

Abstract
Type IV pili are expressed by a wide range of prokaryotes, including the opportunistic pathogen Pseudomonas aeruginosa. These flexible fibres mediate twitching motility, biofilm maturation, surface adhesion, and virulence. The pilus is composed mainly of major pilin subunits while the low abundance minor pilins FimU-PilVWXE and the putative adhesin PilY1 prime pilus assembly and are proposed to form the pilus tip. The minor pilins and PilY1 are encoded in an operon that is positively regulated by the FimS-AlgR two-component system. Independent of pilus assembly, PilY1 was proposed to be a mechanosensory component that—in conjunction with minor pilins—triggers up-regulation of acute virulence phenotypes upon surface attachment. Here, we investigated the link between the minor pilins/PilY1 and virulence. pilW, pilX, and pilY1 mutants had reduced virulence towards Caenorhabditis elegans relative to wild type or a major pilin mutant, implying a role in pathogenicity that is independent of pilus assembly. We hypothesized that loss of specific minor pilins relieves feedback inhibition on FimS-AlgR, increasing transcription of the AlgR regulon and delaying C. elegans killing. Reporter assays confirmed that FimS-AlgR were required for increased expression of the minor pilin operon upon loss of select minor pilins. Overexpression of AlgR or its hyperactivation via a phosphomimetic mutation reduced virulence, and the virulence defects of pilW, pilX, and pilY1 mutants required FimS-AlgR expression and activation. We propose that PilY1 and the minor pilins inhibit their own expression, and that loss of these proteins leads to FimS-mediated activation of AlgR that suppresses expression of acute-phase virulence factors and delays killing. This mechanism could contribute to adaptation of P. aeruginosa in chronic lung infections, as mutations in the minor pilin operon result in the loss of piliation and increased expression of AlgR-dependent virulence factors–such as alginate–that are characteristic of such infections.
Author summary
Pseudomonas aeruginosa causes dangerous infections, including chronic lung infections in cystic fibrosis patients. It uses many strategies to infect its hosts, including deployment of grappling hook-like fibres called type IV pili. Among the components involved in assembly and function of the pilus are five proteins called minor pilins that—along with a larger protein called PilY1—may help the pilus attach to surfaces. In a roundworm infection model, loss of PilY1 and specific minor pilins delayed killing, while loss of other pilus components did not. We traced this effect to increased activation of the FimS-AlgR regulatory system that inhibits the expression of virulence factors used early in infection, while positively regulating chronic infection traits such as alginate production, a phenotype called mucoidy. A disruption in the appropriate timing of FimS-AlgR-dependent virulence factor expression when select minor pilins or PilY1 are missing may explain why those pilus-deficient mutants have reduced virulence compared with others whose products are not under FimS-AlgR control. Increased FimS-AlgR activity upon loss of PilY1 and specific minor pilins could help to explain the frequent co-occurrence of the non-piliated and mucoid phenotypes that are hallmarks of chronic P. aeruginosa lung infections.
Introduction
Pseudomonas aeruginosa is a Gram-negative opportunistic pathogen, recently listed as one of the highest priority antimicrobial-resistant threats by the World Health Organization, due to its intrinsic antibiotic resistance and recalcitrance to therapy [1]. Among its virulence factors are filamentous surface appendages called type IV pili (T4P), sophisticated biological nanomachines that are broadly distributed among bacteria and archaea [2, 3]. In P. aeruginosa, T4P facilitate surface and host cell adhesion, colonization, biofilm maturation, virulence, and twitching, a form of surface-associated motility facilitated by cycles of extension, adhesion, and retraction of T4P fibres [3–11]. T4P are composed of hundreds to thousands of copies of small proteins called major pilins (PilA in P. aeruginosa) along with the low abundance minor pilins (MPs) FimU-PilVWXE [12–16]. The MPs are encoded in a polycistronic operon with the pilY1 gene that codes for a large ~125 kDa non-pilin protein. The operon is positively regulated by the virulence factor regulator Vfr, and the two-component system (TCS) FimS (AlgZ)-AlgR. FimS is a predicted histidine sensor kinase while AlgR is a response regulator that promotes expression of genes important for biofilms and chronic cystic fibrosis (CF) lung infections [17–21]. The N-termini of immature pilins are cleaved and methylated at the cytoplasmic face of the inner membrane by the prepilin peptidase, PilD, while PilY1 may be processed by signal peptidase 1 [22–25]. Mature pilins are polymerized into a T4P fibre via an envelope-spanning assembly machinery, where individual PilA subunits are added or removed at the platform protein, PilC, via action of the ATPases PilB and PilT, respectively [2, 26].
The MPs and PilY1 are required for T4P function in several bacterial species, including P. aeruginosa, Escherichia coli, Neisseria meningitidis, N. gonorrhoeae, and Myxococcus xanthus [12–15, 27–30]. PilY1 and the MPs were originally proposed to oppose pilus retraction, as a few surface pili remain in pilY1 and MP mutants when retraction is blocked via deletion of pilT [23, 28, 29, 31, 32]. We recently showed that when T4P MPs are missing, the equivalent minor pseudopilins of the Xcp type II secretion system can pilus prime extension in the pilT background, and that deletion of both sets of minor components abolishes pilus assembly [24]. We also demonstrated that PilY1 and the MPs are present in sheared pili, and that the loss of PilV, PilW, PilX, or PilY1 excludes the other three components from the pilus [24]. Thus, PilVWXY1 are proposed to form a core assembly-initiation subcomplex, while FimU and PilE are thought to connect this complex to PilA. Initiation of assembly with subsequent addition of multiple PilA subunits would place the MPs at the pilus tip, with PilY1 –the largest component–at the distal position, supporting the hypothesis that PilY1 is a T4P-associated adhesin [31].
PilY1 and the MPs (and their regulators FimS-AlgR) are required for T4P biogenesis, and therefore T4P-mediated functions [12–15, 17, 19]. However, recent studies hinted at more enigmatic roles of PilWXY1 in virulence. Bohn et al. [33] showed that in a non-piliated P. aeruginosa background, subsequent loss of pilY1 reduced virulence in a Caenorhabditis elegans fast killing assay and in a mouse airway infection model, and increased resistance to killing by neutrophils. Thus, PilY1 has a role in virulence that does not require functional pili. Other studies using C. elegans infection models suggested that MP and pilY1 mutants had attenuated virulence relative to WT, and in one case, to a non-piliated mutant [34–37]. Recently, Siryaporn et al. [38] showed that PilWXY1 were required for surface-activated virulence towards amoebae, while other non-piliated mutants had WT virulence. The N-terminal region of PilY1 has weak sequence similarity to the eukaryotic von Willebrand factor A (VWFa) domain, which can be deformed by shear forces [39]. In-frame deletion of this domain from PilY1 allowed normally avirulent planktonic cells to kill amoebae [38]. PilY1 was therefore proposed to be a mechanosensor, where deformation of its VWFa domain upon surface interaction led–by an as-yet unknown mechanism–to increased expression of virulence factors. One important caveat of that study was that an algR mutant (which lacks PilY1 and the MPs) had WT virulence towards amoebae [38].
Deformation of PilA subunits by tensile forces acting upon surface-attached pili was also proposed as a possible way to signal attachment. Detection of partly unfolded pilins by the Pil-Chp chemotaxis system could lead to increased cyclic adenosine monophosphate (cAMP) synthesis via the CyaB adenylate cyclase [40, 41]. cAMP is bound by Vfr, a key transcription factor that promotes expression of virulence factors involved in motility, attachment, and secretion [20, 40, 41]. fimS-algR transcription is activated by Vfr, leading to increased transcription of fimU-pilVWXY1E [40]. PilVWXY1 were proposed to repress their own expression in an AlgR-dependent manner, as the loss of pilV, pilW, pilX, or pilY1 led to elevated expression of the MP operon and fimS-algR [23, 33, 38, 40]. The mechanism of this putative feedback inhibition is largely uncharacterized, but was speculated to involve FimS [40].
Once expression of the MP operon is activated, extracellular PilY1 may sense surface association and transduce this information through the T4P assembly machinery [38, 40]. This signal is thought to activate an inner membrane-localized diguanylate cyclase, SadC, to increase levels of c-di-GMP, promoting expression of genes associated with a biofilm lifestyle, while repressing early-phase virulence traits such as swarming motility [40, 42]. This model was supported by studies demonstrating that loss of pilW, pilX, or pilY1 in a high-c-di-GMP background resulted in hyper-swarming and reduced c-di-GMP levels, as measured by liquid chromatography-mass spectrometry of extracts from surface-grown cells [39, 43]. Rodesney et al. [44] showed that c-di-GMP levels increased in response to shear forces, and that functional T4P were required for this phenomenon, further supporting this hypothesis. However, unlike pilW, pilX, and pilY1 mutants, a sadC mutant had WT virulence towards amoebae, suggesting the PilWXY1-SadC pathway may be important for surface sensing, but not necessarily for surface-activated virulence [38].
Although PilY1 and the MPs clearly influence virulence, the underlying mechanism remains to be established [33–36, 38, 45]. We hypothesized that a subset of these components represses FimS activity, such that loss of pilW, pilX, or pilY1 activates FimS-AlgR, shifting the bacteria to a less pathogenic phenotype typically associated with chronic infection. We found that slow killing (SK) of C. elegans by pilW, pilX, and pilY1 mutants was significantly delayed compared to WT or a pilA mutant, and this delay was dependent on FimS-AlgR, because double mutants had WT killing kinetics. Hyperactivation (via phospho-mimetic point mutation) or overexpression of AlgR alone was sufficient to delay killing. Together, these data are consistent with a model where loss of PilWXY1 relieves feedback inhibition on expression of the AlgR regulon, resulting in dysregulation of virulence factors that are important for C. elegans pathogenesis.
Results
PilWXY1 modulate T4P-independent virulence in PA14 and PAO1
Specific genes in the MP operon were reported to be important for virulence in amoebae, nematodes, and mouse models, but those studies were done using different strains of P. aeruginosa [33–36, 38, 45]. We first confirmed these results in the C. elegans SK model, using two well-studied strains. SK assays were performed using PA14 with deletions of pilA, fimU, pilV, pilW, pilX, pilY1, or pilE (Fig 1A). An E. coli OP50 plate was included as a negative control for pathogenicity; worms began to senesce on these plates around day 7–8, consistent with published data regarding temperature-dependent effects on lifespan [46]. As worms at later time points were at increased risk of death due to ageing in addition to P. aeruginosa infection, statistical significance was assessed using the Gehan-Breslow-Wilcoxon test, which places greater weight on earlier time points [47]. A pilA (major pilin) mutant was slightly less pathogenic than WT; subsequent comparisons were made relative to pilA, since all mutants lack pili. fimU and pilE mutants were more pathogenic than the pilA mutant, similar to WT. In contrast, pilW, pilX, and pilY1 mutants were less pathogenic than the pilA mutant, suggesting that delayed killing was not due to loss of functional T4P. Virulence of the pilV mutant was similar to the pilA mutant. The twitching and virulence defects of pilW, pilX, and pilY1 mutants could be partially complemented by expression of the relevant gene in trans (S1 Fig). The stoichiometry of PilY1 and the MPs is important for optimal T4P function [23], which may explain the lack of full complementation. To verify that these phenotypes were not strain-specific, we tested PAO1 transposon-insertion mutants of pilA, fimU, pilV, pilW, pilX, pilY1, and pilE in the SK assay (Fig 1B). Similar to the results in PA14, PilWXY1 were important for T4P-independent virulence. However, the fimU and pilV mutants killed nematodes more slowly than pilA; the PA14 and PAO1 MPs are divergent (61–75% amino acid similarity), so it is possible that FimU and PilV function slightly differently in PAO1 versus PA14 [48]. To focus on genes that were generally important for virulence of P. aeruginosa, we undertook studies of the mechanism responsible for delayed killing of C. elegans by the pilW, pilX, and pilY1 mutants.
