

Summary
A novel liquid chromatographic method was developed for accurate and precise routine quantification of serotonin in human cerebrospinal fluid. Serotonin is derivatized with an amine-specific reagent leaving its hydroxy group free for electrochemical detection and making it lipophilic enough for efficient extraction into organic solvents. The method is reproducible, linear and free of interferences by endogenous amines other than serotonin. The limit of sensitivity is 100 fmol/ml CSF or 20 parts per trillion. Serotonin concentrations in lumbar cerebrospinal fluid from drug free, depressed patients were in the low nanomolar range.
A relative deficit of central serotonergic neurotransmission has been postulated to have a possible causative role in depression, suicidal behavior, alcoholism and impulsivity (1-4). Clinical studies have so far produced only indirect proof for this hypothesis; a relatively low concentration of 5-hydroxyindoleacetic acid (5HIAA) has been found in the cerebrospinal fluid (CSF) of patients exhibiting these behaviors. Investigators have been forced to quantify CSF 5HIAA, the main metabolite of serotonin, instead of directly measuring the parent amine due to very low serotonin concentrations in the CSF. 5HIAA is, however, transported out of the CSF via an acid transport mechanism (5), whose activity may vary according to the diagnosis of the subjects (6). Furthermore, the CSF concentrations of 5HIAA parallel less closely the brain tissue 5HIAA concentrations than for example CSF homovanillic acid (HVA; the main acid metabolite of dopamine) concentrations parallel the brain tissue HVA concentrations (7). Thus, CSF 5HIAA may be a less than optimal indicator of brain serotonin metabolism.
To enable more thorough investigations of central serotonin metabolism in humans, we have developed a sensitive liquid chromatographic (HPLC) assay for the transmitter itself using a novel derivatization procedure and electrochemical (EC) detection.
Material and Methods
Instrumentation
The HPLC-EC system consisted of an Altex 210 injector, Waters 6000 A pump, Axxion 10 cm, C18 column, with 3 μm particle size, ESA 5011A electrochemical detector, and an LKB single channel recorder.
Chromatographic Conditions
The mobile phase consisted of 0.08 M sodium acetate with 2.3 and 6.3% (v/v) methanol and acetonitrile, respectively. It was adjusted to pH 4.8 with glacial acetic acid and filtered under vacuum through a 0.45 m HA Millipore® filter. The flow rates were varied between 1.4 and 1.6 ml/minute depending on the condition of the column. All chromatography was performed at room temperature. The separations were achieved under isocratic conditions. The detector was operated in the oxidative mode with a voltage setting of 0.30 V.
Reagents
Serotonin and N-acetyl serotonin (NAS) were purchased from Sigma Chemical Co. (St. Louis, M0). Organic solvents were from Fisher Scientific Co. (Silver Spring, Md) and they were HPLC grade. All water used was deionized through a Millipore® deionizer. The derivatization reagent and internal and external standards for quantification of serotonin were synthesized as follows:
N-Succinimidyl propionate (derivatization reagent)
N-hydroxysuccinimide (32.06 g) was suspended in ethyl acetate (l50 ml). Propionic anhydride (37 ml) and triethylamine (39 ml) were added alternately in small portions. A solution formed and the vessel became warm as the reaction progressed. After cooling to room temperature several milliliters of saturated saline were added resulting in the precipitation of a thick crystalline mass (triethyl ammonium propionate). The solid was filtered and washed with ethyl acetate. The combined filtrate was washed with three volumes of saturated saline and one volume of water. Evaporation of the organic layer left a residue of oil, which crystallized upon scratching with a glass rod and trituration with petroleum ether to give 45.36 g of product (95.2% yield). Melting point 44-46°C, analysis (C7H9NO4) calculated 49.62%C, 5.30%H, 8.18%N; found 49.29%C, 5.20%H, 8.19%N.
