram-negative rubber degraders. In agreement with this assumption, we were unable to detect the genes for a complete transmembrane substrate uptake protein system of the YrbE/Mce (mammalian cell entry) type. In contrast, the genomes of the Gram-positive rubber degrading species G. polyisoprenivorans VH2 [7] and N. nova SH22a [8, 9] have genes for multiple YrbE/Mce clusters. One or several of these are thought to be specific for the uptake of rubber degradation products (Table 3). Thus, S. cummioxidans strain 35Y might have evolved a different transport mechanism.
Table 3. Distribution and loci of mammalian cell entry cluster related putative genes in the draft genome of Steroidobacter cummioxidans strain 35Y.
| Gene/Gene product | Locus |
|---|---|
| Mce (Mammalian cell entry protein) | STC_00777, STC_01556, STC_05337 |
| YrbE (ABC transporter permease) | STC_00779, STC_01557, STC_05339 |
A novel ABC transport system for intermembrane phospholipid trafficking, known as the maintenance of bacterial outer membrane lipid asymmetry (Mla) pathway, which maintains lipid asymmetry and is encoded by six genes, (mlaA, mlaB, mlaC, mlaD, mlaE, and mlaF) has been described for Gram-negative bacteria [38]. The entire Mla pathway was predicted to be conserved in S. rubberoxidans strain 35Y (Table D in S1 Text and Fig 3) using comparative genomics. In addition, Gram-negative bacteria also possess two families of substrate-specific outer membrane transporters involved in fatty-acid uptake, namely TonB-dependent outer membrane receptors (TBDRs) and long-chain fatty acid transporters (FadL family) [39]. For the transport of exogenous fatty acids, fadD encoding fatty acyl coenzyme A (CoA) synthetase (FACS) is found in Gram-negative bacteria [40]. It is also suggested that FadL and FACS function together as a fatty acid transport apparatus in Gram-negative bacteria. S. cummioxidans strain 35Y harboured one copy of a putative fadL family transporter gene and eleven copies of putative FACS genes (Table E in S1 Text). In addition, numerous copies of putative TBDR genes were also present in the draft genome. The availability of the draft genome sequence of S. cummioxidans strain 35Y now enables the performance of proteome studies to find experimental evidence if and which of the identified genes are specifically upregulated in cells grown on NR.
Fig 3. Putative gene cluster of maintenance of outer membrane lipid asymmetry pathway in the draft genome of Steroidobacter cummioxidans strain 35Y.
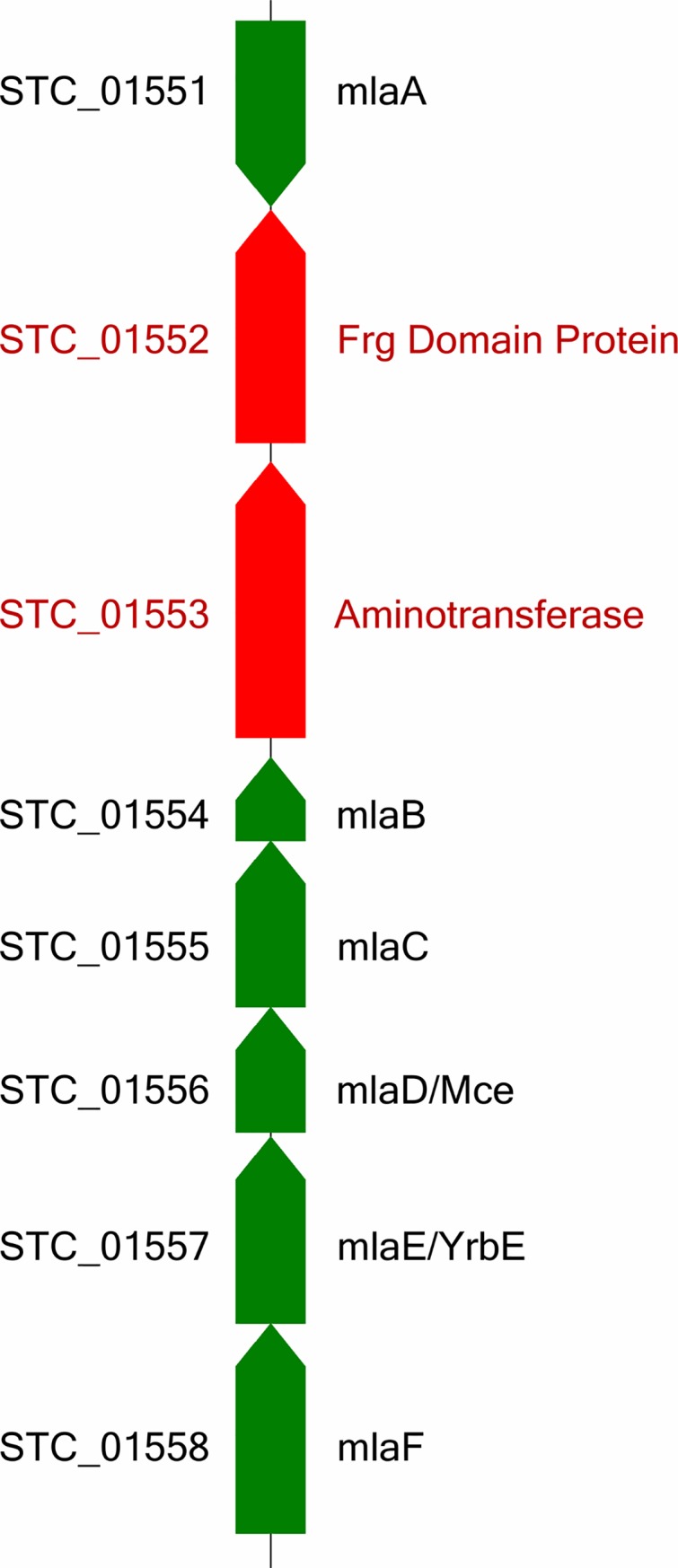
STC_01552 and STC_01553 are intervening genes in this Mla gene cluster (shown in red). Mla: An ABC transport system that maintains lipid asymmetry. For functional description of mla genes, refer to Table D in S1 Text.