Fig 1. PilWXY1 contribute to T4P-independent virulence.

(A) SK assays for PA14 pilA, fimU, pilV, pilW, pilX, pilY1, and pilE mutants. Synchronized L4 worms were seeded onto SK plates and scored for death every 24 h, then plotted as “percent survival” over the course of the assay. “Day” represents the number of days after L4 on which the plates were scored. PA14 fimU and pilE mutants had similar virulence to WT, pilA and pilV mutants were slightly less virulent than WT, and pilW, pilX, and pilY1 mutants killed more slowly than all other strains tested. (B) SK assays for PAO1 pilA, fimU, pilV, pilW, pilX, pilY1, and pilE mutants. The PAO1 pilE mutant had similar virulence to WT, the pilA mutant was slightly less virulent, and fimU, pilV, pilW, pilX, and pilY1 mutants showed significant delays in killing. In (A) and (B), asterisks indicate strains that were significantly different from a pilA mutant by Gehan-Breslow-Wilcoxon test at p = 0.05 (p = 0.00625 with a Bonferroni correction), n = 3 trials.
PilWXY1 promote virulence in a SadC-independent manner
PilWXY1 were previously proposed to increase c-di-GMP production by SadC, such that loss of pilW, pilX, or pilY1 resulted in a biofilm-deficient phenotype, indicative of low intracellular c-di-GMP [39, 40, 43]. Therefore, we hypothesized that biofilm defects of pilW, pilX, and pilY1 might impede their ability to colonize the C. elegans gut, leading to delayed killing. The PA14 and PAO1 parent strains and their cognate pilA, fimU, pilV, pilW, pilX, pilY1, and pilE mutants formed negligible levels of biofilm in liquid SK medium, chosen to approximate the growth conditions used for the SK assay (S2 Fig). To assess the levels of cyclic-di-GMP in these strains, we constructed a luminescence-based cdrA promoter reporter based on an extensively-characterized green fluorescent protein-based reporter system [44, 49–54]. cdrA promoter activity has been positively correlated with c-di-GMP levels, as measured by liquid chromatography-mass spectrometry [49, 51, 53, 54]. We verified that overexpression of SadC led to a ~60-fold increase in cdrA promoter activity, while overexpression of AlgR, which positively regulates genes that promote c-di-GMP production [55, 56], led to a ~2-fold increase in promoter activity that was enhanced to ~4-fold when algR expression was increased with 0.05% L-arabinose (Fig 2A). Deletion of sadC or algR led to a ~2-fold decrease in cdrA promoter activity relative to WT. cdrA promoter activity in WT is expected to be relatively low in liquid media because c-di-GMP levels increase upon surface attachment [43]. Compared to WT, pilW, pilX, and pilY1 had ~3-fold lower cdrA promoter activity, indicative of reduced c-di-GMP (Fig 2B). These results are consistent with reports that PilWXY1 promote c-di-GMP production via SadC [39, 40, 43]. We next investigated whether SadC was required for virulence, as would be predicted if decreased virulence in pilW, pilX, and pilY1 mutants was due to dysregulation of SadC activity. A small decrease in virulence towards C. elegans was previously reported for a PA14 sadC mutant [57]; however, we saw no difference between WT and sadC mutants in the PA14 and PAO1 backgrounds (S3 Fig). Further, overexpression of SadC led to a hyper-biofilm phenotype in vitro in SK medium, but a slight delay in killing, demonstrating that the amount of biofilm formed in vitro does not correlate with virulence in C. elegans (Fig 3). Although the exact mechanisms of P. aeruginosa pathogenesis in C. elegans are not fully understood, biofilms were suggested to be important for establishment of infection [57–59]. Our in vitro data suggests that biofilms may not be a major contributor to P. aeruginosa pathogenesis in this model, but direct visualization and quantification of biofilms in the nematode gut will be required to support this conclusion.
Fig 2. pilW, pilX, and pilY1 mutants have reduced cdrA promoter activity.

(A) cdrA promoter activity in PA14 sadC and algR deletion and overexpression strains. pMS402-PcdrA, containing the lux genes under expression of the cdrA promoter, was introduced into strains of interest, along with pBADGr (vector-only control), pBADGr-sadC, or pBADGr-algR. Assays were set up in technical triplicate in SK media, with or without 0.05% L-arabinose to induce expression of the pBADGr promoter, and measurements were taken every 15 min over 5 h. Loss of sadC or algR led to a subtle decrease in cdrA promoter activity, while SadC overexpression led to a dramatic increase in cdrA promoter activity. Overexpression of AlgR also led to a subtle increase in cdrA promoter activity that was enhanced upon addition of L-arabinose. n = 3 trials. (B) cdrA promoter activity in PA14 pilA, fimU, pilV, pilW, pilX, pilY1, and pilE mutants. Loss of pilW, pilX, or pilY1 led to a decrease in cdrA promoter activity. n = 3 trials.
Fig 3. SadC promotes biofilm formation but is not required for virulence.

(A) Biofilm assays for sadC deletion and overexpression strains. PA14 sadC biofilm levels were similar to WT. Expression of SadC in trans from a multicopy plasmid led to increased biofilm formation relative to WT at 0% (due to leaky promoter) and 0.05% L-arabinose, p < 0.001. Significance was determined by one-way ANOVA followed by Dunnett post-test relative to PA14 + pBADGr, n = 3. (B) SK assays for sadC deletion and overexpression strains. Overexpression of SadC led to a subtle but reproducible delay in killing relative to WT at 0% L-arabinose. A sadC mutant was similar to WT. Asterisks indicate strains that were significantly different from PA14 + pBADGr by Gehan-Breslow-Wilcoxon test at p = 0.05 (p = 0.0125 with a Bonferroni correction), n = 3.
PilVWXY1 repress expression of the MP operon
After ruling out involvement of the SadC pathway, we explored the potential role of FimS-AlgR in PilWXY1-mediated modulation of killing kinetics. Informed by previous work in our laboratory showing that the sensor kinase PilS of the PilSR TCS interacts directly with PilA in the inner membrane to decrease PilR-dependent major pilin expression [60], we hypothesized that FimS interacts with one or more MPs, and that loss of that interaction could lead to activation of AlgR and subsequent upregulation of the MP operon. Bacterial two-hybrid (BACTH) assays were used to identify potential interactions between FimS and PilA, FimU, PilV, PilW, PilX, or PilE (Fig 4A). We also screened for interaction of FimS and AlgR, which has been inferred but never demonstrated [19]. Interactions between FimS and each pilin were identified; however, based on our experience with PilS [60], binding is necessary but not sufficient for inhibition. We also demonstrated interaction of FimS and AlgR (Fig 4A), providing further support for the hypothesis that FimS is the sensor kinase for AlgR.
Fig 4. PilVWXY1 repress their expression via FimS-AlgR.

(A) BACTH assays for FimS, AlgR, PilA, and MPs. Protein fusions with T18 and T25 fragments of the CyaA adenylate cyclase were screened for interactions on MacConkey and LB + X-gal plates. FimS interacted with itself, AlgR, PilA, FimU, PilV, PilW, PilX, and PilE. Positive (+) or negative (-) interactions are indicated below each image, n = 3. (B) fimU promoter activity in PA14 pilA, fimU, pilV, pilW, pilX, pilY1, pilE, fimS, algR, pilY1 fimS, or pilY1 algR mutants. pMS402-PfimU, containing the fimU promoter upstream of the lux genes, was introduced into strains of interest. Loss of pilV, pilW, pilX, or pilY1 led to highly elevated fimU promoter activity. pilA and fimU mutants had moderately increased fimU promoter activity relative to WT. fimS and algR mutants had negligible luminescence, and loss of fimS or algR also reverted fimU promoter activity in the pilY1 mutant to baseline. n = 3 trials.
To decipher which MPs might modulate expression of the operon, we monitored expression from the fimU promoter using a luxCDABE reporter. Compared to WT PA14, there was a ~25-fold increase in luminescence in pilV, pilW, pilX, and pilY1 mutants, which was restored to WT by expressing the corresponding pilin in trans (Fig 4B, S4 Fig). fimU and pilA mutants had ~5-fold increased promoter activity, while that of a pilE mutant was comparable to WT. fimS and algR mutants had low baseline luminescence, ~10-fold lower than WT. To determine whether the increased promoter activity in pilV, pilW, pilX, and pilY1 mutants depended on FimS-AlgR, either fimS or algR was deleted in the pilY1 mutant background. The luminescence was ~10-fold lower than WT in the pilY1 algR double mutant, consistent with AlgR acting as a positive regulator of the MP operon [40]. Loss of fimS in the pilY1 mutant background also abolished fimU promoter activity (~10-fold lower than WT), supporting the idea that FimS may monitor PilVWXY1 and activate AlgR when their levels drop. Based on these data, PilA, FimU, and PilE are unlikely to modulate FimS-AlgR activity even though they can interact with FimS.
PilVWXY1 were previously proposed to form a complex in the inner membrane, such that loss of any one component destabilizes the others [24]. Since PilY1 is thought to be cleaved on the periplasmic side of the inner membrane, it is unlikely to interact directly with the transmembrane domains of FimS [24]. Thus, we suspected that high fimU promoter activity in the pilY1 mutant was due to reduced levels of one or more of the other pilins. To address this, we overexpressed FimU, PilV, PilW, PilX, or PilE in the pilY1 mutant and measured fimU promoter activity. All these strains had luminescence comparable to the pilY1 mutant (S4 Fig). Conversely, distinct effects have been observed in other studies upon overexpression of PilY1 [39, 40, 43]. Therefore, we overexpressed PilY1 in the pilW and pilX (high-luminescence) backgrounds; but PilY1 alone was insufficient to alter fimU promoter activity. Together, the data suggest that no individual component of the PilVWXY1 subcomplex is capable of modulating FimS activity when others are absent.
We also tested whether PilD processing of PilVWX was required for modulation of FimS activity. We constructed a pilD mutant, which lacks twitching motility because unprocessed pilins remain in the inner membrane [23, 61]. The absence of pilD had no impact on fimU promoter activity (S5 Fig), and a pilD mutant had virulence equivalent to a pilA mutant, likely attributable to its lack of T4P. Thus, PilVWX can modulate FimS activity in their unprocessed form.