N-propionyl-5-hydroxytryptamine (external standard)
N-succinimidyl propionate (60 mg, 0.35 mmol) was dissolved in 5 ml methanol and treated with a solution of serotonin creatinine sulfate (136 mg, 0.34 mmol) in 3 ml of 0.2 M sodium phosphate buffer at pH 8. The reaction mixture was stirred for 12 hours under argon, and the methanol was evaporated. The residue was extracted with ethyl acetate. The organic layer was washed with water, dried (Na2S04), and evaporated under vacuum leaving a clear glass. The product yield was 71 mg (96%), it showed mass peaks (CI-NH3) at 250 and 233, and was homogenous by TLC criteria.
5-hydroxy-D,L-tryptophan ethyl and methyl amides (internal standards)
5-hydroxy- D,L-tryptophan ethylester hydrochloride (69 mg) was dissolved in 0.5 ml of an aqueous solution of 40% methylamine or 70% ethylamine and heated on a steam bath at 60° under argon for twelve hours. The solvent was evaporated under a stream of argon. The resulting oil was dissolved in methanol/ether and acidified with gaseous HCl. Insolubles were allowed to precipitate and the solvent decanted. The hydrochloride salt of the product (40 mg) was dried under high vacuum. Mass spectroscopy (CI-NH3) showed a parent ion at m+1 mass units = 248 for ethylamide.
5-hydroxy-L-tryptophan n-propylamide (internal standard)
N-t-butyloxy-carbonyl-5-hydroxy-L-tryptophan (Bachem, Torrance, Ca., 1.03 g) was condensed with n-propylamine (0.26 ml) by the dicyclohexylcarbodiimide/l-hydoxybenzotriazole coupling method (8) in tetrahydrofuran. The reaction mixture was dried and taken up in ethyl acetate. Following an acid/base wash, the dried organic layer was evaporated leaving N-t-butyloxycarbonyl-5-hydroxy-L-try-phophan-n-propylamine (1.12 g, 96% yield) as a glass solid. The material was homogenous by TLC (silica, chloroform: methanol: acetic acid, 85:10:5, Rf = 0.61).
The t-butyloxycarbonyl protecting group was removed using an equivolume mixture of trifluoracetic acid and methylene chloride containing 2% thioanisole. Upon evaporation and trituration of the residue with ether, the title compound was isolated as the trifluoroacetate salt, an amorphous solid. It was reprecipitated from methanol/ether, to give 0.80 g, a 68% yield. It showed a peak at 262 m/z.
NMR data: (D20) 8 ppm 7.44 (1H, J=9 Hz, Ar C6), 7.31 (1H, s, Ar C2), 7.05 (1H, J=2HZ, Ar C4), 6.89 (1H, q, J=2, 9 Hz, Ar C5), 4.18 (1H, q, C), 3.34 (CH2), 3.03 (2H, br m, C), 1.26 (CH2), 0.64 (CH3).
N-propionylation of the product using N-succinimidyl propionate resulted in a product identified as N-propionyl-5-hydroxy-L-tryptophan n-propylamide. Accurate mass (C17H23N3O3) calculated: 317.1738; found: 317.1748.
Stock Solutions
Stock solutions of N-propionyl serotonin and NAS (2 × 10−7 M) were prepared in distilled water, divided into 1 ml aliquots, and stored in tightly capped vials at −70°C. The stock solutions were stable for a minimum of 4 months. Standard solutions were prepared daily and kept on ice until use.
Derivatization Procedure
One ml aliquots of cerebrospinal fluid (CSF) or standard solution containing serotonin (5-HT) and NAS were pipetted into 16 × 125 mm screw cap culture tubes which had been exhaustively silanized. Internal standard was added and each tube was degassed for 5 min under a stream of argon. 25 ul of N-succinimidyl propionate (220 mg in 1.0 ml p-dioxane) and 100 ul of saturated Na2C03 were added. After vortexing, argon degassing was resumed for 5 min. The tube was capped, vortexed and incubated for 10 minutes at 70°C. The reaction was quenched by adding 100 ul of a glycine solution (75 mg in 1.0 ml H2O adjusted to pH 9.7 with NaOH) and agitating at room temperature on a mechanical shaker for 10 minutes.