(3) β-oxidation
β-oxidation is an iterative biological process which is responsible for the breakdown of fatty acids as a part of lipid metabolism. Recently, evidence for the involvement of β-oxidation enzymes in the catabolism of rubber was discussed for the Gram-positive rubber degrading species Streptomyces coelicolor 1A [41], G. polyisoprenivorans VH2 [7], and N. nova SH22a [8, 9]. The entire set of genes involved in the β-oxidation pathway was predicted to be conserved in S. cummioxidans strain 35Y (Table F in S1 Text and Fig 2) using comparative genomics.
(4) Acetyl-CoA and propionyl-CoA metabolism
The cleavage products of NR-degradation have to be channelled into the tricarboxylic acid (TCA) cycle in order to fuel the respiratory chain. The proposed NR-degradation pathway is similar to polyunsaturated fatty acid degradation and released two products, acetyl-CoA and propionyl-CoA [7, 9, 41] (Fig 2). Acetyl-CoA enters directly into the TCA cycle and/or the glyoxylate bypass, whereas propionyl-CoA is channelled indirectly via the methylcitrate pathway and the methylmalonyl-CoA pathway by a cascade of biochemical reactions to convert it into succinic acid. Towards this, the entire set of genes, that could fulfil these functions, was identified in the draft genome of S. cummioxidans strain 35Y (Tables G and H in S1 Text and Fig 2) using comparative genomics.
(5) Anaplerotic reactions and gluconeogenesis
In the absence or limited availability of sugars as the carbon and energy source, microorganisms employ conversion of intermediates of TCA cycle or the glyoxylate bypass to phosphoenolpyruvate (PEP). Based on genome analysis, entire sets of genes for TCA cycle and glyoxylate bypass were predicted in the draft genome of S. cummioxidans strain 35Y (Table I in S1 Text). In addition to this, orthologs of enzymes, including malic enzyme and PEP synthase, which catalyze the alternative routes to PEP biosynthesis [42, 43] were also predicted in the draft genome of S. cummioxidans strain 35Y (Fig 2). PEP acts as a substrate for several enzymatic reactions participating in significant cellular processes, including gluconeogenesis, glyceroneogenesis, amino acid synthesis, and anaplerosis of the TCA or glyoxylate cycle [44]. Phosphoenolpyruvate carboxykinase (PEPCK) is the key enzyme of this step (Fig 2) and catalyzes the guanosine or adenosine mononucleotide-dependent reversible conversion of PEP and oxaloacetic acid. In many bacteria, this enzyme is essential for their growth or survival primarily when grown on organic acids as sole carbon and energy source. Towards this, S. cummioxidans strain 35Y harbours one gene coding for a putative PEP carboxykinase (Table J in S1 Text).