Hyperactivation of AlgR delays killing
Because the results suggested that loss of PilWXY1 relieves feedback inhibition on FimS-AlgR, resulting in AlgR activation, we tested whether hyperactivation of AlgR alone could delay killing of C. elegans. We made chromosomal algRD54E phospho-mimetic point mutants [62] in both PA14 and PAO1 backgrounds. We also made algRD54A point mutants, as AlgR phosphorylation is required for transcription of a subset of genes in its regulon, including the MP operon [17, 62, 63]. We verified that the algRD54A mutant was defective for twitching motility, while the algRD54E mutant had WT twitching (S6 Fig). Unexpectedly, a fimS mutant retained ~50% twitching motility, in contrast to previous reports [18, 62]. In the absence of FimS, AlgR might be phosphorylated by small phosphate donors [64]. Based on the fimS data, we also questioned the assumption that AlgR phosphorylation was necessary for expression from the fimU promoter. When we overexpressed WT AlgR or AlgRD54A in the algR mutant (S6 Fig), its twitching defect was fully complemented by AlgR, and partially complemented (25%) by AlgRD54A. Thus, although it increases binding to the fimU promoter [17, 62], phosphorylation of AlgR is not essential for transcription of the MP operon.
SK assays were then performed for PA14 and PAO1 algRD54A and algRD54E mutants, plus PA14 and PAO1 fimS and algR deletion mutants. PA14 and PAO1 algRD54E mutants killed more slowly than the corresponding WT strains, while fimS, algR and algRD54A mutants had WT virulence (Fig 5A and 5B). Loss of FimS-AlgR decreases expression of the MPs and PilY1 and prevents pilus assembly [17, 40]. Because our data show that loss of FimS-AlgR (and thus MP expression) had no impact, we conclude that delayed killing of nematodes by pilW, pilX, and pilY1 mutants is due to inappropriately timed FimS-AlgR activation.
Fig 5. AlgR hyperactivation delays killing.

SK assays for (A) PA14 and (B) PAO1 fimS, algR, algRD54A, and algRD54E mutants. The fimS, algR, and algRD54A mutants had WT virulence, while the algRD54E mutants showed delays in killing. For (A) and (B), asterisks indicate strains that were significantly different from WT by Gehan-Breslow-Wilcoxon test at p = 0.05 (p = 0.01 with a Bonferroni correction), n = 3 trials.
Overexpression of AlgR delays killing
Increased transcription of fimS-algR in a pilY1 mutant relative to WT has been reported [38], suggesting that delayed killing could arise through expression of increased amounts of the FimS-AlgR TCS, as well as its activation. Therefore, we asked whether increased AlgR levels would attenuate virulence, as previously demonstrated in a mouse infection model [65]. When algR was expressed in trans from a multicopy plasmid in PA14 algR, killing was delayed compared to the vector control (Fig 6A). Because un-phosphorylated AlgR can also affect transcription of a subset of genes [66, 67], we tested the same mutant complemented with AlgRD54A. Complementation of the algR mutant with AlgRD54A resulted in a severe delay in killing relative to the vector-only control. Thus, AlgR hyperactivation and overexpression independently diminish P. aeruginosa virulence towards C. elegans. Lastly, as AlgR is a positive regulator of biofilm formation [17, 55, 56], we performed biofilm assays for PA14 algR complemented with AlgR or AlgRD54A. Expression of either variant led to hyper-biofilm formation (Fig 6B), further emphasizing that the ability of a strain to form biofilms in SK medium does not correlate with virulence in worms. Instead, we suggest that virulence factors repressed by FimS-AlgR are important for C. elegans SK, and an increase in AlgR levels and/or activity at the wrong time delays killing.
Fig 6. AlgR promotes biofilm formation and delays killing.

(A) SK assays for algR deletion and overexpression strains. Loss of algR led to a small increase in virulence, while overexpression of pBADGr-algR or pBADGr-algRD54A delayed killing at 0.05% L-arabinose. Asterisks indicate strains that were significantly different from PA14 + pBADGr by Gehan-Breslow-Wilcoxon test at p = 0.05 (p = 0.00833 with a Bonferroni correction), n = 3 trials. (B) Biofilm assays for algR deletion and overexpression strains. Microtiter plate biofilm assays were performed in liquid SK media over 24 h, in triplicate. Biofilms were stained with 1% crystal violet then solubilized in acetic acid. Loss of algR had no effect on biofilm formation. When grown at 0.05% L-arabinose, overexpression of pBADGr-algR or pBADGr-algRD54A increased biofilm formation, p < 0.001. Significance was determined by one-way ANOVA followed by Dunnett post-test relative to WT, n = 3 trials.
The virulence defects of pilW, pilX and pilY1 mutants are dependent on FimS-AlgR
To provide further support for this model, we asked whether the virulence defects of PA14 pilW, pilX, and pilY1 mutants required FimS-AlgR. We deleted fimS or algR in the pilW, pilX, and pilY1 backgrounds, and tested virulence of the double mutants (Fig 7). We also deleted pilW, pilX, and pilY1 in the algRD54A background, to test if AlgR activation was necessary for the delayed killing by pilW, pilX, and pilY1 mutants. In all cases, the double mutants had WT virulence, equivalent to that of the fimS, algR, or algRD54A single mutants. These results demonstrate that the delay in killing that results from loss of PilWXY1 requires both FimS and AlgR. Although overexpression of AlgRD54A in trans repressed virulence (Fig 6A), the chromosomal mutation was sufficient to alleviate delayed killing by pilW, pilX, and pilY1 mutants, suggesting that AlgR phosphorylation is important for modulation of virulence when PilWXY1 are missing.
Fig 7. Delayed killing by pilW, pilX, and pilY1 mutants is dependent on FimS-AlgR.

SK assays for pilW, pilX, pilY1, fimS, algR, and algRD54A single and double mutants. fimS, algR, and algRD54A mutants have WT virulence. pilW, pilX, and pilY1 killed more slowly relative to WT, fimS, algR, and algRD54A mutants. Combination of pilW, pilX, or pilY1 mutations with fimS, algR, or algRD54A mutations results in killing equivalent to fimS, algR, and algRD54A single mutants, respectively. All graphs represent 1 trial, separated into 3 graphs where strains relevant to (A) pilW, (B) pilX, and (C) pilY1 mutants are included. Asterisks indicate strains that were less virulent than PA14 by Gehan-Breslow-Wilcoxon test at p = 0.05 (p = 0.003125 with a Bonferroni correction), n = 3.
The sigma factor AlgU (AlgT/σ22/σE) acts upstream of FimS-AlgR to promote algR transcription [68–70], thus we tested its potential involvement in modulation of virulence by PilWXY1. An algU mutant killed more rapidly than WT (Fig 8), as previously demonstrated in mouse models [71], while pilW algU, pilX algU, and pilY1 algU double mutants had near-WT virulence (less than an algU mutant, but more than pilW, pilX, and pilY1 single mutants). Although AlgU promotes algR transcription [69], loss of AlgU alone does not prevent AlgR expression [68]. Given the reduced virulence of the pilW algU, pilX algU, and pilY1 algU double mutants relative to algU, PilWXY1 modulation of FimS-AlgR signalling appears to be intact in the algU mutant. These data are consistent with studies showing that mucA and mucD mutants, in which algR and algU are highly transcribed [69, 72–74], are less virulent towards C. elegans [75–77].
Fig 8. PilWXY1-mediated modulation of virulence is not dependent on AlgU.
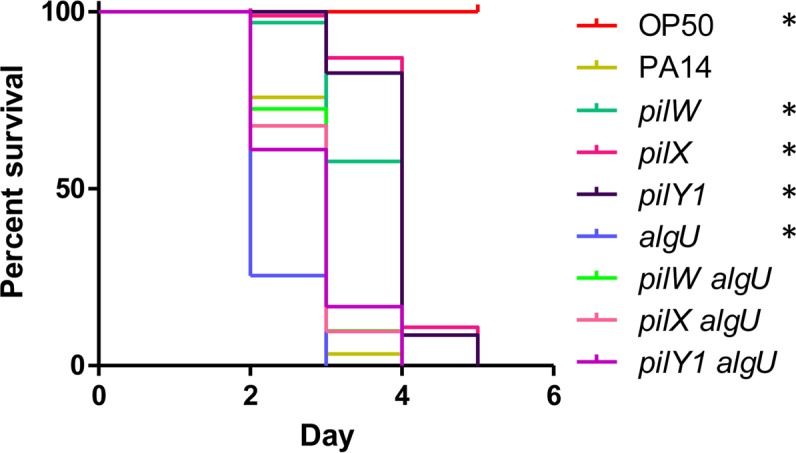
SK assays for PA14 pilW, pilX, pilY1, algU, pilW algU, pilX algU, and pilY1 algU mutants. Loss of algU led to more rapid killing relative to WT, while pilW algU, pilX algU, and pilY1 algU mutants had near-WT virulence. Asterisks indicate strains that were significantly different from PA14 by Gehan-Breslow-Wilcoxon test at p = 0.05 (p = 0.00625 with a Bonferroni correction), n = 3.
Discussion
P. aeruginosa uses T4P to attach to surfaces and host cells, for biofilm maturation, and to move across surfaces via twitching motility [2]. The MPs and PilY1 are important players in T4P biogenesis and function, but also in regulation of swarming motility, surface attachment, mechanosensation, and virulence [38–40, 43]. The MP operon is positively regulated by FimS-AlgR, a TCS implicated in regulation of chronic P. aeruginosa lung infections [17–19]. Here, we explored the connection between loss of PilWXY1 (and thus, loss of T4P) and AlgR activation in virulence towards C. elegans, as summarized in Fig 9. We showed that pilW, pilX, and pilY1 mutants kill nematodes more slowly than WT or a pilA mutant, supporting the idea that PilWXY1 modulate virulence independently of their role in T4P assembly. We confirmed previous reports [23, 33, 40] that in the absence of pilV, pilW, pilX, or pilY1, expression of the MP operon is significantly increased, and that this requires FimS-AlgR. Either hyperactivation or overexpression of AlgR delayed killing, while loss of fimS or algR in pilW, pilX, or pilY1 reverted virulence to WT levels.
Fig 9. Model for regulation of the MPs and virulence by FimS-AlgR.

(A) Loss or inactivation of FimS-AlgR results in sustained WT (acute) virulence towards C. elegans. Under normal conditions, PilVWXY1 suppress FimS activation of AlgR, leading to reduced expression of the MPs and increased expression of acute virulence factors. These phenotypes are mimicked by genetic inactivation of AlgR (D54A) or deletion of fimS or algR. (B) Loss of PilVWXY1 frees FimS to activate AlgR, leading to increased expression of the MPs, reduced expression of acute virulence factors, and delayed nematode killing. Hyperactivating mutations in AlgR (D54E) phenocopy this mechanism. Abbreviations: fimU, U (magenta); pilV, V (orange); pilW, W (teal); pilX, X (pink); pilY1, Y1 (purple); pilE, E (green). Yellow star indicates phosphorylation.
These data–coupled with BACTH data showing that the MPs interact directly with FimS in the inner membrane (Fig 4)–suggest that FimS may act as a molecular thermostat to monitor MP levels, and in their absence, activates AlgR to upregulate expression of the MP operon. A similar inventory control mechanism was recently described for the PilSR TCS, where PilS phosphorylates PilR when PilA levels are low, and dephosphorylates PilR when PilA levels are high [60]. It is not yet clear if FimS responds to changes in levels of the PilVWXY1 subcomplex, thought to prime assembly of T4P [24, 78, 79]. When overexpressed individually in trans, each of the MPs inhibited twitching motility in PAO1 [23], but since the others were still expressed from the chromosome, the exact nature of the signal detected by FimS remains to be determined. When expressed in trans, no single component of the PilVWXY1 subcomplex reduced fimU promoter activity if others were absent (S4 Fig). The specific signal that inhibits FimS activity remains to be deciphered. Whether the FimS-inhibitory signal is the same in PA14 and PAO1 also remains unknown. Though PilWXY1 modulated virulence of PA14 and PAO1, FimU and PilV influenced virulence only in PAO1 (Fig 1A and 1B). Given the MPs are divergent, FimU and PilV may play different roles in PAO1 versus PA14 [48]. It is possible that FimU and PilV are more important for stability of the PilWXY1 subcomplex in PAO1 than in PA14, and/or that PAO1 FimU and PilV can directly modulate FimS activity.