Extraction
A sample was extracted by vortexing for 20 seconds with 3.0 ml of ethyl acetate, which had been passed over a column of activated aluminum oxide immediately prior to use, and centrifuged for 10 minutes. The extraction was repeated once. The organic phase from the combined extracts was evaporated in a Speed Vac (Savant, Hicksville, N.Y.) vacuum evaporator. The product was redissolved in 200 μl of deionized water and vortexed for 10 seconds. An 100 μl aliquot was injected into the HPLC.
Quantitative Analysis
Aliquots of an aqueous mixture of 5 × 10−13 M/mL serotonin and NAS served as external standards for quantitative analysis. They were subjected to preparation procedures identical to the CSF samples. A minimum of two standards were run for every ten CSF samples. The mean results of the standards were used to calculate the analyte concentrations.
Precision
The within day precision of the present method was estimated by adding three different concentrations, bracketing the expected physiological ranges for concentrations of serotonin and NAS, into pools of an artificial CSF solution. The artificial CSF was prepared in deionized water by adding NaH2PO4 0.5 mM, Na2HPO4 0.25 mM, MgCl2 0.4 mM, CaCl2 0.65 mM, KC1 3.0 mM, NaCl 128 mM, and NaHC03 25 mM. Six ml portions were frozen in silanized glass tubes and stored at −20°. On each of 5 days one tube of each concentration was thawed and assayed in five 1.0 ml aliquots for a total of 15 reactions. An aqueous solution of 5-hydroxy-D,L-tryptophan ethylamide to give a final concentration of 3 × 10−9 M was added as an internal standard to each reaction. The between day precision was estimated by analysing a one ml aliquot of the middle concentration of the three pools on each of twenty days.
Exclusion of Interferences
Because the derivatization procedure is specific for amines, and the relatively low oxidation potential makes the detection rather specific for hydroxy groups, we examined the relative retention times of NAS and the N-propionyl derivatives of the following substances: dopamine, epinephrine, metanephrine, 3-methoxytyramine, norepinephrine, normetanephrine, octopamine, serotonin and tyramine.
Sample Sources
Human CSF was collected by lumbar puncture in the lateral decubitus position after an overnight bed rest. Samples were obtained from 20 psychiatric patients, who met the DSM-III criteria for primary, major depression, unipolar (9). The patients had been free of medications on a research ward for a minimum of 21 days prior to sampling. They were maintained on a closely supervised low monoamine diet (10).
Results
Yields of the Derivatization Reaction
Yields of the Derivatization Reaction for serotonin and the internal standard were 53 ± 3% (mean ± SD; n = 12). They were practically identical for the two substances, and were the same for the artificial, human and monkey CSFs (n = 5 each, CV 7% at IO−9 M).
Extraction Efficiencies
Extraction Efficiencies of the serotonin and internal standard derivatives as well as NAS were practically identical at 65 ± 5% (mean ± SD; n = 18).
Chromatographic Quality and Lack of Interference
Chromatograms of standard mixtures of serotonin, NAS and the internal standard as well as a typical CSF are shown in Fig. 1. None of the endogenous monoamines investigated interfered in the chromatographic procedure. Their relative retention times are shown in Table I. Identity of the peaks was confirmed by perfect cochromatography with authentic samples of the N-propionyl derivatives of the amines under two different chromatographic conditions.
FIG. 1.


A Chromatograms of derivatized human CSF using 5-hydroxy-D,L-tryptophan N- propylamide as internal standard. Peaks shown: NAS (N-acetyl serotonin), P5HT (N-propionyl serotonin), P5HTPA (N-propionyl-5-hydroxy-D,L-tryptophan N-propylamide, CSF from a single patient.
B A pool of CSF from several patients spiked with 2 pmol/ml of 5HT. In the absence of spiking the propionyl 5HT peak height was approximately 50% of that shown in the Figure.