Biosynthesis of industrially relevant secondary metabolites
The class Gammaproteobacteria includes several industrially relevant species that produce a wide range of economically-important metabolites, including PHAs, xanthan, xanthomonadin etc. PHA granules are composed of a polymer core which is surrounded by a complex proteinaceous surface layer consisting of at least five types of PHA granule-associated proteins (PGAPs) [45]. S. cummioxidans strain 35Y was predicted to harbour the structural genes, including type III PHA synthase (PhaC-PhaE) and a transcriptional regulator (PhaR), for the biosynthesis of short-chain length PHAs (Table K in S1 Text) using comparative genomics. However, genes for other PHA granule associated proteins PGAPs, such as phasins (PhaPs) [46, 47] and PHB depolymerases [PhaZ(a/d)s] [48, 49], involved in PHA accumulation and homeostasis, were not found. This pointed to the absence of a complete PHA granules assembly pathway and was in agreement with the inability to detect PHA granules upon cultivation of the cells under otherwise PHA-permissive conditions. (Figure Ba in S1 Text). Also, a complete set of genes for the biosynthesis of other carbon storage materials, including granulose (Table L in S1 Text) and cellulose (Table M in S1 Text) were predicted. In addition, a few genes of the xanthan gum biosynthesis pathway were predicted in the draft genome of S. cummioxidans strain 35Y (Table N in S1 Text).
Based on genome analysis, the draft genome of S. cummioxidans strain 35Y was predicted to harbour polyphosphate kinase (ppk) and polyphosphatase (ppx) genes involved in the biosynthesis of polyP containing granules (Table O in S1 Text). The ability to synthesize polyP was confirmed by staining of the cells with DAPI and the appearance of bright fluorescent granules in the cells (Figure Bb in S1 Text).
All genes necessary for the complete biosynthetic pathway of xanthomonadin (Figure C and Table P in S1 Text) were predicted in the draft genome of S. cummioxidans strain 35Y using comparative genomics. Xanthomonadin is a characteristic yellow pigment produced by the members of the genus Xanthomonas. The presence of xanthomonadin had been previously confirmed by solvent-extraction of a yellow compound that revealed an absorption maximum at 441 nm similar to that of xanthomonadin of authentic Xanthomonas species [50]. An m/z value of 560 and a molecular formula of C27H28Br2O3 was determined by mass spectrometry of the thin layer chromatography (TLC) purified pigment. These properties corresponded well with the xanthomonadin pigment group-7 [51] and explain the typical yellow colour of S. cummioxidans strain 35Y colonies on solid media.
Conclusions
This study characterized strain 35Y based on genotypic, physiological, and biochemical features and supported its description as a novel species of the genus Steroidobacter, for which the name Steroidobacter cummioxidans sp. nov., strain 35YT is proposed. However, it has not escaped our notice that some of the characteristic physiological, biochemical, and genomic features of strain 35Y presented in this study can be used as major differentiating features for not only species demarcation but also for the genus demarcation of strain 35Y from the genus Steroidobacter. However, this requires further support from comprehensive taxonomic analysis using additional experimental and in silico data. Further, a comprehensive in silico exploration of the draft genome of S. cummioxidans strain 35Y has led to the elucidation of the putative pathway for the break-down of NR by a Gram-negative rubber degrading species which differs from that of Gram-positive bacteria primarily in the nature of the uptake system for the primary NR-degradation products. The uptake system of S. cummioxidans strain 35Y has to operate with a product of a defined shorter length (acid of tri-isoprenoid cleavage product ODTD) in comparison to the acids of tetra- and higher-oligoisoprenoids of Gram-positive rubber degraders. In addition, S. cummioxidans strain 35Y is also predicted to possess a biosynthetic potential to synthesize secondary metabolites which can have wide industrial applications. In brief, the present study is significant in terms of characterizing a novel species of a Gram-negative bacteria and elucidating its NR-degradation pathway which will aid future studies to exploit the biodegradation of rubber.