Kuchma et al. [39, 43] reported that loss of pilW, pilX, or pilY1 increased swarming motility and decreased biofilm formation, both indicative of low c-di-GMP levels. As biofilms were proposed to contribute to P. aeruginosa pathogenesis in C. elegans, we investigated whether the reduction in virulence in the absence of PilWXY1 was linked to decreased biofilm via loss of SadC activation [57–59, 80]. In our hands, levels of sadC had no impact on virulence even though they clearly modulated the amount of biofilm produced in SK media (Fig 3A and 3B, S3 Fig). Irazoqui et al. [59] examined the C. elegans gut during P. aeruginosa infection and described extracellular material that they suggested might indicate presence of a biofilm. Anti-biofilm compounds reduced P. aeruginosa virulence towards C. elegans, but a mechanism of action for those compounds has not been described [58]. Recently, the small RNA SrbA was shown to modulate both biofilm and virulence towards C. elegans; however, deletion of srbA led to altered transcription of at least 26 other genes that may also affect virulence [81].
Rather than using standard biofilm media, we performed these assays in liquid SK media to more closely mimic the conditions to which bacteria are exposed in the SK assay. To our knowledge, this is the first report to use SK media for biofilm assays. As we found no correlation between biofilm formation and virulence, we suggest that acute-phase virulence factors may be more important for C. elegans pathogenesis in the SK model. However, we recognize that in vitro biofilm assays may not replicate the conditions within the C. elegans gut; direct visualization of bacteria in worms will be needed to clarify the role of biofilm formation.
PilY1 and the MPs have been implicated in surface detection and activation of virulence, via signalling through SadC [38, 40]. Because loss of PilY1 or the MPs prevents T4P assembly and function, it is crucial to distinguish phenotypes resulting from lack of specific proteins versus loss of piliation [24]. Luo et al. [40] suggested that association of PilY1 with surfaces transduces a signal through the T4P machinery to stimulate c-di-GMP production by SadC, while Rodesney et al. [44] showed that loss of pilA, pilY1, or pilT prevents surface-activated c-di-GMP production. Rodesney et al. [44] proposed that both PilY1 and functional T4P are required for mechanosensation; however, it is not possible to delete pilY1 without ablating T4P assembly. Our cdrA promoter reporter data support the idea that PilWXY1 promote cyclic-di-GMP production by SadC, as loss of pilW, pilX, or pilY1 decreased cdrA promoter activity (Fig 2B). However, we argue that the PilWXY1-SadC pathway–though important for c-di-GMP signalling–is not critical for virulence towards C. elegans. Instead, our data show that PilWXY1-FimS-AlgR signalling axis is responsible for T4P-independent changes in virulence of pilW, pilX, and pilY1 mutants. Thus, surface attachment may induce c-di-GMP production via PilWXY1-SadC [40, 43], while the brief trapping of T4P outside the cell upon contact with a surface might transiently deplete PilVWXY1 levels in the inner membrane, resulting in increased FimS-AlgR activity and transition towards a sessile, biofilm lifestyle.
Whether the loss of pilW, pilX, or pilY1 leads to increased amounts of AlgR, its increased phosphorylation via FimS, or both, remains to be clarified. Okkotsu et al. [62] showed that AlgR and AlgRD54E levels are comparable, suggesting that the delay in killing we observed for PA14 algRD54E is attributable to the D54E phospho-mimetic mutation alone. Overexpression of AlgRD54A in trans delayed killing (Fig 6A), but the same mutation on the chromosome reverted virulence of pilW, pilX, and pilY1 mutants to WT levels (Fig 7). Therefore, we suspect that it is primarily AlgR phosphorylation (or lack of AlgR dephosphorylation) that leads to delayed killing. However, it is possible that both increased AlgR protein levels and phosphorylation contribute. Kong et al. [55] showed that AlgR binds fimS-algR, suggesting that the TCS could positively regulate its own transcription in response to reduced PilWXY1 levels.
In addition to being essential for T4P function, FimS and AlgR control alginate production in the context of chronic CF infections, where algR transcription is high [18, 82]. Phosphorylation of AlgR increases binding affinity at some–but not all–of its target sequences [17, 62, 63, 67]. For example, AlgRD54N failed to support twitching motility, but did not affect alginate production [17, 63]. Our twitching motility data suggests that AlgRD54A is capable of binding to the fimU promoter, albeit less efficiently than WT AlgR (S6 Fig). FimS is an unorthodox histidine kinase, with four transmembrane domains instead of the typical two, and lacks both a periplasmic sensing domain and the canonical motif involved in ATP coordination that mediates auto-phosphorylation [19, 83]. Direct interaction and/or phospho-transfer between FimS and AlgR have not been reported. Rather, the idea that FimS acts as a kinase for AlgR comes from this and other studies demonstrating similar phenotypes for fimS, algR, and algRD54N mutants [17, 18, 84]. Here, we demonstrated that FimS and AlgR interact in the BACTH assay (Fig 4) lending further support to this model.
FimS and AlgR promote expression of genes important for production of alginate, biofilms, and c-di-GMP, and inhibit expression of the T3SS, pyocyanin, and quorum sensing [55, 56, 74, 85, 86]. The observation that the loss of algR had no impact on virulence towards amoebae [38] or nematodes (Fig 5A and 5B) suggests that the AlgR-activated genes may not contribute to virulence, although the mechanisms of killing could differ. In mouse models, fimS and algR deletion mutants are attenuated, though overexpression of AlgR also markedly reduces virulence [55, 65, 87]. Further, Little et al. demonstrated that PAO1 algRD54E had WT virulence in Drosophila melanogaster and mouse infection models, while an algRD54A mutant was highly attenuated [87]. The outcomes that result from interaction of P. aeruginosa with different hosts will depend on a combination of factors including host defenses, site of infection, available nutrients, and virulence repertoire of a particular strain. However, our results suggest that changes in the specific repertoire of bacterial virulence factors, or the timing of their production, can tip the balance in the host’s favour.
The subset of AlgR-regulated virulence genes important for C. elegans pathogenesis is not defined. Screening of a PA14 transposon library for loss of virulence implicated several genes encoding regulators rather than individual virulence factors, suggesting that C. elegans pathogenesis is multifactorial [35]. Consistent with this hypothesis, a study of 18 WT P. aeruginosa strains revealed no correlation between pathogenicity and any specific virulence factors [88]. We saw WT or greater levels of virulence for algR and algU mutants, respectively, consistent with a role for AlgRU in repression of acute phase virulence factors (Figs 5 and 8). Factors under positive control of AlgRU (chronic-phase virulence factors) may be important during later stages of infection in more complex mammalian infection models, but not crucial for pathogenesis in nematodes [89, 90]. In support of this hypothesis, past studies have demonstrated that increased mucoidy, via mutation of mucA or mucD, reduced nematode killing [75–77].
While important for the initial stages of infection, T4P are often lost over time in chronic CF lung infections [5, 91, 92]. P. aeruginosa CF isolates frequently become mucoid via activation of AlgR, and production of many virulence factors is reduced [82, 93, 94]. Although the two outcomes are not necessarily temporally or mechanistically linked, mutations that achieve both may be advantageous during chronic CF lung infections. Specifically, loss of PilWXY1 may be adaptive in the context of CF, leading to AlgR activation. To test this idea, it will be interesting to examine the genotypes of mucoid CF isolates for these types of mutations. In conclusion, our results suggest that PilWXY1 promote virulence towards C. elegans by inhibiting FimS-AlgR activation. These data demonstrate how loss of one virulence factor (T4P) may activate others (via AlgR). Because the interplay between virulence factors in P. aeruginosa is complex and dynamic, careful consideration will be required when designing potential anti-virulence therapeutic strategies.
Materials and methods
Bacterial strains and plasmids
Strains and plasmids used in this work are listed in S1 Table. Bacteria were grown at 37°C for 16 h in 5 ml lysogeny broth (LB) Lennox, or on 1.5% agar LB plates, unless otherwise specified. Plasmids were transformed into chemically-competent E. coli by heat-shock, and into P. aeruginosa by electroporation [95]. Where appropriate, gentamicin (Gm) was added at 15 μg/ml for E. coli, and 30 μg/ml for P. aeruginosa. Kanamycin (Kan) was added at 50 μg/ml for E. coli, and 150 μg/ml for P. aeruginosa. Ampicillin (Amp) was added at 100 μg/ml for E. coli. L-arabinose was added at 0.05% where indicated to induce expression from the pBADGr promoter [96].
Cloning procedures
Vectors were constructed using standard cloning procedures, using the primers listed in S2 Table. Deletion constructs were designed to contain 500–1000 bp homology upstream and downstream the gene to be deleted. Deletion constructs for PA14 fimU, pilV, pilW, pilX, pilY1, and pilE were synthesized by Genscript in the pUC57Kan vector. pEX18Gm-sadC was created by amplifying the sadC deletion region from PA14 sadC roeA [42], followed by digestion and ligation into pEX18Gm. pEX18Gm-fimS, pEX18Gm-algRD54A, and pEX18Gm-algRD54E were made by overlap extension PCR [97]. Restriction digestion followed by ligation of the upstream and downstream fragments was used to create the deletion constructs pEX18Gm-algR, pEX18Gm-algU, and pEX18Gm-pilD. pMS402-PfimU and pMS402-PcdrA were created by amplifying and digesting the promoter regions of the PA14 MP operon and cdrA gene, respectively. Digested pBADGr was treated with alkaline phosphatase prior to ligation to avoid re-circularization of the vector. Constructs were verified by Sanger sequencing (MOBIX lab, McMaster, Hamilton, ON).
Mutant generation by allelic exchange
Allelic exchange was used to remove or alter specific genes [98]. pEX18Gm suicide plasmid derivatives (see Cloning procedures and S1 Table) were used to create all mutants in this work. After heat-shock transformation into E. coli SM10 cells, pEX18Gm constructs were conjugated into corresponding PA14 or PAO1 parent strains. Cells were then transferred to Pseudomonas isolation agar (PIA) Gm100 plates and incubated for 18 h at 37°C, to select for integration of pEX18Gm derivatives into the chromosome. Colonies were streaked onto LB/sucrose and incubated at 30°C for 18 h to select against merodiploids. Resultant colonies were patched onto LB and LB Gm30 to identify gentamicin-sensitive colonies. Regions flanking the desired mutations were amplified and sequenced to confirm success.
Twitching motility assays
Twitching motility assays were performed as previously described [99], with the following modifications. Individual colonies were stab-inoculated in triplicate into 1% agar LB solidified in plasma-treated tissue culture-grade plates (Thermo Fisher) and incubated at 30°C for 48 h. Agar was carefully removed and plates were stained with 1% crystal violet for 5 min. Unbound dye was removed by rinsing with water, then stained twitching areas were measured using ImageJ. Twitching zones were normalized to WT (100%).