TABLE I.
Retention Times for N-Propionyl Derivatives
| Parent Compound | Minutes* |
|---|---|
| Norepinephrine | 1.72 |
| Octapamine | 2.40 |
| Normetanephrine | 3.12 |
| Epinephrine | 4.40 |
| Dopamine | 4.40 |
| N-Acetyl Serotonin** | 4.80 |
| Tyramine | 8.25 |
| Serotonin | 8.85 |
| Metanephrine | 10.59 |
| 3-Methoxytyramine | 12.28 |
Conditions: 10 cm Axxion C18 Column 3 μm particle size, 1.4 ml/min. 0.08 M acetate 2.3% Methanol, 6.3% Acetonitrile
Not a propionyl derivative
Linearity of Detector Response and Within Day Precision of the Assay
Linearity of Detector Response and Within Day Precision of the Assay are shown in Fig 3. The between day coefficient of variation of the assay was 9.2% for serotonin and 16.3% for NAS at the concentration of 400 fmol/ml. The limit of sensitivity of the assay was 100 fmol/ml with a signal to noise ratio of 3 to 1.
FIG. 3.

Within run mean ± S.D. of serotonin, 5HT, and N-acetyl serotonin, NAS, determination.
Mean Concentrations in Human CSF Samples
Mean Concentrations in Human CSF Samples were 5.86 + 0.40 and 0.15 + 0.16 pmol/ml N=20 of serotonin and NAS, respectively.
Discussion
The present report describes the first HPLC procedure for quantification of serotonin in human CSF, whose precision and practicality have been rigorously tested and demonstrated to be adequate. Our results are in the same range as those previously reported using a bioassay (11), a direct HPLC procedure (12), and massfragmentography (13). Much higher concentrations of serotonin in human CSF reported by certain authors (14-16) probably represent interferences by various substances in analytical procedures.
A methodological complication we have discovered in our experiments is the instability of serotonin in aqueous solutions at low concentrations similar to those found in the human CSF. When a sample is thawed, approximately 25% of the serotonin content is lost unless the sample contains both ascorbic acid and has an electrolyte concentration similar to CSF.
The principle of our pre-column derivatization is straightforward. It is based on the selective N-acylation of primary and secondary amines in the presence of phenols. The product is both lipophilic enough for extraction into organic solvents, such as ethyl acetate, and electroactive. The reaction yield is somewhat sensitive to extreme changes in electrolyte concentration. Thus, standard curves for CSF analyses have to be produced with samples constituted in artificial, monkey or human CSF, but not water which gives a higher yield by a factor of 1.5 for the internal standard. A series of internal standards, consisting of N-alkylamines of 5-hydroxytryptophan, have been introduced. The choice of which one to use depends on the retention times of interfering peaks. Among methyl, ethyl, and propylamides we have found the longest retained, the propylamide, to be most useful for determining serotonin in human CSF. The procedure yields itself for quantification of very low concentrations of various phenolic amines as demonstrated by the interference study conducted by us. We have initiated formal reproducibility studies to document the applicability of our method for quantification of normetanephrine in human CSF. The goal is to quantify NAS as well as both serotonin and normetanephrine within one run.
Currently, the physiology and pathophysiology of CSF serotonin concentrations in humans is unknown. The present assay allows the quantification of 10 to 20 samples in an 8 hour day. Thus, its application should facilitate the production of the necessary data base for interpreting the meaning of the values.
FIG. 2.