Supporting information
(XLS)
(DOCX)
Acknowledgments
This work was supported by IIT Mandi and the Deutsche Forschungsgemeinschaft (Je152/17). TP acknowledges IIT Mandi and University Stuttgart, Germany for this collaborative project and financial and technical support. VS acknowledges the Ministry of Human Resource Development (MHRD), India for providing the research fellowship. We also thank Bernhard Schink, University of Konstanz, Germany, for helpful advice and discussion on taxonomic nomenclature.
Data Availability
All relevant data are within the paper and its Supporting Information files except for the bacterial genome data which is submitted to the NCBI GenBank database (accession number LSRW00000000) as well as to the Figshare repository (https://doi.org/10.6084/m9.figshare.6226466).
Funding Statement
This work was supported by the Dieter Jendrossek recieved the grant with reference number, Je 152/17-1 from the funding agency "Deutsche Forschungsgemeinschaft (http://www.dfg.de/.)" and Vikas Sharma is recipient of research fellowship from Ministry of Human Resource Development (MHRD), India (http://mhrd.gov.in/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Malaysian Rubber Board. Lembaga Getah Malaysia. Natural Rubber Statistics 2016. Malaysia.
- 2.Ranta PM, Ownby DR. A review of natural-rubber latex allergy in health care workers. Clin Infect Dis. 2004;38:252–256. doi: 10.1086/380789 [DOI] [PubMed] [Google Scholar]
- 3.Jendrossek D, Tomasi G, Kroppenstedt RM. Bacterial degradation of natural rubber: a privilege of actinomycetes? FEMS Microbiol Lett. 1997;150:179–188. [DOI] [PubMed] [Google Scholar]
- 4.Rose K, Tenberge KB, Steinbüchel A. Identification and characterization of genes from Streptomyces sp. strain K30 responsible for clear zone formation on natural rubber latex and poly (cis-1, 4-isoprene) rubber degradation. Biomacromolecules. 2005;6:180–188. doi: 10.1021/bm0496110 [DOI] [PubMed] [Google Scholar]
- 5.Birke J, Röther W, Jendrossek D. Latex clearing protein (Lcp) of Streptomyces sp. strain K30 is a b-type cytochrome and differs from rubber oxygenase A (RoxA) in its biophysical properties. Appl Environ Microbiol. 2015;81:3793–3799. doi: 10.1128/AEM.00275-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Röther W, Austen S, Birke J, Jendrossek D. Cleavage of Rubber by the Latex Clearing Protein (Lcp) of Streptomyces sp. Strain K30: Molecular Insights. Appl Environ Microbiol. 2016;82:6593–6602. doi: 10.1128/AEM.02176-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hiessl S, Schuldes J, Thürmer A, Halbsguth T, Bröker D, Angelov A, et al. Involvement of two latex-clearing proteins during rubber degradation and insights into the subsequent degradation pathway revealed by the genome sequence of Gordonia polyisoprenivorans strain VH2. Appl Environ Microbiol. 2012;78:2874–2887. doi: 10.1128/AEM.07969-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luo Q, Hiessl S, Poehlein A, Steinbüchel A. Microbial gutta-percha degradation shares common steps with rubber degradation by Nocardia nova SH22a. Appl Environ Microbiol. 2013;79:1140–1149. doi: 10.1128/AEM.03016-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luo Q, Hiessl S, Poehlein A, Daniel R, Steinbüchel A. Insights into the microbial degradation of rubber and gutta-percha by analysis of the complete genome of Nocardia nova SH22a. Appl Environ Microbiol. 2014;80:3895–3907. doi: 10.1128/AEM.00473-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watcharakul S, Röther W, Birke J, Umsakul K, Hodgson B, Jendrossek D. Biochemical and spectroscopic characterization of purified Latex Clearing Protein (Lcp) from newly isolated rubber degrading Rhodococcus rhodochrous strain RPK1 reveals novel properties of Lcp. BMC Microbiol. 