Biofilm assays
Biofilm assays were performed as previously described, with modifications [100]. P. aeruginosa cultures were grown for 16 h at 37°C, diluted 1:200 in fresh LB, and grown to OD600 ~0.1. Cultures were then diluted 1:500 in liquid SK media (50 mM NaCl, 0.35% peptone, 1 mM CaCl2, 1 mM MgSO4, 5 μg/ml cholesterol in EtOH, 20 mM KH2PO4, and 5 mM K2HPO4), then 96-well plates were inoculated with 150 μl each strain, in triplicate. Sterility controls (liquid SK media) were included throughout the plate to check for contamination. Plates were covered with peg lids (Nunc) then wrapped in parafilm and incubated at 37°C for 24 h, shaken at 200 rpm. After incubation, the OD600 of the plate was measured to check for uniform growth and lack of contamination. Peg lids were washed for 10 min in 200 μl/well 1X phosphate-buffered saline (PBS), then stained with 200 μl/well 0.1% (w/v) crystal violet for 15 min. Unbound crystal violet was removed by washing lids in 70 ml distilled water 5 times at 10 min intervals. Crystal violet was solubilized from lids in 200 μl/well 33.3% acetic acid, then the absorbance at 600 nm was measured. Optical density and absorbance at 600 nm were plotted for growth and biofilm formation, respectively, then analyzed by one-way ANOVA followed by Dunnett post-test to compare each mutant to the WT control, p = 0.05. Error bars indicate standard error of the mean. Representative wells of acetic acid-solubilized crystal violet were imaged.
Caenorhabditis elegans slow killing assay
SK assays were performed as described previously [101]. SK plates (0.35% peptone, 50 mM NaCl, 2% agar, 1 mM CaCl2, 5 μg/ml cholesterol, 1 mM MgSO4, 20 mM KH2PO4, 5 mM K2HPO4, 100 μM FUDR) were seeded with 100 μl of an overnight culture and incubated overnight at 37°C. The following day, plates were enriched with 1 ml of an overnight culture concentrated to 100 μl. Synchronized L4 worms were collected from E. coli OP50 plates, washed twice in M9 buffer, and then >50 worms were seeded onto each bacterial lawn on individual SK plates. SK plates were incubated at 25°C and scored for dead worms every 24 h. Worms were considered dead when they did not respond to touch, and were removed from the plate. OP50 was included as a negative control for virulence. Percent survival was plotted as a function of time. Survival curves were plotted on GraphPad Prism 5.00 for Windows, then compared using the Gehan-Breslow-Wilcoxon test, p = 0.05. Given that larvae were synchronized at 20°C then transferred at L4 to 25°C for the duration of the assay, worms were at risk of death due to senescence, rather than direct killing by P. aeruginosa, before day 10 [46]. Therefore, the Gehan-Breslow-Wilcoxon test, which gives weight to earlier timepoints, was used in favour of the standard log-rank test (notably, all reported differences were also significant by the standard log-rank test). To correct for multiple analyses, the critical p-value of 0.05 was divided by the number of pairwise comparisons made within an individual trial, as per the Bonferroni method [102]. Each assay was performed at least 3 times, and differences were only considered significant if they were reproducible in the majority of trials. Representative trials are shown; all replicates can be viewed in the Supplemental Material (S1 File).
Luminescent reporter assay
Luminescent reporter assays were performed as previously described, with minor modifications [60]. Various strains harbouring the pMS402-PfimU or pMS402-PcdrA plasmids, encoding the luciferase genes under control of the fimU or cdrA promoters, respectively, were grown for 16 h at 37°C in LB Kan150, then diluted 1:50 in fresh liquid SK media with Kan150, in addition to Gm30 and 0.05% L-arabinose where appropriate. Subsequently, 100 μl of each culture was added to white-walled, clear-bottom 96-well plates (Corning) in triplicate, and incubated with shaking at 37°C in a Synergy 4 microtiter plate reader (BioTek). Luminescence readings were taken every 15 min for 5 h, and normalized to growth (OD600) at each time point. Readings that exceeded the limit of detection (>4 000 000 luminescence units) were discarded. At least 3 individual trials were performed. Error bars indicate standard error of the mean.
Bacterial two-hybrid β-galactosidase activity assay
To test for interactions between FimS and AlgR or individual pilins, BACTH assays were performed as previously described [103]. pUT18C and pKT25 derivatives, encoding the T18 and T25 domains of the Bordetella pertussis CyaA adenylate cyclase fused to the N-terminus of FimS, AlgR, PilA, FimU, PilV, PilW, PilX, or PilE [24, 60, 104], were co-transformed into E. coli BTH 101 to screen for pairwise interactions. Single colonies were inoculated in 5 ml LB Amp100 Kan50 and grown overnight. The following day, 100 μl was inoculated into 5 ml fresh media and grown to OD600 = 0.6, then 5 μl was spotted onto MacConkey plates (1.5% agar, 100μg/ml ampicillin, 50μg/ml kanamycin, 1% (w/v) maltose, 0.5mM isopropyl b-D-thiogalactopyranoside) (Difco) or LB Amp100 Kan50 plates supplemented with 100 μl of 20 mg/ml X-gal. Plates were incubated at 30°C for 24 h. An interaction was considered positive when colonies appeared pink or blue on MacConkey and LB + X-gal plates, respectively. BTH 101 expressing pUT18C-fimS and pKT25-fimS was used as a positive control [49]. Negative controls included BTH 101 expressing the empty vectors pUT18C and pKT25, and BTH 101 expressing pKT25-fimS and pUT18C (empty vector).
Supporting information
(A) Twitching motility assays for complemented PA14 pilW, pilX, and pilY1 mutants. Colonies were stab-inoculated into 1% agar LB plates, in triplicate. Plates were stained with crystal violet after 48 h at 30°C. Complementation of PA14 pilW, pilX, and pilY1 mutants with pBADGr-pilW, pBADGr-pilX, or pBADGr-pilY1, respectively, led to increased TM relative to complementation with pBADGr alone. Numbers indicate percent twitching area relative to WT, n = 3. (B) SK assays for complemented PA14 pilW, pilX, and pilY1 mutants. Complementation of pilW, pilX, and pilY1 mutants with pBADGr-pilW, pBADGr-pilX, or pBADGr-pilY1, respectively, restored virulence to near-WT levels. Asterisks indicate strains that were less virulent than PA14 + pBADGr by Gehan-Breslow-Wilcoxon test at p = 0.05 (p = 0.00833 with a Bonferroni correction), n = 3. Individual graphs represent separate trials.
(TIF)
Biofilm assays for (A) PA14 and (B) PAO1 pilA, fimU, pilV, pilW, pilX, pilY1, and pilE mutants. Very little biofilm formation was detectable in liquid SK media for any strains. There were no differences in biofilm formation as determined by one-way ANOVA followed by Dunnett post-test relative to WT at p = 0.05, n = 3.
(TIF)
SK assays for (A) PA14 and (B) PAO1 sadC mutants. Loss of sadC had no impact on pathogenicity relative to each respective WT strain, as measured by Gehan-Breslow-Wilcoxon test at p = 0.05 (p = 0.025 with a Bonferroni correction), n = 3.
(TIF)
(A) fimU promoter activity of pilV, pilW, pilX, and pilY1 mutants complemented with the respective gene in trans. The high luminescence of each mutant was restored to WT level when pilV, pilW, pilX, and pilY1 were complemented with PilV, PilW, PilX, and PilY1, respectively. (B) fimU promoter activity of a pilY1 mutant expressing each MP in trans. Expression of FimU, PilV, PilW, PilX, or PilE in the pilY1 background had no impact on fimU promoter activity relative to the pilY1 + empty vector control. (C) fimU promoter activity of pilW and pilX mutants overexpressing PilY1. Overexpression of PilY1 had no impact on fimU promoter activity in pilW and pilX backgrounds relative to the respective vector-only controls. Assays in (A), (B), and (C) were carried out in the presence of 0.05% L-arabinose to induce expression of the pBADGr promoter, n = 3.
(TIF)
(A) Twitching motility assays for PA14 pilA and pilD mutants. Loss of pilD resulted in loss of twitching motility. Numbers indicate percent twitching area relative to WT, n = 3. (B) fimU promoter activity of a pilD mutant compared to PA14, pilA, and pilY1. Loss of pilD had no impact on fimU promoter activity relative to WT, n = 3. (C) SK assays for PA14, pilA, pilY1, and pilD mutants. A pilD mutant had equivalent virulence to a pilA mutant; less pathogenic than WT but more pathogenic than a pilY1 mutants. Asterisks represent strains that were significantly different from the pilA mutant by Gehan-Breslow-Wilcoxon test at p = 0.05 (p = 0.0125 with a Bonferroni correction), n = 3.
(TIF)
(A) Twitching motility assays for PA14 pilA, fimS, algR, algRD54A, and algRD54E mutants. Twitching motility was abolished in pilA, algR, and algRD54A mutants, and fully retained in the algRD54E mutant. A fimS mutant twitched to ~50% WT levels. (B) Twitching motility assays for PA14 algR complemented with AlgR or AlgRD54A. An algR mutant was fully complemented by AlgR with and without induction by 0.05% L-arabinose. The AlgRD54A variant supported twitching motility in the algR mutant background in the presence of 0.05% L-arabinose, to ~25% WT levels. In (A) and (B), numbers indicate percent twitching area relative to WT, n = 3.
(TIF)
(DOCX)
Restriction sites are underlined.
(DOCX)
Three independent experiments for Figs 1A, 1B, 3B, 5A, 5B, 6A, 7A–7C and 8, and S1B, S3A, S3B and S5C Figs.