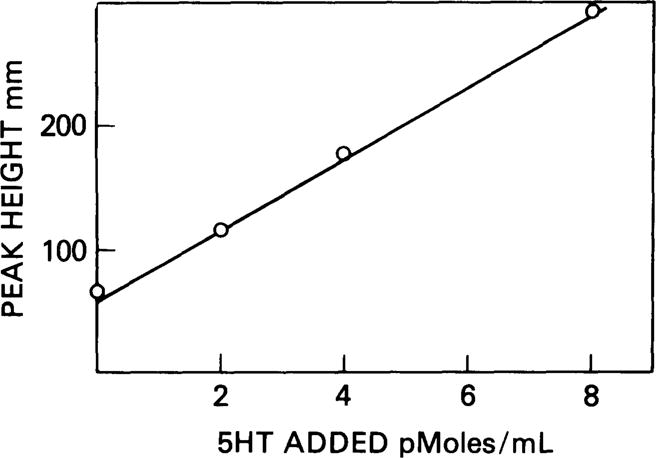
Linearity of method in human CSF. The intercept on the vertical axis represents endogenous 5HT in the human CSF pool.
References
- 1.Asberg M, Thoren P, Traskman L, Bertilsson L, Ringberger V. Science. 1976;191:478–480. doi: 10.1126/science.1246632. [DOI] [PubMed] [Google Scholar]
- 2.Asberg M, Traskman L, Thoren P. Arch Gen Psychiatry. 1976;33:1193–1197. doi: 10.1001/archpsyc.1976.01770100055005. [DOI] [PubMed] [Google Scholar]
- 3.Ballenger JC, Goodwin FK, Major LF, Brown GL. Arch Gen Psychiatry. 1979;36:224–227. doi: 10.1001/archpsyc.1979.01780020114013. [DOI] [PubMed] [Google Scholar]
- 4.Linnoila M, Virkkunen M, Scheinin M, Nuutila A, Rimon R, Goodwin FK. Life Sci. 1983;33:2609–2614. doi: 10.1016/0024-3205(83)90344-2. [DOI] [PubMed] [Google Scholar]
- 5.Neff NH, Tozerand TN, Brodie BB. J Pharmacol Exp Ther. 1967;158:214–218. [PubMed] [Google Scholar]
- 6.Cowdry RW, Ebert MH, van Kammen DP, Post RM, Goodwin FK. Biol Psychiatry. 1983;18:1287–1299. [PubMed] [Google Scholar]
- 7.Mignot E, Laude P, Elghozi JL. J Neurochem. 1984;42:819–825. doi: 10.1111/j.1471-4159.1984.tb02754.x. [DOI] [PubMed] [Google Scholar]
- 8.Konig W, Geiger R. Chem Ber. 1970;103:788–789. doi: 10.1002/cber.19701030319. [DOI] [PubMed] [Google Scholar]
- 9.Diagnostic and Statistical Mannual. 3rd. American Psychiatric Association; Washington, D.C.: 1980. [Google Scholar]
- 10.Muscettola G, Wehr T, Goodwin FK. Am J Psychiatry. 1977;134:914–916. doi: 10.1176/ajp.134.8.914. [DOI] [PubMed] [Google Scholar]
- 11.Turner WJ, Mauss EA. Arch Gen Psychiatry. 1959;1:108–112. doi: 10.1001/archpsyc.1959.03590060108012. [DOI] [PubMed] [Google Scholar]
- 12.Narasimhachari N. J Liq Chromatography. 1984;7:2679–2689. [Google Scholar]
- 13.Markey SP, Colburn RW, Johannesen JN. Biomed Mass Spectrom. 1981;8:301–304. doi: 10.1002/bms.1200080704. [DOI] [PubMed] [Google Scholar]
- 14.Singh KSP, Misra SS, Bhargava KP. J Lab Clin Med. 64:802–807. [PubMed] [Google Scholar]
- 15.Taylor PL, Garrick NA, Burns RS, Tamarkin L, Murphy DL, Markey SP. Life Sci. 1982;31:1993–1999. doi: 10.1016/0024-3205(82)90038-8. [DOI] [PubMed] [Google Scholar]
- 16.Volicer L, Direnfeid LK, Freedman M, Albert ML, Langlais PJ, Bird ED. Arch Neurol. 1985;42:127–129. doi: 10.1001/archneur.1985.04060020037011. [DOI] [PubMed] [Google Scholar]