2016;16:92 doi: 10.1186/s12866-016-0703-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rose K, Steinbüchel A. Biodegradation of natural rubber and related compounds: recent insights into a hardly understood catabolic capability of microorganisms. Appl Environ Microbiol. 2005;71:2803–2812. doi: 10.1128/AEM.71.6.2803-2812.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsuchii A, Takeda K. Rubber-degrading enzyme from a bacterial culture. Appl Environ Microbiol. 1990;56:269–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Imai S, Yoshida R, Endo Y, Fukunaga Y, Yamazoe A, Kasai D, et al. Rhizobacter gummiphilus sp. nov., a rubber-degrading bacterium isolated from the soil of a botanical garden in Japan. J Gen Appl Microbiol. 2013;59:199–205. [DOI] [PubMed] [Google Scholar]
- 14.Kasai D, Imai S, Asano S, Tabata M, Iijima S, Kamimura N, et al. Identification of natural rubber degradation gene in Rhizobacter gummiphilus NS21. Biosci Biotechnol Biochem. 2017;81:614–620. doi: 10.1080/09168451.2016.1263147 [DOI] [PubMed] [Google Scholar]
- 15.Braaz R, Fischer P, Jendrossek D. Novel type of heme-dependent oxygenase catalyzes oxidative cleavage of rubber (poly-cis-1, 4-isoprene). Appl Environ Microbiol. 2004;70:7388–7395. doi: 10.1128/AEM.70.12.7388-7395.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Birke J, Röther W, Jendrossek D. RoxB is a Novel Type of Rubber Oxygenase that Combines Properties of Rubber Oxygenase RoxA and Latex Clearing Protein (Lcp). Appl Environ Microbiol. 2017;AEM-00721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmitt G, Seiffert G, Kroneck PM, Braaz R, Jendrossek D. Spectroscopic properties of rubber oxygenase RoxA from Xanthomonas sp., a new type of dihaem dioxygenase. Microbiology. 2010;156:2537–2548. doi: 10.1099/mic.0.038992-0 [DOI] [PubMed] [Google Scholar]
- 18.Seidel J, Schmitt G, Hoffmann M, Jendrossek D, Einsle O. Structure of the processive rubber oxygenase RoxA from Xanthomonas sp. Proc Natl Acad Sci U S A. 2013;110:13833–13838. doi: 10.1073/pnas.1305560110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Birke J, Röther W, Jendrossek D. Latex clearing protein (Lcp) of Streptomyces sp. strain K30 is a b-type cytochrome and differs from rubber oxygenase A (RoxA) in its biophysical properties. Appl Environ Microbiol. 2015;81:3793–3799. doi: 10.1128/AEM.00275-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ilcu L, Röther W, Birke J, Brausemann A, Einsle O, Jendrossek D. Structural and Functional Analysis of Latex Clearing Protein (Lcp) Provides Insight into the Enzymatic Cleavage of Rubber. Sci Rep. 2017;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yikmis M, Steinbüchel A. Importance of the latex‐clearing protein (Lcp) for poly (cis‐1, 4‐isoprene) rubber cleavage in Streptomyces sp. K30. Microbiol Open. 2012;1:13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chevreux B, Wetter T, Suhai S, editors. Genome sequence assembly using trace signals and additional sequence information. German Conference on Bioinformatics. 1999;99: 45–56. [Google Scholar]
- 23.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–120. [DOI] [PubMed] [Google Scholar]
- 25.Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x [DOI] [PubMed] [Google Scholar]
- 26.Rambaut A: FigTree-v1.4.2. [http://tree.bio.ed.ac.uk/software/figtree/]
- 27.Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57:81–91. doi: 10.1099/ijs.0.64483-0 [DOI] [PubMed] [Google Scholar]