(PDF)
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was funded by an operating grant from the Canadian Institutes of Health Research #MOP 86639 to LLB. VAM held a Canada Graduate Scholarship from CIHR. SLNK held an Ontario Graduate Scholarship from the Government of Ontario. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics [Internet]. World Health Organization 2017. Available from: http://www.who.int/medicines/publications/WHO-PPL-Short_Summary_25Feb-ET_NM_WHO.pdf
- 2.Burrows LL. Pseudomonas aeruginosa twitching motility: type IV pili in action. Annu Rev Microbiol. 2012;66:493–520. doi: 10.1146/annurev-micro-092611-150055 [DOI] [PubMed] [Google Scholar]
- 3.Hospenthal MK, Costa TRD, Waksman G. A comprehensive guide to pilus biogenesis in Gram-negative bacteria. Nat Rev Micro. 2017;15(6):365–79. doi: 10.1038/nrmicro.2017.40 [DOI] [PubMed] [Google Scholar]
- 4.Berry J-L, Pelicic V. Exceptionally widespread nanomachines composed of type IV pilins: the prokaryotic Swiss Army knives. FEMS Microbiol Rev. 2015;39(1):1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mahenthiralingam E, Campbell ME, Speert DP. Nonmotility and phagocytic resistance of Pseudomonas aeruginosa isolates from chronically colonized patients with cystic fibrosis. Infect Immun. 1994;62(2):596–605. PMC186146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chi E, Mehl T, Nunn D, Lory S. Interaction of Pseudomonas aeruginosa with A549 pneumocyte cells. Infect Immun. 1991;59(3):822–8. PMC258333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doig P, Todd T, Sastry PA, Lee KK, Hodges RS, Paranchych W, et al. Role of pili in adhesion of Pseudomonas aeruginosa to human respiratory epithelial cells. Infect Immun. 1988;56(6):1641–6. Epub 1988/06/01. ; PubMed Central PMCID: PMCPMC259449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pier GB, Meluleni G, Neuger E. A murine model of chronic mucosal colonization by Pseudomonas aeruginosa. Infect Immun. 1992;60(11):4768–76. PMC258230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saiman L, Ishimoto K, Lory S, Prince A. The effect of piliation and exoproduct expression on the adherence of Pseudomonas aeruginosa to respiratory epithelial monolayers. J Infect Dis. 1990;161(3):541–8. Epub 1990/03/01. . [DOI] [PubMed] [Google Scholar]
- 10.Zoutman DE, Hulbert WC, Pasloske BL, Joffe AM, Volpel K, Trebilcock MK, et al. The role of polar pili in the adherence of Pseudomonas aeruginosa to injured canine tracheal cells: a semiquantitative morphologic study. Scanning Microsc. 1991;5(1):109–26. Epub 1991/03/01. . [PubMed] [Google Scholar]
- 11.Comolli JC, Hauser AR, Waite L, Whitchurch CB, Mattick JS, Engel JN. Pseudomonas aeruginosa gene products PilT and PilU are required for cytotoxicity in vitro and virulence in a mouse model of acute pneumonia. Infect Immun. 1999;67(7):3625–30. PMC116553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alm RA, Hallinan JP, Watson AA, Mattick JS. Fimbrial biogenesis genes of Pseudomonas aeruginosa: pilW and pilX increase the similarity of type 4 fimbriae to the GSP protein-secretion systems and pilY1 encodes a gonococcal PilC homologue. Mol Microbiol. 1996;22(1):161–73. Epub 1996/10/01. . [DOI] [PubMed] [Google Scholar]
- 13.Alm RA, Mattick JS. Identification of a gene, pilV, required for type 4 fimbrial biogenesis in Pseudomonas aeruginosa, whose product possesses a pre-pilin-like leader sequence. Mol Microbiol. 1995;16(3):485–96. Epub 1995/05/01. . [DOI] [PubMed] [Google Scholar]
- 14.Alm RA, Mattick JS. Identification of two genes with prepilin-like leader sequences involved in type 4 fimbrial biogenesis in Pseudomonas aeruginosa. J Bacteriol. 1996;178(13):3809–17. Epub 1996/07/01. ; PubMed Central PMCID: PMCPMC232641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Russell MA, Darzins A. The pilE gene product of Pseudomonas aeruginosa, required for pilus biogenesis, shares amino acid sequence identity with the N-termini of type 4 prepilin proteins. Mol Microbiol. 1994;13(6):973–85. Epub 1994/09/01. . [DOI] [PubMed] [Google Scholar]
- 16.Hobbs M, Collie ES, Free PD, Livingston SP, Mattick JS. PilS and PilR, a two-component transcriptional regulatory system controlling expression of type 4 fimbriae in Pseudomonas aeruginosa. Mol Microbiol. 1993;7(5):669–82. Epub 1993/03/01. . [DOI] [PubMed] [Google Scholar]
- 17.Belete B, Lu H, Wozniak DJ. Pseudomonas aeruginosa AlgR regulates type IV pilus biosynthesis by activating transcription of the fimU-pilVWXY1Y2E operon. J Bacteriol. 2008;190(6):2023–30. doi: 10.1128/JB.01623-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Whitchurch CB, Alm RA, Mattick JS. The alginate regulator AlgR and an associated sensor FimS are required for twitching motility in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 1996;93(18):9839–43. Epub 1996/09/03. ; PubMed Central PMCID: PMCPMC38516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okkotsu Y, Little AS, Schurr MJ. The Pseudomonas aeruginosa AlgZR two-component system coordinates multiple phenotypes. Front Cell Infect Microbiol. 2014;4:82 doi: 10.3389/fcimb.2014.00082. PMC4064291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolfgang MC, Lee VT, Gilmore ME, Lory S. Coordinate regulation of bacterial virulence genes by a novel adenylate cyclase-dependent signaling pathway. Dev Cell. 2003;4(2):253–63. doi: 10.1016/S1534-5807(03)00019-4 [DOI] [PubMed] [Google Scholar]
- 21.Yu H, Mudd M, Boucher JC, Schurr MJ, Deretic V. Identification of the algZ gene upstream of the response regulator algR and its participation in control of alginate production in Pseudomonas aeruginosa. J Bacteriol. 1997;179(1):187–93. Epub 1997/01/01. ; PubMed Central PMCID: PMCPMC178678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strom MS, Bergman P, Lory S. Identification of active-site cysteines in the conserved domain of PilD, the bifunctional type IV pilin leader peptidase/N-methyltransferase of Pseudomonas aeruginosa. J Biol Chem. 1993;268(21):15788–94. Epub 1993/07/25. . [PubMed] [Google Scholar]
- 23.Giltner CL, Habash M, Burrows LL. Pseudomonas aeruginosa minor pilins are incorporated into type IV pili. J Mol Biol. 2010;398(3):444–61. Epub 2010/03/27. doi: 10.1016/j.jmb.2010.03.028 . [DOI] [PubMed] [Google Scholar]
- 24.Nguyen Y, Sugiman-Marangos S, Harvey H, Bell SD, Charlton CL, Junop MS, et al. Pseudomonas aeruginosa minor pilins prime type IVa pilus assembly and promote surface display of the PilY1 adhesin. J Biol Chem. 2015;290(1):601–11. doi: 10.1074/jbc.M114.616904. PMC4281761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sauvonnet N, Vignon G, Pugsley AP, Gounon P. Pilus formation and protein secretion by the same machinery in Escherichia coli. EMBO J. 2000;19(10):2221–8. doi: 10.1093/emboj/19.10.2221. PMC384360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCallum M, Tammam S, Khan A, Burrows LL, Howell PL. The molecular mechanism of the type IVa pilus motors. Nat Commun. 2017;8:15091 doi: 10.1038/ncomms15091. PMC5424180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramer SW, Schoolnik GK, Wu C-Y, Hwang J, Schmidt SA, Bieber D. The type IV pilus assembly complex: biogenic interactions among the bundle-forming pilus proteins of enteropathogenic Escherichia coli. J Bacteriol. 2002;184(13):3457–65. doi: 10.1128/JB.184.13.3457-3465.2002. PMC135125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Winther-Larsen HC, Wolfgang M, Dunham S, Van Putten JPM, Dorward D, Løvold C, et al. A conserved set of pilin-like molecules controls type IV pilus dynamics and organelle-associated functions in Neisseria gonorrhoeae. Mol Microbiol. 2005;56(4):903–17. doi: 10.1111/j.1365-2958.2005.04591.x [DOI] [PubMed] [Google Scholar]
- 29.Carbonnelle E, Helaine S, Nassif X, Pelicic V. A systematic genetic analysis in Neisseria meningitidis defines the Pil proteins required for assembly, functionality, stabilization and export of type IV pili. Mol Microbiol. 2006;61(6):1510–22. doi: 10.1111/j.1365-2958.2006.05341.x [DOI] [PubMed] [Google Scholar]
- 30.Chang Y-W, Rettberg LA, Treuner-Lange A, Iwasa J, Søgaard-Andersen L, Jensen GJ. Architecture of the type IVa pilus machine. Science. 2016;351(6278). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heiniger RW, Winther-Larsen HC, Pickles RJ, Koomey M, Wolfgang MC. Infection of human mucosal tissue by Pseudomonas aeruginosa requires sequential and mutually dependent virulence factors and a novel pilus-associated adhesin. Cell Microbiol. 2010;12(8):1158–73. doi: 10.1111/j.1462-5822.2010.01461.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wolfgang M, Park H-S, Hayes SF, van Putten JPM, Koomey M. Suppression of an absolute defect in type IV pilus biogenesis by loss-of-function mutations in pilT, a twitching motility gene in Neisseria gonorrhoeae. Proc Natl Acad Sci U S A. 1998;95(25):14973–8. PMC24560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bohn Y-ST, Brandes G, Rakhimova E, Horatzek S, Salunkhe P, Munder A, et al. Multiple roles of Pseudomonas aeruginosa TBCF10839 PilY1 in motility, transport and infection. Mol Microbiol. 2009;71(3):730–47. doi: 10.1111/j.1365-2958.2008.06559.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garvis S, Munder A, Ball G, de Bentzmann S, Wiehlmann L, Ewbank JJ, et al. Caenorhabditis elegans semi-automated liquid screen reveals a specialized role for the chemotaxis gene cheB2 in Pseudomonas aeruginosa virulence. PLOS Pathog. 2009;5(8):e1000540 doi: 10.1371/journal.ppat.1000540. PMC2714965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feinbaum RL, Urbach JM, Liberati NT, Djonovic S, Adonizio A, Carvunis A-R, et al. Genome-wide identification of Pseudomonas aeruginosa virulence-related genes using a Caenorhabditis elegans infection model. PLOS Pathog. 2012;8(7):e1002813 doi: 10.1371/journal.ppat.1002813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lewenza S, Charron-Mazenod L, Giroux L, Zamponi AD. Feeding behaviour of Caenorhabditis elegans is an indicator of Pseudomonas aeruginosa PAO1 virulence. PeerJ. 2014;2:e521 doi: 10.7717/peerj.521. PMC4137669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jansen G, Crummenerl LL, Gilbert F, Mohr T, Pfefferkorn R, Thänert R, et al. Evolutionary transition from pathogenicity to commensalism: global regulator mutations mediate fitness gains through virulence attenuation. Mol Biol Evol. 2015;32(11):2883–96. doi: 10.1093/molbev/msv160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Siryaporn A, Kuchma SL, O’Toole GA, Gitai Z. Surface attachment induces Pseudomonas aeruginosa virulence. Proc Natl Acad Sci U S A. 2014;111(47):16860–5. doi: 10.1073/pnas.1415712111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuchma SL, Ballok AE, Merritt JH, Hammond JH, Lu W, Rabinowitz JD, et al. Cyclic-di-GMP-mediated repression of swarming motility by Pseudomonas aeruginosa: the pilY1 gene and its impact on surface-associated behaviors. J Bacteriol. 