- 28.Richter M, Rosselló-Móra R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A. 2009;106:19126–19131. doi: 10.1073/pnas.0906412106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spiekermann P, Rehm BH, Kalscheuer R, Baumeister D, Steinbüchel A. A sensitive, viable-colony staining method using Nile red for direct screening of bacteria that accumulate polyhydroxyalkanoic acids and other lipid storage compounds. Arch Microbiol. 1999;171:73–80. [DOI] [PubMed] [Google Scholar]
- 30.Brandl H, Gross RA, Lenz RW, Fuller RC. Pseudomonas oleovorans as a source of poly (β-hydroxyalkanoates) for potential applications as biodegradable polyesters. Appl Environ Microbiol. 1988;54:1977–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tijssen J, Beekes H, Van Steveninck J. Localization of polyphosphates in Saccharomyces fragilis, as revealed by 4′, 6-diamidino-2-phenylindole fluorescence. Biochim Biophys Acta. 1982;721:394–398. [DOI] [PubMed] [Google Scholar]
- 32.Kim M, Oh HS, Park SC, Chun J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int J Syst Evol Microbiol. 2014;64:346–351. doi: 10.1099/ijs.0.059774-0 [DOI] [PubMed] [Google Scholar]
- 33.Jyothsna TS, Tushar L, Sasikala C, Ramana CV. Paraclostridium benzoelyticum gen. nov., sp. nov., isolated from marine sediment and reclassification of Clostridium bifermentans as Paraclostridium bifermentans comb. nov. Proposal of a new genus Paeniclostridium gen. nov. to accommodate Clostridium sordellii and Clostridium ghonii. Int J Syst Evol Microbiol. 2016;66:1268–1274. doi: 10.1099/ijsem.0.000874 [DOI] [PubMed] [Google Scholar]
- 34.Cárdenas JP, Ortiz R, Norris PR, Watkin E, Holmes DS. Reclassification of ‘Thiobacillus prosperus’ Huber and Stetter 1989 as Acidihalobacter prosperus gen. nov., sp. nov., a member of the family Ectothiorhodospiraceae. Int J Syst Evol Microbiol. 2015;65:3641–3644. doi: 10.1099/ijsem.0.000468 [DOI] [PubMed] [Google Scholar]
- 35.Meier-Kolthoff JP, Klenk HP, Göker M. Taxonomic use of DNA G+ C content and DNA–DNA hybridization in the genomic age. Int J Syst Evol Microbiol. 2014;64:352–356. doi: 10.1099/ijs.0.056994-0 [DOI] [PubMed] [Google Scholar]
- 36.Jendrossek D, Reinhardt S. Sequence analysis of a gene product synthesized by Xanthomonas sp. during growth on natural rubber latex. FEMS Microbiol Lett. 2003;224:61–65. [DOI] [PubMed] [Google Scholar]
- 37.Stevens JM, Mavridou DA, Hamer R, Kritsiligkou P, Goddard AD, Ferguson SJ. Cytochrome c biogenesis System I. FEBS J. 2011;278:4170–4178. doi: 10.1111/j.1742-4658.2011.08376.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malinverni JC, Silhavy TJ. An ABC transport system that maintains lipid asymmetry in the gram-negative outer membrane. Proc Natl Acad Sci U S A. 2009;106:8009–8014. doi: 10.1073/pnas.0903229106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wiener MC, Horanyi PS. How hydrophobic molecules traverse the outer membranes of Gram-negative bacteria. Proc Natl Acad Sci U S A. 2011;108:10929–10930. doi: 10.1073/pnas.1106927108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Black PN, DiRusso CC. Transmembrane movement of exogenous long-chain fatty acids: proteins, enzymes, and vectorial esterification. Microbiol Mol Biol Rev. 2003;67:454–472. doi: 10.1128/MMBR.67.3.454-472.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bode HB, Zeeck A, Plückhahn K, Jendrossek D. Physiological and chemical investigations into microbial degradation of synthetic poly (cis-1, 4-isoprene). Appl Environ Microbiol. 2000;66:3680–3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riedel C, Rittmann D, Dangel P, Möckel B, Petersen S, Sahm H, Eikmanns, BJ. Characterization of the phosphoenolpyruvate carboxykinase gene from Corynebacterium glutamicum and significance of the enzyme for growth and amino acid production. J Mol Microbiol Biotechnol. 2001;3:573–583. [PubMed] [Google Scholar]
- 43.Hyeon JE, Kang DH, Kim YI, You SK, Han SO. GntR-type transcriptional regulator PckR negatively regulates the expression of phosphoenolpyruvate carboxykinase in Corynebacterium glutamicum. J Bacteriol. 2012;194:2181–2188. doi: 10.1128/JB.06562-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang J, Kalhan SC, Hanson RW. What is the metabolic role of phosphoenolpyruvate carboxykinase?. J Biol Chem. 2009;284:27025–27029. doi: 10.1074/jbc.R109.040543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jendrossek D, Pfeiffer D. New insights in the formation of polyhydroxyalkanoate granules (carbonosomes) and novel functions of poly (3‐hydroxybutyrate). Environ Microbiol. 2014;16:2357–2373. doi: 10.1111/1462-2920.12356 [DOI] [PubMed] [Google Scholar]
- 46.Yoshida K-i, Takemoto Y, Sotsuka T, Tanaka K, Takenaka S. PhaP phasins play a principal role in poly-β-hydroxybutyrate accumulation in free-living Bradyrhizobium japonicum. BMC Microbiol. 2013;13:290 doi: 10.1186/1471-2180-13-290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Steinbüchel A, Aerts K, Liebergesell M, Wieczorek R, Babel W, Föllner C, et al. Considerations on the structure and biochemistry of bacterial polyhydroxyalkanoic acid inclusions. Can J Microbiol. 1995;41:94–105. [DOI] [PubMed] [Google Scholar]
- 48.Brigham CJ, Reimer EN, Rha C, Sinskey AJ. Examination of PHB depolymerases in Ralstonia eutropha: further elucidation of the roles of enzymes in PHB homeostasis. AMB Express. 2012;2:26 doi: 10.1186/2191-0855-2-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jendrossek D, Handrick R. Microbial degradation of polyhydroxyalkanoates. Annual Rev Microbiol. 2002;56:403–432. [DOI] [PubMed] [Google Scholar]
- 50.Kerkhoff K. Ph.D. thesis. Molekularbiologische und biochemische Untersuchungen zum bakteriellen Naturkautschuk-Abbau, sowie Charakterisierung eines dazu behähigten Bakteriums: Niedersächsische Staats-und Universitätsbibliothek; 2000.
- 51.Starr MP, Jenkins CL, Bussey LB, Andrewes AG. Chemotaxonomic significance of the xanthomonadins, novel brominated aryl-polyene pigments produced by bacteria of the genus Xanthomonas. Arch Microbiol. 1977;113:1–9. [DOI] [PubMed] [Google Scholar]
- 52.Gong ZL, Zhang CF, Jin R, Zhang YQ. Steroidobacter flavus sp. nov., a microcystin-degrading Gammaproteobacterium isolated from soil. Antonie Van Leeuwenhoek. 2016;109:1073–1079. doi: 10.1007/s10482-016-0706-5 [DOI] [PubMed] [Google Scholar]
- 53.Sakai M, Hosoda A, Ogura K, Ikenaga M. The growth of Steroidobacter agariperforans sp. nov., a novel agar-degrading bacterium isolated from soil, is enhanced by the diffusible metabolites produced by bacteria belonging to Rhizobiales. Microbes Environ. 2014;29:89–95. doi: 10.1264/jsme2.ME13169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fahrbach M, Kuever J, Remesch M, Huber BE, Kämpfer P, Dott W, et al. Steroidobacter denitrificans gen. nov., sp. nov., a steroidal hormone-degrading gammaproteobacterium. Int J Syst Evol Microbiol. 2008;58:2215–2223. doi: 10.1099/ijs.0.65342-0 [DOI] [PubMed] [Google Scholar]
- 55.Schöner TA, Fuchs SW, Reinhold-Hurek B, Bode HB. Identification and biosynthesis of a novel xanthomonadin-dialkylresorcinol-hybrid from Azoarcus sp. BH72. PloS One. 2014;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLS)
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files except for the bacterial genome data which is submitted to the NCBI GenBank database (accession number LSRW00000000) as well as to the Figshare repository (https://doi.org/10.6084/m9.figshare.6226466).