2010;192(12):2950–64. doi: 10.1128/JB.01642-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luo Y, Zhao K, Baker AE, Kuchma SL, Coggan KA, Wolfgang MC, et al. A hierarchical cascade of second messengers regulates Pseudomonas aeruginosa surface behaviors. mBio. 2015;6(1). doi: 10.1128/mBio.02456-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Persat A, Inclan YF, Engel JN, Stone HA, Gitai Z. Type IV pili mechanochemically regulate virulence factors in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2015;112(24):7563–8. Epub 2015/06/05. doi: 10.1073/pnas.1502025112 ; PubMed Central PMCID: PMCPMC4475988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Merritt JH, Brothers KM, Kuchma SL, O'Toole GA. SadC reciprocally influences biofilm formation and swarming motility via modulation of exopolysaccharide production and flagellar function. J Bacteriol. 2007;189(22):8154–64. Epub 2007/06/26. doi: 10.1128/JB.00585-07 ; PubMed Central PMCID: PMCPMC2168701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuchma SL, Griffin EF, O'Toole GA. Minor pilins of the type IV pilus system participate in the negative regulation of swarming motility. J Bacteriol. 2012;194(19):5388–403. doi: 10.1128/JB.00899-12. PMC3457191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rodesney CA, Roman B, Dhamani N, Cooley BJ, Katira P, Touhami A, et al. Mechanosensing of shear by Pseudomonas aeruginosa leads to increased levels of the cyclic-di-GMP signal initiating biofilm development. Proc Natl Acad Sci U S A. 2017;114(23):5906–11. doi: 10.1073/pnas.1703255114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gallagher LA, Manoil C. Pseudomonas aeruginosa PAO1 kills Caenorhabditis elegans by cyanide poisoning. J Bacteriol. 2001;183(21):6207–14. doi: 10.1128/JB.183.21.6207-6214.2001. PMC100099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang B, Xiao R, Ronan EA, He Y, Hsu A-L, Liu J, et al. Environmental temperature differentially modulates C. elegans longevity through a thermosensitive TRP channel. Cell Rep. 2015;11(9):1414–24. doi: 10.1016/j.celrep.2015.04.066. PMC4758836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Machin D, Cheung YB, Parmar MKB. Comparison of survival curves Survival analysis: a practical approach. 2nd ed: John Wiley & Sons, Ltd; 2006. p. 51–90. [Google Scholar]
- 48.Giltner CL, Rana N, Lunardo MN, Hussain AQ, Burrows LL. Evolutionary and functional diversity of the Pseudomonas type IVa pilin island. Environ Microbiol. 2011;13(1):250–64. doi: 10.1111/j.1462-2920.2010.02327.x [DOI] [PubMed] [Google Scholar]
- 49.Rybtke MT, Borlee BR, Murakami K, Irie Y, Hentzer M, Nielsen TE, et al. Fluorescence-based reporter for gauging cyclic di-GMP levels in Pseudomonas aeruginosa. Appl Environ Microbiol. 2012;78(15):5060–9. doi: 10.1128/AEM.00414-12. PMC3416407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moscoso JA, Jaeger T, Valentini M, Hui K, Jenal U, Filloux A. The diguanylate cyclase SadC is a central player in Gac/Rsm-mediated biofilm formation in Pseudomonas aeruginosa. J Bacteriol. 2014;196(23):4081–8. Epub 2014/09/17. doi: 10.1128/JB.01850-14 ; PubMed Central PMCID: PMCPMC4248864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bouffartigues E, Moscoso JA, Duchesne R, Rosay T, Fito-Boncompte L, Gicquel G, et al. The absence of the Pseudomonas aeruginosa OprF protein leads to increased biofilm formation through variation in c-di-GMP level. Front Microbiol. 2015;6:630 doi: 10.3389/fmicb.2015.00630. PMC4477172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li K, Yang G, Debru AB, Li P, Zong L, Li P, et al. SuhB regulates the motile-sessile switch in Pseudomonas aeruginosa through the Gac/Rsm pathway and c-di-GMP signaling. Front Microbiol. 2017;8(1045). doi: 10.3389/fmicb.2017.01045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nair HAS, Periasamy S, Yang L, Kjelleberg S, Rice SA. Real time, spatial, and temporal mapping of the distribution of c-di-GMP during biofilm development. J Biol Chem. 2017;292(2):477–87. doi: 10.1074/jbc.M116.746743. PMC5241725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Valentini M, Laventie B-J, Moscoso J, Jenal U, Filloux A. The diguanylate cyclase HsbD intersects with the HptB regulatory cascade to control Pseudomonas aeruginosa biofilm and motility. PLoS Genet. 2016;12(10):e1006354 doi: 10.1371/journal.pgen.1006354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kong W, Zhao J, Kang H, Zhu M, Zhou T, Deng X, et al. ChIP-seq reveals the global regulator AlgR mediating cyclic di-GMP synthesis in Pseudomonas aeruginosa. Nucleic Acids Res. 2015;43(17):8268–82. doi: 10.1093/nar/gkv747. PMC4787818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morici LA, Carterson AJ, Wagner VE, Frisk A, Schurr JR, Honer zu Bentrup K, et al. Pseudomonas aeruginosa AlgR represses the Rhl quorum-sensing system in a biofilm-specific manner. J Bacteriol. 2007;189(21):7752–64. Epub 2007/09/04. doi: 10.1128/JB.01797-06 ; PubMed Central PMCID: PMCPMC2168728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang L, Fritsch M, Hammond L, Landreville R, Slatculescu C, Colavita A, et al. Identification of genes involved in Pseudomonas aeruginosa biofilm-specific resistance to antibiotics. PLOS ONE. 2013;8(4):e61625 doi: 10.1371/journal.pone.0061625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Tilburg Bernardes E, Charron-Mazenod L, Reading DJ, Reckseidler-Zenteno SL, Lewenza S. Exopolysaccharide-repressing small molecules with antibiofilm and antivirulence activity against Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2017. doi: 10.1128/aac.01997-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Irazoqui JE, Troemel ER, Feinbaum RL, Luhachack LG, Cezairliyan BO, Ausubel FM. Distinct pathogenesis and host responses during infection of C. elegans by P. aeruginosa and S. aureus. PLOS Pathog. 2010;6:e1000982 Epub 2010/07/10. doi: 10.1371/journal.ppat.1000982 ; PubMed Central PMCID: PMCPMC2895663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kilmury SLN, Burrows LL. Type IV pilins regulate their own expression via direct intramembrane interactions with the sensor kinase PilS. Proc Natl Acad Sci U S A. 2016;113(21):6017–22. doi: 10.1073/pnas.1512947113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Strom MS, Lory S. Amino acid substitutions in pilin of Pseudomonas aeruginosa. Effect on leader peptide cleavage, amino-terminal methylation, and pilus assembly. J Biol Chem. 1991;266(3):1656–64. Epub 1991/01/25. . [PubMed] [Google Scholar]
- 62.Okkotsu Y, Tieku P, Fitzsimmons LF, Churchill ME, Schurr MJ. Pseudomonas aeruginosa AlgR phosphorylation modulates rhamnolipid production and motility. J Bacteriol. 2013;195(24):5499–515. Epub 2013/10/08. doi: 10.1128/JB.00726-13 ; PubMed Central PMCID: PMCPMC3889618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Whitchurch CB, Erova TE, Emery JA, Sargent JL, Harris JM, Semmler AB, et al. Phosphorylation of the Pseudomonas aeruginosa response regulator AlgR is essential for type IV fimbria-mediated twitching motility. J Bacteriol. 2002;184(16):4544–54. Epub 2002/07/27. doi: 10.1128/JB.184.16.4544-4554.2002 ; PubMed Central PMCID: PMCPMC135261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Deretic V, Leveau JH, Mohr CD, Hibler NS. In vitro phosphorylation of AlgR, a regulator of mucoidy in Pseudomonas aeruginosa, by a histidine protein kinase and effects of small phospho-donor molecules. Mol Microbiol. 1992;6(19):2761–7. Epub 1992/10/01. . [DOI] [PubMed] [Google Scholar]
- 65.Lizewski SE, Lundberg DS, Schurr MJ. The transcriptional regulator AlgR is essential for Pseudomonas aeruginosa pathogenesis. Infect Immun. 2002;70(11):6083–93. doi: 10.1128/IAI.70.11.6083-6093.2002. PMC130412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stacey SD, Williams DA, Pritchett CL. The Pseudomonas aeruginosa two-component regulator AlgR directly activates rsmA expression in a phosphorylation independent manner. J Bacteriol. 2017. doi: 10.1128/jb.00048-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ma S, Selvaraj U, Ohman DE, Quarless R, Hassett DJ, Wozniak DJ. Phosphorylation-independent activity of the response regulators AlgB and AlgR in promoting alginate biosynthesis in mucoid Pseudomonas aeruginosa. J Bacteriol. 1998;180(4):956–68. Epub 1998/02/24. ; PubMed Central PMCID: PMCPMC106978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pritchett CL, Little AS, Okkotsu Y, Frisk A, Cody WL, Covey CR, et al. Expression analysis of the Pseudomonas aeruginosa AlgZR two-component regulatory system. J Bacteriol. 2015;197(4):736–48. doi: 10.1128/JB.02290-14. PMC4334192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wozniak DJ, Ohman DE. Transcriptional analysis of the Pseudomonas aeruginosa genes algR, algB, and algD reveals a hierarchy of alginate gene expression which is modulated by algT. J Bacteriol. 1994;176(19):6007–14. PMC196818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Falcone M, Ferrara S, Rossi E, Johansen HK, Molin S, Bertoni G. The small RNA ErsA of Pseudomonas aeruginosa contributes to biofilm development and motility through post-transcriptional modulation of AmrZ. Front Microbiol. 2018;9(238). doi: 10.3389/fmicb.2018.00238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yu H, Boucher JC, Hibler NS, Deretic V. Virulence properties of Pseudomonas aeruginosa lacking the extreme-stress sigma factor AlgU (sigmaE). Infect Immun. 1996;64(7):2774–81. PMC174138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Damron FH, Yu HD. Pseudomonas aeruginosa MucD regulates the alginate pathway through activation of MucA degradation via MucP proteolytic activity. J Bacteriol. 2011;193(1):286–91. doi: 10.1128/JB.01132-10. PMC3019965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Firoved AM, Deretic V. Microarray analysis of global gene expression in mucoid Pseudomonas aeruginosa. J Bacteriol. 2003;185(3):1071–81. doi: 10.1128/JB.185.3.1071-1081.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jones AK, Fulcher NB, Balzer GJ, Urbanowski ML, Pritchett CL, Schurr MJ, et al. Activation of the Pseudomonas aeruginosa AlgU regulon through mucA mutation inhibits cyclic AMP/Vfr signaling. J Bacteriol. 2010;192(21):5709–17. Epub 2010/09/08. doi: 10.1128/JB.00526-10 ; PubMed Central PMCID: PMCPMC2953679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yorgey P, Rahme LG, Tan MW, Ausubel FM. The roles of mucD and alginate in the virulence of Pseudomonas aeruginosa in plants, nematodes and mice. Mol Microbiol. 2001;41(5):1063–76. doi: 10.1046/j.1365-2958.2001.02580.x [DOI] [PubMed] [Google Scholar]
- 76.Reddy KC, Hunter RC, Bhatla N, Newman DK, Kim DH. Caenorhabditis elegans NPR-1–mediated behaviors are suppressed in the presence of mucoid bacteria. Proc Natl Acad Sci U S A. 2011;108(31):12887–92. doi: 10.1073/pnas.1108265108. PMC3150904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rau MH, Hansen SK, Johansen HK, Thomsen LE, Workman CT, Nielsen KF, et al. Early adaptive developments of Pseudomonas aeruginosa after the transition from life in the environment to persistent colonization in the airways of human cystic fibrosis hosts. Environ Microbiol. 2010;12(6):1643–58. doi: 10.1111/j.1462-2920.2010.02211.x [DOI] [PubMed] [Google Scholar]
- 78.Korotkov KV, Hol WG. Structure of the GspK-GspI-GspJ complex from the enterotoxigenic Escherichia coli type 2 secretion system. Nat Struct Mol Biol. 2008;15(5):462–8. Epub 2008/04/29. doi: 10.1038/nsmb.1426 . [DOI] [PubMed] [Google Scholar]
- 79.Cisneros DA, Bond PJ, Pugsley AP, Campos M, Francetic O. Minor pseudopilin self-assembly primes type II secretion pseudopilus elongation. EMBO J. 2012;31(4):1041–53. doi: 10.1038/emboj.2011.454. PMC3280553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jenal U, Malone J. Mechanisms of cyclic-di-GMP signaling in bacteria. Annu Rev Genet. 2006;40:385–407. doi: 10.1146/annurev.genet.40.110405.090423 [DOI] [PubMed] [Google Scholar]
- 81.Taylor PK, Van Kessel ATM, Colavita A, Hancock REW, Mah T-F. A novel small RNA is important for biofilm formation and pathogenicity in Pseudomonas aeruginosa. PLOS ONE. 2017;12(8):e0182582 doi: 10.1371/journal.pone.0182582. PMC5542712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Darzins A, Chakrabarty AM. Cloning of genes controlling alginate biosynthesis from a mucoid cystic fibrosis isolate of Pseudomonas aeruginosa. J Bacteriol. 1984;159(1):9–18. Epub 1984/07/01. ; PubMed Central PMCID: PMCPMC215585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim D, Forst S. Genomic analysis of the histidine kinase family in bacteria and archaea. Microbiology. 2001;147(Pt 5):1197–212. Epub 2001/04/26. doi: 10.1099/00221287-147-5-1197 . [DOI] [PubMed] [Google Scholar]
- 84.Cody WL, Pritchett CL, Jones AK, Carterson AJ, Jackson D, Frisk A, et al. Pseudomonas aeruginosa AlgR controls cyanide production in an AlgZ-dependent manner. J Bacteriol. 2009;191(9):2993–3002. Epub 2009/03/10. doi: 10.1128/JB.01156-08 ; PubMed Central PMCID: PMCPMC2681793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lizewski SE, Schurr JR, Jackson DW, Frisk A, Carterson AJ, Schurr MJ. Identification of AlgR-regulated genes in Pseudomonas aeruginosa by use of microarray analysis. J Bacteriol. 2004;186(17):5672–84. doi: 10.1128/JB.186.17.5672-5684.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Intile PJ, Diaz MR, Urbanowski ML, Wolfgang MC, Yahr TL. The AlgZR two-component system recalibrates the RsmAYZ posttranscriptional regulatory system to inhibit expression of the Pseudomonas aeruginosa type III secretion system. J Bacteriol. 2014;196(2):357–66. doi: 10.1128/JB.01199-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Little AS, Okkotsu Y, Reinhart AA, Damron FH, Barbier M, Barrett B, et al. Pseudomonas aeruginosa AlgR phosphorylation status differentially regulates pyocyanin and pyoverdine production. mBio. 2018;9(1). doi: 10.1128/mBio.02318-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee DG, Urbach JM, Wu G, Liberati NT, Feinbaum RL, Miyata S, et al. Genomic analysis reveals that Pseudomonas aeruginosa virulence is combinatorial. Genome Biol. 2006;7(10):R90 doi: 10.1186/gb-2006-7-10-r90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Moradali MF, Ghods S, Rehm BHA. Pseudomonas aeruginosa lifestyle: a paradigm for adaptation, survival, and persistence. Front Cell Infect Microbiol. 2017;7:39 doi: 10.3389/fcimb.2017.00039. PMC5310132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lebeaux D, Chauhan A, Rendueles O, Beloin C. From in vitro to in vivo models of bacterial biofilm-related infections. Pathogens. 2013;2(2):288–356. doi: 10.3390/pathogens2020288. PMC4235718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kus JV, Tullis E, Cvitkovitch DG, Burrows LL. Significant differences in type IV pilin allele distribution among Pseudomonas aeruginosa isolates from cystic fibrosis (CF) versus non-CF patients. Microbiology. 2004;150(5):1315–26. doi: 10.1099/mic.0.26822–0 [DOI] [PubMed] [Google Scholar]
- 92.Pasloske BL, Joffe AM, Sun Q, Volpel K, Paranchych W, Eftekhar F, et al. Serial isolates of Pseudomonas aeruginosa from a cystic fibrosis patient have identical pilin sequences. Infect Immun. 1988;56(3):665–72. PMC259343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schurr MJ, Yu H, Martinez-Salazar JM, Hibler NS, Deretic V. Biochemical characterization and posttranslational modification of AlgU, a regulator of stress response in Pseudomonas aeruginosa. Biochem Biophys Res Commun. 1995;216(3):874–80. Epub 1995/11/22. . [DOI] [PubMed] [Google Scholar]
- 94.Martin DW, Schurr MJ, Mudd MH, Govan JR, Holloway BW, Deretic V. Mechanism of conversion to mucoidy in Pseudomonas aeruginosa infecting cystic fibrosis patients. Proc Natl Acad Sci U S A. 1993;90(18):8377–81. Epub 1993/09/15. ; PubMed Central PMCID: PMCPMC47359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dower WJ, Miller JF, Ragsdale CW. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 1988;16(13):6127–45. Epub 1988/07/11. ; PubMed Central PMCID: PMCPMC336852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Asikyan ML, Kus JV, Burrows LL. Novel proteins that modulate type IV pilus retraction dynamics in Pseudomonas aeruginosa. J Bacteriol. 2008;190(21):7022–34. Epub 2008/09/09. doi: 10.1128/JB.00938-08 ; PubMed Central PMCID: PMCPMC2580705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77(1):51–9. [DOI] [PubMed] [Google Scholar]
- 98.Hmelo LR, Borlee BR, Almblad H, Love ME, Randall TE, Tseng BS, et al. Precision-engineering the Pseudomonas aeruginosa genome with two-step allelic exchange. Nat Protoc. 2015;10(11):1820–41. Epub 2015/10/23. doi: 10.1038/nprot.2015.115 ; PubMed Central PMCID: PMCPMC4862005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gallant CV, Daniels C, Leung JM, Ghosh AS, Young KD, Kotra LP, et al. Common β-lactamases inhibit bacterial biofilm formation. Molec Microbiol. 2005;58(4):1012–24. doi: 10.1111/j.1365-2958.2005.04892.x. PMC3097517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wenderska IB, Chong M, McNulty J, Wright GD, Burrows LL. Palmitoyl-DL-carnitine is a multitarget inhibitor of Pseudomonas aeruginosa biofilm development. ChemBioChem. 2011;12(18):2759–66. Epub 2011/11/03. doi: 10.1002/cbic.201100500 . [DOI] [PubMed] [Google Scholar]
- 101.Tan MW, Mahajan-Miklos S, Ausubel FM. Killing of Caenorhabditis elegans by Pseudomonas aeruginosa used to model mammalian bacterial pathogenesis. Proc Natl Acad Sci U S A. 1999;96(2):715–20. Epub 1999/01/20. ; PubMed Central PMCID: PMCPMC15202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57(1):289–300. [Google Scholar]
- 103.Buensuceso RNC, Nguyen Y, Zhang K, Daniel-Ivad M, Sugiman-Marangos SN, Fleetwood AD, et al. The conserved tetratricopeptide repeat-containing C-terminal domain of Pseudomonas aeruginosa FimV is required for its cyclic AMP-dependent and -independent functions. J Bacteriol. 2016;198(16):2263–74. doi: 10.1128/JB.00322-16. PMC4966435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nguyen Y, Harvey H, Sugiman-Marangos S, Bell SD, Buensuceso RNC, Junop MS, et al. Structural and functional studies of the Pseudomonas aeruginosa minor pilin, PilE. J Biol Chem. 2015;290(44):26856–65. doi: 10.1074/jbc.M115.683334 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Twitching motility assays for complemented PA14 pilW, pilX, and pilY1 mutants. Colonies were stab-inoculated into 1% agar LB plates, in triplicate. Plates were stained with crystal violet after 48 h at 30°C. Complementation of PA14 pilW, pilX, and pilY1 mutants with pBADGr-pilW, pBADGr-pilX, or pBADGr-pilY1, respectively, led to increased TM relative to complementation with pBADGr alone. Numbers indicate percent twitching area relative to WT, n = 3. (B) SK assays for complemented PA14 pilW, pilX, and pilY1 mutants. Complementation of pilW, pilX, and pilY1 mutants with pBADGr-pilW, pBADGr-pilX, or pBADGr-pilY1, respectively, restored virulence to near-WT levels. Asterisks indicate strains that were less virulent than PA14 + pBADGr by Gehan-Breslow-Wilcoxon test at p = 0.05 (p = 0.00833 with a Bonferroni correction), n = 3. Individual graphs represent separate trials.
(TIF)
Biofilm assays for (A) PA14 and (B) PAO1 pilA, fimU, pilV, pilW, pilX, pilY1, and pilE mutants. Very little biofilm formation was detectable in liquid SK media for any strains. There were no differences in biofilm formation as determined by one-way ANOVA followed by Dunnett post-test relative to WT at p = 0.05, n = 3.
(TIF)
SK assays for (A) PA14 and (B) PAO1 sadC mutants. Loss of sadC had no impact on pathogenicity relative to each respective WT strain, as measured by Gehan-Breslow-Wilcoxon test at p = 0.05 (p = 0.025 with a Bonferroni correction), n = 3.
(TIF)
(A) fimU promoter activity of pilV, pilW, pilX, and pilY1 mutants complemented with the respective gene in trans. The high luminescence of each mutant was restored to WT level when pilV, pilW, pilX, and pilY1 were complemented with PilV, PilW, PilX, and PilY1, respectively. (B) fimU promoter activity of a pilY1 mutant expressing each MP in trans. Expression of FimU, PilV, PilW, PilX, or PilE in the pilY1 background had no impact on fimU promoter activity relative to the pilY1 + empty vector control. (C) fimU promoter activity of pilW and pilX mutants overexpressing PilY1. Overexpression of PilY1 had no impact on fimU promoter activity in pilW and pilX backgrounds relative to the respective vector-only controls. Assays in (A), (B), and (C) were carried out in the presence of 0.05% L-arabinose to induce expression of the pBADGr promoter, n = 3.
(TIF)
(A) Twitching motility assays for PA14 pilA and pilD mutants. Loss of pilD resulted in loss of twitching motility. Numbers indicate percent twitching area relative to WT, n = 3. (B) fimU promoter activity of a pilD mutant compared to PA14, pilA, and pilY1. Loss of pilD had no impact on fimU promoter activity relative to WT, n = 3. (C) SK assays for PA14, pilA, pilY1, and pilD mutants. A pilD mutant had equivalent virulence to a pilA mutant; less pathogenic than WT but more pathogenic than a pilY1 mutants. Asterisks represent strains that were significantly different from the pilA mutant by Gehan-Breslow-Wilcoxon test at p = 0.05 (p = 0.0125 with a Bonferroni correction), n = 3.
(TIF)
(A) Twitching motility assays for PA14 pilA, fimS, algR, algRD54A, and algRD54E mutants. Twitching motility was abolished in pilA, algR, and algRD54A mutants, and fully retained in the algRD54E mutant. A fimS mutant twitched to ~50% WT levels. (B) Twitching motility assays for PA14 algR complemented with AlgR or AlgRD54A. An algR mutant was fully complemented by AlgR with and without induction by 0.05% L-arabinose. The AlgRD54A variant supported twitching motility in the algR mutant background in the presence of 0.05% L-arabinose, to ~25% WT levels. In (A) and (B), numbers indicate percent twitching area relative to WT, n = 3.
(TIF)
(DOCX)
Restriction sites are underlined.
(DOCX)
Three independent experiments for Figs 1A, 1B, 3B, 5A, 5B, 6A, 7A–7C and 8, and S1B, S3A, S3B and S5C Figs.
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.